

Summary
Enantiomers of the tobacco-specific carcinogen metabolite NNAL, found in the urine of virtually all tobacco users as well as people exposed to secondhand tobacco smoke, are powerful lung carcinogens in rats and form pulmonary DNA adducts throughout the treatment period.
Abstract
4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) is metabolized to enantiomers of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL), found in the urine of virtually all people exposed to tobacco products. We assessed the carcinogenicity in male F-344 rats of (R)-NNAL (5 ppm in drinking water), (S)-NNAL (5 ppm), NNK (5 ppm) and racemic NNAL (10 ppm) and analyzed DNA adduct formation in lung and pancreas of these rats after 10, 30, 50 and 70 weeks of treatment. All test compounds induced a high incidence of lung tumors, both adenomas and carcinomas. NNK and racemic NNAL were most potent; (R)-NNAL and (S)-NNAL had equivalent activity. Metastasis was observed from primary pulmonary carcinomas to the pancreas, particularly in the racemic NNAL group. DNA adducts analyzed were O 2-[4-(3-pyridyl)-4-oxobut-1-yl]thymidine (O 2-POB-dThd), 7-[4-(3-pyridyl)-4-oxobut-1-yl]guanine(7-POB-Gua),O 6-[4-(3-pyridyl)-4-oxobut-1-yl]deoxyguanosine(O 6-POB-dGuo),the 4-(3-pyridyl)-4-hydroxybut-1-yl(PHB)adductsO 2-PHB-dThd and 7-PHB-Gua, O 6-methylguanine (O 6-Me-Gua) and 4-hydroxy-1-(3-pyridyl)-1-butanone (HPB)-releasing adducts. Adduct levels significantly decreased with time in the lungs of rats treated with NNK. Pulmonary POB-DNA adducts and O 6-Me-Gua were similar in rats treated with NNK and (S)-NNAL; both were significantly greater than in the (R)-NNAL rats. In contrast, pulmonary PHB-DNA adduct levels were greatest in the rats treated with (R)-NNAL. Total pulmonary DNA adduct levels were similar in (S)-NNAL and (R)-NNAL rats. Similar trends were observed for DNA adducts in the pancreas, but adduct levels were significantly lower than in the lung. The results of this study clearly demonstrate the potent pulmonary carcinogenicity of both enantiomers of NNAL in rats and provide important new information regarding DNA damage by these compounds in lung and pancreas.
Introduction
Lung and pancreatic cancer are two of the most deadly common cancers worldwide, with average survival times measured in months. There were 1589800 deaths from lung cancer and 330372 deaths from pancreatic cancer worldwide in 2012 (1). Cigarette smoking is estimated to cause ~71% of global lung cancer deaths and ~30% of pancreatic cancer mortality (2,3). As addictive and carcinogenic tobacco products are effectively marketed worldwide by multinational companies with seemingly unlimited resources, it is not likely that this major cause of fatal cancer will disappear or even diminish significantly in the near future. There are currently ~1.25 billion cigarette smokers in the world (4). It is crucial to understand mechanisms of tobacco carcinogenesis in order to develop effective approaches to cancer prevention in smokers.
Among the multiple carcinogens in cigarette smoke, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK, Figure 1A) is widely regarded as one of the most important due to its potent carcinogenicity in laboratory animals and presence in all tobacco products, sometimes in relatively high amounts. NNK readily induces adenoma and adenocarcinoma of the lung in rats, mice, hamsters and ferrets; pancreatic tumors as well as tumors of the nasal mucosa and liver have also been observed in rats (5), and pancreatic tumors have been reported in offspring of Syrian golden hamsters treated with NNK or NNK plus ethanol (6,7). NNK and the related carcinogenic nitrosamine N′-nitrosonornicotine (NNN), which always occur together in tobacco products, are considered ‘carcinogenic to humans’ by the International Agency for Research on Cancer (8). NNK is converted in varying amounts to its metabolite 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL, Figure 1A) in virtually all biological systems, both in vitro and in vivo (5). NNAL and NNK have similar carcinogenic activity in rats, while NNK is somewhat more potent than NNAL in A/J mice (5). Virtually all people who use tobacco products, as well as non-users exposed to secondhand tobacco smoke, have NNAL and its glucuronide metabolites in their urine (9–12). Three prospective epidemiologic studies have demonstrated a relationship to lung cancer of urinary levels of NNAL and its glucuronides, even after correction for number of cigarettes smoked per day and number of years of smoking (13–15).
Fig. 1.

(A) Overview of NNK and NNAL metabolism leading to DNA adduct formation. (B) Structures of the DNA adducts quantified in this study.
NNAL, with a chiral center at its 1-position, exists in enantiomeric forms. Several studies have investigated the chirality of NNAL formed from NNK in vitro in rodent and human subcellular fractions in reactions which are catalyzed by aldo-keto reductases, carbonyl reductases, 11-β-hydroxysteroid dehydrogenase and other enzymes (16–19). (S)-NNAL was the major enantiomer formed in most experiments with rat and mouse liver and lung microsomes and cytosol (17,18). The main enantiomer produced by human liver microsomes depended on the study, while cytosol converted NNK predominantly to (S)-NNAL (17,18). Human lung microsomes and normal human bronchial epithelial cells gave mainly (R)-NNAL while (S)-NNAL was the main product in cytosol (17,20). Human pancreatic microsomes and cytosol incubated with NNK gave varying amounts of the two NNAL enantiomers (19). Both enantiomers were present in similar quantities in the urine of smokers (21), and ratios of (S)-NNAL:(R)-NNAL and (S)-NNAL-glucuronide:(R)-NNAL-glucuronide were significantly higher 7 days after cessation of tobacco use than at baseline in both smokeless tobacco users and smokers, indicating selective retention of (S)-NNAL in the body (22). In view of these diverse results and the paucity of data on the carcinogenicity of NNAL enantiomers (23), it is difficult to evaluate the relative importance of (S)- and (R)-NNAL in carcinogenesis by NNK.
Both NNK and NNAL require metabolic activation to pro-mutagenic DNA adducts to exert their carcinogenic effects (5). These reactions, which are illustrated in Figure 1A, are catalyzed by cytochrome P450 enzymes. NNK undergoes α-hydroxylation to produce the unstable intermediates 3 and 4, which spontaneously yield the DNA alkylating agents 7 and 9, resulting in the formation of pyridyloxobutyl (POB)-DNA adducts and methyl-DNA adducts. The POB-DNA adducts can be converted to 4-hydroxy-1-(3-pyridyl)-1-butanone (HPB, 12) by acid hydrolysis. Similarly, NNAL is metabolized by α-hydroxylation to intermediates 5 and 6, which ultimately produce methyl-DNA adducts and pyridylhydroxybutyl (PHB)-DNA adducts via intermediates 9 and 11 (5). The structures of the DNA adducts investigated in this study are shown in Figure 1B.
In the study reported here, we assessed the carcinogenicity of the NNAL enantiomers and NNK, administered in the drinking water to male F-344 rats. Subgroups of rats were euthanized at 10, 30, 50 and 70 weeks of treatment for investigation of DNA adduct formation in the lung and pancreas. Previous studies of NNAL enantiomer–DNA adduct formation in rats have involved treatment for a maximum of only 20 weeks (24–26). Furthermore, there are relatively few studies in the literature that have investigated DNA adduct formation from any carcinogen during chronic treatment for periods >1 month (27–37). The results of this study therefore have the potential to determine the relative importance of the NNAL enantiomers in carcinogenesis by NNK, and to clarify the role of specific DNA adducts in lung and pancreatic carcinogenesis by these compounds.
Materials and methods
Test compounds
NNK was purchased from Toronto Research Chemicals. Racemic NNAL, (S)-NNAL and (R)-NNAL were prepared essentially as described and characterized by 1H-NMR and MS (38,39). Chemical purities as assessed by HPLC with UV detection were (S)-NNAL, 98.2%; (R)-NNAL, 99.1% and racemic NNAL, 99%. Enantiomeric purities determined as described previously (21) were (S)-NNAL, 99% and (R)-NNAL, 98% (Note: the assignments of (S)-NNAL and (R)-NNAL in reference [38] are reversed; all descriptions of these enantiomers in the current manuscript use the correct assignments).
Rat carcinogenicity and DNA binding study
This study was approved by the University of Minnesota Institutional Animal Care and Use Committee. Male F-344 rats, age 6 weeks, were obtained from Charles River Laboratories (Kingston, NY), housed 2 per cage with Harlan-irradiated corncob bedding (Harlan, Indianapolis, IN), and allowed to acclimate to the Research Animal Resources Facility, University of Minnesota, for 1 week. The rats were maintained under standard conditions (20–24°C, 29–32% relative humidity and 14/10 light/dark cycle). They were fed Harlan Teklad 7022 (NIH-07) diet. The rats were divided into groups as follows: NNK, (60 rats); (S)-NNAL, 60 rats; (R)-NNAL, 60 rats; racemic NNAL (positive control), 15 rats and negative control, 60 rats. Test compounds were added to the drinking water as follows: NNK, (S)-NNAL and (R)-NNAL, 5 ppm; racemic NNAL, 10 ppm; negative control, no addition. Aqueous stock solutions of the test compounds were prepared weekly and stored at 4°C. Stock solutions were analyzed using an Agilent 1100 capillary flow HPLC with a diode array UV detector set at 254nm (Agilent Technologies, Palo Alto, CA). A 4.6mm × 25cm Luna 5 μm C18 column (Phenomenex, Torrance, CA) was used with a gradient from 5 to 40% CH3OH in H2O over the course of 35min at a flow rate of 10 μl/min. Based on the concentration of the stock solutions, the appropriate dilutions were performed to prepare the drinking water solutions, and the concentrations of the drinking water solutions were confirmed by HPLC. Drinking water was also analyzed after administration to the rats. The average concentrations measured for the four solutions over the course of the study were 5±1 ppm for (S)-NNAL, (R)-NNAL and NNK and 10±2 ppm for racemic NNAL. Drinking water was changed three times weekly and consumption was measured. Rats were inspected daily and weighed monthly. Doses were calculated from the amount of water consumed in each rat cage and the average weights of the rats in that cage.
Nine rats each from the NNK, (S)-NNAL, (R)-NNAL and control groups were humanely euthanized using CO2 from a tank at 10, 30, 50 and 70 weeks following the beginning of the experiment. Tissues from these rats were excised for DNA adduct analyses. The remaining rats were followed closely throughout and humanely euthanized when moribund. A total of 22 rats from the control and the (S)-NNAL groups, 24 rats from the (R)-NNAL and NNK groups and 15 from the racemic-NNAL group were humanely euthanized at the end of the study (90 weeks) and necropsied. A summary of the total number of animals used in the study is provided in Supplementary Table S1, available at Carcinogenesis Online.
DNA was isolated from lung and pancreas harvested at the various time points from the NNK, (S)-NNAL, (R)-NNAL and control groups. DNA isolation was performed following the modified Puregene DNA isolation protocol (Qiagen) as reported (24,26). Lung and pancreas DNA were isolated from three rats at each time point. O 6-Me-Gua, POB adducts, PHB adducts and HPB-releasing adducts were analyzed by isotope dilution liquid chromatography–electrospray ionization–tandem mass spectrometry (LC-ESI-MS/MS) methods as reported previously (26,37,40,41). Further details of the protocols and the LC-ESI-MS/MS analyses are summarized in the Supplementary Material, available at Carcinogenesis Online.
Necropsy and histopathology
A complete necropsy was performed on all animals with close attention paid to the lungs, pancreas and nasal cavity since these are known target organs for NNK and NNAL. All tumors, major organs and gross lesions were fixed in 10% formalin. The head was decalcified using a commercially prepared decalcifying solution consisting of dilute HCl and ethylenediamine tetraacetic acid (Newcomer Supply, Middleton, WI). Fixed tissue specimens were processed into paraffin blocks using standard histology techniques, sectioned to 4 µm thickness and stained with hematoxylin and eosin. Histology slides were evaluated using light microscopy by ACVP-board certified pathologists (CSJ, with review by RCK and MGO’S).
Prior to embedding in paraffin, all fixed lungs were evaluated grossly and given a lung lesion score on a 1–4 scale based on the number and size of visible tumors. Histological sections of the nasal cavity, pancreas, liver, kidney and lung (including representative tumors involving lung and thoracic cavity, where present) were evaluated from each animal.
Statistical analyses
Tumor incidence was analyzed using the chi-square test. Gross lung lesion scores were analyzed using the chi-square test for ordered contingency tables (two-sided P value). Statistical analysis of adduct levels was performed using Stata software (StataIC 11, College Station, TX). Comparison of adduct levels generated by NNK, (S)-NNAL and (R)-NNAL in the lung and the pancreas were performed using analysis of variance (ANOVA) with Bonferroni correction for multiple comparisons. This includes comparisons of adducts generated at each time point by each nitrosamine/enantiomer as well as overall adduct levels over all time points in the pancreas or lung. P values <0.05 were considered statistically significant. When comparing levels of adducts formed in the lung with levels formed in the pancreas, a two sample t-test with equal variances was performed and P values of <0.05 were considered statistically significant (two-sided P value).
Results
On the basis of measured water consumption, average calculated total doses of NNK, (S)-NNAL and (R)-NNAL at 10, 30, 50, 70 and 90 weeks were as follows in mg (mg/kg body weight): 7 (19), 21 (44), 33 (68), 47 (97) and 61 (131), respectively. The average calculated dose of racemic NNAL was 114mg (261mg/kg body weight) at 90 weeks when the study ended. Further details are presented in Supplementary Table S2, available at Carcinogenesis Online. Weight curves are presented in Supplementary Figure S1, available at Carcinogenesis Online.
The major tumors encountered involved the lungs and pancreas; only one nasal adenocarcinoma was observed (racemic NNAL group). Typical F344 rat strain-related tumors, for example large granular lymphoma, interstitial cell tumors and mesothelioma were also observed. Lung and pancreatic lesion incidence in the rats not euthanized for the DNA adduct studies is summarized in Table IA. One-hundred and seven of these 111 rats came to necropsy; the remaining 4 were lost to cannibalism. NNK, racemic NNAL and both NNAL enantiomers were significantly carcinogenic to the lung (P < 0.0001 compared with controls). Nearly all rats treated with these compounds had lung tumors, but none were observed in control animals. Representative gross pathology images of lung tumors and photomicrographs of tumors involving lung and pancreas are shown in Figure 2 and the types of tumors are summarized in Table IA. Based on gross lung lesion scores (Table IB), NNK was significantly more potent than either (S)-NNAL or (R)-NNAL (P < 0.001), and there was no significant difference between (S)-NNAL and (R)-NNAL. Based on carcinoma incidence (Table IA), NNK was more carcinogenic than (R)-NNAL (P = 0.0015), but the differences between NNK and (S)-NNAL, and between (R)-NNAL and (S)-NNAL, were not significant (P = 0.08). Racemic NNAL (the positive control compound administered at twice the dose of the individual NNAL enantiomers or NNK) was significantly more potent than both (S)-NNAL and (R)-NNAL (P = 0.001) with respect to carcinomas, and all rats in this group had pulmonary adenomas and carcinomas.
Table I.
Lung and pancreatic lesions in male F-344 rats given (S)-NNAL, (R)-NNAL, NNK or racemic NNAL in their drinking water
| Control | (R)-NNAL, 5 ppm | (S)-NNAL, 5 ppm | NNK, 5 ppm | Racemic NNAL, 10 ppm | ||||
|---|---|---|---|---|---|---|---|---|
| A. Tumor incidence | ||||||||
| No. of rats necropsied | 22 | 24 | 22 | 24 | 15 | |||
| No. of rats with histologically confirmed lung lesions | ||||||||
| Hyperplasia | 0 | 24 | 22 | 24 | 15 | |||
| Adenoma | 0 | 23 | 22 | 24 | 15 | |||
| Carcinoma | 0 | 6a | 10a | 17a,b | 15a | |||
| Tumor incidence | 0 | 23c | 22c | 24c | 15c | |||
| No. of rats with thoracic cavity tumors | 0 | 2 | 2 | 6 | 9 | |||
| No. of rats with metastasis to pancreas | 0 | 1d | 2e | 3f | 4g,h | |||
| Gross lung lesion score | Number of rats with lesions | Number of rats evaluated | ||||||
| 0 | 1 | 2 | 3 | 4 | ||||
| B. Gross lung lesion scores | ||||||||
| Control | 21 | 0 | 0 | 0 | 0 | 0 | 21i | |
| (R)-NNALj | 0 | 6 | 16 | 1 | 0 | 23 | 23 | |
| (S)-NNAL | 0 | 2 | 17 | 3 | 0 | 22 | 22 | |
| NNKj | 0 | 0 | 9 | 11 | 3k | 23 | 23 | |
| Racemic NNAL | 0 | 0 | 4 | 8 | 3 | 15 | 15 | |
a P < 0.01 compared with control.
bGreater than (R)-NNAL, P = 0.0015.
c P < 0.0001 compared with control.
dOne rat with two tumors.
eOne rat with one tumor and one rat with two tumors.
fEach rat had one tumor.
gOne rat with one tumor; one with three tumors, one with four tumors and one with six tumors. A total of 14 pancreatic tumors were observed in this group.
h P < 0.05 versus control.
iThe control group had 22 animals, of which one was excluded because of lung lesions attributable to large granular lymphocyte leukemia.
jOne animal in the (R)-NNAL and NNK groups was not scored due to prior sectioning of lungs for histology.
kOverall gross lung lesion score significantly greater than (R)-NNAL (P < 0.0001) and (S)-NNAL (P = 0.0006).
Fig. 2.

Gross images of fresh or fixed lungs with tumors illustrating examples of gross lung lesion scores 1–4: (panels A through F, bar = 1cm), and photomicrographs of carcinomas involving lung and pancreas (panels G, H). Panel A: control lung, Panel B: grade 1 from a rat treated with (R)-NNAL, Panel C: grade 2, (S)-NNAL, Panel D: grade 3, racemic NNAL, Panel E: grade 4, NNK, Panel F: Extrapulmonary masses, racemic NNAL. Panel G: Pulmonary carcinoma, racemic NNAL, H&E stain. Panel H: Metastatic carcinoma involving pancreas, NNK, H&E stain.
Pulmonary lesions were categorized as bronchoalveolar hyperplasia, adenoma, or carcinoma by light microscopy. Adenomas were generally well demarcated, displayed no or minimal cellular atypia, and slightly compressed the adjacent normal tissue. Some adenomas had poorly delineated borders, and foci of minimal to moderate cellular atypia. Carcinomas were expansile and invasive, and often had a significant degree of cellular atypia. The predominant morphological patterns observed were papillary or tubular, sometimes with a squamous component.
Two of the (S)-NNAL-treated rats, two of the (R)-NNAL-treated rats, nine of the racemic NNAL-treated rats and six of the NNK-treated rats had thoracic cavity tumors (Table IA). These tumors were firm, tan or white, and expansile multinodular masses which variably filled the mediastinum and thoracic cavity (Figure 2, panel F). Some tumors compressed or merged with tumors involving the adjacent pulmonary parenchyma and others completely encircled and entrapped the lungs and/or seeded the thoracic parietal pleura. The thoracic cavity and pulmonary tumors were histologically similar; for example, tumors in both locations had a tubulopapillary pattern and/or evidence of squamous differentiation. The thoracic cavity tumors were interpreted to be carcinomas that originated from pulmonary carcinomas by direct extension or by seeding of thoracic cavity and pleural surfaces.
Pancreatic tumors were only detected histologically and ranged from ~1 to 8mm in diameter. These tumors comprised moderately well-demarcated, nodular masses that compressed the adjacent tissue and were frequently associated with a moderate to marked scirrhous reaction. Further characterization of these tumors indicated that they are metastatic lesions arising from primary pulmonary carcinomas. Two of the (S)-NNAL-treated rats, one of the (R)-NNAL-treated rats, three of the NNK-treated rats and four of the racemic NNAL-treated rats had pancreatic tumors. Pancreatic tumor incidence was highest in the racemic NNAL group and was significant compared with controls (P < 0.05). Some rats had multiple tumors; most notably 14 tumors were observed in the racemic NNAL-treated group (Table IA). Considering all groups, a total of 22 pancreatic tumors were observed. Despite careful gross and histological examination of all livers and kidneys only one other suspect metastatic tumor was observed, in the kidney of one animal treated with racemic NNAL.
Six DNA adducts (Figure 1B)—7-POB-Gua, O 2-POB-dThd, O 6-POB-dGuo, 7-PHB-Gua, O 2-PHB-dThd and O 6-Me-Gua—were quantified by conventional LC-ESI-MS/MS in macroscopically normal lung tissue after 10, 30, 50 and 70 weeks of treatment. None of these adducts was detected in DNA of control rats. Typical chromatograms obtained upon analysis of lung DNA are illustrated in Supplementary Figures S2–S5, available at Carcinogenesis Online. 7-POB-Gua, O 2-POB-dThd, 7-PHB-Gua, O 2-PHB-dThd and O 6-Me-Gua were readily detected and quantified in these samples, but O 6-POB-dGuo and O 6-PHB-dGuo were not. Since the chromatograms obtained in the analysis of O 6-POB-dGuo (but not O 6-PHB-dGuo) indicated its possible presence, although not clearly resolved from interfering peaks, high resolution LC-ESI-MS/MS analysis was performed. This produced readily quantifiable chromatograms as illustrated in Supplementary Figure S4, available at Carcinogenesis Online. We did not attempt to quantify dCyd adducts (data not shown).
Levels of these six DNA adducts in lung DNA over the course of the study are illustrated in Figure 3A–F and Supplementary Tables S3–S6, available at Carcinogenesis Online. Considering first the POB-DNA adducts—7-POB-Gua, O 2-POB-dThd, and O 6-POB-dGuo (Figure 3A–C)—the highest adduct levels at 10 weeks of treatment were observed in the DNA of the rats treated with NNK; for 7-POB-Gua these were significantly greater (P < 0.03) than the amounts seen in the lungs of the rats treated with either (S)-NNAL or (R)-NNAL. Levels of these adducts in the lung DNA of the rats treated with (R)-NNAL were lower than those from NNK or (S)-NNAL throughout the study, significantly so for 7-POB-Gua and O 2-POB-dThd (P < 0.0001, Supplementary Table S4, available at Carcinogenesis Online). A consistent finding was a decrease in the adduct levels in the lung tissue of NNK-treated rats over the course of the study: 7-POB-Gua (1000 versus 300fmol/mg DNA, P < 0.0001); O 2-POB-dThd (3600 versus 2000fmol/mg DNA, P = 0.004); and O 6-POB-dGuo (34 versus 4fmol/mg DNA, non-significant, P = 0.093). Little or no decrease, or in the case of O 2-POB-dThd an increase, in these adducts was observed during the same time period in the lungs of rats treated with (S)-NNAL, while levels from (R)-NNAL remained low.
Fig. 3.

Levels of DNA adducts formed in pulmonary tissue of rats treated with 5 ppm NNK (●), (S)-NNAL (□) or (R)-NNAL (×) in their drinking water for 10, 30, 50 or 70 weeks. DNA was isolated and hydrolyzed to deoxyribonucleosides or nucleobases which were analyzed by liquid chromatography–tandem mass spectrometry. Panel A, 7-POB-Gua; Panel B, O 2-POB-dThd; Panel C, O 6-POB-dGuo; Panel D, 7-PHB-Gua; Panel E, O 2-PHB-dThd; Panel F, O 6-Me-Gua, Panel G, total POB-DNA adducts; Panel H, total PHB-DNA adducts; Panel I, POB + PHB-DNA adducts.
The highest levels of O 6-Me-Gua were found in the lungs of the rats treated with NNK or (S)-NNAL compared with the (R)-NNAL-treated rats (Figure 3F). Levels of the adducts from NNK or (S)-NNAL decreased throughout the study, and the overall decrease was significant for NNK (P = 0.028).
A completely different picture was observed in the case of the PHB-DNA adducts—7-PHB-Gua and O 2-PHB-dThd (Figure 3D and E). In both cases, (R)-NNAL produced significantly higher adduct levels (P < 0.0001) than either NNK or (S)-NNAL throughout the study. 7-PHB-Gua levels ranged from ~400 to 900fmol/mg DNA in the lung DNA of rats treated with (R)-NNAL and O 2-PHB-dThd levels ranged from 4000 to 6500fmol/mg DNA.
Levels of total POB-DNA adducts and total PHB-DNA adducts in lung DNA are summarized in Figure 3G–I and Supplementary Tables S4 and S5, available at Carcinogenesis Online. Total POB-DNA adducts were significantly higher at all time points in the lungs of rats treated with NNK or (S)-NNAL compared with those treated with (R)-NNAL (P < 0.003) while the opposite was observed for total PHB-DNA adducts (P < 0.005). POB-DNA adducts plus PHB-DNA adducts (Figure 3I and Supplementary Table S6, available at Carcinogenesis Online) were not significantly different among the three compounds at most time points.
As shown in Figure 1A, acid hydrolysis of POB-DNA adducts produces HPB (12). Specifically, O 2-POB-dThd and O 6-POB-dGuo are converted to HPB under these conditions, while it is estimated that about 50% of 7-POB-dGuo is hydrolyzed to HPB under neutral thermal conditions (42). Levels of HPB released from pulmonary DNA in this study were quantified. The results are summarized in Figure 4 and Supplementary Table S7, available at Carcinogenesis Online. Consistent with the POB-DNA adduct data, levels of HPB released were greatest in the rats treated with NNK or (S)-NNAL, with significantly lower levels in the rats treated with (R)-NNAL (P < 0.0001, Supplementary Table S7, available at Carcinogenesis Online).
Fig. 4.
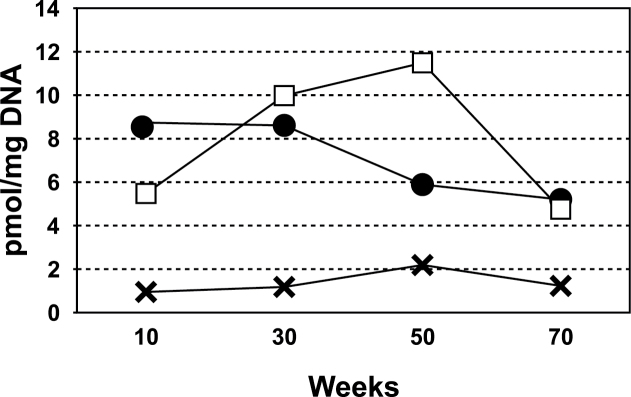
Levels of HPB-releasing DNA adducts formed in pulmonary tissue of rats treated with 5 ppm NNK (●), (S)-NNAL (□), or (R)-NNAL (×) in their drinking water for 10, 30, 50 or 70 weeks.
Analyses of pancreatic DNA adducts are summarized in Figure 5A–D and Supplementary Tables S3–S6, available at Carcinogenesis Online. 7-POB-Gua, O 2-POB-dThd, 7-PHB-Gua and O 2-PHB-dThd were detected and quantified, but amounts of O 6-POB-dGuo, O 6-PHB-dGuo and O 6-Me-Gua were below the limits of quantitation. The same pattern of formation of these DNA adducts as in the lung—relatively higher amounts of POB-DNA adducts from NNK and (S)-NNAL and higher amounts of PHB-DNA adducts from (R)-NNAL—was observed, but the overall amounts of each adduct were significantly less than in the lung DNA (P < 0.0001), except in the case of 7-PHB-Gua from NNK and (S)-NNAL. We also analyzed pancreatic DNA from rats treated with 14 ppm in the drinking water of (S)-NNN or (R)-NNN for 10, 30, 50 or 70 weeks (37) for 7-POB-Gua, O 2-POB-dThd and O 6-POB-dGuo; only trace amounts of O 2-POB-dThd were detected in the rats treated with (S)-NNN.
Fig. 5.

Levels of DNA adducts formed in pancreatic tissue of rats treated with 5 ppm NNK (●), (S)-NNAL (□), or (R)-NNAL (×) in their drinking water for 10, 30, 50 or 70 weeks. DNA was isolated and hydrolyzed to deoxyribonucleosides or nucleobases which were analyzed by liquid chromatography–mass spectrometry. Panel A, 7-POB-Gua; B, O 2-POB-dThd; C, 7-PHB-Gua; D, O 2-PHB-dThd.
Discussion
Virtually all cigarette smokers and smokeless tobacco users, and many people exposed to secondhand tobacco smoke, have NNAL in their urine (9,11,12). Only limited data are available on the enantiomeric composition of this NNAL, but both forms have been identified in human urine samples (22). The results presented here demonstrate that both NNAL enantiomers are powerful pulmonary carcinogens in F-344 rats, with activity nearly as great as their parent compound, the tobacco smoke constituent NNK. This potent systemic activity in rats, observed upon administration of the compounds in the drinking water, is distinctive among constituents of tobacco products and their metabolites. The DNA adduct studies presented here are consistent with the carcinogenicity data and provide a rational basis for the observations as well as some new insights on mechanisms of carcinogenicity by these compounds.
This is the first study to investigate the carcinogenicity of the NNAL enantiomers in rats. In the one previous report of racemic NNAL carcinogenicity in male F-344 rats, the compound was administered in the drinking water at a dose of 5 ppm, identical to that used here for each enantiomer (43). The study was terminated after 112 weeks. In a group of 30 rats, 26 had lung tumors, 5 with adenoma, 12 with adenocarcinoma and 9 with adenosquamous carcinoma. In terms of overall carcinogenicity to the lung, the results are quite comparable since the enantiomers examined here and racemic NNAL in both studies all produced a high incidence of lung tumors. NNK, given at 5 ppm in the drinking water in both studies, also caused a high incidence of malignant lung tumors in each. Considering that these studies of NNAL and NNK carcinogenicity were performed in different institutions with different personnel, with more than 20 years intervening, the consistency of the results is quite remarkable and serves to highlight the potent pulmonary carcinogenicity of NNAL and NNK. It is worth noting that chemical induction of lung tumors in F-344 rats is relatively uncommon. In the United States National Toxicology Program, ‘clear evidence’ or ‘some evidence’ of pulmonary carcinogenicity was reported in studies of only 38 of 574 (6.6%) compounds tested in F-344 rats (44).
With respect to histopathological findings in the lung, the results of this study are in substantial agreement with the previous study of NNK and NNAL in rats (43). We also report here the universal presence of bronchoalveolar epithelial hyperplasia in the NNK and NNAL groups. A notable finding in this study that was not previously reported was the presence of extensive extrapulmonary masses involving the thoracic cavity and mediastinum that were interpreted to arise from aggressive pulmonary carcinomas. Despite careful examination we initially only observed one metastatic lesion (involving kidney) at typical sites for metastasis, but further characterization of the pancreatic tumors encountered in this study indicated that they are metastatic lesions arising from primary lung carcinomas, and that the pancreas is the main organ to which the pulmonary carcinomas metastasize. Full details of this observation will be reported separately. The significant 27% incidence of pancreatic tumors observed here in the racemic NNAL group, treated with 10 ppm in the drinking water, is consistent with our previous report, although the effect was somewhat less in this study (43). The previous study also reported a significant 27% incidence of pancreatic tumors, but the dose of racemic NNAL was only 5 ppm. We note, however, that DNA adduct formation in the pancreas was quite low compared with the lung, and to other tissues examined in previous studies (24–26).
One previous study reported the tumorigenicity of the NNAL enantiomers in A/J mice, a strain highly susceptible to lung tumor development (23). (S)-NNAL induced 25.6 lung tumors per mouse, 16 weeks after a single intraperitoneal dose of 20 µmol, activity similar to that of NNK, while (R)-NNAL caused 8.2 lung tumors per mouse. Tumorigenic activity in this system is closely linked to the formation of the DNA adduct O 6-Me-Gua and consequent mutations in the k-ras gene (45,46). Metabolism studies demonstrated that (S)-NNAL was more likely to activate this pathway than was (R)-NNAL in the A/J mouse lung (23).
Few previous studies have examined DNA adduct formation throughout the course of a long-term carcinogenicity experiment (27–37). Therefore, the adduct data presented here provide a unique opportunity to relate DNA damage to carcinogenicity. This time course data could be important not only with respect to mechanisms of carcinogenicity by these compounds but also for considering the potential use of DNA adducts as biomarkers of exposure and metabolic activation of NNK/NNAL in smokers. We have shown in previous studies that DNA adduct formation in rats treated with NNK is higher in lung than in most other tissues including liver when the NNK doses are relatively low, comparable with those used here (24,47). Consistent with the powerful pulmonary carcinogenicity of NNK observed in this study, initial levels of POB-DNA adducts and O 6-Me-Gua in lung tissue of NNK-treated rats were relatively high. O 2-POB-dThd, O 6-POB-dGuo and O 6-Me-Gua all have miscoding properties (48–51). As the study progressed, adduct levels in the NNK-treated rats decreased. Total POB-DNA plus PHB-DNA adduct levels in lung tissue were similar throughout the study in the rats treated with (S)-NNAL and (R)-NNAL, consistent with their similar carcinogenic activities (Figure 3I). The slightly higher carcinogenicity of (S)-NNAL than (R)-NNAL might be related to its production of significantly (P = 0.012) higher levels of pulmonary O 6-MeGua throughout the study. Overall, our results are consistent with the critical role of miscoding DNA adducts as causes of cancer.
The levels of pulmonary POB-DNA adducts in the rats treated with NNK, (S)-NNAL, or (R)-NNAL were consistent with observations in our previous 20 week study, considering that the dose was twice as high in that one (24). The results of that study raised the possibility that NNK or NNAL-DNA adducts in the lung might decrease with time, and we did indeed observe decreases in NNK-DNA adducts as the current study progressed. Thus, O 2-POB-dThd was maximal at 30 weeks and decreased thereafter while 7-POB-Gua, O 6-POB-dGuo and O 6-MeGua were maximal at 10 weeks (Figure 3). While total POB-DNA adducts and O 6-MeGua decreased significantly with time of treatment with NNK, this was not the case in the rats treated with (S)-NNAL. Levels of POB-DNA adducts and O 6-MeGua were low throughout in the lung tissue of rats treated with (R)-NNAL. The decreases in adduct levels in the lung tissue of the NNK-treated rats contrasts markedly with the results of our recent study of POB-DNA adducts in tissues of rats treated chronically with 14 ppm of NNN in their drinking water for 75 weeks (37). In that study, levels of O 2-POB-dThd and 7-POB-Gua remained constant throughout in all tissues examined, although the amounts in lung tissue were considerably lower (~300–800fmol/mg DNA) than observed here. This suggests that the decrease in DNA adduct levels observed here is specific to biological properties of NNK, rather than to a general response to the presence of POB-DNA adducts.
There are at least two plausible explanations for the decrease in lung DNA adduct levels with time in the rats treated with NNK. The first pertains specifically to the decrease in levels of O 6-Me-Gua. We have previously observed a decrease in pulmonary α-methylene hydroxylation of NNK to intermediate 4 (Figure 1), as evidenced by decreased microsomal formation of keto aldehyde 8 and levels of O 6-Me-Gua in rats during treatment with multiple subcutaneous injections of NNK over a period of 20 weeks (52,53). We hypothesize that keto aldehyde 8 can inhibit NNK metabolism by binding to the active site of the relevant cytochrome P450 enzyme. The observed decrease in O 6-Me-Gua in the lungs of the rats treated with (S)-NNAL, although not significant, probably is an ultimate consequence of its reconversion to NNK, followed by inhibition of α-methylene hydroxylation.
The second explanation involves the possible role of inflammation as an inhibitor of NNK metabolism in the rat lung. Wu et al. (54) have recently demonstrated that treatment of humanized mice with the bacterial endotoxin lipopolysaccharide and the pro-inflammatory cytokine IL-6 can suppress CYP2A13 messenger RNA and protein levels in the lung. Based on their observation, it is plausible that there could be a corresponding decrease in levels of rat CYP2A3 messenger RNA and protein levels in the lungs of the NNK-treated rats during this carcinogenicity study. Cytochrome P450 2A3 is a major catalyst of NNK metabolic activation in the rat lung; cytochrome P450s 2A13 and 2A3 are closely related in structure and activity toward NNK (55). This phenomenon would be expected to affect all NNK-DNA adducts, as we observed. NNAL is a poorer substrate for P450 2A3 than NNK, so the effect may be less pronounced; other P450s as well as glucuronidation (see below) may be involved in DNA adduct formation by NNAL. Further studies are required to investigate the possible role of inflammation in a time-dependent decrease of NNK metabolic activation in the rat lung.
HPB-releasing DNA adducts have been detected in the lungs of smokers and lung cancer patients and have been discussed as possible biomarkers of NNK and NNN metabolic activation (56,57). Our results do not support the use of pulmonary HPB-releasing DNA adducts as biomarkers because they vary with time, a consequence of their derivation from POB-DNA adducts. Thus, measurement of HPB-releasing DNA adducts or POB-DNA adducts in lung tissue (frequently obtained at surgery for lung cancer) at a single time point may not be a reliable indicator of NNK metabolic activation in a given individual because levels of these adducts may change over time, at least based on the studies carried out here in rats, and those of Wu et al. (54) on expression of CYP2A13. PHB-DNA adducts may suffer from similar limitations with respect to their use as biomarkers of pulmonary DNA damage.
As in our previous studies, we observed clearly distinct DNA adduct patterns in the rats treated with (R)-NNAL versus (S)-NNAL. Those from (S)-NNAL in many respects mirrored those from NNK, while those from (R)-NNAL were different (25,26). Thus, POB-DNA adducts, HPB-releasing DNA adducts, and O 6-Me-Gua were generally significantly higher in lung and pancreas of rats treated with NNK or (S)-NNAL compared with (R)-NNAL. In contrast, PHB-DNA adducts were significantly higher in lung and pancreas of rats treated with (R)-NNAL compared with NNK or (S)-NNAL. The similarity in adduct formation between NNK and (S)-NNAL is probably a consequence of the established equilibrium between these two compounds. NNK is readily and extensively converted to (S)-NNAL in rat tissues, and (S)-NNAL is reconverted to NNK significantly more readily than is (R)-NNAL (18). Thus, administration of NNK to a rat is similar to administration of (S)-NNAL, and the consequences are similar. (R)-NNAL on the other hand is not readily reconverted to NNK in rats, but does undergo extensive metabolism to its glucuronide metabolites (58). In rat lung microsomes, (S)-NNAL is a significantly better substrate for α-hydroxylation to intermediates 5 and 6 than is (R)-NNAL (18); this is also indicated by the O 6-Me-Gua levels in lung observed here. How, then, do we get such high levels of PHB-DNA adducts in the lung tissue of rats treated with (R)-NNAL, but not (S)-NNAL? We hypothesize that this is due to transport of the α-hydroxymethyl glucuronide of (R)-6 from the liver to the lung, where it is deconjugated by β-glucuronidase to release 6, which then spontaneously yields 11 and the PHB-DNA adducts. In support of this hypothesis, rates of microsomal metabolism of (R)-NNAL and (S)-NNAL to 5 and 6 are similar in rat liver (18), in contrast to lung. Further, an α-hydroxymethyl glucuronide of NNK has been characterized previously in rats (59), and glucuronidation of the 1-hydroxy group of (R)-NNAL is favored compared with (S)-NNAL (58). We have previously discussed possible reasons for low PHB-DNA adduct formation from (S)-NNAL in the lung, possibly involving sequestration followed by rapid conversion to NNK (25).
The results of the analyses of pancreatic DNA for POB- and PHB-DNA adducts are fully consistent with our previous study of NNK and the NNAL enantiomers administered at 10 ppm in the drinking water for 20 weeks (26). As in the lung DNA adduct studies discussed above, POB-DNA adducts predominated in the rats treated with NNK or (S)-NNAL while PHB-DNA adducts were most extensively formed in the pancreas of the rats treated with (R)-NNAL. The levels of these DNA adducts were generally significantly lower than in the lung. The relative specificity of pancreatic metabolic activation of NNK and NNAL is indicated by the generally undetectable levels of POB-DNA adducts in pancreatic DNA of rats treated with 14 ppm (S)-NNN or (R)-NNN, which do not induce pancreatic tumors in rats. The potential role of low levels of pancreatic DNA adducts in facilitating metastasis to this tissue requires further study.
This is the first study to compare levels of HPB-releasing DNA adducts and individual POB-DNA adducts in vivo in any system. Acid hydrolysis of O 2-POB-dThd and O 6-POB-dGuo releases HPB while hydrolysis of 7-POB-dGuo is estimated to produce HPB and 7-POB-Gua in approximately equal amounts (42). Based on this, and the amounts of total O 2-POB-dThd and 7-POB-Gua versus HPB released from lung DNA of NNK-treated rats at the four time points, unknown POB-DNA adducts account for ~12–56% of released HPB. The other likely sources of HPB-releasing DNA adducts are phosphate adducts, deoxyadenosine adducts and deoxycytidine adducts, which were not quantified here.
In summary, this study clearly demonstrates the potent carcinogenicity to the rat lung of both enantiomers of NNAL, metabolites of NNK present in the urine of people exposed to tobacco products. The powerful carcinogenic effect of NNK in the rat lung confirms the results of our earlier study, suggesting little safety margin for exposure to this tobacco-specific nitrosamine. These findings underline the urgent need for immediate regulation of human exposure to NNK and NNAL, achievable by controlling the levels of NNK in tobacco, as recently suggested (60). Regulation of NNK has the potential to reduce lung cancer mortality. The study reported here also provides unique and valuable DNA adduct measurements over the course of the carcinogenicity study. The persistence of O 2-PHB-dThd in pulmonary DNA throughout the study is particularly notable, suggesting its potential value as a biomarker of NNK and NNAL carcinogenesis in people exposed to tobacco products.
Supplementary material
Supplementary Tables S1–S7 and Figures S1–S5 can be found at http://carcin.oxfordjournals.org/
Funding
United States National Cancer Institute (CA-81301).
Supplementary Material
Acknowledgements
The authors thank Josh Parker and Paula Overn for technical assistance, and Bob Carlson for editorial assistance.
Conflict of Interest Statement: None declared.
Glossary
Abbreviations:
- HPB
4-hydroxy-1-(3-pyridyl)-1-butanone
- NNAL
4-(meth ylnitrosamino)-1-(3-pyridyl)-1-butanol
- NNK
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone
- NNN
N′-nitrosonornicotine
- PHB
pyridylhydroxybutyl
- POB
pyridyloxobutyl.
References
- 1. Stewart B.W., et al. (2014). World Cancer Report 2014. IARC, Lyon, FR. [Google Scholar]
- 2. World Health Organization. Cancer Fact Sheet N°297. http://www.who.int/mediacentre/factsheets/fs297/en/index.html (15 January 2014, date last accessed). [Google Scholar]
- 3. World Health Organization. Tobacco Free Initiative (TFI). http://www.who.int/tobacco/research/cancer/en/ (15 January 2014, date last accessed). [Google Scholar]
- 4. Shafey O., et al. (2009). The Tobacco Atlas. 3rd edn. American Cancer Society and World Lung Foundation, Atlanta, GA. [Google Scholar]
- 5. Hecht S.S. (1998). Biochemistry, biology, and carcinogenicity of tobacco-specific N-nitrosamines. Chem. Res. Toxicol., 11, 559–603. [DOI] [PubMed] [Google Scholar]
- 6. Schüller H.M., et al. (1994). Transplacental carcinogenicity of low doses of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone administered subcutaneously or intratracheally to hamsters. J. Cancer Res. Clin. Oncol., 120, 200–203. [DOI] [PubMed] [Google Scholar]
- 7. Schüller H.M., et al. (1993). Transplacental induction of pancreas tumors in hamsters by ethanol and the tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Cancer Res., 53, 2498–2501. [PubMed] [Google Scholar]
- 8. International Agency for Research on Cancer. (2012). Personal habits and indoor combustions. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, v. 100E. IARC, Lyon, FR, pp. 319–331. [PMC free article] [PubMed] [Google Scholar]
- 9. Xia Y., et al. (2011). Tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL) in smokers in the United States: NHANES 2007–2008. Biomarkers, 16, 112–119. [DOI] [PubMed] [Google Scholar]
- 10. Roethig H.J., et al. (2009). Population estimates for biomarkers of exposure to cigarette smoke in adult U.S. cigarette smokers. Nicotine Tob. Res., 11, 1216–1225. [DOI] [PubMed] [Google Scholar]
- 11. Vogel R.I., et al. (2011). The ratio of a urinary tobacco-specific lung carcinogen metabolite to cotinine is significantly higher in passive than in active smokers. Biomarkers, 16, 491–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Carmella S.G., et al. (2013). High throughput liquid and gas chromatography–tandem mass spectrometry assays for tobacco-specific nitrosamine and polycyclic aromatic hydrocarbon metabolites associated with lung cancer in smokers. Chem. Res. Toxicol., 26, 1209–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Church T.R., et al. (2009). A prospectively measured serum biomarker for a tobacco-specific carcinogen and lung cancer in smokers. Cancer Epidemiol. Biomarkers Prev., 18, 260–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yuan J.M., et al. (2009). Urinary levels of tobacco-specific nitrosamine metabolites in relation to lung cancer development in two prospective cohorts of cigarette smokers. Cancer Res., 69, 2990–2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yuan J.M., et al. (2011). Urinary levels of cigarette smoke constituent metabolites are prospectively associated with lung cancer development in smokers. Cancer Res., 71, 6749–6757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Finckh C., et al. (2001). Expression and NNK reducing activities of carbonyl reductase and 11beta-hydroxysteroid dehydrogenase type 1 in human lung. Chem. Biol. Interact., 130–132, 761–773. [DOI] [PubMed] [Google Scholar]
- 17. Breyer-Pfaff U., et al. (2004). Enantioselectivity of carbonyl reduction of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone by tissue fractions from human and rat and by enzymes isolated from human liver. Drug Metab. Dispos., 32, 915–922. [PubMed] [Google Scholar]
- 18. Upadhyaya P., et al. (2000). Formation and metabolism of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol enantiomers in vitro in mouse, rat and human tissues. Carcinogenesis, 21, 1233–1238. [PubMed] [Google Scholar]
- 19. Trushin N., et al. (2008). The tobacco carcinogen NNK is stereoselectively reduced by human pancreatic microsomes and cytosols. Langenbecks Arch. Surg., 393, 571–579. [DOI] [PubMed] [Google Scholar]
- 20. Yang Y., et al. (2011). Metabolic study of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone to the enantiomers of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol in vitro in human bronchial epithelial cells using chiral capillary electrophoresis. J. Chromatogr. A, 1218, 6505–6510. [DOI] [PubMed] [Google Scholar]
- 21. Carmella S.G., et al. (1999). Stereochemistry of metabolites of a tobacco-specific lung carcinogen in smokers’ urine. Cancer Res., 59, 3602–3605. [PubMed] [Google Scholar]
- 22. Hecht S.S., et al. (2002). Quantitation of metabolites of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone after cessation of smokeless tobacco use. Cancer Res., 62, 129–134. [PubMed] [Google Scholar]
- 23. Upadhyaya P., et al. (1999). Tumorigenicity and metabolism of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol enantiomers and metabolites in the A/J mouse. Carcinogenesis, 20, 1577–1582. [DOI] [PubMed] [Google Scholar]
- 24. Lao Y., et al. (2007). Formation and accumulation of pyridyloxobutyl DNA adducts in F344 rats chronically treated with 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and enantiomers of its metabolite, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol. Chem. Res. Toxicol., 20, 235–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Upadhyaya P., et al. (2008). Quantitation of pyridylhydroxybutyl-DNA adducts in liver and lung of F-344 rats treated with 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and enantiomers of its metabolite 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol. Chem. Res. Toxicol., 21, 1468–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang S., et al. (2009). Analysis of pyridyloxobutyl and pyridylhydroxybutyl DNA adducts in extrahepatic tissues of F344 rats treated chronically with 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and enantiomers of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol. Chem. Res. Toxicol., 22, 926–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Beland F.A., et al. (1982). Persistence of DNA adducts in rat liver and kidney after multiple doses of the carcinogen N-hydroxy-2-acetylaminofluorene. Cancer Res., 42, 1348–1354. [PubMed] [Google Scholar]
- 28. Gupta R.C., et al. (1988). Analysis of DNA adducts in putative premalignant hepatic nodules and nontarget tissues of rats during 2-acetylaminofluorene carcinogenesis. Cancer Res., 48, 5270–5274. [PubMed] [Google Scholar]
- 29. Poirier M.C., et al. (1984). DNA adduct formation, removal and persistence in rat liver during one month of feeding 2-acetylaminofluorene. Carcinogenesis, 5, 1591–1596. [DOI] [PubMed] [Google Scholar]
- 30. Herron D.C., et al. (1982). DNA methylation during chronic administration of 1,2-dimethylhydrazine in a carcinogenic regimen. Carcinogenesis, 3, 857–860. [DOI] [PubMed] [Google Scholar]
- 31. Swenberg J.A., et al. (1987). The molecular dosimetry of DNA adducts formed by continuous exposure of rats to alkylating hepatocarcinogens. Prog. Exp. Tumor Res., 31, 42–51. [DOI] [PubMed] [Google Scholar]
- 32. Buss P., et al. (1990). Linear dose-response relationship for DNA adducts in rat liver from chronic exposure to aflatoxin B1. Carcinogenesis, 11, 2133–2135. [DOI] [PubMed] [Google Scholar]
- 33. Yamashita K., et al. (1990). DNA adducts formed by 2-amino-3,8-dimethylimidazo[4,5-f]quinoxaline in rat liver: dose-response on chronic administration. Jpn. J. Cancer Res., 81, 470–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zaidi N.H., et al. (1993). Tissue and cell specific methylation, repair and synthesis of DNA in the upper gastrointestinal tract of Wistar rats treated with N-methyl-N′-nitro-N-nitrosoguanidine via the drinking water. Carcinogenesis, 14, 1991–2001. [DOI] [PubMed] [Google Scholar]
- 35. Talaska G., et al. (1996). Chronic, topical exposure to benzo[a]pyrene induces relatively high steady-state levels of DNA adducts in target tissues and alters kinetics of adduct loss. Proc. Natl. Acad. Sci. U. S. A., 93, 7789–7793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yu H.S., et al. (2012). The effect of ethanol on the formation of N2-ethylidene-dG adducts in mice: implications for alcohol-related carcinogenicity of the oral cavity and esophagus. Biomarkers, 17, 269–274. [DOI] [PubMed] [Google Scholar]
- 37. Zhao L., et al. (2013). Quantitation of pyridyloxobutyl-DNA adducts in tissues of rats treated chronically with (R)- or (S)-N′-nitrosonornicotine (NNN) in a carcinogenicity study. Chem. Res. Toxicol., 26, 1526–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hecht S.S., et al. (1997). Absolute configuration of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol formed metabolically from 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Carcinogenesis, 18, 1851–1854. [DOI] [PubMed] [Google Scholar]
- 39. Hecht S.S., et al. (1980). Metabolism in the F344 rat of 4-(N-methyl-N-nitrosamino)-1-(3-pyridyl)-1-butanone, a tobacco-specific carcinogen. Cancer Res., 40, 4144–4150. [PubMed] [Google Scholar]
- 40. Upadhyaya P., et al. (2009). Comparative levels of O 6-methylguanine, pyridyloxobutyl-, and pyridylhydroxybutyl-DNA adducts in lung and liver of rats treated chronically with the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Drug Metab. Dispos., 37, 1147–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Stepanov I., et al. (2013). Analysis of 4-hydroxy-1-(3-pyridyl)-1-butanone (HPB)-releasing DNA adducts in human exfoliated oral mucosa cells by liquid chromatography–electrospray ionization–tandem mass spectrometry. Chem. Res. Toxicol., 26, 37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang M., et al. (2003). Identification of adducts formed by pyridyloxobutylation of deoxyguanosine and DNA by 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanone, a chemically activated form of tobacco specific carcinogens. Chem. Res. Toxicol., 16, 616–626. [DOI] [PubMed] [Google Scholar]
- 43. Rivenson A., et al. (1988). Induction of lung and exocrine pancreas tumors in F344 rats by tobacco-specific and Areca-derived N-nitrosamines. Cancer Res., 48, 6912–6917. [PubMed] [Google Scholar]
- 44. National Toxicology Program. (2014) Chemicals Associated with Site-Specific Neoplasia. http://ntp.niehs.nih.gov/?objectid=DA65BAEA-CF6B-CDFF-6213E6AE0FD281EB (15 January 2014, date last accessed). [Google Scholar]
- 45. Peterson L.A., et al. (1991). O 6-Methylguanine is a critical determinant of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone tumorigenesis in A/J mouse lung. Cancer Res., 51, 5557–5564. [PubMed] [Google Scholar]
- 46. Ronai Z.A., et al. (1993). G to A transitions and G to T transversions in codon 12 of the Ki-ras oncogene isolated from mouse lung tumors induced by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) and related DNA methylating and pyridyloxobutylating agents. Carcinogenesis, 14, 2419–2422. [DOI] [PubMed] [Google Scholar]
- 47. Murphy S.E., et al. (1990). Dose-response study of DNA and hemoglobin adduct formation by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in F344 rats. Cancer Res., 50, 5446–5452. [PubMed] [Google Scholar]
- 48. Gowda A.S., et al. (2012). Low fidelity bypass of O2-(3-pyridyl)-4-oxobutylthymine, the most persistent bulky adduct produced by the tobacco specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone by model DNA polymerases. Chem. Res. Toxicol., 25, 1195–1202. [DOI] [PubMed] [Google Scholar]
- 49. Jasti V.P., et al. (2011). Tobacco-specific nitrosamine-derived O 2-alkylthymidines are potent mutagenic lesions in SOS-induced Escherichia coli. Chem. Res. Toxicol., 24, 1833–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pauly G.T., et al. (2002). Mutagenesis by O6-[4-oxo-4-(3-pyridyl)butyl]guanine in Escherichia coli and human cells. Chem. Res. Toxicol., 15, 165–169. [DOI] [PubMed] [Google Scholar]
- 51. Delaney J.C., et al. (2008). Biological properties of single chemical-DNA adducts: a twenty year perspective. Chem. Res. Toxicol., 21, 232–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Staretz M.E., et al. (1997). Effects of long term dietary phenethyl isothiocyanate on the microsomal metabolism of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol in F344 rats. Carcinogenesis, 18, 1715–1722. [DOI] [PubMed] [Google Scholar]
- 53. Staretz M.E., et al. (1997). Evidence for an important role of DNA pyridyl oxobutylation in rat lung carcinogenesis by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone: effects of dose and phenethyl isothiocyanate. Cancer Res., 57, 259–266. [PubMed] [Google Scholar]
- 54. Wu H., et al. (2013). Transcriptional suppression of CYP2A13 expression by lipopolysaccharide in cultured human lung cells and the lungs of a CYP2A13-humanized mouse model. Toxicol. Sci., 135, 476–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jalas J.R., et al. (2005). Cytochrome P450 enzymes as catalysts of metabolism of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, a tobacco specific carcinogen. Chem. Res. Toxicol., 18, 95–110. [DOI] [PubMed] [Google Scholar]
- 56. Schlöbe D., et al. (2008). 4-Hydroxy-1-(3-pyridyl)-1-butanone-releasing DNA adducts in lung, lower esophagus and cardia of sudden death victims. Toxicology, 245, 154–161. [DOI] [PubMed] [Google Scholar]
- 57. Foiles P.G., et al. (1991). Mass spectrometric analysis of tobacco-specific nitrosamine-DNA adducts in smokers and nonsmokers. Chem. Res. Toxicol., 4, 364–368. [DOI] [PubMed] [Google Scholar]
- 58. Zimmerman C.L., et al. (2004). Stereoselective metabolism and tissue retention in rats of the individual enantiomers of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL), metabolites of the tobacco-specific nitrosamine, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK). Carcinogenesis, 25, 1237–1242. [DOI] [PubMed] [Google Scholar]
- 59. Murphy S.E., et al. (1995). Glucuronidation of 4-(hydroxymethyl nitrosamino)-1-(3-pyridyl)-1-butanone, a metabolically activated form of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, by phenobarbital-treated rats. Chem. Res. Toxicol., 8, 772–779. [DOI] [PubMed] [Google Scholar]
- 60. Hecht S.S. (2014). It is time to regulate carcinogenic tobacco-specific nitrosamines in cigarette tobacco. Cancer Prev. Res. (Phila)., 7, 639–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.