

Highlights
-
•
Mithramycin (MTR) binds to core histones but not to linker histone H1.
-
•
Unlike MTR–DNA interaction, MTR–histone association is metal independent.
-
•
MTR alters H3K18 acetylation in vitro and ex vivo.
-
•
MTR is a dual binder (binds to both DNA and histones) in the chromatin context.
Abbreviations: MTR, mithramycin; HD, Huntington’s disease; NIH, National Institutes of Health; EWS-FLI1, transcription factor with a DNA binding domain FLI1 and a transcription enhancer domain EWS; HAT, histone acetyltransferase; CBP, CREB-binding protein; H3K18Ac, histone H3 lysine 18 acetylation; SGR, sanguinarine; ITC, isothermal titration calorimetry; CD, circular dichroism; EM, electron microscopy; FACS, fluorescence activated cell sorting; BAC, benzalkonium chloride; TCA, trichloroacetic acid; TBST, Tris-buffered saline Tween-20; MTT, 3-(4-5 dimethylthiazol-2-yl) 2-5diphenyl-tetrazolium bromide; PBS, phosphate-buffered saline; BSA, bovine serum albumin; PTM, post-translational modification; M2+, bivalent metal ion such as Mg2+
Keywords: Mithramycin, Core histones, Dual binding mode, Epigenetic modulator, H3K18 acetylation
Abstract
Mithramycin (MTR) is a clinically approved DNA-binding antitumor antibiotic currently in Phase 2 clinical trials at National Institutes of Health for treatment of osteosarcoma. In view of the resurgence in the studies of this generic antibiotic as a human medicine, we have examined the binding properties of MTR with the integral component of chromatin – histone proteins – as a part of our broad objective to classify DNA-binding molecules in terms of their ability to bind chromosomal DNA alone (single binding mode) or both histones and chromosomal DNA (dual binding mode). The present report shows that besides DNA, MTR also binds to core histones present in chromatin and thus possesses the property of dual binding in the chromatin context. In contrast to the MTR–DNA interaction, association of MTR with histones does not require obligatory presence of bivalent metal ion like Mg2+. As a consequence of its ability to interact with core histones, MTR inhibits histone H3 acetylation at lysine 18, an important signature of active chromatin, in vitro and ex vivo. Reanalysis of microarray data of Ewing sarcoma cell lines shows that upon MTR treatment there is a significant down regulation of genes, possibly implicating a repression of H3K18Ac-enriched genes apart from DNA-binding transcription factors. Association of MTR with core histones and its ability to alter post-translational modification of histone H3 clearly indicates an additional mode of action of this anticancer drug that could be implicated in novel therapeutic strategies.
1. Introduction
Mithramycin, MTR (Fig. 1) is a DNA-binding antitumor antibiotic of the aureolic acid group isolated from soil borne bacteria like Streptomyces plicatus [1]. At physiological pH, it exists in anionic form (pKa = 5.0). It inhibits tumor cell growth without affecting normal cells [2] via inhibition of Sp1 gene. This is attributed to the ability of MTR to cross the nuclear membrane, bind to GC rich DNA sequences and block the Sp1 transcription factor from its binding sites on the promoters of oncogenes such as Myc to inhibit their expression [3,4]. Its neuroprotective ability was tested in vitro and in vivo employing different model systems [5–9]. The major hurdle for drugs to be used in brain targeting is their ability to cross the blood–brain barrier which MTR can achieve [6]. It was reported to improve altered nucleosome homeostasis in HD mice, normalizing the chromatin modification pattern [10]. One of these studies showed that it has the ability to rebalance epigenetic histone modifications like acetylation and methylation [10] and has been used in preclinical trials in Huntington’s disease (HD). MTR is currently in Phase 2 clinical trial at NIH Clinical Center for treatment of osteosarcoma where it presumably functions via inhibition of EWS-FLI1 pathway [11]. Earlier it had been employed in the treatment of various neoplastic diseases like chronic myelogenous leukemia, testicular carcinoma, pancreatic cancer and prostate cancer [12–19]. Anionic form of MTR has the ability to bind bivalent cations and form high-affinity antibiotic–metal ion complexes (1:1 and 1:2 stoichiometry in terms of metal:antibiotic), which bind to the chromosomal DNA in GC selective manner via the minor groove at and above physiological pH [20,21]. Thus MTR–metal complex acts as the substrate for DNA binding [21–25]. Its antitumor activity is ascribed to its GC specific recognition that permits MTR to bind to numerous promoter regions, thereby regulating the expression of downstream genes. It effectively inhibits the expression of proto-oncogenes like c-myc, c-Ha-ras and c-myb [26–29].
Fig. 1.
Chemical structure of mithramycin (MTR).
Current studies from our laboratory have shown the histone binding potential of DNA binders and their ability to alter histone post-translational modifications (PTMs) [30,31]. Hence it is likely, that MTR may have additional interacting partners in chromatin. Taking a cue from the above studies, we have examined the binding properties of MTR with other prospective cellular targets namely histone proteins with an objective to classify small DNA binding molecules in terms of their ability to bind chromosomal DNA alone (single binding mode) or both histones and chromosomal DNA (dual binding mode) [30–33].
The current study reports the association of MTR with core histones and its functional consequence. The interactions are enthalpy driven with apparent dissociation constants of micromolar order. The noteworthy feature of this interaction is that unlike MTR–DNA interaction, bivalent metal ion such as Mg2+ is not a requirement for the association of MTR with core histones. We have picked up few histone modifications related to transcription regulation and have found that MTR is a potent inhibitor of K18 acetylation mark of histone H3, both in vitro and ex vivo. Taken together, our results indicate that MTR exhibits a dual binding mode to both DNA and core histones and can specifically modulate epigenetic signature implicated in transcription [6,34]. Thus this study opens up a new aspect of the mode of action of the antibiotic, MTR.
2. Materials and methods
2.1. Materials
Chemicals were purchased from Sigma Chemical Company, USA, unless otherwise specified. Recombinant histones H10, H2A, H2B, H3.3, H4, H2A–H2B dimer and H3.1–H4 tetramer were from New England Biolabs. Anti-acetyl-Histone H3 (Lys18 and Lys 27) antibodies were purchased from Abcam and anti-acetyl-Histone H3 (Lys 9) antibody was from Millipore. The secondary anti-rabbit antibody was from Abcam. Chemiluminescent substrates were from Thermo Scientific (SuperSignal West Pico Substrate) and the developer and fixer solutions were from Kodak. Histone acetyltransferase CBP and GCN5 were from Enzo Life Sciences. The commercial histones were dialyzed extensively against 10 mM Tris–HCl, pH 7.0 containing 150 mM NaCl prior to experiments.
2.2. Methods
2.2.1. Preparation of MTR solution
Stock solution was prepared in Milli Q water and the concentration was determined spectrophotometrically using the molar extinction coefficient (ε = 10,000 M−1 cm−1) [35] at 400 nm. The ratio of absorbance of MTR at 400–440 nm was checked to be above 3.3 to rule out any bivalent metal contamination.
2.2.2. Preparation of chromatin samples
Chromatin and chromatosomes were isolated from liver of male albino Sprague–Dawley rats following standard protocol [36]. Internationally recognized guidelines were followed for all experiments using rat. The experiments were performed with the approval for ethical clearance from Institutional Animal Ethics Committee, IAEC (Approval No.: IAEC/BI/09/2012), Bose Institute, Kolkata. In brief, rat liver nuclei were isolated [31,36,37] and chromatin was prepared by partial digestion with micrococcal nuclease. Long fragments and chromatosomes were separately collected by centrifugation through a 5–25% linear sucrose density gradient. All samples, prior to experiment were dialyzed extensively against 10 mM Tris–HCl, pH 7.0 containing 15 mM NaCl and mononucleotide concentrations of the samples were determined spectrophotometrically using the molar extinction coefficient (ε260) of 6600 M−1 cm−1.
Core octamer was prepared from chicken erythrocytes using the method described by Peterson and Hansen [38]. Prior to experiments, the core octamer was dialyzed against 10 mM Tris–HCl, pH 7.0, 2 M NaCl. Concentration of core octamer was determined using the molar extinction coefficient (ε230) of 507,233 M−1 cm−1.
2.2.3. Steady state fluorescence measurements
The fluorescence emission spectra of MTR were monitored in presence of recombinant histones, core octamer, H2A–H2B dimer, H3–H4 tetramer, chromatin/chromatosome and N-terminal histone peptides where MTR/protein ratio was gradually increased. The samples were excited at 470 nm in order to avoid photodegradation of MTR [25]. The excitation and emission slit widths were kept at 10 nm and the scan speed was 200 nm/min. An incubation time of 3 min was maintained before each reading. The measurements were performed in a Perkin Elmer LS 55 Luminescence Spectrometer at 25 °C.
2.2.4. Isothermal titration calorimetry
MTR was titrated against histones H2A, H2B, H3.3 and H4 in a Microcal ITC 200 microcalorimeter at 25 °C with a constant stirring speed of 300 rpm. Control experiment was performed for each system to subtract the background heat of dilution. The resulting thermogram was fitted employing one set of binding sites model of Levenberg–Marquardt nonlinear least squares curve fitting algorithm, inbuilt in the MicroCal LLC software.
2.2.5. Circular dichroism study
The circular dichroism measurements were performed in a Spectropolarimeter from Bio Logic Science Instruments, France and data were analyzed with inbuilt Bio-Kine 32 V4.49-1 software. The measurements were performed at 25 °C. The molar ellipticity was plotted against the wavelength. Acquisition duration was fixed at 2 s and the required wavelength range was scanned at 0.5 nm intervals. Each spectrum was an average of 4 accumulations and the corrected spectra were obtained by subtraction of the appropriate control.
2.2.6. Electron microscopy
Chromatin was extensively dialyzed against HEGN buffer (10 mM Hepes, pH 7.5; 0.25 mM EDTA; 10% glycerol; 15 mM NaCl). It was treated with MTR at a ligand:DNA base ratio of 0.2 and incubated for 3 h at 25 °C in a water bath. Treated and untreated samples were processed for electron microscopy study as detailed in the Supplemental methods section.
2.2.7. Dynamic light scattering
Dynamic light scattering was used as a tool to examine MTR-induced alteration of chromatin structure, if any. The parameter monitored was the hydrodynamic diameter of untreated and MTR-treated chromatin. The detailed procedure is provided in the Supplementary section.
2.2.8. Histone acetyltransferase (HAT) assay
Standard HAT assay protocol was followed as reported previously [39]. 2 μg of recombinant histone H3 was incubated in HAT assay buffer containing 50 mM Tris–HCl, pH 8.0, 10% (v/v) glycerol, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, 0.1 mM EDTA, pH 8.0, 0.1 mM sodium butyrate at 30 °C for 10 min in the presence and absence of MTR followed by addition of 1 μL of acetyl-CoA and further incubated for another 10 min at 30 °C. The final reaction volume was 30 μL. The acetylation reaction was terminated by chilling. Histone was precipitated by trichloroacetic acid (TCA) and analyzed on a 15% SDS page followed by Western blot analysis. The acetylation marks probed were H3K18 (dilution 1:5000 in 3% skim milk prepared in TBST), H3K9 (dilution 1:10,000) and H3K27 (dilution 1:1000). Signals were generated using chemiluminescent substrates in a dark room on X-ray films providing very short exposures of ∼2 s and the blots were developed using developer and fixer solutions.
2.2.9. Flow cytometry analysis
HeLa cells (untreated or treated with 10 μM MTR for 9 h) were subjected to flow cytometry analysis as described in the Supplemental methods section.
2.2.10. Cell viability assay (MTT assay)
MTT assay was performed following standard protocol [40] to test the cell viability upon MTR treatment. The method is detailed in the Supplementary section.
2.2.11. Cell culture and MTR treatment
HeLa cells were maintained in Dulbecco’s modified Eagle’s medium (Gibco) supplemented with 10% fetal bovine serum (Gibco) and penstrep (10 μL/mL of medium, Gibco) at 37 °C in 5% (v/v) CO2.
For MTR treatment, HeLa cells were plated in 90 mm dishes (Nunc) at a density of 1.2 million cells per dish. The cells were treated with 10 μM MTR for 9 h. After treatment, the cells were observed under light microscope and processed for immunoblot analysis.
2.2.12. Immunoblot analysis
HeLa cells were harvested after rinsing with 1× PBS and whole cell extracts were made in Laemmli buffer (4% SDS, 20% Glycerol, 120 mM Tris, pH 6.8) as per standard protocol [41]. Total protein extracts were boiled in 5× SDS buffer and analyzed in a 15% gel followed by Western blotting. The blot was probed with H3K18Ac primary antibody (dilution 1:10,000).
2.2.13. Confocal microscopy
For immunofluorescence staining, HeLa cells were grown in 35 mm tissue culture dish (Nunc) with a cover slip at a density of 0.3 million cells per dish. They were treated with different concentrations (0.1–10 μM) of MTR for 9 h. The cells were rinsed with 1× PBS and then fixed with 4% PBS at room temperature for 10 min. The cells were subsequently permeabilized in 1% Triton X-100 for 10 min at room temperature. Cells were blocked with 3% BSA in PBS, incubated with anti-rabbit-H3K18Ac antibody (Active Motif) at a dilution of 1:1000, followed by Alexa 568 conjugated anti-rabbit–antiserum at a dilution of 1:1000 for 1hour each at room temperature. The cover slips were mounted onto a glass slide with a commercial mounting medium (Invitrogen). The slides were visualized in Andor Spinning Disk Confocal Microscope and multiple images were taken at different fields. Higher magnification images were taken in a super resolution microscopy N-SIM (Nikon) equipped with Nikon Imaging Software (NIS). Detailed statistical analysis was done to quantify the extent of repression of H3K18 acetylation upon increasing concentration of MTR treatment, using Image J software.
2.2.14. Gene expression analysis upon MTR treatment
In order to understand the basal level effect of MTR on gene expression, we used the publically available microarray data of MTR treated sarcoma cell line from NCBI GEO with series accession number GSE25127. The data consist of two cell lines TC71 and TC32, which were treated with or without MTR and differential gene expression analysis was performed in 12 arrays (three biological replicates per cell line/treatment). The detailed procedure for data analysis is described in the Supplementary section.
3. Results
3.1. Evidence for physical association of MTR with core histones and the associated energetics
Interactions of MTR:M2+ complexes (M2+: bivalent metal ion such as Mg2+) with DNA or chromatin have been well-characterized [20–22,24,25,42]. Since core histones are integral components of chromatin, MTR could have a potential histone interacting ability in the cellular context. However, there is no report yet on the interaction of anionic MTR with histone(s) at or near physiological pH. We have characterized here the association of MTR with histones by means of steady state fluorescence and circular dichroism spectroscopy. Histone concentration dependent decrease in fluorescence emission intensity of MTR upon addition of individual core histone proteins suggests their complex formation. The fluorescence spectrum of MTR (λex = 470 nm) [25], is progressively quenched in presence of increasing concentrations of histone(s) H2A, H2B, H3 and H4 (Fig. 2A and Fig. S1). The plot of emission intensity at peak (540 nm) against histone concentration is characteristic of a linear binding isotherm originating from the association between the histone(s) and MTR. Curve fitting analysis [24,42,43] shows that MTR binds to histones with micromolar dissociation constant (Table 1). Conformational alteration of MTR in presence of core histones was probed by CD spectroscopy. The molar ellipticity of MTR decreased in presence of core histones supporting the complex formation (Fig. 2B for histone H3 and Fig. S2 for the rest). The core histones are assembled under in vivo conditions as an octamer [44]. At physiological ionic strength (150 mM NaCl) histone octamer stripped off DNA exists as dimer–tetramer equilibrium [45]. Under in vitro conditions such octamer is formed at high salt concentration of 2 M NaCl [45]. We, therefore, studied the association of MTR with hierarchical levels of octamer assembly, viz. individual core histones (Fig. 2 and Fig. S1), H2A–H2B dimer, H3–H4 tetramer (Fig. S3) and octamer (Fig. 3A) by means of fluorescence to check if there is any difference in the nature of interaction. In case of octamer, the band around 550 nm in the emission spectrum of free MTR gives way to a red-shifted new band around 600 nm. The equilibrium between free and octamer bound MTR was indicated from the binding isotherm obtained from a plot of the emission intensity of MTR at 600 nm against the input concentration of the octamer. The significant red shift of the emission peak of MTR can be ascribed to alteration in the local physical environment of the chromophore in MTR which is possibly caged into the assembled octamer. The contribution of other parameters such as electrostatic interaction of the anionic MTR with positively charged amino acid in the N-terminal tails in the histone also appears to be a plausible factor. The dissociation constant value of 0.60 ± 0.01 μM is relatively lower than the corresponding values for the individual histone(s) implying stronger association when the core histones form octamer. The concentration dependent variation in molar ellipticity of core histone octamer with gradual addition of MTR (Fig. 3B) is another evidence for MTR-octamer association. The possibility of interaction of MTR with the tail peptides, if any, was also checked. The emission intensity of MTR decreases upon progressive addition of H3 tail peptide (residues 1–21, Fig. 2D). Fig. S4 shows the percentage change in fluorescence intensity of MTR at the emission maximum as a function of input concentration of the N-terminal tail peptides (of histone H2A, H2B, H3 and H4). The results suggest a preferential interaction between MTR and the N-terminal tail (1–21 residues) of H3.
Fig. 2.
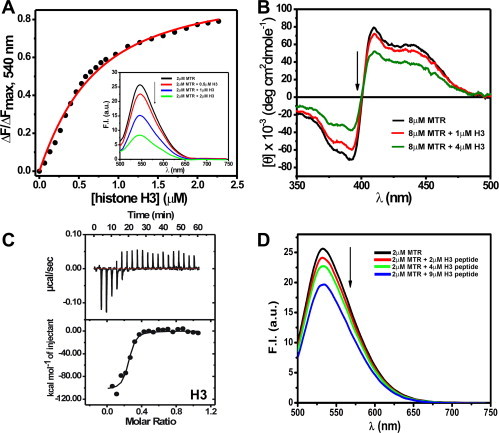
(A) Binding isotherm for the interaction of MTR with histone H3 in 10 mM Tris–HCl, pH 7.0 containing 150 mM NaCl at 25 °C obtained from steady state fluorescence spectroscopy. Inset shows the emission spectra of 2 μM MTR in absence (black) and presence of increasing concentrations (0.5 μM, red; 1 μM, blue; 2 μM, green) of human recombinant histone H3. λex = 470 nm. (B) Circular dichroism spectra of 8 μM MTR in 10 mM Tris–HCl, pH 7.0 containing 150 mM NaCl at 25 °C in absence (black) and presence of 1 μM (red) and 4 μM (green) histone H3 in the visible range. (C) ITC profile for the association of MTR with histone H3 at 25 °C in 10 mM Tris–HCl, pH 7.0 containing 150 mM NaCl. The lower panel contains the background heat subtracted fitted isotherm. Emission spectra of 2 μM MTR in 10 mM Tris–HCl, pH 7.0 containing 150 mM NaCl at 25 °C in absence (black) and in presence of (2 μM, red; 4 μM, green; 9 μM, blue) N-terminal tail peptide H3 (residues 1–21). λex = 470 nm. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Table 1.
Dissociation constants of MTR–histone interactions at 25 °C as obtained from spectrofluorimetric analysis.
| Histones | Kd (μM) |
|---|---|
| H2A | 0.96 ± 0.02 |
| H2B | 1.7 ± 0.05 |
| H3.3 | 0.66 ± 0.02 |
| H4 | 1.25 ± 0.05 |
| H2A–H2B dimer | 0.78 ± 0.03 |
| H3.1–H4 tetramer | 0.77 ± 0.03 |
Fig. 3.
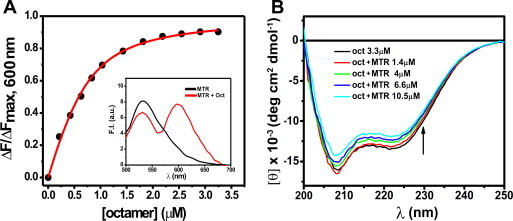
(A) Interaction of MTR with core octamer in 10 mM Tris–HCl, pH 7.0 containing 2 M NaCl at 25 °C: Binding isotherm obtained using non-linear curve fitting analysis. Inset: Emission spectra of MTR (2 μM) in absence (black) and in presence (red) of 3.3 μM core octamer. λex = 470 nm. (B) Near UV CD spectra of 3.3 μM histone octamer in 10 mM Tris–HCl, pH 7.0 containing 2 M NaCl at 25 °C in absence (black) and in presence of increasing MTR concentrations. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Association of MTR with core histone(s) and the energetics of the interactions were quantitated by means of ITC. Representative thermograms for the titration of MTR with histones, H2A, H2B, H3, and H4 are shown in Fig. 2C and Fig. S5. The thermodynamic parameters obtained by ITC are summarized in Table 2. The association with histone(s) has also been carried out at high salt concentration to eliminate the possibility of non-specific association (Fig. S6 and Table S2). The interactions are enthalpy driven and enthalpy–entropy compensation accounts for the overall free energy change for the reactions. In order to investigate the ability of MTR to interact with linker histones (if any), ITC and fluorescence measurements were performed. No significant interaction between MTR and linker histone H1 was scored by either of the two methods (Figs. S1D and S5D). The interaction of MTR with core histones might induce structural alterations like compaction/decompaction of chromatin as caused by other DNA binding small molecules [30,37]. The possibility was examined using DLS and EM. The hydrodynamic diameter of chromatin was monitored in absence and presence of different concentrations of MTR by DLS. The Zav diameter of chromatin was found to be 132.8 nm in absence of MTR and the same was noted to be 131.8 nm at a ligand: DNA base ratio of 0.25 (Fig. S7A). EM images of untreated and MTR-treated chromatin (Fig. S7B) suggest that MTR causes no significant alteration of chromatin structure. The chromatin network is retained in presence of MTR and no change in the compactness of chromatin was observed upon MTR treatment. These data rule out the possibility of H1 eviction by MTR.
Table 2.
Thermodynamic parameters of MTR–histone interactions in 10 mM Tris–HCl, pH 7.0 containing 150 mM NaCl as obtained by ITC at 25 °C.
| Histone | Kd (μM) | ΔH (kcal mol−1) | ΔS (e.u.) | ΔG (kcal mol−1) |
|---|---|---|---|---|
| H2A | 0.05 ± 0.01 | −14.8 ± 0.7 | −16.2 | −9.97 |
| H2B | 1.4 ± 0.05 | −14.1 ± 1.2 | −20.4 | −8.02 |
| H3.3 | 0.05 ± 0.01 | −24.3 ± 0.96 | −49.0 | −9.70 |
| H4 | 0.051 ± 0.03 | −24.3 ± 0.65 | −48.1 | −9.99 |
3.2. Metal-independent binding of MTR to chromatin
It was previously proposed that MTR binds to chromatin only in presence of bivalent metal ions like Mg2+ via minor groove of DNA [42]. Here we have shown that the interaction of MTR with chromatin occurs even in absence of Mg2+. In contrast to the interaction with free histones, the fluorescence intensity of MTR at 540 nm increases (Fig. 4, panels A and D) with addition of aliquots of chromatin/chromatosome and eventually reaches a plateau. The dissociation constant is in the micromolar range (Table S1). Notable feature is that the association in all cases described above is stronger than that was reported in case of chromatin with the MTR:Mg2+ complexes [21,42]. Chiro-optical properties of MTR in the visible region were monitored to examine its association with chromatosome and chromatin. The CD spectral behavior of MTR in presence of chromatin (Fig. 4C) and chromatosome (Fig. 4F) is similar to that of interaction of MTR with individual histones corroborating the proposal that MTR interacts with histones in chromatin/chromatosome.
Fig. 4.
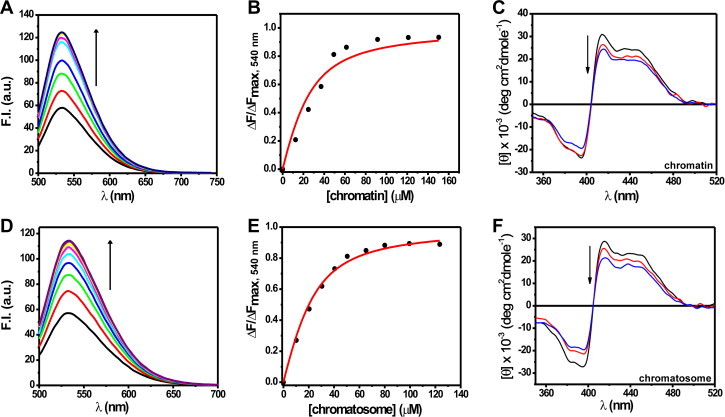
Emission spectra of 20 μM MTR in 10 mM Tris–HCl, pH 7.0 containing 15 mM NaCl at 25 °C in presence of increasing concentrations of chromatin (A) and chromatosome (D). λex = 470 nm. Panels (B) and (E) show corresponding binding isotherms for MTR–chromatin/chromatosome interactions. Circular dichroism spectra of 15 μM MTR monitored at 25 °C in absence (black) and presence of chromatin (C) and chromatosome (F). Red curves represent 45 μM and blue curves represent 120 μM of chromatin/chromatosome respectively. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.3. Role of MTR as an epigenetic modulator
The cell modulates nucleosome mobility and turnover via chemical modifications of histones. We have demonstrated that MTR binds to core histones and we have also observed an interaction between MTR and histone H3 tail (1–21 amino acids). N-terminal tails of the core histones undergo PTMs and impact chromatin structure and function. Taking a cue from literature, where histone interacting small molecules are found to modulate its PTMs (such as histone acetylation) [30,31,46] we went on to check whether MTR can alter histone acetylation. We have picked up few commonly studied N-terminal acetylation marks of histones H3 and H4. H3K18Ac, an important transcription activation mark, is altered in presence of MTR both in vitro and ex vivo. In vitro HAT assay using CBP HAT domain, showed that MTR inhibits H3K18Ac (Fig. 5A). Acetylation levels at two other acetylation sites H3K9Ac (by GCN5) and H3K27Ac (by CBP) [47] were not altered in presence of MTR (Fig. 5B and C). Neither did we notice any change in the acetylation levels of H4K5Ac and H4K8Ac (by CBP) (Fig. S8). Since H3K18Ac was altered in vitro, we further investigated the modulation of H3K18Ac mark in cellular context. It was found that MTR inhibits H3K18Ac in a dose dependent and time dependent manner in HeLa cells by immunoblot analysis (Fig. S9). Reduction in acetylation level was prominent upon 10 μM MTR treatment for 9 h (Fig. 5D, compare lanes 1 and 2) and almost total inhibition was found upon 20 μM MTR treatment for 12 h (Fig. 5D, compare lanes 1 and 3). The variation in H3K18 acetylation status was further monitored by confocal microscopy. Titration of different ligand concentrations in HeLa cells (Fig. S10) showed a prominent repression (∼50%) in H3K18Ac level upon treatment with 10 μM MTR (Fig. 5E).
Fig. 5.
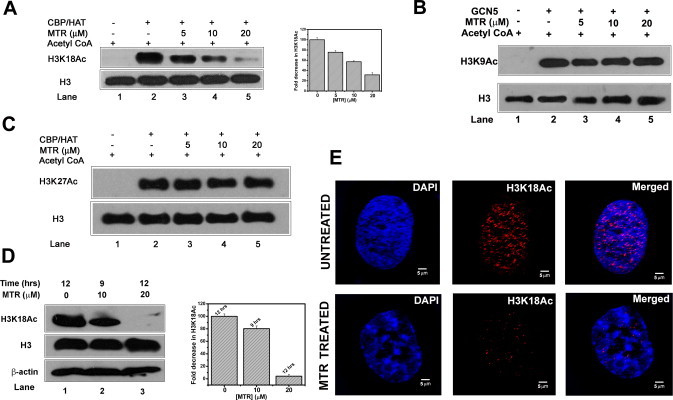
In vitro HAT assay to examine alteration of H3K18 acetylation (A), H3K9 acetylation (B) and H3K27 acetylation (C) by MTR in recombinant histone H3. (D) Modulation of H3K18 acetylation, ex vivo, probed by Western blot analysis of MTR-treated HeLa cells. Extent of MTR-induced repression of H3K18Ac has been quantified by Image J software. (E) Inhibition of H3K18 acetylation in HeLa cells by MTR monitored by confocal microscopy. The scale bar is 10 μm.
There could be a possibility that the reduction in H3K18Ac is an indirect effect through the cytotoxic ability of MTR. In order to ascertain whether the dosage and time course of MTR treatment induce apoptosis in HeLa cells, MTT assays were performed. However, no significant cell death was observed even after 15 h of MTR treatment (Fig. S11D). To reconfirm this observation and additionally monitor if MTR can at all influence cell cycle stages, flow cytometry analysis was carried out. There was no significant change in the stages of cell cycle upon MTR treatment (Fig. S11A).
Literature report shows that SGR, a histone interacting small molecule capable of modulating histone PTMs can affect global gene expression [30]. Since MTR binds to core histones and alters an important epigenetic signature in HeLa cells, we were interested to do a gene expression analysis as a sequel to the ligand treatment. From available literature data [48] we have seen that MTR can alter the global gene expression in sarcoma cell lines (TC32 and TC71). Further, a subset of genes that were differentially expressed also harbored H3K18Ac. These two separate observations prompted us to make a reanalysis of microarray data (with statistical significance) and also compare the H3K18Ac enrichments in the gene sets which were differentially expressed. We wanted to check whether MTR exhibits a basal level activity irrespective of the cell lines. In our reanalysis we observed genome-wide repression of gene expression upon treatment with MTR in TC32 (870 genes were down regulated and 288 genes were up regulated) and TC71 (532 genes were down regulated and 115 genes were up regulated) cell lines. Unsupervised hierarchical clustering (Fig. 6) showed high degree of similarity in differential expression in both the cell lines. We have also generated Biological Analysis Networks (BAN) which shows the key cellular pathways influenced by MTR (Fig. S12). Thus MTR is a regulator of histone H3K18Ac in vitro and ex vivo which could be modulating global gene expression.
Fig. 6.
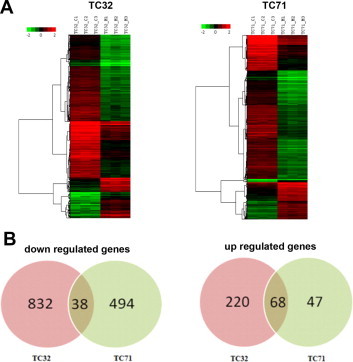
Unsupervised hierarchical clustering of differentially expressed genes (A) using Pearson uncentered algorithm with average distance matrix shows distinct patterns of up and down regulated genes upon treatment in comparison to untreated samples. Venn diagram representation (B) of up and down regulated genes between two cell lines indicates less number of genes that are commonly regulated by MTR.
4. Discussion
Current report demonstrates the physical association of MTR with core histone(s) in the absence of a bivalent metal ion. This interaction occurs even at high ionic strength thereby suggesting the specific nature of the recognition among the histone(s) and MTR and ruling out the possibility of non-specific association of the anionic MTR with positively charged histone(s). Till date the transcription inhibitory potential of MTR has been ascribed to its DNA binding ability [3,4]. Earlier reports by our group had shown that the binding of DNA and chromatin with MTR occurs only in the presence of bivalent metal ions like Mg2+ [20,21,25,42]. The interaction of MTR:M2+ complexes with chromatin was proposed to be via the minor groove of DNA with the histone proteins having a negative impact on the binding interactions between antibiotic:Mg2+ complexes (complex I, 1:1 in terms of MTR:Mg2+ and complex II 2:1 in terms of MTR:Mg2+) and DNA [42]. So, the present report of the association of MTR with histone proteins indicates an additional intracellular mode of action of MTR. It places MTR among the list of DNA binding small molecules exhibiting dual binding mode in the chromatin context [30–33]. It is noteworthy that the association of MTR with histones does not require the presence of bivalent metal ion like Mg2+.
Steady state fluorescence spectroscopy characterizes an association of MTR with core histone components of chromatin. The difference in the trend of fluorescence change of the chromophore in MTR upon association with chromatin, individual histones and core octamer can be ascribed to the altered electronic environment and other parameters such as the local polarity of the histone bound MTR. Literature reports show that there is an enhancement in the fluorescence intensity when MTR binds to DNA in presence of Mg2+ [25] whereas fluorescence intensity decreases upon binding of MTR to bivalent metal ions, like Mg2+, Zn2+, etc. [20,49]. The physical environment in case of DNA, chromatin, free histones and assembled octamer are different. Hence the peak position and the trend in variation of emission intensity of the chromophore in MTR differ in these scenarios.
Since we have observed that binding affinity of MTR follows the order: core histones > naked DNA > DNA/histone complex, it might be proposed that MTR binding to DNA is partially precluded by core histones. MTR binds to chromatin/chromatosome even in absence of Mg2+ and the apparent dissociation constants obtained suggest that the major mode of binding of MTR in chromatin differs in presence and absence of the bivalent metal ion Mg2+ (Table S1).
Absence of any radical change in the CD spectral shape of MTR upon association with histones implies that the binding could originate from surface complementarity followed by favorable non-covalent interactions. It is worth-mentioning that CD spectroscopy is a frequently used tool to examine ligand–macromolecule interaction [42,30,50] and was employed to study the association of daunomycin (minor groove binder) and mitoxantrone (intercalator) with histones [51,52].
The thermodynamic scenario of MTR–histone(s) interactions was obtained from ITC. Negative enthalpy changes might be attributed to the stacking interactions and hydrogen bonding between the potential loci of MTR and its counterparts in histone, both amide backbone and side chains. Additionally, electrostatic interactions of the anionic MTR with positively charged side chains of the histones possibly contribute to the favorable negative enthalpy changes. The negative entropy values might originate from the ordering of the water molecules at the surface of the histones and MTR as a sequel to the association.
The dissociation constant values characterizing histone–MTR association are not same by the two methods, fluorescence and ITC. Such disagreement of the two methodologies is not counter-intuitive and was justified earlier in case of polymer-small ligand association [53]. It has been attributed to higher concentration of the polymer and ligand necessary to obtain measurable enthalpy change.
To throw light on the specificity of MTR–core histone(s) interactions, experiments were designed to check if MTR interacts with the linker histone. Results from both steady state fluorescence spectroscopy and ITC provide direct evidence that MTR has no binding affinity towards linker histone H1. It may also be noted that MTR does not induce gross structural alteration of chromatin by histone H1 eviction as observed through EM and DLS measurements. Thus the hierarchical level of chromatin folding is not adversely affected in presence of MTR.
We have also examined whether MTR, like other dual binders [30,31,46], modulate histone modifications. MTR could selectively alter H3K18Ac but not H3K9Ac/H3K27Ac. Acetylation levels of histone H4 at lysine K5 and K8 were also unaffected in presence of MTR. These observations indicate that probably MTR has a site-specific interaction to histone H3 tail and could be sequestering the key residues which are modified by the HAT enzyme. Thus various sites modified by the same HAT are differentially affected by MTR, indicating probably the mechanism is not through enzyme active site targeting.
H3K18Ac is also altered in presence of MTR in the cellular context. The ligand concentration chosen had no effect on cell survivability as observed through MTT assay and flow cytometry analysis. Ability of MTR to bind DNA and core histones and selectively modulate histone post-translational modification implicates its role in regulating global gene expression. Genome-wide perturbation of gene expression upon treatment with MTR in sarcoma cell lines (TC32 and TC71) observed from reanalysis of microarray data reveals that apart from the genes involved in DNA binding (eg. Zinc finger transcription factors) those involved in regulation of chromatin function including histone modification (Supplementary_Doc2, Table I) were differentially expressed. The alteration of H3K18Ac upon MTR treatment in HeLa cells is possibly a basal activity of the ligand as genes harboring H3K18Ac have been found to be down-regulated in MTR treated other cell lines. Thus, although speculative, the altered gene expression could be following a similar trend in HeLa cells. Earlier studies on chromatin binding molecules capable of altering histone modifications have reported alteration in the gene expression upon ligand treatment [30]. In case of SGR, there is no preference for up-regulated or down-regulated genes. Since MTR represses the expression of H3K18Ac, a transcription activation signature, we speculated a global down-regulation of gene expression upon MTR treatment. In accordance, we indeed find a repression of gene expression upon MTR treatment by reanalyzing the microarray results.
In summary, we have identified the dual binding potential of MTR in the chromatin context and have shown that MTR modulates H3K18Ac status, an epigenetic signature of active chromatin. Its histone binding potential suggests an additional mode of action of MTR. Further, its ability to affect global gene expression indicates a complex mechanism of action of MTR in biological context where DNA and core histones exist as competing binding substrates. This new finding could have a broader biological implication and lends a new dimension to the role of MTR in the cellular context.
Acknowledgements
We acknowledge Srijan Halder of Biophysics and Structural Genomics Division of Saha Institute of Nuclear Physics for helping us with the FACS experiments.
This work was supported by intramural Grants from the Molecular Mechanism of Disease and Drug Action (MMDDA) project (Grant 11-R&D-SIN-5.04) and Biomolecular Assembly, Recognition and Dynamics (BARD) project (Grant 12-R&D-SIN-5.04-0103) from the Department of Atomic Energy (DAE), Government of India. CD acknowledges Ramalingaswami Fellowship. SR acknowledges Ramanujan Fellowship.
The author(s) declare that they have no competing interests.
AB, CD, DD planned experiments, analyzed data and wrote the manuscript. AB, SS performed experiments. KK analyzed microarray data. CD, KJ, SR, DD provided certain facilities mentioned in the paper.
Contributor Information
Chandrima Das, Email: chandrima.das@saha.ac.in.
Dipak Dasgupta, Email: dipak.dasgupta@saha.ac.in.
Appendix A. Supplementary data
Supplementary Fig. S1.
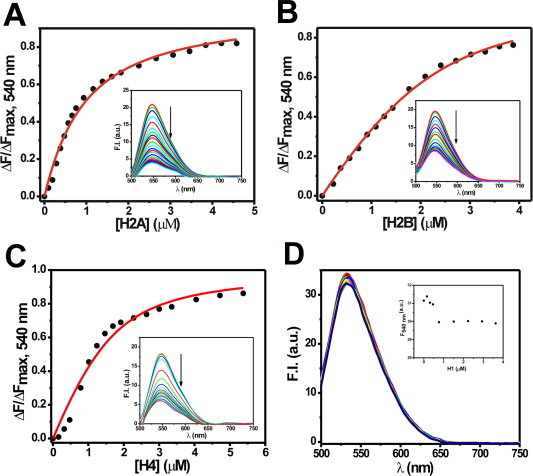
Binding isotherms obtained from fluorimetric titration of MTR with human recombinant histones H2A (A), H2B (B) H4 (C) at 25 °C. The insets show the emission spectra of 2 μM MTR in 10 mM Tris–HCl, pH 7.0 containing 150 mM NaCl in presence of increasing concentration of the histones. λex = 470 nm. (D) Emission spectra of 2 μM MTR in presence of increasing concentrations of linker histone H1. Inset shows the variation of fluorescence intensity of MTR at emission maximum with increasing concentration of H1.
Supplementary Fig. S2.
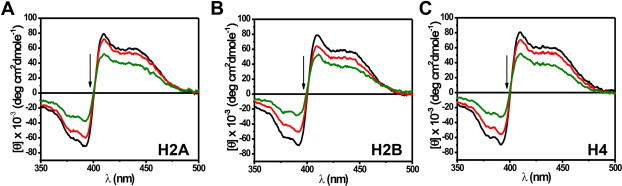
CD spectra of 8 μM MTR in 10 mM Tris–HCl, pH 7.0 containing 150 mM NaCl at 25 °C in absence (black) and in presence of 1 μM (red); 4 μM (green) histones H2A (A), H2B (B) and H4 (C) in the visible range.
Supplementary Fig. S3.
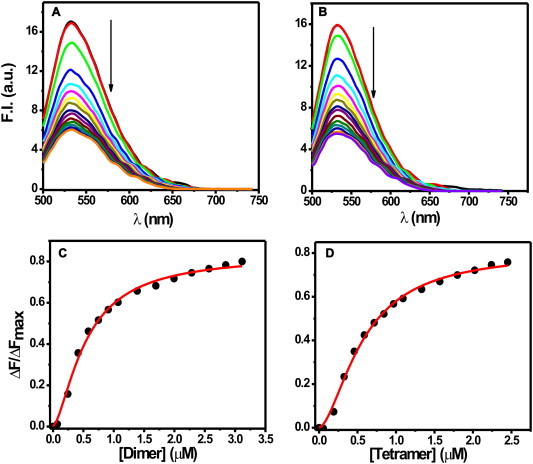
Fluorimetric titration of MTR with histone dimer/tetramer. Emission spectra of 2 μM MTR in 10 mM Tris–HCl, pH 7.0 containing 150 mM NaCl at 25 °C in presence of H2A-H2B dimer (A) and H3–H4 tetramer (B). λex = 470 nm. Panels (C), (D) show the corresponding fitted isotherms obtained by non-linear curve fitting analysis.
Supplementary Fig. S4.
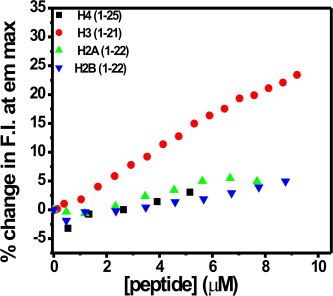
The percentage change in emission intensity at the emission maximum of MTR (540 nm) upon interaction with H2A, H2B, H3 and H4 tail peptides at 25 °C.
Supplementary Fig. S5.
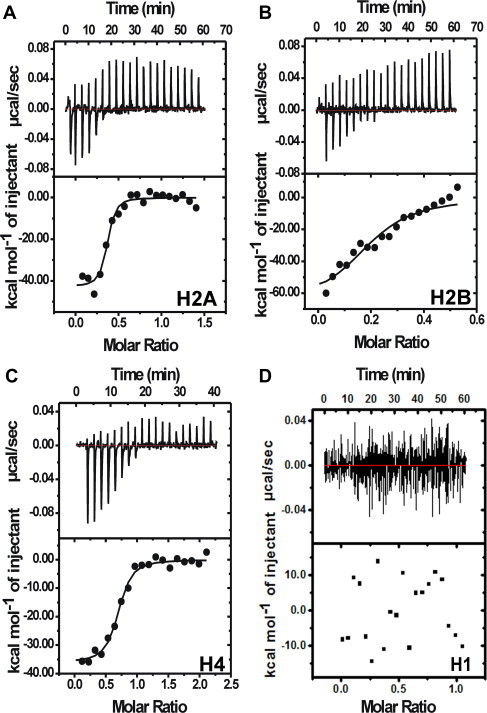
ITC profiles for the interaction of MTR with histones H2A (A), H2B (B), H4 (C) and H1 (D) at 25 °C in 10 mM Tris–HCl, pH 7.0 containing 150 mM NaCl. The lower panels contain the background heat subtracted fitted isotherms. H1 does not interact with MTR, so binding isotherm could not be obtained in (D).
Supplementary Fig. S6.
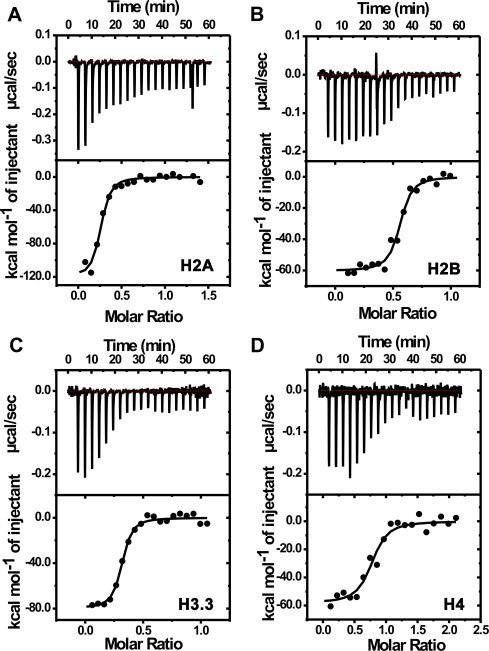
ITC to examine MTR–histone interactions at high salt. Thermograms for the interaction of MTR with histones H2A (A), H2B (B), H3 (C) and H4 (D) at 25 °C in 10 mM Tris–HCl, pH 7.0 containing 1 M NaCl. The lower panels contain the background heat subtracted fitted isotherms.
Supplementary Fig. S7.
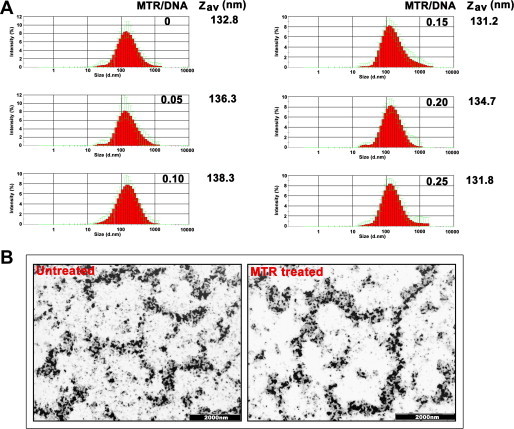
(A) Dynamic Light Scattering measurements to study the effect of MTR on the hydrodynamic diameter (Zav) of chromatin. Intensity statistics of 10 measurements each are plotted for each sample. Error bars indicate standard deviations. Standard deviations of Zav values obtained from three independent sets of experiment are within 5%. (B) Electron Microscopy images of untreated and MTR treated chromatin. The scale bar is 2000 nm. Chromatin was treated with MTR for 3 h at MTR/DNA base ratio of 0.2
Supplementary Fig. S8.
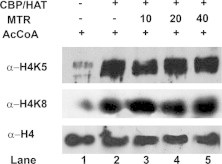
In vitro HAT assay to examine the effect of MTR on H4 acetylation at K5 and K8.
Supplementary Fig. S9.
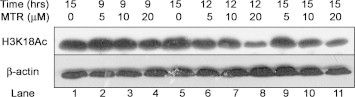
Time dependence and dose dependence Western blot analysis of MTR-treated HeLa cells. β-Actin has been used as loading control.
Supplementary Fig. S10.
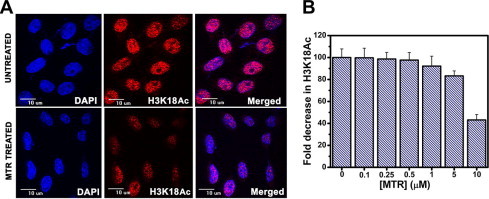
MTR-induced repression of H3K18 acetylation in HeLa cells as monitored by confocal microscopy. (A) Representative population of untreated and 10 μM MTR-treated HeLa cells for 9 h. The scale bar is 10 μM. (B) Quantification of the extent of repression of H3K18 acetylation upon increasing concentrations of MTR treatment (0.1–10 μM), using Image J software. Statistical analyses were performed over 300 cells in each case.
Supplementary Fig. S11.
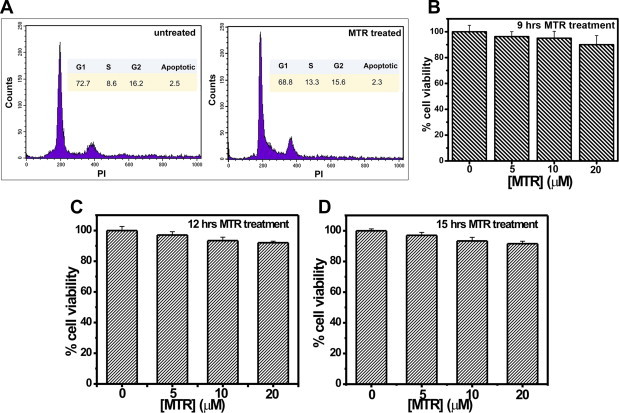
(A) Flow cytometry analysis of MTR-treated and untreated HeLa cells ([MTR] = 10 μM, treatment duration = 9 h). (B–D) MTT assay to assess HeLa cell viability upon 10 μM MTR treatment for 9, 12 and 15 h respectively.
Supplementary Fig. S12.
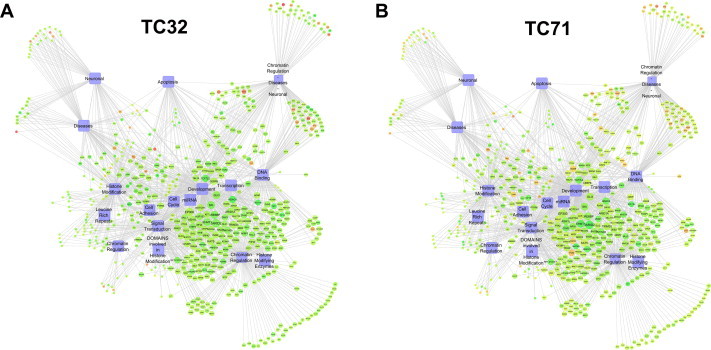
BAN (Biological Analysis Network) of differentially expressed genes and biological processes. Network of differentially expressed genes and biological processes shows high degree of down regulation in (A) TC32 cell line treated with MTR, as compared to (B) TC71. Biological processes are colored in (violet blocks) and genes are colored (green for down regulated and orange for up regulated) by their fold change upon treatment compared with untreated cell line. Among them a good number of differentially expressed genes are associated with chromatin regulation, DNA binding, histone modification, cell cycle, development, diseases, miRNA, neuronal, signal transduction and transcription.
Reanalyzed microarray data showing the significant biology.
List of differentially expressed genes depicting statistical and biological significance in TC32 and TC71 Ewing Sarcoma cell lines.
Differentially expressed genes targeting miRNA.
List of differentially expressed genes depicting statistical and biological significance.
Dissociation constants of interaction of MTR and MTR:Mg2+ (complex 1) with chromatin/ chromatosome at 25 °C as obtained from Spectrofluorimetric Analysis.
Thermodynamic parameters of MTR–histone interactions as obtained by ITC at 25 °C in 10 mM Tris–HCl, pH 7.0 containing 1 M NaCl.
References
- 1.Lombo F., Menendez N., Salas J.A., Mendez C. The aureolic acid family of antitumor compounds: structure, mode of action, biosynthesis, and novel derivatives. Appl. Microbiol. Biotechnol. 2006;73:1–14. doi: 10.1007/s00253-006-0511-6. [DOI] [PubMed] [Google Scholar]
- 2.Torrance C.J., Agrawal V., Vogelstein B., Kinzler K.W. Use of isogenic human cancer cells for high-throughput screening and drug discovery. Nat. Biotechnol. 2001;19:940–945. doi: 10.1038/nbt1001-940. [DOI] [PubMed] [Google Scholar]
- 3.Sleiman S.F., Langley B.C., Basso M., Berlin J., Xia L., Payappilly J.B., Kharel M.K., Guo H., Marsh J.L., Thompson L.M., Mahishi L., Ahuja P., Maclellan W.R., Geschwind D.H., Coppola G., Rohr J., Ratan R.R. Mithramycin is a gene-selective Sp1 inhibitor that identifies a biological intersection between cancer and neurodegeneration. J. Neurosci. 2011;31:6858–6870. doi: 10.1523/JNEUROSCI.0710-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sleiman S.F., Berlin J., Basso M., Karuppagounder S., Rohr J.R., Ratan R.R. Histone deacetylase inhibitors and mithramycin A impact a similar neuroprotective pathway at a crossroad between cancer and neurodegeneration. Pharmaceuticals. 2011;4:1183–1195. doi: 10.3390/ph4081183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chatterjee S., Zaman K., Ryu H., Conforto A., Ratan R.R. Sequence-selective DNA binding drugs mithramycin A and chromomycin A3 are potent inhibitors of neuronal apoptosis induced by oxidative stress and DNA damage in cortical neurons. Ann. Neurol. 2001;49:345–354. [PubMed] [Google Scholar]
- 6.Ferrante R.J., Ryu H., Kubilus J.K., D’mello S., Sugars K.L., Lee J., Lu P., Smith K., Browne S., Beal M.F., Kristal B.S., Stavrovskaya I.G., Hewett S., Rubinsztein D.C., Langley B., Ratan R.R. Chemotherapy for the brain: the antitumor antibiotic mithramycin prolongs survival in a mouse model of Huntington’s disease. J. Neurosci. 2004;24:10335–10342. doi: 10.1523/JNEUROSCI.2599-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qiu Z., Norflus F., Singh B., Swindell M.K., Buzescu R., Bejarano M., Chopra R., Zucker B., Benn C.L., Dirocco D.P. Sp1 is up-regulated in cellular and transgenic models of Huntington disease, and its reduction is neuroprotective. J. Biol. Chem. 2006;281:16672–16680. doi: 10.1074/jbc.M511648200. [DOI] [PubMed] [Google Scholar]
- 8.Ryu H., Lee J., Hagerty S.W., Soh B.Y., Mcalpin S.E., Cormier K.A., Smith K.M., Ferrante R.J. ESET/SETDB1 gene expression and histone H3 (K9) trimethylation in Huntington’s disease. Proc. Natl. Acad. Sci. 2006;103:19176–19181. doi: 10.1073/pnas.0606373103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Voisine C., Varma H., Walker N., Bates E.A., Stockwell B.R., Hart A.C. Identification of potential therapeutic drugs for Huntington’s disease using Caenorhabditis elegans. PLoS One. 2007;2:e504. doi: 10.1371/journal.pone.0000504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stack E.C., Del Signore S.J., Luthi-Carter R., Soh B.Y., Goldstein D.R., Matson S., Goodrich S., Markey A.L., Cormier K., Hagerty S.W., Smith K., Ryu H., Ferrante R.J. Modulation of nucleosome dynamics in Huntington’s disease. Hum. Mol. Genet. 2007;16:1164–1175. doi: 10.1093/hmg/ddm064. [DOI] [PubMed] [Google Scholar]
- 11.Gaspar N., Di Giannatale A., Geoerger B., Redini F., Corradini N., Enz-Werle N., Tirode F., Marec-Berard P., Gentet J.C., Laurence V., Piperno-Neumann S., Oberlin O., Brugieres L. Bone sarcomas: from biology to targeted therapies. Sarcoma. 2012;2012:301975. doi: 10.1155/2012/301975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sampi K., Hozumi M., Kumai R., Honma Y., Sakurai M. Differentiation of blasts from patients in myeloid crisis of chronic myelogenous leukemia by in-vivo and in-vitro plicamycin treatment. Leuk. Res. 1987;11:1089–1092. doi: 10.1016/0145-2126(87)90161-5. [DOI] [PubMed] [Google Scholar]
- 13.Ueki K., Tsuchida A., Murakami H., Karasawa M., Kobayashi N., Omine M., Naruse T. Myeloid crisis of chronic myelogenous leukemia showing dramatic response to mithramycin and hydroxyurea combination. Rinsho Ketsueki. 1989;30:1840–1842. [PubMed] [Google Scholar]
- 14.Brown J.H., Kennedy B.J. Mithramycin in the treatment of disseminated testicular neoplasms. N. Engl. J. Med. 1965;272:111–118. doi: 10.1056/NEJM196501212720301. [DOI] [PubMed] [Google Scholar]
- 15.Kennedy B.J., Griffen W.O., Jr., Lober P. Specific effect of mithramycin on embryonal carcinoma of the testis. Cancer. 1965;18:1631–1636. doi: 10.1002/1097-0142(196512)18:12<1631::aid-cncr2820181216>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 16.Kennedy B.J., Torkelson J.L. Long-term follow-up of stage III testicular carcinoma treated with mithramycin (plicamycin) Med. Pediatr. Oncol. 1995;24:327–328. doi: 10.1002/mpo.2950240511. [DOI] [PubMed] [Google Scholar]
- 17.Jia Z., Gao Y., Wang L., Li Q., Zhang J., Le X., Wei D., Yao J.C., Chang D.Z., Huang S., Xie K. Combined treatment of pancreatic cancer with mithramycin A and tolfenamic acid promotes Sp1 degradation and synergistic antitumor activity. Cancer Res. 2010;70:1111–1119. doi: 10.1158/0008-5472.CAN-09-3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yuan P., Wang L., Wei D., Zhang J., Jia Z., Li Q., Le X., Wang H., Yao J., Xie K. Therapeutic inhibition of Sp1 expression in growing tumors by mithramycin a correlates directly with potent antiangiogenic effects on human pancreatic cancer. Cancer. 2007;110:2682–2690. doi: 10.1002/cncr.23092. [DOI] [PubMed] [Google Scholar]
- 19.Malek A., Nunez L.E., Magistri M., Brambilla L., Jovic S., Carbone G.M., Moris F., Catapano C.V. Modulation of the activity of Sp transcription factors by mithramycin analogues as a new strategy for treatment of metastatic prostate cancer. PLoS One. 2012;7:e35130. doi: 10.1371/journal.pone.0035130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aich P., Dasgupta D. Role of Mg++ in the mithramycin–DNA interaction: evidence for two types of mithramycin–Mg++ complex. Biochem. Biophys. Res. Commun. 1990;173:689–696. doi: 10.1016/s0006-291x(05)80090-7. [DOI] [PubMed] [Google Scholar]
- 21.Mir M.A., Majee S., Das S., Dasgupta D. Association of chromatin with anticancer antibiotics, mithramycin and chromomycin A3. Bioorg. Med. Chem. 2003;11:2791–2801. doi: 10.1016/s0968-0896(03)00211-6. [DOI] [PubMed] [Google Scholar]
- 22.Huang H., Li D., Cowan J. Biostructural chemistry of magnesium. Regulation of mithramycin–DNA interactions by Mg2+ coordination. Biochimie. 1995;77:729–738. doi: 10.1016/0300-9084(96)88190-9. [DOI] [PubMed] [Google Scholar]
- 23.Chakraborty H., Devi P.G., Sarkar M., Dasgupta D. Multiple functions of generic drugs: future perspectives of aureolic acid group of anti-cancer antibiotics and non-steroidal anti-inflammatory drugs. Mini Rev. Med. Chem. 2008;8:331–349. doi: 10.2174/138955708783955944. [DOI] [PubMed] [Google Scholar]
- 24.Majee S., Sen R., Guha S., Bhattacharyya D., Dasgupta D. Differential interactions of the Mg2+ complexes of chromomycin A3 and mithramycin with poly (dG–dC)·poly (dC–dG) and poly (dG)·poly (dC) Biochemistry. 1997;36:2291–2299. doi: 10.1021/bi9613281. [DOI] [PubMed] [Google Scholar]
- 25.Aich P., Dasgupta D. Role of magnesium ion in mithramycin–DNA interaction: binding of mithramycin–Mg2+ complexes with DNA. Biochemistry. 1995;34:1376–1385. doi: 10.1021/bi00004a032. [DOI] [PubMed] [Google Scholar]
- 26.Snyder R.C., Ray R., Blume S., Miller D.M. Mithramycin blocks transcriptional initiation of the c-myc P1 and P2 promoters. Biochemistry. 1991;30:4290–4297. doi: 10.1021/bi00231a027. [DOI] [PubMed] [Google Scholar]
- 27.Hardenbol P., Van Dyke M.W. In vitro inhibition of c-myc transcription by mithramycin. Biochem. Biophys. Res. Commun. 1992;185:553–558. doi: 10.1016/0006-291x(92)91660-i. [DOI] [PubMed] [Google Scholar]
- 28.Campbell V.W., Davin D., Thomas S., Jones D., Roesel J., Tran-Patterson R., Mayfield C.A., Rodu B., Miller D.M., Hiramoto R.A. The G-C specific DNA binding drug, mithramycin, selectively inhibits transcription of the C-MYC and C-HA-RAS genes in regenerating liver. Am. J. Med. Sci. 1994;307:167–172. doi: 10.1097/00000441-199403000-00002. [DOI] [PubMed] [Google Scholar]
- 29.Vigneswaran N., Thayaparan J., Knops J., Trent J., Potaman V., Miller D.M., Zacharias W. Intra- and intermolecular triplex DNA formation in the murine c-myb proto-oncogene promoter are inhibited by mithramycin. Biol. Chem. 2001;382:329–342. doi: 10.1515/BC.2001.040. [DOI] [PubMed] [Google Scholar]
- 30.Selvi B.R., Pradhan S.K., Shandilya J., Das C., Sailaja B.S., Shankar G.N., Gadad S.S., Reddy A., Dasgupta D., Kundu T.K. Sanguinarine interacts with chromatin, modulates epigenetic modifications, and transcription in the context of chromatin. Chem. Biol. 2009;16:203–216. doi: 10.1016/j.chembiol.2008.12.006. [DOI] [PubMed] [Google Scholar]
- 31.Banerjee A., Majumder P., Sanyal S., Singh J., Jana K., Das C., Dasgupta D. The DNA intercalators ethidium bromide and propidium iodide also bind to core histones. FEBS Open Bio. 2014;4:251–259. doi: 10.1016/j.fob.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ghosh S., Majumder P., Pradhan S.K., Dasgupta D. Mechanism of interaction of small transcription inhibitors with DNA in the context of chromatin and telomere. Biochim. Biophys. Acta. 2010;1799:795–809. doi: 10.1016/j.bbagrm.2010.06.008. [DOI] [PubMed] [Google Scholar]
- 33.Dasgupta D., Majumder P., Banerjee A. A revisit of the mode of interaction of small transcription inhibitor with genomic DNA. J. Biosci. 2012;37:475–481. doi: 10.1007/s12038-012-9211-9. [DOI] [PubMed] [Google Scholar]
- 34.Daujat S., Bauer U.M., Shah V., Turner B., Berger S., Kouzarides T. Crosstalk between CARM1 methylation and CBP acetylation on histone H3. Curr. Biol. 2002;12:2090–2097. doi: 10.1016/s0960-9822(02)01387-8. [DOI] [PubMed] [Google Scholar]
- 35.Lahiri S., Devi P.G., Majumder P., Das S., Dasgupta D. Self-association of the anionic form of the DNA-binding anticancer drug mithramycin. J. Phys. Chem. B. 2008;112:3251–3258. doi: 10.1021/jp710503g. [DOI] [PubMed] [Google Scholar]
- 36.Blobel G., Potter V.R. Nuclei from rat liver: isolation method that combines purity with high yield. Science. 1966;154:1662–1665. doi: 10.1126/science.154.3757.1662. [DOI] [PubMed] [Google Scholar]
- 37.Majumder P., Dasgupta D. Effect of DNA groove binder distamycin A upon chromatin structure. PLoS One. 2011;6:e26486. doi: 10.1371/journal.pone.0026486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peterson C.L., Hansen J.C. Chicken erythrocyte histone octamer preparation. CSH Protoc. 2008:2008. doi: 10.1101/pdb.prot5112. (pdb prot5112) [DOI] [PubMed] [Google Scholar]
- 39.Balasubramanyam K., Altaf M., Varier R.A., Swaminathan V., Ravindran A., Sadhale P.P., Kundu T.K. Polyisoprenylated benzophenone, garcinol, a natural histone acetyltransferase inhibitor, represses chromatin transcription and alters global gene expression. J. Biol. Chem. 2004;279:33716–33726. doi: 10.1074/jbc.M402839200. [DOI] [PubMed] [Google Scholar]
- 40.Sinha M., Ghose J., Bhattarcharyya N.P. Micro RNA -214, -150, -146a and-125b target Huntingtin gene. RNA Biol. 2011;8:1005–1021. doi: 10.4161/rna.8.6.16035. [DOI] [PubMed] [Google Scholar]
- 41.Gong W., Russell M., Suzuki K., Riabowol K. Subcellular targeting of p33ING1b by phosphorylation-dependent 14-3-3 binding regulates p21WAF1 expression. Mol. Cell. Biol. 2006;26:2947–2954. doi: 10.1128/MCB.26.8.2947-2954.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mir M.A., Dasgupta D. Interaction of antitumor drug, mithramycin, with chromatin. Biochem. Biophys. Res. Commun. 2001;280:68–74. doi: 10.1006/bbrc.2000.4075. [DOI] [PubMed] [Google Scholar]
- 43.Chakrabarti S., Roy P., Dasgupta D. Interaction of the antitumor antibiotic chromomycin A3 with glutathione, a sulfhydryl agent, and the effect upon its DNA binding properties. Biochem. Pharmacol. 1998;56:1471–1479. doi: 10.1016/s0006-2952(98)00249-4. [DOI] [PubMed] [Google Scholar]
- 44.Wolffe, A. (1998) Chromatin: structure and function. Access Online via Elsevier.
- 45.Feng H.P., Scherl D.S., Widom J. Lifetime of the histone octamer studied by continuous-flow quasielastic light scattering: test of a model for nucleosome transcription. Biochemistry. 1993;32:7824–7831. doi: 10.1021/bi00081a030. [DOI] [PubMed] [Google Scholar]
- 46.Khan S.N., Yennamalli R., Subbarao N., Khan A.U. Mitoxantrone induced impediment of histone acetylation and structural flexibility of the protein. Cell Biochem. Biophys. 2011;60:209–218. doi: 10.1007/s12013-010-9141-9. [DOI] [PubMed] [Google Scholar]
- 47.Jin Q., Yu L.R., Wang L., Zhang Z., Kasper L.H., Lee J.E., Wang C., Brindle P.K., Dent S.Y., Ge K. Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. EMBO J. 2011;30:249–262. doi: 10.1038/emboj.2010.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grohar P.J., Woldemichael G.M., Griffin L.B., Mendoza A., Chen Q.R., Yeung C., Currier D.G., Davis S., Khanna C., Khan J., Mcmahon J.B., Helman L.J. Identification of an inhibitor of the EWS-FLI1 oncogenic transcription factor by high-throughput screening. J. Natl Cancer Inst. 2011;103:962–978. doi: 10.1093/jnci/djr156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Devi P.G., Pal S., Banerjee R., Dasgupta D. Association of antitumor antibiotics, mithramycin and chromomycin, with Zn(II) J. Inorg. Biochem. 2007;101:127–137. doi: 10.1016/j.jinorgbio.2006.08.018. [DOI] [PubMed] [Google Scholar]
- 50.Majumder P., Banerjee A., Shandilya J., Senapati P., Chatterjee S., Kundu T.K., Dasgupta D. Minor groove binder distamycin remodels chromatin but inhibits transcription. PLoS One. 2013;8:e57693. doi: 10.1371/journal.pone.0057693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rabbani A., Finn R.M., Thambirajah A.A., Ausio J. Binding of antitumor antibiotic daunomycin to histones in chromatin and in solution. Biochemistry. 2004;43:16497–16504. doi: 10.1021/bi048524p. [DOI] [PubMed] [Google Scholar]
- 52.Hajihassan Z., Rabbani-Chadegani A. Interaction of mitoxantrone, as an anticancer drug, with chromatin proteins, core histones and H1, in solution. Int. J. Biol. Macromol. 2011;48:87–92. doi: 10.1016/j.ijbiomac.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 53.Haq I., Ladbury J.E., Chowdhry B.Z., Jenkins T.C., Chaires J.B. Specific binding of hoechst 33258 to the d(CGCAAATTTGCG)2 duplex: calorimetric and spectroscopic studies. J. Mol. Biol. 1997;271:244–257. doi: 10.1006/jmbi.1997.1170. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Reanalyzed microarray data showing the significant biology.
List of differentially expressed genes depicting statistical and biological significance in TC32 and TC71 Ewing Sarcoma cell lines.
Differentially expressed genes targeting miRNA.
List of differentially expressed genes depicting statistical and biological significance.
Dissociation constants of interaction of MTR and MTR:Mg2+ (complex 1) with chromatin/ chromatosome at 25 °C as obtained from Spectrofluorimetric Analysis.
Thermodynamic parameters of MTR–histone interactions as obtained by ITC at 25 °C in 10 mM Tris–HCl, pH 7.0 containing 1 M NaCl.