

Abstract
Objective
Better understanding of the pathophysiology of critical illness has led to an increase in clinical trials designed to improve the clinical care and outcomes of patients with life threatening illness. Knowledge of basic principles of clinical trial design and interpretation will assist the clinician in better applying the results of these studies into clinical practice.
Data Sources
We review selected clinical trials to highlight important design features that will improve understanding of the results of critical care clinical trials.
Main Results
Trial design features such as patient selection, sample size calculation, and primary outcome measure may influence the results of a critical care clinical study. In conjunction with trial design knowledge, understanding the size of the anticipated treatment effect, the importance of any clinical endpoint achieved, and whether patients in the trial are representative of typical patients with the illness will assist the reader in determining whether the results should be applied to usual clinical practice.
Conclusions
Better understanding of important aspects of trial design and interpretation, such as whether patients enrolled in both intervention arms were comparable and whether the primary outcome of the trial is clinically important, will assist the reader in determining whether to apply the findings from the clinical study into clinical practice.
Keywords: Clinical Trial, Critical Care, Adult, Outcomes
Introduction
In the past 50 years, critical care has evolved from a discipline present in few institutions to a widely prevalent specialty (1, 2). This rapid growth has been accompanied with an increased understanding of the pathophysiology of critical illness. For example, many of the laboratory advances in the understanding of sepsis and acute lung injury (ALI) have led to clinical trials. In addition, a number of national and international critical care trials groups (Canadian Critical Care Trials Group; www.ccctg.ca, The Australian and New Zealand Intensive Care Society, www.anzics.com.au, the United States National Institutes of Health ARDS Network, www.ardsnet.org, The United States Critical Illness and Injury Trials Group (public.wudosis.wustl.edu/USCIITG/default.aspx) have been created to advance new treatments for critically ill patients.
Critical care trials present special challenges. For example, many illnesses that prompt intensive care unit (ICU) admission are syndromes rather than diseases and thus suffer from heterogeneity between patients and lack a biological diagnostic test (“biomarker”) to facilitate early identification of patients for interventions. Additionally, the short time window and inability to obtain consent directly leads to a relatively low ratio of screened to enrolled patients, raising concerns about the generalization of completed clinical trials in routine clinical practice (external validity). This review is intended to highlight important trial design and interpretation concepts, and to provide clinicians and clinical or translational researchers the tools to better understand critical care clinical trials. In doing so, we will use recent examples from the critical care literature. A recent supplement in this Journal addressed in-depth issues regarding trial design that may be of particular interest to critical care researchers. (3) We have divided this review into sections discussing trial design, selection of experimental subjects and controls, outcome measures, bias, sample size calculation, and interpretation of results that highlight specific trial design concepts that have implications for understanding recent clinical trials.
Study Design
Clinical studies are generally divided into two primary groups according to study design: observational and experimental studies. Observational studies include case-control, cross-sectional and cohort studies. In contrast, the randomized, controlled clinical trial is the archetypal example of an experimental study. The primary difference between these two designs is that observational trials “observe” the results of exposures or interventions in study subjects as a method to ascertain associations between these exposures and an outcome of interest, while experimental studies assign subjects to an exposure or intervention and prospectively evaluate its effect on an outcome of interest. Participants enrolled into experimental trials are often more highly selected than participants enrolled into observational trials.(4)
Case-control studies (Figure 1) involve identification of study subjects with (cases) and without (controls) an outcome of interest and then examining retrospectively the potential exposures or interventions (predictor variables) associated with the outcome of interest (5). Case-control studies are the most efficient study design for rare diseases or infrequent outcomes, but they are subject to multiple potential biases, can only examine one chosen outcome, and are at high risk for confounding (5). Confounding occurs when a third factor is associated with both the exposure or intervention and the outcome of interest. A confounder may distort the relationship between the exposure and outcome under study. For example, in a case-control study examining the costs of MRSA infection in the ICU, matching between cases and controls was performed on age, co-morbidity, severity of illness, and the number of organ failures.(6) Despite these adjustments, other factors likely differ between those patients who developed nosocomial MRSA infections and those who did not: operative intervention, duration of ICU stay, immune function, among other variables could not be considered in this study.(6) (6) Any of these variables may be a confounder since any of them might be present more frequently in one group than the other and could reasonable be associated with the presence of MRSA.
Figure 1.

Illustration of a case-control study. At the time of starting the study (depicted as “the present”) the investigator selects a sample of cases from subjects with the outcome of interest and selects a sample of controls from those without the outcome of interest. Exposures potentially associated with the outcome of interest (shaded areas) are then assessed retrospectively, in the past, as denoted by the dark reverse arrows.
Cohort studies may be undertaken to describe the occurrence of certain outcomes or to analyze associations between exposures (predictors) and outcomes, respectively (5). Prospective cohort studies (Figure 2a) involve selection of a group of study subjects for enrollment, collection of data regarding exposures (predictor variables) and then observing the subjects over time with standardized assessment of outcomes. Retrospective cohort studies (Figure 2b) also start with selection of a group of subjects, but in this case selection is from a historical or theoretical group of patients that existed in the past, and then collecting data on both exposures and outcomes, often at the same time. Both prospective and retrospective cohort studies are subject to potential confounding and have limitations in assigning causality in the observed associations between exposure and outcome (5) (7). Prospective cohorts have the ability to determine temporal relationships between exposure and outcome while retrospective studies are more time and resource efficient. For example, a prospective cohort study sought to determine long-term outcomes after ARDS and thus identified a cohort of ARDS survivors in hospital who were then prospectively evaluated at 3, 6 and 12 months after ICU discharge.(8) Despite limitations in sample size, a single geographic location and heterogeneity among ARDS survivors, they identified predictors of physical function to inform future studies and clinical care.
Figure 2.
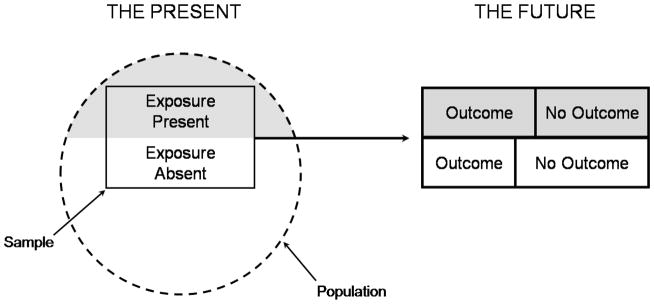
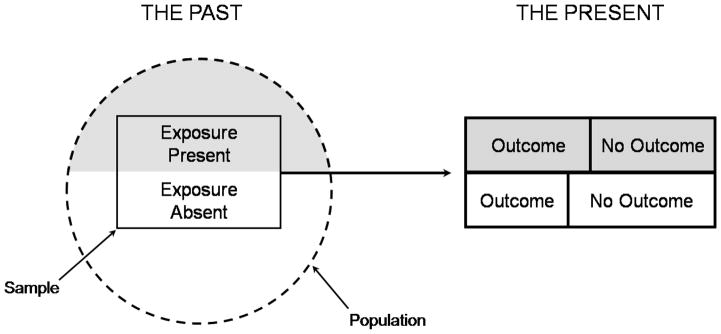
Figures 2a and 2b. Illustration of a prospective and retrospective cohort study. In a prospective cohort study (2a), at the outset of the study the investigator selects a sample from the population and measures the exposures of interest (shaded areas) before following the patient over time to determine the occurrence of relevant outcomes and their association with the exposures (following the dark forward arrow). In a retrospective cohort study (2b), at the outset of the study the investigator identifies a cohort of subjects that existed in the past and assesses exposure retrospectively from that group (shaded areas), and then assesses the relevant outcomes in the present (dark forward arrow).
Randomized clinical trials are the best study design for measuring the efficacy of an intervention in causing the outcome of interest (7, 9, 10). The use of random allocation of study subjects has the potential to substantially reduce or theoretically eliminate confounding by randomly distributing all known and unknown predictor variables, except the intervention being tested, evenly between the intervention and control groups. In a randomized, controlled trial, (Figure 3) study subjects are selected prospectively according to eligibility criteria, and after informed consent, they are randomly allocated to a study group (e.g. treatment or control) (5). Blinding, or masking, is critical in clinical trials to prevent bias in application of the randomized intervention or co-interventions, or assessment of the relevant outcomes, particularly when the outcomes are subjective (10). Blinding is important if the outcome measured is subjective, there is a possibility that the investigators may consciously or unconsciously use different co-interventions if they are aware of the intervention.(5, 10) For interventions that may be relevant to a single patient population but do not overlap or interfere with each other, factorial randomization may be used to test both interventions in a single study (10). For example, in ALI patients, the ARDS Network compared outcomes with pulmonary artery catheterization versus central venous catheterization, and also compared a fluid liberal versus a fluid conservative strategy (11) (12). In this trial, the investigators reported no difference based upon catheter randomization but a significant benefit in the fluid conservative vs. fluid liberal management group. (11, 12) Similar study designs using factorial randomization have reported significant differences in both randomized interventions, such as when comparing intravenous fluid solutions (crystalloids vs. starch solutions) along with glucose control (intensive vs. conventional insulin management).(13) Some critical care interventions (use of high frequency oscillation versus conventional mechanical ventilation) do not lend themselves to blinding. (14) Blinding, or concealment is important if the outcome measured is subjective, there is a possibility that the investigators may consciously or unconsciously use different co- interventions if they are aware of the intervention.(5) The primary advantage of a clinical trial, stronger evidence for a cause-effect relationship for the intervention and outcome, is balanced against the greater cost, complexity and time required for study completion. (7) In addition, interventions in which physicians lack equipoise or uncertainty about the effect of the intervention cannot be tested in a randomized trial.(15, 16)
Figure 3.
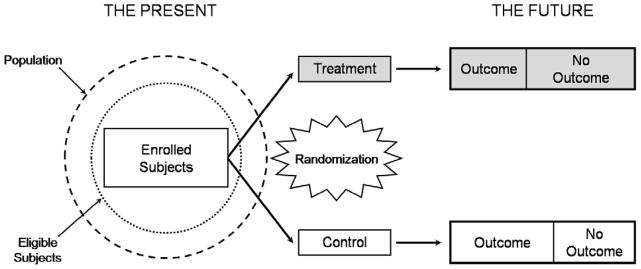
Illustration of a randomized, controlled trial. In a randomized, controlled clinical trial, the investigator identifies a group of eligible subjects (dotted circle) from an appropriate patient population (hashed circle), and then applies the defined inclusion and exclusion criteria to enroll subjects. At the start of the study the investigator measures baseline variables and randomly allocates subjects to intervention groups (“treatment” in gray, or “control”). Outcomes are measured prospectively during the follow-up period. Ideally, allocation, intervention and outcome assessment should be blinded.
Randomized controlled trials generally rank more highly in terms of ability to draw valid causal inferences regarding the intervention and the outcome. (7, 10, 17) One example that illustrates the advantages of randomized controlled trials is the publication of a cohort study asserting an increased risk of death with the use of pulmonary artery catheterization in critically ill patients.(18) At the time, clinicians lacked equipoise to permit an effective randomized study of pulmonary artery catheterization. (19) However, after the publication of this cohort study, physician investigators were able to complete several randomized, controlled clinical trials to determine the effect of pulmonary artery catheters on clinical outcomes.(12, 20–22) Of note, the findings of an increased risk of death in observational studies was not confirmed in these subsequent RCTs, a conflicting result between observational studies and randomized trials that occurs up to 33% of the time. (23) In table 1, we summarize pros and cons of observational versus clinical trials.
Table 1.
Comparison Between Observational and Controlled Studies
| Observational Studies | Controlled Studies |
|---|---|
Strengths:
|
Strengths:
|
Weaknesses:
|
Weaknesses:
|
Selection of Experimental Subjects and Controls
The designation and identification of the desired participant (critically ill patient) for enrollment is a crucial part of most trial designs. Ideally the study investigators want to select the patient who would benefit most from a planned intervention. The importance of understanding the most appropriate patients for enrollment is highlighted by the publication of two clinical trials testing the use of glucocorticoids in septic shock patients. Annane and colleagues tested the use of corticosteroids in patients with a systolic pressure < 90 despite fluid resuscitation and vasopressor therapy. (24) The CORTICUS trial enrolled patients who had a systolic pressure < 90 mm Hg or the need for vasopressor therapy after volume resuscitation. (25) Thus the patients enrolled in the two studies may have been chosen from different patient populations; one with refractory shock and the other without refractory shock. (24, 25) While there were other differences between the two studies, it is possible that the differing effect of glucocorticoids on mortality observed in these two studies may be related to differences in the severity of septic shock of the patients enrolled.
Outcome Measures
Surrogate Endpoints
Critical care trials are difficult and expensive to implement. One way to limit both the expense and the scope of a clinical trial is to pick an easily measured endpoint that is associated with the outcome of interest. While death is often used as the “gold standard” clinical outcome, it may not be feasible to design a small, exploratory trial that will show a statistically significant mortality benefit. Surrogate outcome measures may be chosen because fewer patients are required to show a treatment effect, leading to a simpler and less expensive trial (26) A common example of a surrogate endpoint outside of critical care is the use of CD4+ cell count in clinical trials of antiretroviral therapies. Since many drugs that raise CD4+ counts will also improve mortality rates in patients with HIV, the use of CD4+ count as a surrogate endpoint has been widespread. (27, 28)
Mean arterial pressure (MAP) may appear to be a logical surrogate endpoint for trials of therapies for patients with septic shock. Since patients with septic shock develop hypotension as a result of distributive shock, and therapy for septic shock includes giving drugs to raise systemic blood pressure, there is biologic plausibility for the use of MAP as a surrogate endpoint. (29) However, a non-specific nitric oxide synthase inhibitor that effectively raises blood pressure was shown to increase mortality rates in patients with septic shock. (30, 31) Thus, at least in this study, MAP was not a useful surrogate marker for patient mortality.
An ideal surrogate outcome marker is causally or pathogenetically related to the disease (and thus, to the outcome) and accurately linked with the outcome. Many surrogate outcomes such as MAP in septic shock and oxygenation in acute lung injury have been used in critical care.(31–34) Unfortunately, to date no individual surrogate prognostic marker has been found to be sufficiently robust to accurately predict mortality in critically-ill subjects. (35)
Combined Endpoints
Combined or composite endpoints represent another potential method to decrease the number of patients required to show an effect size in critical care trials (36). The use of a combined endpoint requires that the combined endpoints be clinically important and of equal importance to the patient, potentially biologically distinct and have direct interpretation (10, 36). Some investigators also advocate that the frequency of occurrence of each component in the combined endpoint should be similar and should have similar risk reductions in response to an effective intervention.(37) If these conditions are not met, a composite endpoint may provide misleading information (37). One example of a composite endpoint is the ventilator free days score. This score combines the number of days in which a patient is both alive and free of mechanical ventilation over a specific time period, usually 28 days.(38, 39) The use of these combined endpoints may allow more efficient trials of specific therapies in critically ill patients in which a mortality benefit and a decrease in the number of days of mechanical ventilation among those who are alive is expected. It is important to note that the value of these scores must be placed into the context of the other risks and benefits of effective therapies (36). Methods for competing risks, if used in the proper setting, may be an adequate alternative to the use of combined endpoints for the evaluation of interventions in critical care trials. (40) Specifically, mixtures of survival distributions may be particularly useful to model outcomes that are competing and mutually exclusive such as designs in which participants are followed until inhospital death or unassisted breathing among those who are eventually discharged home alive (40).
Sample size calculation
Determination of sample size is an important aspect of planning for any clinical study, and sample size calculations should be described in detail in any published report.(41, 42) The goal of this section is to provide critical care researchers with an overview to sample size calculations for Phase III randomized, controlled trials with two treatment groups and a well defined, patient-centered clinical endpoint such as 60-day mortality. Formulae and software to assist with sample size calculation are found elsewhere.(9, 43–45)
The calculation of sample size for any clinical investigation requires a priori specification of the population, hypotheses under study, and explicit definition of the primary outcome. Hypotheses make statements about a population “parameter,” such as the proportion of critically-ill patients who died within 60 days. The goal of a hypothesis test is to decide, based on a sample from the population under study, which of the two complementary hypotheses is true (10). In any clinical investigation, hypothesis testing risks two fundamental errors. First, a single experiment may lead to investigators to believe that two treatments differ when, in fact, they do not, leading to a false-positive result (type I error rate, defined as α) (5). Second, a single experiment may lead investigators to believe that two treatments do not differ when, in fact, they do, leading to a false-negative result (type II error rate, defined as β). (5, 10)
The factors that contribute to the calculation of sample size when comparing a primary outcome between two interventions include: 1) the “test statistic” used to compare a difference in the primary outcome measure between two interventions; and, 2) the error rates associated with the hypothesis test (α and β (26) The “test statistic” depends on both the expected difference in the primary outcome between two treatment groups and on estimate of the variance of the primary outcome measure, and are usually based on data from sets of smaller clinical trials or observational studies or preferably obtained from the same institution or hospital centers where the clinical trial is to be conducted. (26) Different assumptions about α and β will have a direct effect on sample size. Ordinarily, the value of α is set at 0.05 but it can be set at a smaller value to decrease the chance of a false-positive result. The value for β is usually set between 0.05 and 0.20.(5, 10, 26) The complement of β is statistical power (1 – β), which represents the probability of declaring that two interventions are different when in, in fact, they are truly different. (5, 10) Other factors that directly affect sample size include: clustering in the responses of participants as in the case of cluster randomized trials, allocation ratio of participants other than 1:1 to each intervention, early stopping rules and testing three or more interventions in one trial. (44, 46–48)
Early stopping rules are intended to protect participants in clinical trials from unsafe interventions, hasten the implementation of beneficial interventions or to stop trials that are unlikely to show treatment differences between groups. Methods for stopping trials by design have been available for more than fifty years, but recent advances in statistical methodology and better computer software have made these methods more accessible to trialists (47). In general, an initial sample size still needs to be calculated based on the principles discussed above, but the overall sample size calculation is then penalized (increased) by the number of interim analyses planned.(49) Examples of stopping clinical trials by design include the ARDS Network studies, e.g., the lower tidal volume trial stopped early at 861 patients (of out an originally proposed sample size of 1000) after the fourth interim analysis showed that lower tidal volumes were significantly more beneficial than higher tidal volumes.(39) In general, both a convincing treatment effect and a large number of patients enrolled should be present to stop a trial early.(49)
To illustrate all of the above factors in play, we discuss the sample size calculation from the CORTICUS trial.(25) In this trial, the investigators calculated that a sample size of 800 participants (400 per group) was needed to achieve a statistical power of 80% (β = 0.20) with a maximum overall type I error rate of 5% (α = 0.05) to detect an absolute decrease in mortality of 10% for an overall expected 28-day mortality of 50% in the control group. (25) Indeed, the overall 28-day mortality in their study population was 33% (164/499). Ignoring interim analyses, the expected sample size for a mortality of 33% and a difference of 10% between groups would have been lower at 670 participants (335 per group).(25) The investigators had planned stopping rules, in which they penalized the type I error rate using the O’Brien and Fleming method as follows: p = 0.0006 for the first interim analysis, p = 0.005 for the second interim analysis, and p = 0.047 for the final analysis.(25, 50) However, the trial was administratively stopped with 499 patients enrolled for multiple reasons.(25) Despite a lower overall 28-day mortality in the treatment group, the investigators were probably still underpowered to detect a mortality difference of 10%.
Internal and External Validity
There are many potential reasons why a clinical trial might show benefit. Whether the treatment is responsible for the clinical benefit underlies the concept of internal validity. The NIH ARDS Network low tidal volume trial provides a useful example of a study with good internal validity. To minimize treatment variation, the ventilatory practices used in the study were mandated by protocol, and the coordinating center randomly audited selected ventilator settings for compliance with the protocol. Moreover, it is important to ensure the use of similar treatment protocols for other relevant interventions associated with the primary outcome across different centers to minimize the risk that an unmeasured or unintended factor was responsible for the differences seen between treatment groups. (39)
Whether study results can be generalized to patients who are not enrolled in the study reflects the concept of external validity. Many critical care trials require the screening of 10 patients for each patient enrolled.(11, 51) Thus, it is unknown whether patients who were not enrolled in these trials because of co-morbidities and other reasons would have similar or different responses to the tested interventions compared with enrolled patients.
Bias in Clinical Studies
Bias results from a systematic error in study design or execution leading to differences between study groups. (10, 52) It can generally be divided into two categories: selection bias and information bias. Selection bias refers to an unrecognized difference between treatment and control groups, while information bias refers to an incorrect assessment or measurement of exposure or outcome, or both.(5, 52) Selection bias includes errors in choosing the individuals or groups to take part in a scientific study, and sampling bias, in which some members of the population are more likely to be included than others. Information bias includes errors in diagnosis or exposure assessment, including misclassification of the outcome or exposure of interest. (52) If the subjects in one group are misclassified more frequently than the others, then “differential” misclassification may lead to invalid conclusions while non-differential misclassification increases “noise” and may obscure true results, i.e., bias towards the null hypothesis. (5, 52) While bias is more common in observational research, even randomized, controlled trials may have bias, most often resulting from selection bias related to unconcealed allocation, unblinding and exclusions after randomization. (52)
Misclassification bias refers to the possibility of incorrectly identifying patients with (or without) an illness. (5) If participants are enrolled into a trial who are more or less likely to be influenced by the therapy, this will alter the results compared to the “average” patient and their susceptibility. This is particularly challenging for conditions without a perfect diagnosis, such as syndromic conditions such as sepsis and acute lung injury, thus making misclassification bias an important consideration in many clinical critical care trials.
One often cited example of misclassification bias centers around the consensus definition for the acute respiratory distress syndrome (ARDS). The American European Consensus Definition of ARDS includes 4 criteria including acute onset, diffuse bilateral infiltrates consistent with pulmonary vascular congestion, a PaO2/FiO2 ratio ≤ 200 and no evidence of left atrial hypertension. (53) A study of chest radiographs of ARDS patients suggested that there was heterogeneity of opinions among expert clinicians regarding whether a specific radiograph met the criteria for ARDS. (54) In addition, a recent trial examining whether the use of a pulmonary artery catheter improved outcomes in patients with acute lung injury (ALI) demonstrated that 29% of the patients enrolled into the trial had pulmonary capillary wedge pressures > 18 cm H2O, and thus potentially could have been excluded from the trial if these results had been available prior. (12)
Interpretation of results
When reviewing the results of a controlled clinical trial, several factors will help the reader determine whether the results are valid and clinically important.(37, 55) First, the reader must determine if patients enrolled in either intervention arm were comparable. Comparability suggests that randomization worked appropriately. Second, the reader must determine primary outcome in the manuscript clinically important and consistent with that reported in the methods and a clinical trial registry. A trial that shows an improvement in mortality or in the number of organ failures would obviously be more compelling than one that shows a change in a non patient-centered outcome such as a laboratory test. (37) There are several ways in which investigators may present the overall effect size of a clinical trial. We summarize the most commonly used measures of effect size when mortality is used as the outcome in Table 2.
Table 2.
Common Formulae To Calculate Treatment Effect Size
| Measure of effect size | Formula | Interpretation | |
|---|---|---|---|
| Death rate in experimental group | πexperimental | Proportion of deaths (number of participants who died over total enrolled) in the experimental arm. | |
| Death rate in control group | πcontrol | Proportion of deaths (number of participants who died over total enrolled) in the control arm. | |
| Odds of death |
|
Ratio of the death to survival rates in either the experimental or control arms. | |
| Relative risk (RR) |
|
Ratio of mortality rate between experimental and control arm. RR < 1 indicates benefit favoring the experimental approach, whereas RR > 1 indicates harm. | |
| Absolute risk (AR) | πexperimental – πcontrol | Absolute difference in mortality rates between experimental and control arms. AR < 0 indicates benefit favoring the experimental approach, whereas AR > 0 indicates harm. | |
| Odds ratio (OR) |
|
Ratio of odds of death between the experimental and control arms. OR < 1 indicates benefit favoring the experimental approach whereas OR > 1 indicates harm. | |
| Relative risk reduction (RRR) or Relative risk increase (RRI) |
|
The extent to which the experimental approach reduces death relative to the control approach. Values < 0 indicate benefit favoring the experimental group (RRR), values > 0 indicate harm (RRI). Usually presented as a percentage. | |
| Numbers needed to treat (NNT) or harm (NNH) |
|
Values > 0 indicate NNT; values < 0 indicate NNH. The number of patients who need to be treated in order to prevent a death (NNT) or result in death (NNH). The higher the NNT (or NNH), the less effective (or less harmful) is the experimental approach. |
Some clinical trials have shown benefits in specific subgroups of patients, but not in the primary patient population.(56–59) In general, subgroup analyses not defined by design a priori should be avoided, because any post-randomization differences (e.g. bias) will undermine the original randomization assignment. Subgroup analyses may be considered hypothesis generating for an additional study, but should not be considered to be practice changing.(59–61) Finally, the reader should evaluate whether the study population is similar to their population of patients or to those for whom the intervention may be applied. (10)
It is important to distinguish between results that are statistically different and clinically important. For example, in the SAFE trial comparing albumin versus saline resuscitation, the baseline central venous pressure in the albumin versus saline group was 9.0±4.7 vs. 8.6±4.6 mm Hg, a difference that was statistically different, but probably not clinically important. (62) A clinically relevant and objective endpoint, such as mortality, is likely to garner more attention than a more subjective endpoint.(10, 36) In addition, information about the size of the treatment effect is also important in determining whether a trial result should change practice.(63) Other factors that may influence whether a trial should change practice include whether the trial is multicenter, has an adequate sample size, and is repeated with similar results. (64–66)
Conclusion
Critically ill patients present unique challenges to the investigator charged with testing a new therapy. An understanding of the important basic concepts of trial design and interpretation will assist the critical care clinician to decide whether the results of a critical care clinical trial would and should apply to the patient in front of them. The size of the trial and the treatment effect, in addition to the patient population enrolled should help the clinician determine whether to choose a particular therapy for a specific patient based on a clinical trial.
Acknowledgments
JES is supported by K23 GM O71399, WC is supported by K99HL096955 and a Clinician Scientist Award of the Johns Hopkins University, GSM is supported by R01 FD-003440, P50 AA-013757 and U54 RR-024380.
Footnotes
No reprints will be ordered
References
- 1.SAFAR P, DEKORNFELD TJ, PEARSON JW, et al. The intensive care unit. A three year experience at baltimore city hospitals. Anaesthesia. 1961;16:275–284. doi: 10.1111/j.1365-2044.1961.tb13827.x. [DOI] [PubMed] [Google Scholar]
- 2.Vincent JL, Fink MP, Marini JJ, et al. Intensive care and emergency medicine: Progress over the past 25 years. Chest. 2006;129:1061–1067. doi: 10.1378/chest.129.4.1061. [DOI] [PubMed] [Google Scholar]
- 3.Reade MC, Angus DC. The clinical research enterprise in critical care: What’s right, what’s wrong, and what’s ahead? Crit Care Med. 2009;37:S1–9. doi: 10.1097/CCM.0b013e318192074c. [DOI] [PubMed] [Google Scholar]
- 4.Phua J, Badia JR, Adhikari NK, et al. Has mortality from acute respiratory distress syndrome decreased over time?: A systematic review. Am J Respir Crit Care Med. 2009;179:220–227. doi: 10.1164/rccm.200805-722OC. [DOI] [PubMed] [Google Scholar]
- 5.Szklo M, Nieto FJ. Epidemiology. 2. Sudbury: Jones and Bartlett; 2007. [Google Scholar]
- 6.Chaix C, Durand-Zaleski I, Alberti C, et al. Control of endemic methicillin-resistant staphylococcus aureus: A cost-benefit analysis in an intensive care unit. JAMA. 1999;282:1745–1751. doi: 10.1001/jama.282.18.1745. [DOI] [PubMed] [Google Scholar]
- 7.Concato J, Shah N, Horwitz RI. Randomized, controlled trials, observational studies, and the hierarchy of research designs. N Engl J Med. 2000;342:1887–1892. doi: 10.1056/NEJM200006223422507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Herridge MS, Cheung AM, Tansey CM, et al. One-year outcomes in survivors of the acute respiratory distress syndrome. N Engl J Med. 2003;348:683–693. doi: 10.1056/NEJMoa022450. [DOI] [PubMed] [Google Scholar]
- 9.Piantadosi S. Clinical trials: A methodologic perspective. 2. Hoboken: John Wiley and Sons; 2005. [Google Scholar]
- 10.Meinert C. Clinical trials: Design, conduct, and analysis. New York: Oxford University Press; 1986. [Google Scholar]
- 11.National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network. Wiedemann HP, Wheeler AP, et al. Comparison of two fluid-management strategies in acute lung injury. N Engl J Med. 2006;354:2564–2575. doi: 10.1056/NEJMoa062200. [DOI] [PubMed] [Google Scholar]
- 12.National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network. Wheeler AP, Bernard GR, et al. Pulmonary-artery versus central venous catheter to guide treatment of acute lung injury. N Engl J Med. 2006;354:2213–2224. doi: 10.1056/NEJMoa061895. [DOI] [PubMed] [Google Scholar]
- 13.Brunkhorst FM, Engel C, Bloos F, et al. Intensive insulin therapy and pentastarch resuscitation in severe sepsis. N Engl J Med. 2008;358:125–139. doi: 10.1056/NEJMoa070716. [DOI] [PubMed] [Google Scholar]
- 14.Derdak S, Mehta S, Stewart TE, et al. High-frequency oscillatory ventilation for acute respiratory distress syndrome in adults: A randomized, controlled trial. Am J Respir Crit Care Med. 2002;166:801–808. doi: 10.1164/rccm.2108052. [DOI] [PubMed] [Google Scholar]
- 15.Sackett DL. Equipoise, a term whose time (if it ever came) has surely gone. CMAJ. 2000;163:835–836. [PMC free article] [PubMed] [Google Scholar]
- 16.Shapiro SH, Glass KC. Why sackett’s analysis of randomized controlled trials fails, but needn’t. CMAJ. 2000;163:834–835. [PMC free article] [PubMed] [Google Scholar]
- 17.Benson K, Hartz AJ. A comparison of observational studies and randomized, controlled trials. N Engl J Med. 2000;342:1878–1886. doi: 10.1056/NEJM200006223422506. [DOI] [PubMed] [Google Scholar]
- 18.Connors AF, Jr, Speroff T, Dawson NV, et al. The effectiveness of right heart catheterization in the initial care of critically ill patients. SUPPORT investigators. JAMA. 1996;276:889–897. doi: 10.1001/jama.276.11.889. [DOI] [PubMed] [Google Scholar]
- 19.Guyatt G. A randomized control trial of right-heart catheterization in critically ill patients. ontario intensive care study group. J Intensive Care Med. 1991;6:91–95. doi: 10.1177/088506669100600204. [DOI] [PubMed] [Google Scholar]
- 20.Sandham JD, Hull RD, Brant RF, et al. A randomized, controlled trial of the use of pulmonary-artery catheters in high-risk surgical patients. N Engl J Med. 2003;348:5–14. doi: 10.1056/NEJMoa021108. [DOI] [PubMed] [Google Scholar]
- 21.Richard C, Warszawski J, Anguel N, et al. Early use of the pulmonary artery catheter and outcomes in patients with shock and acute respiratory distress syndrome: A randomized controlled trial. JAMA. 2003;290:2713–2720. doi: 10.1001/jama.290.20.2713. [DOI] [PubMed] [Google Scholar]
- 22.Harvey S, Harrison DA, Singer M, et al. Assessment of the clinical effectiveness of pulmonary artery catheters in management of patients in intensive care (PAC-man): A randomised controlled trial. Lancet. 2005;366:472–477. doi: 10.1016/S0140-6736(05)67061-4. [DOI] [PubMed] [Google Scholar]
- 23.LeLorier J, Gregoire G, Benhaddad A, et al. Discrepancies between meta-analyses and subsequent large randomized, controlled trials. N Engl J Med. 1997;337:536–542. doi: 10.1056/NEJM199708213370806. [DOI] [PubMed] [Google Scholar]
- 24.Annane D, Sebille V, Charpentier C, et al. Effect of treatment with low doses of hydrocortisone and fludrocortisone on mortality in patients with septic shock. JAMA. 2002;288:862–871. doi: 10.1001/jama.288.7.862. [DOI] [PubMed] [Google Scholar]
- 25.Sprung CL, Annane D, Keh D, et al. Hydrocortisone therapy for patients with septic shock. N Engl J Med. 2008;358:111–124. doi: 10.1056/NEJMoa071366. [DOI] [PubMed] [Google Scholar]
- 26.Piantadosi S. Clinical trials. Hoboken: John Wiley& Sons; 2005. [Google Scholar]
- 27.Cowles MK. Bayesian estimation of the proportion of treatment effect captured by a surrogate marker. Stat Med. 2002;21:811–834. doi: 10.1002/sim.1057. [DOI] [PubMed] [Google Scholar]
- 28.Kalish LA, McIntosh K, Read JS, et al. Evaluation of human immunodeficiency virus (HIV) type 1 load, CD4 T cell level, and clinical class as time-fixed and time-varying markers of disease progression in HIV-1-infected children. J Infect Dis. 1999;180:1514–1520. doi: 10.1086/315064. [DOI] [PubMed] [Google Scholar]
- 29.Practice parameters for hemodynamic support of sepsis in adult patients in sepsis. task force of the american college of critical care medicine, society of critical care medicine. Crit Care Med. 1999;27:639–660. doi: 10.1097/00003246-199903000-00049. [DOI] [PubMed] [Google Scholar]
- 30.Bakker J, Grover R, McLuckie A, et al. Administration of the nitric oxide synthase inhibitor NG-methyl-L-arginine hydrochloride (546C88) by intravenous infusion for up to 72 hours can promote the resolution of shock in patients with severe sepsis: Results of a randomized, double-blind, placebo-controlled multicenter study (study no. 144-002) Crit Care Med. 2004;32:1–12. doi: 10.1097/01.CCM.0000105118.66983.19. [DOI] [PubMed] [Google Scholar]
- 31.Lopez A, Lorente JA, Steingrub J, et al. Multiple-center, randomized, placebo-controlled, double-blind study of the nitric oxide synthase inhibitor 546C88: Effect on survival in patients with septic shock. Crit Care Med. 2004;32:21–30. doi: 10.1097/01.CCM.0000105581.01815.C6. [DOI] [PubMed] [Google Scholar]
- 32.Taccone P, Pesenti A, Latini R, et al. Prone positioning in patients with moderate and severe acute respiratory distress syndrome: A randomized controlled trial. JAMA. 2009;302:1977–1984. doi: 10.1001/jama.2009.1614. [DOI] [PubMed] [Google Scholar]
- 33.Taylor RW, Zimmerman JL, Dellinger RP, et al. Low-dose inhaled nitric oxide in patients with acute lung injury: A randomized controlled trial. JAMA. 2004;291:1603–1609. doi: 10.1001/jama.291.13.1603. [DOI] [PubMed] [Google Scholar]
- 34.Abroug F, Ouanes-Besbes L, Elatrous S, et al. The effect of prone positioning in acute respiratory distress syndrome or acute lung injury: A meta-analysis. areas of uncertainty and recommendations for research. Intensive Care Med. 2008;34:1002–1011. doi: 10.1007/s00134-008-1062-3. [DOI] [PubMed] [Google Scholar]
- 35.Ware LB, Koyama T, Billheimer DD, et al. Prognostic and pathogenetic value of combining clinical and biochemical indices in patients with acute lung injury. Chest. 2009 doi: 10.1378/chest.09-1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tomlinson G, Detsky AS. Composite end points in randomized trials: There is no free lunch. JAMA. 2010;303:267–268. doi: 10.1001/jama.2009.2017. [DOI] [PubMed] [Google Scholar]
- 37.Guyatt GH, editor. Users’ guides to the medical literature. Columbus, Ohio: Mcgraw Hill; 2008. [Google Scholar]
- 38.Brower RG, Lanken PN, MacIntyre N, et al. Higher versus lower positive end-expiratory pressures in patients with the acute respiratory distress syndrome. N Engl J Med. 2004;351:327–336. doi: 10.1056/NEJMoa032193. [DOI] [PubMed] [Google Scholar]
- 39.The Acute Respiratory Distress Syndrome Network. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med. 2000;342:1301–1308. doi: 10.1056/NEJM200005043421801. [DOI] [PubMed] [Google Scholar]
- 40.Checkley W, Brower R, Munoz A. Inference for mutually exclusive competing events through a mixture of generalized gamma distributions. Epidemiology. 2010 doi: 10.1097/EDE.0b013e3181e090ed. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moher D, Schulz KF, Altman D. The CONSORT statement: Revised recommendations for improving the quality of reports or parallel group trials. Lancet. 2001;357:1191–94. [PubMed] [Google Scholar]
- 42.von Elm E, Altman DG, Egger M, Pocock SJ, Gøtzsche PC, Vandenbroucke JP. STROBE initiative. the strengthening the reporting of observational studies in epidemiology (STROBE) statement: Guidelines for reporting observational studies. Lancet. 2007;370:1453–7. doi: 10.1016/S0140-6736(07)61602-X. [DOI] [PubMed] [Google Scholar]
- 43.Meinert CL. Clinical trials: Design, conduct, and analysis. New York: Oxford University Press; 1986. [Google Scholar]
- 44.Wittes J. Sample size calculations for randomized controlled trials. Epidemiol Rev. 2002;24:39–53. doi: 10.1093/epirev/24.1.39. [DOI] [PubMed] [Google Scholar]
- 45.Dupont WD, Plummer WD. Power and sample size calculations for studies involving linear regression. Controlled Clinical Trials. 1998;19:589–601. doi: 10.1016/s0197-2456(98)00037-3. [DOI] [PubMed] [Google Scholar]
- 46.Hayes RJ, Moulton LH. Cluster randomized trials. Chapman & Hall; CRC Press; 2008. [Google Scholar]
- 47.Whitehead J. The design and analysis of sequential clinical trials. 2. Chichester: John Wiley and Sons; 1997. [Google Scholar]
- 48.Day SJ, Graham DF. Sample size estimation for comparing two or more treatment groups in clinical trials. Stat Med. 1991;10:33–43. doi: 10.1002/sim.4780100109. [DOI] [PubMed] [Google Scholar]
- 49.Pocock S, White I. Trials stopped early: Too good to be true? Lancet. 1999;353:943–944. doi: 10.1016/S0140-6736(98)00379-1. [DOI] [PubMed] [Google Scholar]
- 50.O’Brien PC, Fleming TR. A multiple testing procedure for clinical trials. Biometrics. 1979;35:549–556. [PubMed] [Google Scholar]
- 51.Russell JA, Walley KR, Singer J, et al. Vasopressin versus norepinephrine infusion in patients with septic shock. N Engl J Med. 2008;358:877–887. doi: 10.1056/NEJMoa067373. [DOI] [PubMed] [Google Scholar]
- 52.Grimes DA, Schulz KF. Bias and causal associations in observational research. Lancet. 2002;359:248–252. doi: 10.1016/S0140-6736(02)07451-2. [DOI] [PubMed] [Google Scholar]
- 53.Bernard GR, Artigas A, Brigham KL, et al. The american-european consensus conference on ARDS. definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Respir Crit Care Med. 1994;149:818–824. doi: 10.1164/ajrccm.149.3.7509706. [DOI] [PubMed] [Google Scholar]
- 54.Rubenfeld GD, Caldwell E, Granton J, et al. Interobserver variability in applying a radiographic definition for ARDS. Chest. 1999;116:1347–1353. doi: 10.1378/chest.116.5.1347. [DOI] [PubMed] [Google Scholar]
- 55.Guyatt GH, Sinclair J, Cook DJ, et al. Users’ guides to the medical literature: XVI. how to use a treatment recommendation. JAMA. 1999;281:1836–1843. doi: 10.1001/jama.281.19.1836. [DOI] [PubMed] [Google Scholar]
- 56.Dhainaut JF, Tenaillon A, Hemmer M, et al. Confirmatory platelet-activating factor receptor antagonist trial in patients with severe gram-negative bacterial sepsis: A phase III, randomized, double-blind, placebo-controlled, multicenter trial. BN 52021 sepsis investigator group. Crit Care Med. 1998;26:1963–1971. doi: 10.1097/00003246-199812000-00021. [DOI] [PubMed] [Google Scholar]
- 57.Opal SM, Fisher CJ, Jr, Dhainaut JF, et al. Confirmatory interleukin-1 receptor antagonist trial in severe sepsis: A phase III, randomized, double-blind, placebo-controlled, multicenter trial. the interleukin-1 receptor antagonist sepsis investigator group. Crit Care Med. 1997;25:1115–1124. doi: 10.1097/00003246-199707000-00010. [DOI] [PubMed] [Google Scholar]
- 58.Knaus WA, Harrell FE, Jr, LaBrecque JF, et al. Use of predicted risk of mortality to evaluate the efficacy of anticytokine therapy in sepsis. the rhIL-1ra phase III sepsis syndrome study group. Crit Care Med. 1996;24:46–56. doi: 10.1097/00003246-199601000-00010. [DOI] [PubMed] [Google Scholar]
- 59.Natanson C, Esposito CJ, Banks SM. The sirens’ songs of confirmatory sepsis trials: Selection bias and sampling error. Crit Care Med. 1998;26:1927–1931. doi: 10.1097/00003246-199812000-00001. [DOI] [PubMed] [Google Scholar]
- 60.Cook DI, Gebski VJ, Keech AC. Subgroup analysis in clinical trials. Med J Aust. 2004;180:289–291. doi: 10.5694/j.1326-5377.2004.tb05928.x. [DOI] [PubMed] [Google Scholar]
- 61.Cook D, Crowther M. Targeting anemia with erythropoietin during critical illness. N Engl J Med. 2007;357:1037–1039. doi: 10.1056/NEJMe078150. [DOI] [PubMed] [Google Scholar]
- 62.The SAFE Study Investigators. A comparison of albumin and saline for fluid resuscitation in the intensive care unit. N Engl J Med. 2004;350:2247–2256. doi: 10.1056/NEJMoa040232. [DOI] [PubMed] [Google Scholar]
- 63.Pocock SJ, Ware JH. Translating statistical findings into plain english. The Lancet. 2009;373:1926–1928. doi: 10.1016/S0140-6736(09)60499-2. [DOI] [PubMed] [Google Scholar]
- 64.Bellomo R, Warrillow SJ, Reade MC. Why we should be wary of single-center trials. Crit Care Med. 2009;37:3114–3119. doi: 10.1097/CCM.0b013e3181bc7bd5. [DOI] [PubMed] [Google Scholar]
- 65.Sweeney DA, Danner RL, Eichacker PQ, et al. Once is not enough: Clinical trials in sepsis. Intensive Care Med. 2008;34:1955–1960. doi: 10.1007/s00134-008-1274-6. [DOI] [PubMed] [Google Scholar]
- 66.Levy MM. Facilitating knowledge transfer with single-center trials. Crit Care Med. 2009;37:3120–3123. doi: 10.1097/CCM.0b013e3181bdd9ae. [DOI] [PubMed] [Google Scholar]