

A new size exclusion chromatography and mass spectrometry-based method was developed for proteomic analysis of endogenous protein complexes. It is predicted that about one-third of the detected cytosolic proteins exist in stable oligomeric complexes under optimal growth conditions. The method can be adapted to predict the composition and dynamics of protein complexes under different conditions.
Abstract
Global analyses of protein complex assembly, composition, and location are needed to fully understand how cells coordinate diverse metabolic, mechanical, and developmental activities. The most common methods for proteome-wide analysis of protein complexes rely on affinity purification-mass spectrometry or yeast two-hybrid approaches. These methods are time consuming and are not suitable for many plant species that are refractory to transformation or genome-wide cloning of open reading frames. Here, we describe the proof of concept for a method allowing simultaneous global analysis of endogenous protein complexes that begins with intact leaves and combines chromatographic separation of extracts from subcellular fractions with quantitative label-free protein abundance profiling by liquid chromatography-coupled mass spectrometry. Applying this approach to the crude cytosolic fraction of Arabidopsis thaliana leaves using size exclusion chromatography, we identified hundreds of cytosolic proteins that appeared to exist as components of stable protein complexes. The reliability of the method was validated by protein immunoblot analysis and comparisons with published size exclusion chromatography data and the masses of known complexes. The method can be implemented with appropriate instrumentation, is applicable to any biological system, and has the potential to be further developed to characterize the composition of protein complexes and measure the dynamics of protein complex localization and assembly under different conditions.
INTRODUCTION
Protein complexes, defined as the quaternary structure of multiple polypeptides that physically associate with one another, are the cornerstones of cellular control mechanisms (Alberts, 1998; Srere, 2000). Invariably, plant cells use combinations of protein complexes to adjust their metabolism (Winkel, 2004), growth (Szymanski, 2005; Ingram and Waites, 2006), and physiology (Yi and Deng, 2005) as a function of ever-changing developmental states and environmental conditions. In this context, protein complexes can be considered as fundamental building blocks of cell biology that enable precise control within and between cellular pathways (Hartwell et al., 1999; Good et al., 2011). This view is widely supported by genetic data, in which null alleles of different subunits of a protein complex often display identical phenotypes, and reveal genes and proteins that function as part of a common pathway. Therefore, in order to gain broad insight into the function and integration of cellular pathways, deep knowledge about protein complex formation and dynamics is needed.
This is a tall order to fill. In Arabidopsis thaliana leaves, roughly ∼16,000 genes are expressed in specific cell types (Marks et al., 2009) and there are no unifying rules for protein oligomerization: The complexes are diverse in terms of composition, stability, and function (Nooren and Thornton, 2003). For example, many protein complexes assemble to do mechanical work at a specific place and time. The highly organized long-distance intracellular transport system of the cell has, at its core, protein complexes containing myosin motors that link regulated cargo selection to ATP-dependent transport on another protein complex, the actin cytoskeleton. Many other protein complexes use ATP to function in the context of protein complex remodeling (Kressler et al., 2008) or protein turnover (Pickart and Cohen, 2004). In other instances, complex mechanical tasks are executed by evolutionarily conserved protein complexes that cooperate, for example, to drive specific organelle fusion events (Ohya et al., 2009; Stroupe et al., 2009), generate transport vesicles at specialized subdomains of organelle surfaces (Barlowe et al., 1994; Kaksonen et al., 2003), or segregate replicated chromosomes during cell division (Karsenti and Vernos, 2001; Masoud et al., 2013). Metabolic pathways and enzyme systems are strongly affected by protein complex formation. Homo-oligomeric protein complexes can create new intermolecular interfaces and highly efficient enzyme complexes with clustered active sites (Marianayagam et al., 2004; Chen et al., 2011). Hetero-oligomeric protein complexes that contain sequential enzymes in a metabolic pathway can shield metabolites from the bulk cytosol and promote metabolite flux into specific final products (Srere, 1987). In other instances, protein complex formation can negatively regulate an enzyme, alter its substrate specificity, or dictate its subcellular localization (Winkel, 2004).
The biological significance of protein complex formation is perhaps most frequently considered in the context of signal transduction. The protein complex hardware for information flow assembles through regulated physical interactions among subunits that give the complex stability, yet allow conformational or compositional changes in response to ligand binding or other types of input signals. During signaling, protein complexes function dynamically as coincidence detectors to efficiently couple signal inputs from multiple pathways into a specific output behavior in the cell (Stradal and Scita, 2006; Pawson, 2007). Distribution of shared subunits among protein complexes with distinct functions is another mechanism to coordinate information flow and cellular activities (Krause et al., 2004; Huberts and van der Klei, 2010). The highly specific, contingent, and plastic behaviors of protein complexes generate robust signaling systems that could not be constructed with monomeric proteins and posttranslational modifications.
Protein complexes are typically assembled through distinct binary interactions between pairs of subunits. Binding of two proteins cannot be effectively gleaned from genome sequencing and bioinformatics-based protein predictions alone. Consequently, extensive effort has been directed toward generating proteome-wide maps of protein-protein interactions in model organisms from all kingdoms of life. Clever and effective techniques for large-scale detection of pairwise protein-protein interactions have been developed, such as phage display (Jacobsson and Frykberg, 1996), protein chips (Zhu et al., 2001), and the yeast two-hybrid system (Fields and Sternglanz, 1994; Jansen et al., 2003). Successful large-scale yeast two-hybrid screens of soluble proteins have been conducted in the model plant Arabidopsis (Yu et al., 2008; Mukhtar et al., 2011); however, there are technical limitations to the yeast two-hybrid system (Jansen and Gerstein, 2004; Wodak et al., 2009), and it is only capable of detecting binary interactions. Tandem affinity purification (TAP) (Rigaut et al., 1999) and related affinity purification strategies employ trans-gene expression technology to introduce affinity tags that allow biochemical isolation of all proteins in a stable complex via a single uniform protocol followed by mass spectrometry (MS) to identify copurifying proteins (Gavin et al., 2006). In plants, TAP has been used to efficiently discover protein complexes and analyze important aspects of cell cycle control, 14-3-3 client proteins, and other basic cellular processes (Braun et al., 2013). A disadvantage of TAP is that each tagged protein must be analyzed individually, and broad proteome coverage would require the generation of thousands of transgenic lines and greater numbers of parallel purifications and MS analyses. The expression level of the tagged proteins and the effect of the affinity tag on protein function are important technical considerations for this technology (Wodak et al., 2009). Importantly, TAP is not possible in most crop plants because it is not feasible to generate the enormous number of high-quality transgenic lines that would be needed.
An alternative approach to protein complex analysis is to capitalize on the parallel protein detection and quantification capability of liquid chromatography-tandem mass spectrometry (LC-MS/MS) to analyze endogenous protein complexes (Dong et al., 2008; Liu et al., 2008). In plants, the potential benefits of label-free MS protein quantification have been discussed (Thelen and Peck, 2007), and these methods have been used to analyze the size and putative composition of endogenous protein complexes in the chloroplast stroma that were separated by native gel electrophoresis (Peltier et al., 2006) and size exclusion chromatography (SEC) (Olinares et al., 2010). This powerful approach requires only a well-annotated genome sequence and retains useful information on subcellular localization. In order to use profiling-based methods to predict protein complex composition from a cell fraction, hundreds of LC-MS/MS samples from different column fractions would be needed to sufficiently reduce the confounding effects of chance coelution. Such strategies are being developed in human cell culture systems, in which MS-based abundance profiling of native complexes has been coupled with different types of column chromatography and bioinformatic analyses to make global predictions about protein complex composition (Havugimana et al., 2012; Kristensen et al., 2012) or analyze how protein complexes can change as a function of growth factor treatment (Kristensen et al., 2012) or posttranscriptional control (Kirkwood et al., 2013). However, at present, the extent to which LC-MS/MS-based profiling methods are sufficient to accurately predict the interactome is not known, and similar methods have not been applied to an intact organ, which could provide a useful physiological system to analyze protein complex behaviors.
We seek to develop a protein complex analysis technology for plants that couples cell fractionation and multidimensional chromatographic separation of protein complexes with label-free LC-MS/MS protein abundance profiling across all chromatography fractions. As a first step toward this goal, we begin with intact leaves of Arabidopsis and report on the use of LC-MS/MS to measure the relative abundance profiles and apparent masses of soluble proteins following their separation by SEC. The cytosol is an important intracellular compartment for cellular organization and metabolism and has been previously subjected to a proteomic analysis in Arabidopsis protoplasts (Ito et al., 2011). Application of our method to the crude cytosolic fraction from leaf extracts revealed hundreds of proteins with apparent masses indicative of stable multimeric complexes, many of which appear to be novel. This article demonstrates the feasibility of using label-free quantitative MS to simultaneously analyze the oligomerization state and localization of endogenous protein complexes and sets the stage for the addition of other chromatographic steps that should allow the determination of protein complex composition.
RESULTS
Overview of the Proteomic Pipeline
The aim of this study was to develop a LC-MS/MS-based proteomics workflow that would allow us to globally monitor protein complexes. We implemented and assessed the method by analyzing the extent to which Arabidopsis leaf cytosol includes a system of proteins assembled into stable multimeric complexes. The overall approach is outlined in Figure 1. A crude cytosol fraction was prepared from 21 d after germination plants under native condition by differential centrifugation and the final 200,000g supernatant was defined as the crude cytosolic fraction. Protein complexes in the crude cytosol were separated using SEC. The proteins in each fraction were analyzed by LC-MS/MS in a gel-free workflow and identified by Mascot database searches (Figure 1). The column was calibrated and the apparent masses of the endogenous proteins (Mapp) were determined based on the elution peaks. To estimate whether or not a protein existed as part of a stable complex, the ratio of Mapp to the predicted monomeric mass (Mmono) was calculated. This ratio, which we call Rapp, should reflect the oligomerization state of most proteins (Figure 1). Thirty-four SEC fractions were analyzed, but only the first 20 contained intact proteins. Therefore, fractions 1 through 20 were used to determine apparent masses. Two independent cytosol preparations designated as biological replicate 1 (Biol1) and biological replicate 2 (Biol2) were analyzed in this manner.
Figure 1.

Schematic Overview of the Experimental Workflow.
Proteins were extracted from intact Arabidopsis leaves under native conditions by Polytron grinding and cytosolic proteins were enriched by differential centrifugation. After separation by SEC, proteins were identified by LC-MS/MS and MASCOT database searches. Relative abundances of proteins were estimated using spectral counts, and the peak elution fraction was used to calculate apparent molecular masses, from which multimeric protein assemblies were inferred. Mapp, apparent molecular mass; Mmono, predicted molecular mass of monomer; Rapp, the ratio of the Mapp to the Mmono; Biol1, biological replicate 1; Biol2, biological replicate 2. See Methods for additional details.
Cell Fractionation and Analysis of Cytosol Purity
The initial step of leaf grinding unavoidably leads to chloroplast breakage and contamination, but we chose this method because we wanted to analyze the cytosolic protein complex network under near-normal growth conditions. A cytosolic fraction with greater purity can be obtained from cell wall digestion, protoplast isolation, and gentle cell lysis (Ito et al., 2011); however, the hours of wall digestion causes a cell stress response that could alter protein complex composition. In addition, no method can produce a truly “pure cytosol,” and there always is a need to filter out contaminants. Therefore, we chose to accept the reduced purity of our sample and use reliable knowledge-based data sets and bioinformatic tools to remove contaminating proteins.
To assess the extent of contamination of our cytosol preparations by proteins from other cellular compartments, we analyzed cell fractions by protein gel blot using antibodies to a variety of organelle markers. The 200,000g supernatant was enriched for cytosolic proteins, as expected, because known cytosolic enzymes such as PHOSPHOENOL-PYRUVATE CARBOXYLASE (PEPC) and ENOLASE were primarily detected in this fraction (Figure 2A). Glycolytic enzymes are known to associate with organelles (Giegé et al., 2003), and our detection of ENOLASE also in the 10,000g and 200,000g pellets was consistent with some association with membranes and other organelles (Figure 2A). A peripheral membrane protein of the Golgi, the COPI coat protein SEC21, was present in the cytosolic fraction but was partitioned more extensively into the microsomal fractions. Integral membrane marker proteins from the plasma membrane (H+-ATPase), endoplasmic reticulum (ER; SEC12), or prevacuolar compartment (SYP22) were not detected in the cytosolic fraction, suggesting minimal contamination from the membranes of these compartments. However, we found evidence for contamination of the cytosol fraction with the luminal contents of broken organelles. The major source of cytosol contamination came from broken chloroplasts. Figure 2B shows a Coomassie blue-stained gel, with a prominent band that was identified by LC-MS/MS analysis as the large subunit of RIBULOSE BISPHOSPHATE CARBOXYLASE/OXYGENASE (RBCL), a highly abundant, chloroplast-encoded protein that is released into the soluble fraction from lysed chloroplasts. The ASPARTIC PROTEINASE (ASP) accumulates in the lumen of the vacuole. The detection of a fraction of the ASP pool in the soluble fraction could reflect either artifactual release of a luminal pool during homogenization or the existence of a cytosolic pool that has been reported previously (Ito et al., 2011). We also detected a small cytosolic pool of the ER lumen chaperone BIP (Figure 2A). It is possible that cytosolic pools of luminal proteins exist in vivo, either through alternative transcription start sites or mRNA processing that removes the signal sequences that target them to the secretory pathway. However, for the purpose of this work, we took the conservative approach of removing all proteins from our list of cytosolic proteins if the gene model includes a signal sequence or membrane-spanning domain identified using established bioinformatic methods (see below). Taken together, we conclude that our leaf homogenization and differential centrifugation strategy provides an enriched but highly contaminated cytosolic fraction. However, prior knowledge of protein localization and bioinformatics can be used to remove contaminants and enable a targeted analysis of the cytosol. Although we have focused on the cytosol here, the same approach should be suitable for other organelles.
Figure 2.

Purity of the Cytosol Preparation from Intact Leaves.
(A) Protein gel blots of different subcellular fractions separated by differential centrifugation. The fractions were blotted for proteins that localize to specific cellular compartments. SEC12, BIP, H+-ATPase, SEC21, SYP22, and ASP were used as markers for ER, plasma membrane (PM), Golgi, prevacuolar compartment (PVC), and vacuole lumen (VAC), respectively. PEPC and ENOLASE were used as cytosol (Cyt) markers. P, pellet; S, supernatant; CHL, chloroplast.
(B) Coommassie blue-stained SDS-PAGE gel showing distribution of RBCL in different subcellular fractions.
SEC Separation of Soluble Proteins and Reproducibility of LC-MS/MS Analysis
SEC effectively separated the soluble leaf proteins. The absorbance chromatogram and SDS-PAGE analysis of SEC fractions from one cytosolic preparation are shown in Supplemental Figure 1. SDS-PAGE revealed many proteins whose masses, based on electrophoretic mobility, were greatly exceeded by the apparent mass of their respective SEC peak fractions based on column calibration with standard globular proteins. This indicated that many stable oligomeric complexes were present in the sample.
The two biological replicates were used to test the reproducibility of our LC-MS/MS method with respect to protein identification and quantitation across SEC fractions. Mass spectra from each of the 34 SEC fractions were searched against the TAIR10 Arabidopsis protein database for protein identification. Decoy database searches and peptide score thresholds were set to yield a false discovery rate (FDR) of 1.5% at the protein level. The relative abundances of proteins across the SEC fractions were determined using spectral counts. Spectral counts are the total number of MS/MS spectra of all the peptides mapped to a protein during the entire LC-MS/MS run, and this number correlates with its abundance (Liu et al., 2004). Assigning spectral counts to a protein is complicated by the presence of multiple isoforms in the cell. Arabidopsis has undergone two genome duplication events (Blanc et al., 2003), and small gene families and protein isoforms are common. To account for protein isoforms, the total spectral counts among isoforms were divided into shared and unique peptide counts. The shared counts were then distributed among the isoforms according to the proportion of unique counts (Zybailov et al., 2006; Olinares et al., 2010). For simplicity, these adjusted spectral counts were subsequently defined as “spectral counts” in this article, and these values were used for quantification and comparisons between the biological replicates. The resulting analysis generated spectral counts for each protein across the 34 SEC fractions. There were 1601 proteins identified in Biol1 and 1575 in Biol2, of which 1283 proteins (∼80% of the total proteins identified in Biol1 and Biol2) were identified in both (Figure 3A), which is a typical level of reproducibility for an MS-based proteomic analysis. The set of proteins identified in both replicates was used for further analyses. Detailed information on the peptides mapping to these proteins is provided in Supplemental Data Set 1. Additional detailed information on identified proteins is available in Supplemental Data Sets 2 to 4, as further described below.
Figure 3.

Reproducibility of the LC-MS/MS Analyses.
(A) A Venn diagram showing the overlap of identified proteins between the two replicates.
(B) Matrix representation of Pearson correlation coefficients of relative protein abundances (spectral counts) between all possible pairs of SEC fractions in the two biological replicates. The correlation matrix was generated using Data Analysis and Extension Tool (DAnTE) (Polpitiya et al., 2008). Proteins identified in the first 20 SEC fractions in both the replicates that were within the mass resolution of the column were used. Color scale indicates the range from lowest correlation (dark blue) to highest correlation (dark red) coefficient.
Figure 3B is a heat map of the Pearson correlation coefficients of the spectral counts for the full set of identified proteins between Biol1 and Biol2 as a function of SEC fraction number. The highest correlation coefficients clearly fell along the diagonal, which indicated the protein elution peaks were similar between the biological replicates. We conclude that protein identification and spectral count quantification are consistent across the column fractions and our overall approach is reproducible. The slightly elevated correlation signal in the lower left quadrant of the graph reflected protein degradation, which was detected in low apparent mass fractions of both biological replicates, but was more evident in Biol1.
Identification and Characterization of Cytosolic Proteins
We sought to restrict our detailed analyses to the subset of soluble proteins that were likely to be cytosolic. Therefore, we applied several filtering criteria to remove likely noncytosolic contaminants from our protein list, despite the risk that many proteins that function in other organelles may also exist as cytosol-localized pools with alternate functions. We first searched for and eliminated proteins encoded by the chloroplast and mitochondrial genomes. Only 4 of the 90 chloroplast-encoded proteins and none of the 124 mitochondrion-encoded proteins were found. Next, TargetP 1.1 (Nielsen et al., 1997; Emanuelsson et al., 2007) was used to identify proteins with likely secretory and organelle-targeting signals, resulting in removal of 166 putative secreted proteins, 106 putative chloroplast-targeted proteins, and 33 putative mitochondria-targeted proteins. An additional 242 proteins from a previously published chloroplast proteome (Olinares et al., 2010) that contain chloroplast-targeting signals and were identified in our data set were also removed. Finally, 19 predicted transmembrane domain (TMD)-containing proteins that are unlikely to exist in soluble cytosolic form were removed. Several of these proteins contain multiple predicted TMDs. For example, cation efflux family protein (AT2G04620) is predicted to contain 14 TMDs, ATP binding cassette subfamily B4 (AT2G47000), nine TMDs, and CELLULOSE SYNTHASE-LIKE B1 (AT2G32610), eight TMDs. Many of these putative integral membrane proteins are not detected as low mass degraded proteins that could have been artifactually released into the cytosol fraction, and the mechanism by which they are partitioned into the soluble phase is not known. In Supplemental Data Set 2, we included the spectral count profiles and estimated apparent mass for all the identified proteins in both biological replicates; however, further analyses were restricted to the cytosolic proteins.
Filtering contaminants from our data set left us with 713 predicted cytosolic proteins (Supplemental Data Set 2) that were subjected to further analysis. The total number of cytosolic proteins detected in this study is comparable to a previous report (Ito et al., 2011), with 353 proteins identified in both studies. Based on MapMan classification (Thimm et al., 2004b), our list includes proteins with a diverse array of functions, including protein degradation pathways (20% of proteins), signaling (8%), stress (6%), cell organization (7%), amino acid metabolism (6%), and secondary metabolism (4%) (Supplemental Figure 2). To estimate the relative abundance of the detected cytosolic proteins, the spectral counts across all fractions were summed and divided by amino acid number to normalize for protein size (Paoletti et al., 2006; Schmidt et al., 2007). The normalized counts were rescaled to the range of 1 to 1000. These abundance scores are included in Supplemental Data Set 3 and were reproducible between the biological replicates. More than half of our cytosolic proteins were enzymes based on the AraCyc database of Arabidopsis biochemical pathways (http://www.arabidopsis.org/tools/aracyc) and included components of glycolysis, the pentose phosphate pathway, and nucleotide and hormone biosynthesis. Our coverage of most pathways was sparse, which was not surprising, because when the same contaminant removal criteria are applied to the entire Arabidopsis proteome (TAIR10), ∼7700 cytosolic proteins were predicted. There is considerable room to improve proteome coverage with further protein fractionation, increased mass spectrometer speed and sensitivity, and the use of RBCL depletion methods (Cellar et al., 2008; Aryal et al., 2012). Of the 713 cytosolic proteins, 648 proteins were detected in the first 20 fractions and were analyzed for their oligomerization states (see below).
Validation of Global SEC Peak Determinations
As a first step toward the eventual identification of novel protein complex subunits, we performed a distance-based clustering analysis of the spectral count profiles. After removal of degraded proteins with Rapp values of < 0.5, the clustering result was plotted as a heat map (Figure 4A). A high-resolution image of Figure 4A that can be searched by locus IDs is available as Supplemental Data Set 5. Most proteins displayed a single major SEC peak, allowing effective clustering based on the profiles. A minority of proteins exhibited multiple peaks or broad elution profiles that may represent both monomeric and/or a variety of oligomeric forms. Deconvolution of the chromatogram into its constituent peaks was not possible in this study due to many low spectral count fractions and the highly abundant RBCL protein that caused obvious signal suppression of the coeluting proteins during the LC-MS/MS runs and hence generated a large number of artifactual peaks. Therefore, for this study, we only used the global maximum of the spectral counts for each identified protein as the peak for Mapp calculation.
Figure 4.

Validation of Spectral Count-Based Protein Abundance Profiling.
(A) Hierarchical clustering of protein elution profiles. Proteins identified in both replicates were clustered based on the similarity of their spectral count elution profiles, which are displayed as a heat map of relative protein abundance.
(B) and (C) ENOLASE (B) and PEPC (C) antibodies were used for protein immunoblot validation of SEC elution peaks. The arrow indicates peak elution fraction based on LC-MS/MS spectral counts, which was used to determine the Mapp. NF, nonfractionated input.
(D) and (E) Elution profiles of 20S core and fully assembled 26S proteasomes that includes lower abundance regulatory subunits of the proteasome.
(F) Strong agreement between the LC-MS/MS-measured apparent masses and previously published values using SEC. The correlation was strong for both replicates. Only replicate 2 is shown.
(G) Comparison of apparent masses from this study with masses of solved structures obtained from the PDB.
To assess the accuracy of this protein identification and quantitation strategy, we compared the spectral count-based abundance profiles for two proteins, ENOLASE and PEPC, to those obtained using an orthogonal technique, protein gel blotting (Figures 4B and 4C). There was good agreement between the methods in assigning the peak signal for both proteins. However, both protein quantification methods are somewhat noisy, leading to some unexpected fluctuations in signal intensity. Variability of transfer efficiency is a major source of noise for the protein gel blots, and this signal fluctuation is exacerbated by the low dynamic range of the film compared with the greater dynamic range of the mass spectrometer. Nonetheless, the elution peaks were consistently identified and the Mapp of ENOLASE and PEPC was 105 and 377 kD, respectively. These values are close to the published values of 80 to 100 kD for ENOLASE (depending on the isoforms; Pancholi, 2001) and 440 kD for PEPC (O’Leary et al., 2009). Therefore, the Rapp values of 2.2 for ENOLASE and 3.4 for PEPC were close to their known dimeric (Pal-Bhowmick et al., 2004) and tetrameric (O’Leary et al., 2009) forms, respectively.
Subunits of known protein complexes should coelute on the SEC column. Therefore, to further validate our profiling strategy, we tested for coelution of the known subunits of the proteasome. The proteasome is responsible for the majority of regulated protein degradation in eukaryotic cells (Finley, 2009). The highly abundant catalytic 20S core particle (CP) is a heteromeric assembly of 28 α- and β-subunits, all of roughly 25 to 30 kD size, and has a predicted mass of 700 kD. Consistent with this, we identified 16 abundant CP subunits that coeluted as a single peak in fraction 3 (Figure 4D), with an estimated mass of 577 kD. The 26S proteasome holoenzyme consists of two regulatory particles (RPs) capping each end of the barrel-shaped 20S CP and is less abundant than the free 20S CP (Tanahashi et al., 2000). We identified three RP subunits (Figure 4E) that were of much lower abundance compared with the CP subunits. The three RP subunits eluted a fraction earlier than the CP subunit peak, consistent with the larger size of the 26S holoenzyme. These results further demonstrate the accuracy of our global method for protein complex characterization.
We next compared our Mapp values with published apparent masses of 25 proteins in our data set (Supplemental Table 1). A plot of Mapp against masses reported in the literature revealed strong agreement, with an r2 = 0.83 (Figure 4F). Finally, we compared our results with mass values of protein complexes derived from crystal structures present in the Protein Data Bank (PDB). Since PDB contains 172 Arabidopsis proteins (Tier 1), we included homologs from human, yeast, worm, and fruit fly as long as complete conservation of subunit composition was evident between plant and non-plant complexes (Tier 2, 3693 proteins). The deduced masses of these protein complexes in the PDB database correlated well with our Mapp values (r2 of 0.66 in replicate 1 and 0.72 in replicate 2; Figure 4G; Supplemental Data Set 4). However, in many instances, our deduced masses were greater than those obtained from PDB, suggesting that in many cases the solved complex, usually derived from recombinant proteins, does not capture the complete composition of the endogenous complex.
Quantitative Metrics for Protein Complex Prediction
Having established the validity of the spectral count-based relative abundance profiling method for estimating functional protein masses, we wanted to make predictions about which cytosolic proteins are likely to assemble into stable protein complexes. There was general support for the existence of protein complexes in our LC-MS/MS data set because the distribution of predicted monomer masses is concentrated in a much narrower and lower mass range than the distribution of measured apparent masses (Figure 5A). We used Rapp as a metric to predict whether or not a given protein existed in an oligomeric state. A value of 2 for Rapp has been previously used as an indicator of protein complex formation (Liu et al., 2008), reflecting a substantial deviation from the expected mobility of a monomeric, globular protein. Rapp values greater than 2 are unlikely to be generated by inconsistent or inaccurate mass determinations because this would correspond to a 4 fraction shift in our chromatography system. Errors of such magnitude were very uncommon in our data set. The Rapp scores for the cytosolic proteins were graphically compared between replicates in Figure 5B. The distinct linear patterns in the data are a consequence of the discrete mass values assigned to the finite number of SEC fractions. About 30% of the detected proteins fell on the diagonal and had an identical elution peak in the two biological replicates. Forty percent of the proteins had a one-fraction shift, and 22% had a two fractions shift between the biological replicates. Only 8% exhibited highly variable elution peaks. Despite the overall accuracy of the method, Rapp does have limitations and any cutoffs are somewhat arbitrary and will lead to some level of incorrect predictions. For example, false positives will occur when a monomeric protein has an elongated shape and its increased hydrodynamic radius leads to overestimation of apparent mass. False negatives will occur when large proteins associate with much smaller partners that do not sufficiently increase the hydrodynamic radius to generate an Rapp greater than 2. For these reasons, we referred to the proteins that exceed our Rapp cutoff criteria as putative protein complex subunits. The scatterplots in Figure 5C shows the scoring schemes of all 648 cytosolic proteins based on their Rapp values and associated apparent molecular masses in Biol2 compared with the monomers. Rapp values ranged from 40 to 0.05 in replicate 1 and from 54 to 0.07 in replicate 2 (Supplemental Data Set 3). However, in borderline cases in which Rapp values were very close to 2, we applied a rule in which a protein is considered to have quaternary structure if the Rapp was 2 or greater in one biological replicate and at least 1.5 in the second biological replicate. Using these criteria, 41% of the cytosolic proteins detected in both biological replicates were predicted to be oligomeric. These proteins were functionally diverse and included enzymes, signaling proteins, and putative nuclear factors, and 42 protein complexes that contain proteins of no known function. We saw no evidence for protein aggregation-driven complex formation because when we plotted the protein abundance estimates against Rapp for the predicted oligomeric forms there was no correlation (r2 = 0.00074 for replicate 1 and r2 = 0.0005 for replicate 2; Supplemental Figure 3 and Supplemental File 1). Therefore, the probability of a protein being detected in an oligomeric state was not related to its abundance. The magnitude of Rapp also was not driven by the monomeric mass of the detected protein because the mean monomeric masses of the total population and the subset with Rapp > 2 were not significantly different in both the biological replicates according to a Mann-Whitney test (P = 0.65 for Biol1 and 0.63 for Biol2). A subset of the proteins appeared to exist in a highly oligomeric state, with Rapp > 5 in both the replicates (Supplemental Table 2). This list contains several examples of proteins that are not known to exist as large multimeric complexes. This highlights the power of the approach to identify previously unknown protein assemblies.
Figure 5.

LC-MS/MS-Based Abundance Profiling Can Be Used to Predict the Protein Complex Formation.
(A) Histogram showing the different distributions of the monomeric (blue) and experimentally determined apparent masses (green and red) of the cytosolic proteins that were quantified in both biological replicates.
(B) Scatterplot showing Rapp distribution of 648 cytosolic proteins. Circles represent Rapp values. The proteins (∼78%) were identified with reproducible Rapp scores between the two replicates within 0 to 2 fraction shifts in SEC elution profiles. Inset: zoomed view of the boxed region of the plot indicating in gray the Rapp thresholds that were used in borderline cases to predict protein complex formation.
(C) Graphical display of oligomerization state of cytosolic proteins. Scatterplots of the apparent and the monomer masses of proteins in replicate 2 (inset: zoomed view of the boxed region of the plot).
DISCUSSION
Plant cells respond to endogenous signals and changing environmental conditions as a system in which the activities of thousands of protein complexes are modulated to generate an adaptive response. Accordingly, there is a strong need to develop new methods to measure the size, location, and composition of protein complexes, as well as monitor their dynamics under different conditions. The ability of mass spectrometry to simultaneously detect and quantify thousands of proteins makes it an appealing technology to address this need. For example, it has already been shown that organelle purification, coupled with native gel electrophoresis and quantitative protein MS, can identify proteins that reside in large, megadalton complexes in the chloroplast stroma (Olinares et al., 2010). In this article, we began with intact leaves and combined cell fractionation, SEC, and LC-MS/MS to globally analyze the oligomeric state of cytosolic proteins. This method is reliable and unbiased because it monitors the assembly status of endogenous protein complexes on an SEC column and only requires a well-annotated genome to enable accurate protein identification. Therefore, it is easily applicable to many species and subcellular fractions other than the cytosol.
Our protein complex analysis platform provides reliable estimates for the mass of soluble proteins. The technical advance is that the apparent masses of ∼1300 leaf-expressed proteins were determined in parallel, using a gel-free, label-free, LC-MS/MS workflow. This analysis provided cytosol localization and oligomerization data for 83 proteins of unknown function, many of which appear to be in a protein complex. Although we used spectral counts to quantify MS signal and define chromatographic profiles, other label-free quantitative approaches, including MS1 extracted ion chromatogram integration, or new data-independent acquisition, will likely provide better quantification. Overall, our measurements of protein mass were reproducible and accurate. Our method was validated by comparing the LC-MS/MS-derived mass estimations with those obtained by protein immunoblots (Figures 4B and 4C) and those reported in previous studies (Figures 4F and 4G). Using the proteasome as an example, we demonstrated the precise cofractionation of subunits of a large complex and the ability to distinguish distinct forms of a complex (Figures 4D and 4E). Importantly, we demonstrated the ability to use a clustering algorithm to identify those proteins with highly similar chromatographic elution profiles. This analysis will be critical as we further develop this technology to identify the components of novel complexes.
We used our apparent mass measurements of proteins in the soluble cytosolic fraction to predict whether or not each exists in a stable protein complex using the Rapp. Based on comparisons of our Rapp values to the subunit composition of known complexes, we demonstrated it to be a reasonable metric for oligomeric state, and it allowed us to define a population of proteins that putatively exist in stable multimeric complexes (those with an Rapp greater than 2.0). Furthermore, because an Rapp value of 2 is associated with a roughly 4 fraction shift in SEC mobility compared with its monomeric form, we believe these measurements and assignments are influenced negligibly by variation in the SEC and our LC-MS/MS analyses. We acknowledge that use of Rapp has limitations, and in some cases, we are likely to misidentify large proteins as monomeric in cases where they interact with proteins that are relatively small. However, overall our Rapp predictions agree very well with the oligomerization state of known protein complexes, and provide a useful benchmark for more directed studies.
Our proteomic pipeline detected many large, apparently novel, protein complexes (Supplemental Table 2), a few of which will be highlighted below. In some instances, we detected proteins with broad elution profiles and multiple SEC peaks. For example, the glycolytic enzymes ALDOLASE and GAPC are distinct compared with other glycolytic enzymes in that their elution profiles are very broad and complex (Figure 6B). ALDOLASE and GAPC are known moonlighting proteins (Lee et al., 2002; Huberts and van der Klei, 2010; Tunio et al., 2010), and their complex elution profiles might reflect the existence of compositionally and functionally distinct protein complexes. We may have also uncovered moonlighting functions for proteins involved in protein translation. There are 20 cytosolic aminoacyl t-RNA synthetases/ligases in Arabidopsis, and we detected 11 of them in our study (Supplemental Figure 4), all of relatively low abundance. This result is expected because the primary function of the enzymes, the specific aminoacylation of tRNAs, occurs in the nucleus (Lund and Dahlberg, 1998). Of the 11 tRNA ligases, two had an obvious quaternary structure. GLUTAMINYL-tRNA LIGASE/AT5G26710 is an 81-kD protein that was reproducibly detected in high mass fractions with Rapp values of 7.1 and 8.8 (Supplemental Table 2 and Supplemental Figure 4). ISOLEUCINE-tRNA LIGASE/AT4G10320 is a 135-kD protein and is the only other tRNA ligase that had a peak abundance in high mass fractions. In many organisms, tRNA ligases have multiple functions that are independent of protein synthesis (Jahn et al., 1992; Martinis et al., 1999; Park et al., 2005), and perhaps that is also the case in Arabidopsis.
Figure 6.

Oligomerization State of Enzymes in the Cytosolic Glycolytic Pathway.
(A) Overview of the cytosolic glycolytic pathway showing the enzymes and their substrates. Adjacent to each enzyme is the Rapp score (blue) and the enzymes relative abundance (red). The enzymes catalyzing each reaction and detected in this study are illustrated by symbols as follows: PGM, PHOSPHOGLUCOMUTASE; SIS/GPI, SUGAR ISOMERASE/GLUCOSE-6-PHOSPHATE ISOMERASE; PFK, PHOSPHOFRUCTOKINASE; FBA, FRUCTOSE BISPHOSPHATE ALODOLASE; TPI, TRIOSEPHOSPHATE ISOMERASE; GAPC, GLYCERALDEHYDE-3-PHOSPHATE DEHYDROGANSE C SUBUNIT; ALDH/GAPN, ALDEHYDE DEHYDROGENASE/NADP-DEPENDENT GLYCERALDEHYDE-3-PHOSPHATE DEHYDROGENASE; PGK, PHOSPHOGLYCERATE KINASE; GPM, PHOSPHOGLYCERATE MUTASE; ENOS, ENOLASE; PEPC, PHOSPHOENOLPYRUVATE CARBOXYLASE; PK, PYRUVATE KINASE. The abbreviations are as follows: Glu-1-P, glucose-1-phosphate; Glu-6-P, glucose-6-phosphate; Fru-6-P, fructose-6-phosphate; Fru-1,6-P2, fructose-1,6-bisphosphate; DHAP, dihydroxyacetone phosphate; G-3-P, glyceraldehyde-3-phosphate; 1,3-DPGA, 1,3-diphosphateglycerate; 3-PGA, 3-phosphoglycerate; 2-PGA, 2-phosphoglycerate; PEP, phosphoenolpyruvate; OAA, oxaloacetate. Arrows indicate the direction of the reaction. → Indicates physically irreversible reaction, or ⇌ indicates physically reversible reaction. Numbers in parentheses represent the Rapp values for replicate 2 (blue) and the scaled relative abundance values (red). Rel. Abun., relative abundance.
(B) Elution profiles of detected glycolytic enzymes shown in (A). Profiles are organized in columns to reflect their order in the pathway.
The dehydrin proteins are classic transcriptional markers for signaling output of pathways that allow plants to effectively adapt to the osmotic and oxidative stresses associated with drought (Saavedra et al., 2006). The dehydrins are a group of small, highly charged proteins present during normal growth conditions and that may interact with membranes during stress conditions. The dehydrin COR47 is a classic marker for osmotic stress (Welin et al., 1995), and like other dehydrins, it is almost always analyzed in terms of its transcriptional upregulation in response to cold or osmotic stress. We detected the 30-kD COR47/AT1G20440 protein, a relatively abundant protein with reproducible elution peaks in the ∼200 kD mass fraction (Supplemental Table 2 and Supplemental Figure 5). We also detected the Arabidopsis homolog of maize (Zea mays) TANGLED, which is an important cytoskeletal control protein that positions a specialized microtubule structure termed the preprophase band to define the future plane of cell division and location of new cell wall formation during cytokinesis (Walker et al., 2007). The 49-kD TANGLED was weakly detected, but reproducibly peaked in high mass fractions in the two biological replicates (Supplemental Table 2 and Supplemental Figure 5). Despite the variation in apparent mass, our data indicated that TANGLED exists in a relatively large, stable, oligomeric cytosolic complex. TANGLED is recruited from a cytosolic pool to the preprophase band in a cell-cycle-dependent manner (Rasmussen et al., 2011). It will be important to determine how the oligomerization state of TANGLED and its cytosolic pool relates to its cortical functions during cell division.
Our experimental system is expected to detect proteins that are partitioned between the cytosol and the nucleoplasm; indeed, there were a number of proteins with known nuclear functions as apparent subunits of large cytosolic complexes. We cannot rule out the possibility that this is an artifact of contamination; however, we did not detect abundant nuclear proteins such as histones, high mobility group chromatin-associated proteins, or multiple subunits of RNA polymerase complexes. We therefore propose that our data set reveals nuclear proteins that may either be regulated by cytosolic localization or they may have independent functions in the cytosol. We may have uncovered a cytosolic function for a domesticated transposase. We detected the hAT-family transposase named DAYSLEEPER/AT3G42170 in a large cytosolic complex (Supplemental Table 2 and Supplemental Figure 6). It is a domesticated transposase that is present in angiosperms and has a proven importance for plant development in Arabidopsis (Bundock and Hooykaas, 2005; Knip et al., 2012). We detected the 30-kD SC35-LIKE SPLICING FACTOR 30A/SCL30A as a component of a large complex greater than 700 kD in both biological replicates that may have cytosolic functions (Supplemental Table 2). We also detected putative transcription factors in our analysis of cytosolic protein complexes. An 86-kD myb-like TRF class putative transcription factor (AT1G58220) was detected in a high mass complex greater than 700 kD in both replicates (Supplemental Table 2 and Supplemental Figure 6). The expression of AT1G58220 is induced by jasmonic acid and auxin (Yanhui et al., 2006). Perhaps the nuclear localization and function of AT1G58220 is conditional and its subcellular partitioning is mediated by this large cytosolic protein complex. All of these complexes merit further study.
One immediate application of our method is to analyze the oligomerization state of enzymes that function within a specific metabolic pathway and how it might change under different growth conditions. For example, glycolysis is perhaps the most ancient and conserved pathway in primary metabolism and in many instances enzyme activity is related to oligomerization (Plaxton, 1996). There is experimental evidence in plant and microbial systems for physical associations among enzymes in the pathway that could mediate substrate channeling and/or efficient delivery of pyruvate, the end product of the pathway, into mitochondria (Brandina et al., 2006; Gavin et al., 2002). In the hypothetical extreme case in which substrate channeling is an inherent and stable property of all enzymes in the glycolytic pathway, one would expect to detect coelution of sequential enzymes. This overly simplistic regulatory scheme is not supported by our experimental data (Figure 6). For example, PHOSPHOGLUCOMUTASE (PGM), SUGAR ISOMERASE (SIS), and PHOSPHOFRUCTOKINASE (PGK) are sequential enzymes in the early steps of the pathway and they all have distinct elution peaks with PGM and SIS detected as monomers (Figure 6B). Along similar lines, the later acting glycolytic enzymes, PHOSPHOGLYCERATE KINASE (PGK), PHOSPHOGLYCERATE MUTASE (GPM), and ENOLASE2 (ENOS2) have distinct elution profiles and therefore do not appear to exist primarily as stable subunits of a hetero-oligomeric complex. However, higher order protein complexes containing sequential enzymes may exist as transient complexes that are not detected using our method, or they may assemble more efficiently on organelle surfaces (Giegé et al., 2003; Graham et al., 2007).
In conclusion, the work described here provides a strong starting point for the parallel analysis of protein complexes using gel-free, quantitative MS. This particular SEC-based application provides a new way to globally analyze the oligomerization state of a system of protein complexes. Our data set points to the existence of many new, unexplored, protein complexes. We hope that these biochemical road maps to protein complexes will be used by the research community to accelerate the analysis of specific proteins of interest. This protein complex analysis method can also be expanded to analyze the dynamics of a large population of cytosolic proteins under different developmental or environmental conditions. The application also needs to be adapted to include the many plant protein complexes that associate with membrane surfaces. These microsomal fractions were discarded in this study. We already have developed robust methods for protein complex solubilization and SEC separation (Basu et al., 2008; Kotchoni et al., 2009), and we will expand this technology to include organelle-associated proteins.
A major challenge in the field is to use LC-MS/MS profiling to globally predict protein complex composition (Havugimana et al., 2012; Kristensen et al., 2012; Kirkwood et al., 2013). In the future, we will combine orthogonal chromatographic separations (either serially or in parallel) with LC-MS/MS profiling to extend the resolution space for stable complexes. Development of clustering analyses for the multidimensional chromatographic separations, combined with the quantitative spectral information and bioinformatic tools is expected to allow precise definition of subunit composition in novel protein assemblies. Other improvements will include RUBISCO depletion methods (Aryal et al., 2012) to minimize suppression of cofractionating proteins, more extensive proteome coverage using improved analytical instrumentation (liquid chromatography and MS), and improved accuracy of quantitative elution profiles by replacement of spectral counting with MS1 peak integration of detected peptides. These efforts will provide powerful new tools to broadly analyze the composition, localization, and dynamics of protein complex systems in the cell.
METHODS
Plant Growth and Leaf Cytosolic Protein Extraction
Arabidopsis thaliana (Columbia-0) seeds were imbibed and cold-treated at 4°C for 3 d and grown under sterile conditions at 22°C and continuous light using 0.5× Murashige and Skoog mineral salts with BactoAgar. After 10 d of growth, seedlings were transferred to new plates with fresh media at low density for 10 more days prior to leaf harvest. Young leaves including the petiole and midrib were collected from several seedlings of the same plate to prepare cell extracts.
Cell Fractionation
Two grams of fresh leaf material was homogenized by Polytron grinding (Brinkmann Instrument) at 4°C in homogenization buffer [50 mM HEPES-KOH, pH 7.5, 250 mM sorbitol, 50 mM KOAc, 2 mM Mg(OAc)2, 1 mM EDTA, 1 mM EGTA, and 1 mM DTT]. Prior to Polytron grinding, phenyl methyl sulfonylfluoride and protease inhibitor cocktail (160 μg/mL benzamidine-HCl, 12 μg/mL phenanthroline, 0.1 mg/mL aprotinin, 100 μg/mL leupeptin, and 0.1 mg/mL pepstatin A) was added to the homogenization buffer at a final concentrations of 2 mM and 1% (v/v), respectively. The slurry was centrifuged for 10 min at ∼500g (Juan Precision CR 42; Lab Care America) to remove cell debris. Centrifugation of supernatant at successively higher speeds at 4°C yielded the following fractions: crude nuclear and chloroplast fraction at 1000g for 15 min (1k pellet), mitochondria at 10,000g for 15 min (10k pellet), and microsomes at 200,000g for 45 min (220k pellet). The final supernatant (200k S) was the crude cytosolic fraction. Protein concentration was determined using the Bradford reagent (Bio-Rad). The cytosolic fraction was concentrated in 10-kD Amicon Microcon centrifugal filters (Millipore) to obtain a final volume of 1 mL.
SEC and Gel Electrophoresis
The two biological replicates were processed identically. Cytosol (0.5 mL, ∼1.8 mg) was loaded onto a Superdex 200 10/300 GL column (GE Healthcare) using an ÄKTA FPLC system (Amersham Biosciences). SEC elution was performed with 50 mM HEPES/KOH, pH 7.8, 100 mM NaCl, 10 mM MgCl2, and 5% glycerol at a flow rate of 0.3 mL/min and absorbance was measured at 280 nm. The column was calibrated using protein standards (MWGF1000; Sigma-Aldrich) covering a mass range from 669 to 29 kD. The void volume was measured with blue dextran.
SEC separation was performed at 6°C, and 34,500-μL fractions were collected. Proteins in each fraction were denatured in 8 M urea and concentrated to 50 μL using 10-kD Amicon Microcon centrifugal filters. A portion of each SEC fraction was mixed with 5× sample buffer (0.5 M Tris-HCl, pH 6.8, 5% SDS, 20% glycerol, and 0.05% bromophenol blue) and separated using SDS-PAGE. Proteins were visualized using Bio-Safe Coomassie Blue stain (Bio-Rad), and gels were scanned using an Epson Perfection 1240U photo scanner.
Proteolysis and Reverse-Phase Capillary LC-MS/MS Analysis
For LC-MS/MS analysis, 30 μL of the total 50 μL of protein sample in each SEC fraction was reduced by 10 mM freshly prepared dithiothreitol at 60°C for 45 min and cysteines alkylated with 20 mM iodoacetamide at room temperature in the dark for 45 min. After dilution with 50 mM NH4HCO3 to 200 μL, CaCl2 was added to a final concentration of 1 mM and proteins were digested with sequencing-grade modified porcine trypsin (Sigma-Aldrich) at a 1:50 (w/w) enzyme-to-substrate ratio for 5 h at 37°C. The resulting peptides were desalted using Pierce C18 spin columns (Pierce Biotechnology) following the manufacturer’s protocol, and reconstituted in 20 μL of 25 mM fresh NH4HCO3. One microliter was analyzed using an Agilent 1100 capillary HPLC system coupled on-line to a linear ion trap (LTQ)-Orbitrap mass spectrometer (ThermoFisher Scientific) via a nano-electrospray ionization source. Peptides were separated using Zorbax 300SB- C18 column (Agilent technologies). Peptides were first loaded into a 5 mm long × 0.3 mm inner diameter trapping column packed with 5-μm C18 particle size and eluted through a 150 mm long × 75 μm inner diameter analytical column packed with 3.5-μm C18 particles size with a 115 min gradient from 0 to 70% acetonitrile in 0.2% formic acid at a flow rate of 400 nL/min. The Orbitrap was operated in positive ion mode. Precursor ion spectra were acquired in profile mode and product ion spectra in centroid mode. Each MS survey scan (m/z 400 to 2000) was followed by collision-induced MS/MS spectra (normalized collision energy setting of 35%) for the 10 most abundant ions.
Data Analysis and Relative Protein Abundance Profiling
Acquired MS/MS .RAW files were converted into .DTA files and merged into .MGF in Mascot Daemon (Matrix Science) using Extract_msn by ThermoFinnigan LCQ/DECA RAW file data import filter. MS/MS spectra were searched against the Arabidopsis protein database downloaded from The Arabidopsis Information Resource (TAIR10; 35,386 protein sequences) using Mascot (http://www.matrixscience.com). To control the FDR, spectra were also searched against the corresponding reverse sequence database by selecting the decoy option. The search parameters were as follows: (1) mass tolerances of ± 10 ppm and ± 0.5 D for peptide and fragment ions, respectively, (2) carbamidomethylation of cysteine as a fixed modification and oxidation of methionine as a variable modification, and (3) one missed cleavage allowed. Peptide matches were accepted if the significance scores of their match had P value < 0.05. All of the Mascot search results were merged and exported to Microsoft Access 2007 and filtered to accept peptides with rank 1 and ion scores >4 to obtain a combined FDR ≤1.5% at the protein level for all fractions. Protein identification generally required a minimum of two unique peptides and a minimum of one unique peptide if other matched peptides were shared among multiple protein isoforms. Relative protein abundance distributions across SEC fractions were determined by spectral counts. To account for the presence of protein isoforms, the raw spectral count data were classified into total spectral counts, shared spectral counts, and adjusted spectral counts. The adjusted spectral count is the sum of unique spectral counts and a fraction of the spectral counts from peptides shared among multiple proteins and distributed in proportion to their unique spectral counts (Zybailov et al., 2006; Olinares et al., 2010). Adjusted spectral counts were determined by the following equation:
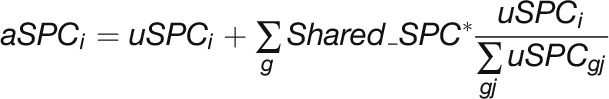 |
where uSPCi is the unique spectral count for i-th protein in the sample, and  is the sum of all the unique spectral counts of proteins with shared peptides defined as a group (g). The computer code and an example data form used to calculate aSPC is available at DRYAD. The SEC elution peak of each protein was defined as the fraction with maximum adjusted spectral counts.
is the sum of all the unique spectral counts of proteins with shared peptides defined as a group (g). The computer code and an example data form used to calculate aSPC is available at DRYAD. The SEC elution peak of each protein was defined as the fraction with maximum adjusted spectral counts.
To estimate the relative abundance of all identified cytosolic proteins, we first normalized the summed adjusted spectral counts across SEC fractions to protein length by using the following equation as described (Paoletti et al., 2006):
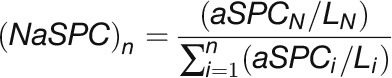 |
where NaSPC is the normalized adjusted spectral counts. In this equation, the total number of tandem MS spectra matching to protein N across SEC fractions (aSPCN) was divided by protein length (LN) in amino acid residues, then divided by the sum of aSPCN/LN for all proteins to determine a normalized scaled abundance. Values were scaled from 1 to 1000 using the equation given below.
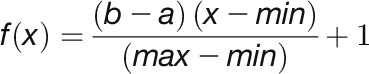 |
In this equation, a and b are the maximum and the minimum ranges of the scale, which in our case is 1000 and 1, respectively. The variable x is the NaSPC of a protein, and min and max are minimum and maximum NaSPC values in all identified cytosolic proteins, so [(b-a)/(max-min)] provides the scaling factor between the new range and the range of the NaSPC data. Spectral counts data were loaded into Data Analysis Tool Extension (DAnTE) version 1.2 (Polpitiya et al., 2008) and log2 transformed to generate correlation plots. We performed clustering analysis of all identified proteins using their adjusted spectral count profiles across SEC separations. Proteins were clustered based on similarities in these abundance profiles so that proteins in the same cluster were more similar than those in the other clusters. For each protein, the adjusted spectral counts across SEC fractions were first normalized between 0 (lowest spectral counts) and 1 (maximum spectral counts). Profile similarity between a protein pair was measured by the Euclidean distance. We then implemented hierarchical clustering using the statistical software R and plotted protein abundance as a heat map.
Removal of Cytosol Contaminants and Protein Functional Classification
To discriminate likely contaminants from true cytosolic proteins, the common set of identified proteins was systematically filtered using the following step-by-step process: (1) Any proteins encoded by the chloroplast and mitochondrial genomes were removed. (2) Chloroplast-targeted proteins were filtered by comparing with the published chloroplast proteome database (Olinares et al., 2010). (3) Remaining proteins were analyzed using TargetP (v1.1) (http://www.cbs.dtu.dk/services/TargetP/) (Emanuelsson et al., 2007), and proteins having reliability class of 1 or a specificity ≥0.7 to mitochondria, chloroplast, or secretory pathways were identified as likely contaminants and removed from the list. (4) Finally, proteins containing likely transmembrane domains were determined using the TMHMM program (v2.0) (Tusnády and Simon, 1998) and removed. All identified proteins were sorted into functional groups using MapMan classification (Thimm et al., 2004a) in the Classification SuperViewer Tool w/bootstrap (http://bar.utoronto.ca/ntools/cgi-bin/ntools_classification_superviewer.cgi).
Protein Immunoblot Analysis
Equal proportions of each total, 1000g pellet, 10,000g pellet, 200,000g pellet, and 200,000g supernatant fractions were electrophoresed on 10% SDS-PAGE gels in a Tris-glycine buffer system at a constant 120 V/gel for 1.5 h on ice. Proteins were transferred to a nitrocellulose membrane (GE Healthcare) for 2 h at room temperature in a Bio-Rad Mini Protein Tetra System (Bio-Rad Laboratories) according to the manufacturer’s instructions. Blots were probed with primary antibodies against an ER marker SEC12 (Bar-Peled and Raikhel, 1997) at a 1:2000 dilution, an ER marker BIP (Agrisera antibodies) at a 1:2000 dilution, a Golgi marker SEC21 (Agrisera antibodies) at a 1:2000 dilution, prevacuolar compartment marker SYP22 (Sanderfoot et al., 1999) at a 1:1000 dilution, plasma membrane marker H+-ATPase (Agrisera antibodies) at a 1:10,000 dilution, and vacuole lumen marker ASP (Rose Biotechnology) at a 1:1000 dilution. ENOLASE and PEPC antibodies (Agrisera antibodies) were used as positive controls at 1:2000 dilutions. Antibodies were detected with secondary anti-rabbit antibodies conjugated with horseradish peroxidase and enhanced chemoluminescence reagent (Amersham Pharmacia Biotech).
Comparison of Mapp Values with PDB Entries
To assess accuracy of the apparent masses of protein complexes determined based on their SEC elution profiles, we compared them to masses derived from the solved crystal structures of protein complexes obtained from the PDB database. As there are only a few Arabidopsis proteins in the PDB, we generated two benchmark data sets for comparison. The first data set (Tier 1) contained the binary interactions of proteins extracted from the known structures of Arabidopsis proteins in the PDB, and the second level (Tier 2) consists of Arabidopsis homologs in worm (Caenorhabditis elegans), fruit fly (Drosophila melanogaster), yeast (Saccharomyces cerevisiae), and human (Homo sapiens) (Li et al., 2003). The Tier 1 data set contains 172 protein pairs, while the Tier 2 data set contains 3693 protein pairs (55 proteins from worm, 119 proteins from fruit fly, 3226 proteins from human, and 293 proteins from yeast).
Supplemental Data
The following materials are available in the online version of this article.
Supplemental Figure 1. SDS-PAGE Analysis of SEC Fractions Obtained from Leaf Cytosol.
Supplemental Figure 2. Functional Categories of Total, Predicted Cytosolic Proteins, and Our Experimentally Analyzed Cytosolic Proteins.
Supplemental Figure 3. Scatterplots of Rapp and Relative Abundances of Putative Protein Complexes in Biological Replicates 1 and 2.
Supplemental Figure 4. Elution Profiles of Aminoacyl t-RNA Synthetases.
Supplemental Figure 5. Elution Profiles of Dehydrin COR47 (AT1G20440) and TANGLED (AT3G05330).
Supplemental Figure 6. Elution Profiles of hAT-Family Transposase DAYSLEER (AT3G42170) and myb-Like TRF Transcription Factor (AT1G58220).
Supplemental Table 1. Comparison of the Apparent Protein Masses (Mapp) Determined from This Study with the Published Complex Masses in the Literature (Mref) Determined Based on Gel Filtration Chromatography.
Supplemental Table 2. Proteins Predicted to Be Subunits of Large Protein Complexes with an Rapp > 5 in Both Biological Replicates.
Supplemental Data Set 1. All Peptides with Mapped Proteins, Experimental and Calculated m/z, Molecular Weight, Charge State, Peptide Counts, and Peptide Ion Scores.
Supplemental Data Set 2. List of 1283 Proteins Identified in Both the Biological Replicates with Spectral Count Distribution across SEC Fractions.
Supplemental Data Set 3. List of 713 Cytosolic Proteins from Supplemental Data Set 2 Detected in All SEC Fractions.
Supplemental Data Set 4. Comparison of the Apparent Masses Determined from This Study (Mapp) with the Reported Mass Values of Protein Complexes Derived from the Crystal Structures Present in Protein Data Bank (PDB; MPDB).
Supplemental Data Set 5. Hierarchical Clustering of Protein Elution Profiles (High Resolution pdf File).
The following materials have been deposited in the DRYAD repository under accession number http://dx.doi.org/10.5061/dryad.3r8p4.
Supplemental File 1. Computer Code and Example Data Form Used to Calculate aSPC.
Supplementary Material
Acknowledgments
We thank Sung Min Lee and Christy Reick for help with protein gel blot analyses and Donglai Chen for cluster analyses. We also thank members of Szymanski lab for input during the course of this research. This work was supported by a Purdue University Discovery Park Seed Grant and the National Science Foundation Award IOS-1127027 to D.B.S.
AUTHOR CONTRIBUTIONS
D.B.S. conceived the study. U.K.A., M.C.H., and D.B.S. designed the experiments. U.K.A. performed the experiments. U.K.A., Y.X., J.X., Z.M., D.K., and D.B.S. analyzed the data. U.K.A., M.C.H., and D.B.S. wrote the article. All authors revised the article.
Glossary
- TAP
tandem affinity purification
- MS
mass spectrometry
- LC-MS/MS
liquid chromatography-tandem mass spectrometry
- SEC
size exclusion chromatography
- ER
endoplasmic reticulum
- FDR
false discovery rate
- TMD
transmembrane domain
- CP
core particle
- RP
regulatory particle
- PDB
Protein Data Bank
Footnotes
Online version contains Web-only data.
Articles can be viewed online without a subscription.
References
- Alberts B. (1998). The cell as a collection of protein machines: preparing the next generation of molecular biologists. Cell 92: 291–294. [DOI] [PubMed] [Google Scholar]
- Aryal U.K., Krochko J.E., Ross A.R. (2012). Identification of phosphoproteins in Arabidopsis thaliana leaves using polyethylene glycol fractionation, immobilized metal-ion affinity chromatography, two-dimensional gel electrophoresis and mass spectrometry. J. Proteome Res. 11: 425–437. [DOI] [PubMed] [Google Scholar]
- Bar-Peled M., Raikhel N.V. (1997). Characterization of AtSEC12 and AtSAR1. Proteins likely involved in endoplasmic reticulum and Golgi transport. Plant Physiol. 114: 315–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlowe C., Orci L., Yeung T., Hosobuchi M., Hamamoto S., Salama N., Rexach M.F., Ravazzola M., Amherdt M., Schekman R. (1994). COPII: a membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell 77: 895–907. [DOI] [PubMed] [Google Scholar]
- Basu D., Le J., Zakharova T., Mallery E.L., Szymanski D.B. (2008). A SPIKE1 signaling complex controls actin-dependent cell morphogenesis through the heteromeric WAVE and ARP2/3 complexes. Proc. Natl. Acad. Sci. USA 105: 4044–4049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc G., Hokamp K., Wolfe K.H. (2003). A recent polyploidy superimposed on older large-scale duplications in the Arabidopsis genome. Genome Res. 13: 137–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandina I., Graham J., Lemaitre-Guillier C., Entelis N., Krasheninnikov I., Sweetlove L., Tarassov I., Martin R.P. (2006). Enolase takes part in a macromolecular complex associated to mitochondria in yeast. Biochim. Biophys. Acta 1757: 1217–1228. [DOI] [PubMed] [Google Scholar]
- Braun P., Aubourg S., Van Leene J., De Jaeger G., Lurin C. (2013). Plant protein interactomes. Annu. Rev. Plant Biol. 64: 161–187. [DOI] [PubMed] [Google Scholar]
- Bundock P., Hooykaas P. (2005). An Arabidopsis hAT-like transposase is essential for plant development. Nature 436: 282–284. [DOI] [PubMed] [Google Scholar]
- Cellar N.A., Kuppannan K., Langhorst M.L., Ni W., Xu P., Young S.A. (2008). Cross species applicability of abundant protein depletion columns for ribulose-1,5-bisphosphate carboxylase/oxygenase. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 861: 29–39. [DOI] [PubMed] [Google Scholar]
- Chen H.C., Li Q., Shuford C.M., Liu J., Muddiman D.C., Sederoff R.R., Chiang V.L. (2011). Membrane protein complexes catalyze both 4- and 3-hydroxylation of cinnamic acid derivatives in monolignol biosynthesis. Proc. Natl. Acad. Sci. USA 108: 21253–21258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong M., Yang L.L., Williams K., Fisher S.J., Hall S.C., Biggin M.D., Jin J., Witkowska H.E. (2008). A “tagless” strategy for identification of stable protein complexes genome-wide by multidimensional orthogonal chromatographic separation and iTRAQ reagent tracking. J. Proteome Res. 7: 1836–1849. [DOI] [PubMed] [Google Scholar]
- Emanuelsson O., Brunak S., von Heijne G., Nielsen H. (2007). Locating proteins in the cell using TargetP, SignalP and related tools. Nat. Protoc. 2: 953–971. [DOI] [PubMed] [Google Scholar]
- Fields S., Sternglanz R. (1994). The two-hybrid system: an assay for protein-protein interactions. Trends Genet. 10: 286–292. [DOI] [PubMed] [Google Scholar]
- Finley D. (2009). Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem. 78: 477–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavin A.C., et al. (2006). Proteome survey reveals modularity of the yeast cell machinery. Nature 440: 631–636. [DOI] [PubMed] [Google Scholar]
- Gavin A.C., et al. (2002). Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 415: 141–147. [DOI] [PubMed] [Google Scholar]
- Giegé P., Heazlewood J.L., Roessner-Tunali U., Millar A.H., Fernie A.R., Leaver C.J., Sweetlove L.J. (2003). Enzymes of glycolysis are functionally associated with the mitochondrion in Arabidopsis cells. Plant Cell 15: 2140–2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good M.C., Zalatan J.G., Lim W.A. (2011). Scaffold proteins: hubs for controlling the flow of cellular information. Science 332: 680–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham J.W., Williams T.C., Morgan M., Fernie A.R., Ratcliffe R.G., Sweetlove L.J. (2007). Glycolytic enzymes associate dynamically with mitochondria in response to respiratory demand and support substrate channeling. Plant Cell 19: 3723–3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwell L.H., Hopfield J.J., Leibler S., Murray A.W. (1999). From molecular to modular cell biology. Nature 402 (suppl.): C47–C52. [DOI] [PubMed] [Google Scholar]
- Havugimana P.C., et al. (2012). A census of human soluble protein complexes. Cell 150: 1068–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huberts D.H., van der Klei I.J. (2010). Moonlighting proteins: an intriguing mode of multitasking. Biochim. Biophys. Acta 1803: 520–525. [DOI] [PubMed] [Google Scholar]
- Ingram G.C., Waites R. (2006). Keeping it together: co-ordinating plant growth. Curr. Opin. Plant Biol. 9: 12–20. [DOI] [PubMed] [Google Scholar]
- Ito J., Batth T.S., Petzold C.J., Redding-Johanson A.M., Mukhopadhyay A., Verboom R., Meyer E.H., Millar A.H., Heazlewood J.L. (2011). Analysis of the Arabidopsis cytosolic proteome highlights subcellular partitioning of central plant metabolism. J. Proteome Res. 10: 1571–1582. [DOI] [PubMed] [Google Scholar]
- Jacobsson K., Frykberg L. (1996). Phage display shot-gun cloning of ligand-binding domains of prokaryotic receptors approaches 100% correct clones. Biotechniques 20: 1070–1076, 1078, 1080–1081. [DOI] [PubMed] [Google Scholar]
- Jahn D., Verkamp E., Söll D. (1992). Glutamyl-transfer RNA: a precursor of heme and chlorophyll biosynthesis. Trends Biochem. Sci. 17: 215–218. [DOI] [PubMed] [Google Scholar]
- Jansen R., Gerstein M. (2004). Analyzing protein function on a genomic scale: the importance of gold-standard positives and negatives for network prediction. Curr. Opin. Microbiol. 7: 535–545. [DOI] [PubMed] [Google Scholar]
- Jansen R., Yu H., Greenbaum D., Kluger Y., Krogan N.J., Chung S., Emili A., Snyder M., Greenblatt J.F., Gerstein M. (2003). A Bayesian networks approach for predicting protein-protein interactions from genomic data. Science 302: 449–453. [DOI] [PubMed] [Google Scholar]
- Kaksonen M., Sun Y., Drubin D.G. (2003). A pathway for association of receptors, adaptors, and actin during endocytic internalization. Cell 115: 475–487. [DOI] [PubMed] [Google Scholar]
- Karsenti E., Vernos I. (2001). The mitotic spindle: a self-made machine. Science 294: 543–547. [DOI] [PubMed] [Google Scholar]
- Kirkwood K.J., Ahmad Y., Larance M., Lamond A.I. (2013). Characterization of native protein complexes and protein isoform variation using size-fractionation-based quantitative proteomics. Mol. Cell. Proteomics 12: 3851–3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knip M., de Pater S., Hooykaas P.J. (2012). The SLEEPER genes: a transposase-derived angiosperm-specific gene family. BMC Plant Biol. 12: 192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotchoni S.O., Zakharova T., Mallery E.L., Le J., El-Assal Sel.-D., Szymanski D.B. (2009). The association of the Arabidopsis actin-related protein2/3 complex with cell membranes is linked to its assembly status but not its activation. Plant Physiol. 151: 2095–2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause R., von Mering C., Bork P., Dandekar T. (2004). Shared components of protein complexes—versatile building blocks or biochemical artefacts? BioEssays 26: 1333–1343. [DOI] [PubMed] [Google Scholar]
- Kressler D., Roser D., Pertschy B., Hurt E. (2008). The AAA ATPase Rix7 powers progression of ribosome biogenesis by stripping Nsa1 from pre-60S particles. J. Cell Biol. 181: 935–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensen A.R., Gsponer J., Foster L.J. (2012). A high-throughput approach for measuring temporal changes in the interactome. Nat. Methods 9: 907–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H., Guo Y., Ohta M., Xiong L., Stevenson B., Zhu J.-K. (2002). LOS2, a genetic locus required for cold-responsive gene transcription encodes a bi-functional enolase. EMBO J. 21: 2692–2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Stoeckert C.J. Jr., Roos D.S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13: 2178–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F., Ni W., Griffith M.E., Huang Z., Chang C., Peng W., Ma H., Xie D. (2004). The ASK1 and ASK2 genes are essential for Arabidopsis early development. Plant Cell 16: 5–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Yang W.C., Gao Q., Regnier F. (2008). Toward chromatographic analysis of interacting protein networks. J. Chromatogr. A 1178: 24–32. [DOI] [PubMed] [Google Scholar]
- Lund E., Dahlberg J.E. (1998). Proofreading and aminoacylation of tRNAs before export from the nucleus. Science 282: 2082–2085. [DOI] [PubMed] [Google Scholar]
- Marianayagam N.J., Sunde M., Matthews J.M. (2004). The power of two: protein dimerization in biology. Trends Biochem. Sci. 29: 618–625. [DOI] [PubMed] [Google Scholar]
- Marks M.D., Wenger J.P., Gilding E., Jilk R., Dixon R.A. (2009). Transcriptome analysis of Arabidopsis wild-type and gl3-sst sim trichomes identifies four additional genes required for trichome development. Mol. Plant 2: 803–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinis S.A., Plateau P., Cavarelli J., Florentz C. (1999). Aminoacyl-tRNA synthetases: a family of expanding functions. EMBO J. 18: 4591–4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masoud K., Herzog E., Chabouté M.E., Schmit A.C. (2013). Microtubule nucleation and establishment of the mitotic spindle in vascular plant cells. Plant J. 75: 245–257. [DOI] [PubMed] [Google Scholar]
- Mukhtar M.S., Carvunis A.R., Dreze M., Epple P., Steinbrenner J., Moore J., Tasan M., Galli M., Hao T., Nishimura M.T., Pevzner S.J., Donovan S.E., et al. European Union Effectoromics Consortium (2011). Independently evolved virulence effectors converge onto hubs in a plant immune system network. Science 333: 596–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen H., Engelbrecht J., Brunak S., von Heijne G. (1997). Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng. 10: 1–6. [DOI] [PubMed] [Google Scholar]
- Nooren I.M., Thornton J.M. (2003). Diversity of protein-protein interactions. EMBO J. 22: 3486–3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohya T., Miaczynska M., Coskun U., Lommer B., Runge A., Drechsel D., Kalaidzidis Y., Zerial M. (2009). Reconstitution of Rab- and SNARE-dependent membrane fusion by synthetic endosomes. Nature 459: 1091–1097. [DOI] [PubMed] [Google Scholar]
- O’Leary B., Rao S.K., Kim J., Plaxton W.C. (2009). Bacterial-type phosphoenolpyruvate carboxylase (PEPC) functions as a catalytic and regulatory subunit of the novel class-2 PEPC complex of vascular plants. J. Biol. Chem. 284: 24797–24805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olinares P.D.B., Ponnala L., van Wijk K.J. (2010). Megadalton complexes in the chloroplast stroma of Arabidopsis thaliana characterized by size exclusion chromatography, mass spectrometry, and hierarchical clustering. Mol. Cell. Proteomics 9: 1594–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal-Bhowmick I., Sadagopan K., Vora H.K., Sehgal A., Sharma S., Jarori G.K. (2004). Cloning, over-expression, purification and characterization of Plasmodium falciparum enolase. Eur. J. Biochem. 271: 4845–4854. [DOI] [PubMed] [Google Scholar]
- Pancholi V. (2001). Multifunctional alpha-enolase: its role in diseases. Cell. Mol. Life Sci. 58: 902–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti A.C., Parmely T.J., Tomomori-Sato C., Sato S., Zhu D., Conaway R.C., Conaway J.W., Florens L., Washburn M.P. (2006). Quantitative proteomic analysis of distinct mammalian Mediator complexes using normalized spectral abundance factors. Proc. Natl. Acad. Sci. USA 103: 18928–18933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S.G., Ewalt K.L., Kim S. (2005). Functional expansion of aminoacyl-tRNA synthetases and their interacting factors: new perspectives on housekeepers. Trends Biochem. Sci. 30: 569–574. [DOI] [PubMed] [Google Scholar]
- Pawson T. (2007). Dynamic control of signaling by modular adaptor proteins. Curr. Opin. Cell Biol. 19: 112–116. [DOI] [PubMed] [Google Scholar]
- Peltier J.B., Cai Y., Sun Q., Zabrouskov V., Giacomelli L., Rudella A., Ytterberg A.J., Rutschow H., van Wijk K.J. (2006). The oligomeric stromal proteome of Arabidopsis thaliana chloroplasts. Mol. Cell. Proteomics 5: 114–133. [DOI] [PubMed] [Google Scholar]
- Pickart C.M., Cohen R.E. (2004). Proteasomes and their kin: proteases in the machine age. Nat. Rev. Mol. Cell Biol. 5: 177–187. [DOI] [PubMed] [Google Scholar]
- Plaxton W.C. (1996). The organization and regulation of plant glycolysis. Annu. Rev. Plant Physiol. Plant Mol. Biol. 47: 185–214. [DOI] [PubMed] [Google Scholar]
- Polpitiya A.D., Qian W.J., Jaitly N., Petyuk V.A., Adkins J.N., Camp D.G. II, Anderson G.A., Smith R.D. (2008). DAnTE: a statistical tool for quantitative analysis of -omics data. Bioinformatics 24: 1556–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen C.G., Sun B., Smith L.G. (2011). Tangled localization at the cortical division site of plant cells occurs by several mechanisms. J. Cell Sci. 124: 270–279. [DOI] [PubMed] [Google Scholar]
- Rigaut G., Shevchenko A., Rutz B., Wilm M., Mann M., Séraphin B. (1999). A generic protein purification method for protein complex characterization and proteome exploration. Nat. Biotechnol. 17: 1030–1032. [DOI] [PubMed] [Google Scholar]
- Saavedra L., Svensson J., Carballo V., Izmendi D., Welin B., Vidal S. (2006). A dehydrin gene in Physcomitrella patens is required for salt and osmotic stress tolerance. Plant J. 45: 237–249. [DOI] [PubMed] [Google Scholar]
- Sanderfoot A.A., Kovaleva V., Zheng H., Raikhel N.V. (1999). The t-SNARE AtVAM3p resides on the prevacuolar compartment in Arabidopsis root cells. Plant Physiol. 121: 929–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M.W., Houseman A., Ivanov A.R., Wolf D.A. (2007). Comparative proteomic and transcriptomic profiling of the fission yeast Schizosaccharomyces pombe. Mol. Syst. Biol. 3: 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srere P.A. (1987). Complexes of sequential metabolic enzymes. Annu. Rev. Biochem. 56: 89–124. [DOI] [PubMed] [Google Scholar]
- Srere P.A. (2000). Macromolecular interactions: tracing the roots. Trends Biochem. Sci. 25: 150–153. [DOI] [PubMed] [Google Scholar]
- Stradal T.E., Scita G. (2006). Protein complexes regulating Arp2/3-mediated actin assembly. Curr. Opin. Cell Biol. 18: 4–10. [DOI] [PubMed] [Google Scholar]
- Stroupe C., Hickey C.M., Mima J., Burfeind A.S., Wickner W. (2009). Minimal membrane docking requirements revealed by reconstitution of Rab GTPase-dependent membrane fusion from purified components. Proc. Natl. Acad. Sci. USA 106: 17626–17633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szymanski D.B. (2005). Breaking the WAVE complex: the point of Arabidopsis trichomes. Curr. Opin. Plant Biol. 8: 103–112. [DOI] [PubMed] [Google Scholar]
- Tanahashi N., Murakami Y., Minami Y., Shimbara N., Hendil K.B., Tanaka K. (2000). Hybrid proteasomes. Induction by interferon-gamma and contribution to ATP-dependent proteolysis. J. Biol. Chem. 275: 14336–14345. [DOI] [PubMed] [Google Scholar]
- Thelen J.J., Peck S.C. (2007). Quantitative proteomics in plants: choices in abundance. Plant Cell 19: 3339–3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thimm O., Bläsing O., Gibon Y., Nagel A., Meyer S., Krüger P., Selbig J., Müller L.A., Rhee S.Y., Stitt M. (2004a). MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 37: 914–939. [DOI] [PubMed] [Google Scholar]
- Thimm O., Bläsing O., Gibon Y., Nagel A., Meyer S., Krüger P., Selbig J., Müller L.A., Rhee S.Y., Stitt M. (2004b). MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 37: 914–939. [DOI] [PubMed] [Google Scholar]
- Tunio S.A., Oldfield N.J., Berry A., Ala’Aldeen D.A., Wooldridge K.G., Turner D.P. (2010). The moonlighting protein fructose-1, 6-bisphosphate aldolase of Neisseria meningitidis: surface localization and role in host cell adhesion. Mol. Microbiol. 76: 605–615. [DOI] [PubMed] [Google Scholar]
- Tusnády G.E., Simon I. (1998). Principles governing amino acid composition of integral membrane proteins: application to topology prediction. J. Mol. Biol. 283: 489–506. [DOI] [PubMed] [Google Scholar]
- Walker K.L., Müller S., Moss D., Ehrhardt D.W., Smith L.G. (2007). Arabidopsis TANGLED identifies the division plane throughout mitosis and cytokinesis. Curr. Biol. 17: 1827–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welin B.V., Olson A., Palva E.T. (1995). Structure and organization of two closely related low-temperature-induced dhn/lea/rab-like genes in Arabidopsis thaliana L. Heynh. Plant Mol. Biol. 29: 391–395. [DOI] [PubMed] [Google Scholar]
- Winkel B.S. (2004). Metabolic channeling in plants. Annu. Rev. Plant Biol. 55: 85–107. [DOI] [PubMed] [Google Scholar]
- Wodak S.J., Pu S., Vlasblom J., Séraphin B. (2009). Challenges and rewards of interaction proteomics. Mol. Cell. Proteomics 8: 3–18. [DOI] [PubMed] [Google Scholar]
- Yanhui C., et al. (2006). The MYB transcription factor superfamily of Arabidopsis: expression analysis and phylogenetic comparison with the rice MYB family. Plant Mol. Biol. 60: 107–124. [DOI] [PubMed] [Google Scholar]
- Yi C., Deng X.W. (2005). COP1 - from plant photomorphogenesis to mammalian tumorigenesis. Trends Cell Biol. 15: 618–625. [DOI] [PubMed] [Google Scholar]
- Yu H., et al. (2008). High-quality binary protein interaction map of the yeast interactome network. Science 322: 104–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H., et al. (2001). Global analysis of protein activities using proteome chips. Science 293: 2101–2105. [DOI] [PubMed] [Google Scholar]
- Zybailov B., Mosley A.L., Sardiu M.E., Coleman M.K., Florens L., Washburn M.P. (2006). Statistical analysis of membrane proteome expression changes in Saccharomyces cerevisiae. J. Proteome Res. 5: 2339–2347. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.