

Abstract
Background
Few, if any, protozoan parasites are reported to exhibit extreme organ tropism like the flagellate Tritrichomonas foetus. In cattle, T. foetus infects the reproductive system causing abortion, whereas the infection in cats results in chronic large bowel diarrhoea. In the absence of a T. foetus genome, we utilized a de novo approach to assemble the transcriptome of the bovine and feline genotype to identify host-specific adaptations and virulence factors specific to each genotype. Furthermore, a subset of orthologs was used to characterize putative druggable targets and expose complications of in silico drug target mining in species with indefinite host-ranges.
Results
Illumina RNA-seq reads were assembled into two representative bovine and feline transcriptomes containing 42,363 and 36,559 contigs, respectively. Coding and non-coding regions of the genome libraries revealed striking similarities, with 24,620 shared homolog pairs reduced down to 7,547 coding orthologs between the two genotypes. The transcriptomes were near identical in functional category distribution; with no indication of selective pressure acting on orthologs despite differences in parasite origins/host. Orthologs formed a large proportion of highly expressed transcripts in both genotypes (bovine genotype: 76%, feline genotype: 56%). Mining the libraries for protease virulence factors revealed the cysteine proteases (CP) to be the most common. In total, 483 and 445 bovine and feline T. foetus transcripts were identified as putative proteases based on MEROPS database, with 9 hits to putative protease inhibitors. In bovine T. foetus, CP8 is the preferentially transcribed CP while in the feline genotype, transcription of CP7 showed higher abundance. In silico druggability analysis of the two genotypes revealed that when host sequences are taken into account, drug targets are genotype-specific.
Conclusion
Gene discovery analysis based on RNA-seq data analysis revealed prominent similarities between the bovine and feline T. foetus, suggesting recent adaptation to their respective host/niche. T. foetus represents a unique case of a mammalian protozoan expanding its parasitic grasp across distantly related host lineages. Consequences of the host-range for in silico drug targeting are exposed here, demonstrating that targets of the parasite in one host are not necessarily ideal for the same parasite in another host.
Electronic supplementary material
The online version of this article (doi:10.1186/1471-2164-15-955) contains supplementary material, which is available to authorized users.
Background
The protozoan flagellate Tritrichomonas foetus belongs to the phylum Parabasalia, which includes the human parasite; Trichomonas vaginalis [1] . Originally described as a nasal and gastrointestinal commensal of pigs, T. foetus infects the urogenital tract of cattle resulting in disease known as trichomoniasis [2–4]. Transmission of the disease to female cows occurs during coitus with infected bulls, which can result in abortion of the foetus [5–8]. Recently, T. foetus has been identified as the etiological agent of gastrointestinal infection of domestic cats, where infection results in chronic large bowel diarrhoea [9–11]. The disease in both hosts is very difficult to treat. Bovine trichomoniasis is currently untreatable and the only control measure available to farmers is to cull infected bulls or remove them from the breeding herd. Although treatment options for feline trichomoniasis do exist, they are becoming increasingly ineffective due to evolving parasite drug resistance and issues associated with host drug toxicity [12–14].
Tritrichomonas foetus represents an intriguing model to study host-parasite interactions. There has been much conjecture as to the origins of the bovine and feline isolates. In particular, are they different parasites or merely closely related genotypes? Given that both infections are caused by T. foetus, it is not surprising that historically they have been assumed to be the same parasite. The adaptation of parasites to different hosts is nothing new, however, the extreme host-organ tropism of the bovine isolate (vagina) and feline isolate (gastrointestinal tract) suggest that they are distinct genotypes that, over the course of their respective evolutions, have preferentially adapted to their respective host/niche [4, 15]. Further evidence as to their distinctiveness (albeit limited) has been demonstrated experimentally when the bovine genotype is introduced into the feline intestinal tract, and vice versa. Although both genotypes are capable of establishing infection in their non-typical host, the pathology is markedly less than on their preferred hosts [10, 16]. Successful delineation of the genotypes would enable a more precise estimation of trichomonas species richness and permit a better epidemiological understanding of the pathological basis of these diseases. Currently, artificial insemination and culling of infected animals ensures that a low infection level is maintained in intensively-managed cattle industries [17, 18]. Infections, however, are still prevalent in extensive farming systems [17, 18]. Moreover, current evaluation of the infection in domestic cats indicated a high prevalence, especially in young (6–12 month old) pedigree catteries [19].
Although it has recently been established that the porcine T. foetus (formerly known as T. suis) and the bovine T. foetus are synonymous [2, 4], the relationship between the bovine and feline T. foetus is proving harder to elucidate. Evidence of the limited genetic distinctness between the bovine and the feline isolates is apparent when highly conserved species-level nucleotide sequences for the internal transcribed spacer 2 (ITS2) and elongation factor 1 alpha (EF-1α) of the two genotypes are compared [4, 15, 20]. As the original diagnostic marker, a single nucleotide polymorphism in ITS2 between the bovine and feline T. foetus amounts to a sequence difference of only 0.3% [15, 20]. More recent studies demonstrated genetic difference between the two genotypes by analysing the cysteine protease multigene family [4, 21]. This family of genes is known to play a key role in parasite virulence [22–25]. It has been suggested, however, that these minor sequence differences between genotypes may merely represent intraspecific variation and not have any significant phenotypic consequences [26]. Nevertheless, ambiguity arises when single gene assays are used in an attempt to compare very closely related genotypes with broad host ranges, such as the bovine and feline T. foetus.
There is the need for a more comprehensive cell-wide approach to enable further elucidation of the relationship between the bovine and feline T. foetus genotypes. This is further confounded by a lack of T. foetus genome data which has undoubtedly hindered our understanding of host-switching and search for novel drug targets in these parasites. Therefore, in the absence of a genome, we have used RNA-seq to sequence the transcriptome of bovine and feline T. foetus genotypes in an attempt to provide a blueprint of functional capacity of each of the host/niche adapted T. foetus genotypes. This study represents the first cell-wide comparative analysis of T. foetus genotypes, enabling us to determine the extent to which differences between host/niche is reflected in their transcriptomes. In addition to investigating T. foetus host-specific biological and virulence mechanisms, we utilised our transcriptomic libraries to explore the usefulness of in silico techniques for the identification of potential parasite drug targets, taking into account their expanded host-range.
Results
Transcriptome
Illumina sequencing and transcriptome assembly
A total of 64,744,882 and 64,009,804 100 bp paired-end Illumina reads were obtained following the sequencing of total RNA isolated from bovine and feline Tritrichomonas foetus isolates, respectively. Assessment of read quality by FastQC revealed good quality reads (data not shown). Raw reads were mapped onto a small, previously published bovine T. foetus EST library and visually assessed to confirm a non-biased and even distribution of sequenced reads. Paired-end sequencing reads from each genotype were assembled using Trinity [27] into two transcriptomes consisting of 42,363 and 36,559 contigs representing the bovine and feline genotypes respectively (Table 1). A mean contig length of 895.2 bp was obtained in the bovine T. foetus transcriptome, with minimum and maximum contig lengths of 201 bp and 14,314 bp respectively. The feline T. foetus transcriptome had a mean contig length of 806.6 bp with a minimum length of 201 bp and a maximum of 17,195 bp in length.
Table 1.
Summary of sequenced reads and the assembled transcriptomes
| Feature | BovineT. foetus | FelineT. foetus |
|---|---|---|
| Total number of reads | 64744882 | 64009804 |
| Total base pairs (bp) | 6539233082 | 6464990204 |
| Average read length (bp) | 101 | 101 |
| Total number of contigs | 42363 | 36559 |
| Total assembled bases | 37882427 | 29525551 |
| Mean length of contigs (bp) | 895.25 | 806.61 |
| Median contig length (bp) | 653 | 562 |
| % GC content in transcriptome | 34.62 | 34.87 |
| Minimum contig length (bp) | 201 | 201 |
| Maximum contig length (bp) | 14314 | 17195 |
| Contig N50 | 1259 | 1178 |
Ortholog identification
Homologous transcript pairs between the bovine and the feline genotypes were identified using a reciprocal blast. This method identified 24,620 pairs of homologous transcripts which were further subjected to a BlastX search against the Swissport database (e-value 1 × 106) to remove putative paralogous genes [28, 29]. Orthologous pairs of coding regions were defined by comparing corresponding sequence pairs originating from each genotype and only those found to have identical top-hit in the BlastX results were selected. A total of 7,547 transcript pairs were selected using this method and were thus considered strong orthologs of the bovine and feline T. foetus (Figure 1).
Figure 1.
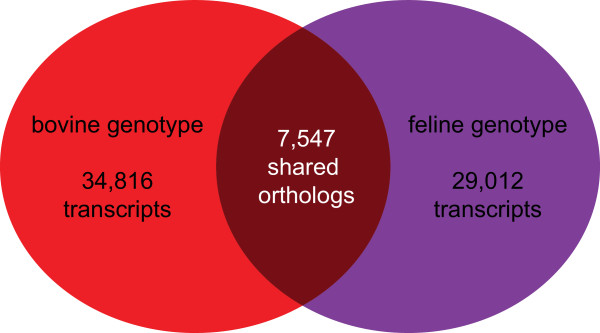
Distribution of shared transcripts between the bovine and feline T. foetus genotype. Venn diagram illustrating the shared bovine and feline transcriptome obtained by de novo assembly of Illumina RNA-seq sequenced data. A total of 7,547 transcripts were identified as true orthologs shared between the two genotypes.
A local version of the full-lengther next (FLN) algorithm [30] was implemented to identify putative coding regions in the orthologs using the invertebrate database, as this resulted in superior protein annotation results. Approximately 4,600 transcripts were protein annotated by the algorithm, of which, 1,511 pairs of ortholog pairs were found to be full length transcripts (i.e. the presence of both a start and stop codon). The coding regions of the 1,511 ortholog pairs were isolated using bash scripting.
Functional annotation and highly transcribed genes
Functional annotation at the BlastX and Gene Ontology level of the assembled transcriptomes were carried out using a combination of locally implemented BlastX searches and Blast2GO [28, 31]. Whole transcriptome annotations and annotations of only orthologs revealed a similar distribution of functional categories between the bovine and feline genotypes (Figures 2, 3). The difference in size of the two assembled transcriptomes can account for the slight variation in the number of transcripts obtained per functional category. The absence of four functional categories from the feline genotype that were identified in the bovine genotype is solely a limitation of the threshold of reported sequences set for presenting GO categories.
Figure 2.
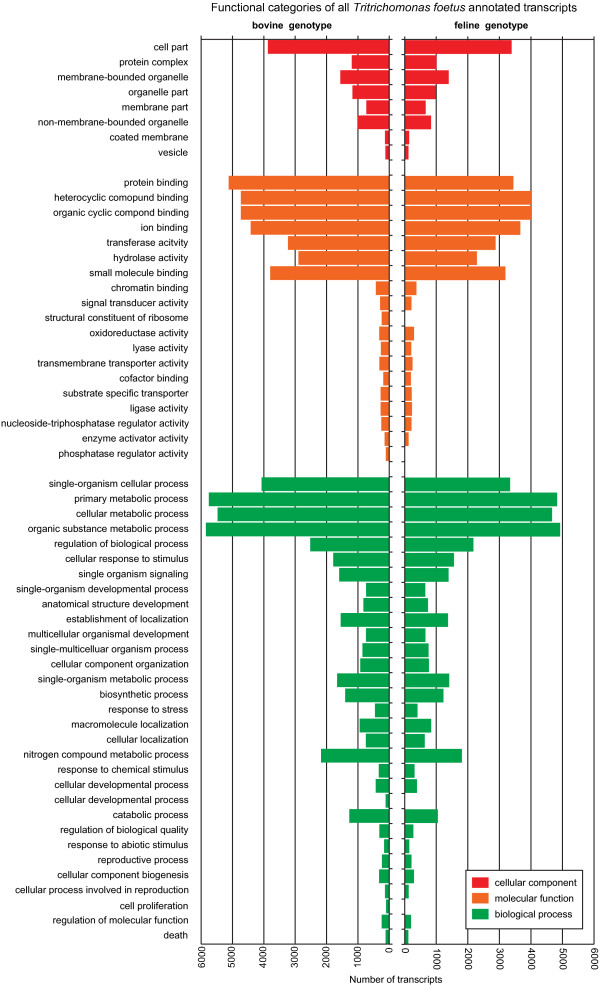
Top ranked GO categories of the bovine and feline Tritrichomonas foetus whole transcriptomes. Functional characterisation of the bovine (left) and feline (right) expressed genome based on Gene Ontology categories showing top ranked categories for cellular component, molecular function and biological process. Categories presented represent a minimum threshold filter value of 100 sequences.
Figure 3.
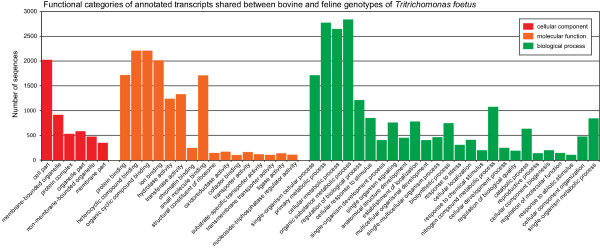
Representative functional annotation of shared orthologs between the bovine and feline T. foetus genotypes. Representative Gene Ontology functional categories of the bovine and feline shared orthologous gene pairs for cellular component, molecular function and biological process.
To identify the top 100 transcribed genes from each transcriptome, raw sequencing reads were mapped back onto the assembled transcriptome of each genotype and counts were normalized to RPKM (reads per kilobase per million of mapped reads). The top 100 transcripts with the highest RPKM values were selected from the bovine and feline transcriptomes. The top 100 RKPM values for the bovine genotype ranged from 21,107 to 1,119, whereas the RPKM range for the top 100 transcribed genes in the feline transcriptome was from 18,670 to 1,277. Blast annotations obtained previously were used to extract the putative functions of the top 100 bovine and feline transcripts. Within this list, 56 feline transcripts and 76 bovine transcripts were identified as ortholog genes (Figure 4), however, only 29 were orthologs pairs common to both genotypes (Figure 5). The common ortholog genes included mainly genes involved in metabolic activity, oxygen scavenging and regulation of homeostasis; all of which are expected in trichomonads. The remaining 24 and 44 non-ortholog, but highly-expressed transcripts from the bovine and feline genotypes were annotated as ribosomal-related proteins (data not shown).
Figure 4.

Annotation of the most highly expressed ortholog genes in the bovine and feline T. foetus.BlastX functional annotation of ortholog transcripts present within the top 100 highly expressed transcripts in the bovine (left, A) and feline (right, B) T. foetus genotypes after RPKM normalization of reads counts. The graphs show the 76 and 56 orthologous transcripts of the bovine and feline genotypes, respectively. Non-orthologous transcripts are not shown. Green bars represent the orthologs pairs that are highly expressed in both genotypes.
Figure 5.
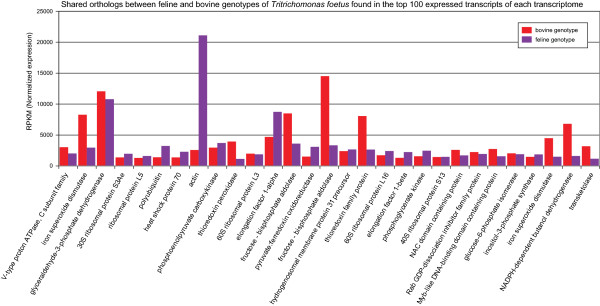
Normalized expression values of 29 highly expressed orthologous transcripts. Normalized read counts (RPKM) and BlastX annotation of 29 pairs of orthologous transcripts present within the top 100 highly expressed transcripts in the bovine and feline T. foetus genotypes.
UTR analysis
Un-translated regions (UTRs) of mRNA flank either end of the coding region and contain regulatory elements that dictate translation of genes [32–34]. Only the 1,511 full-length ortholog transcripts were used to compare the UTR lengths and regulatory content of the two T. foetus genotypes. The average length of 5′UTRs were 64.2 and 73.3 nucleotides (nt) for the bovine and feline genotypes, respectively. The longest bovine and feline 5′UTRs were 4,325 nt and 4,253 nt in length. The 1 – 25 nt length range contained approximately 67% of 5′UTRs from both genotypes (Figure 6). Longer 5′UTRs (>1,000 nt) were more prominent in the feline genotype with an additional nine feline sequences within this length category compared to that of the bovine genotype. Length analysis of 3′UTRs revealed that 41.8% and 38.2% of bovine and feline sequences, respectively, were found within the 51 – 200 nt length range. On average, 3′UTRs were longer than 5′UTRs with the mean length amounting to 77.7 nt for the bovine T. foetus and 70 nt for the feline genotype. The maximum length of 3′UTRs were, however, shorter than the longest 5′UTR with the lengths reaching to 1,360 nt and 1,331 nt for the bovine and feline genotypes, respectively.
Figure 6.
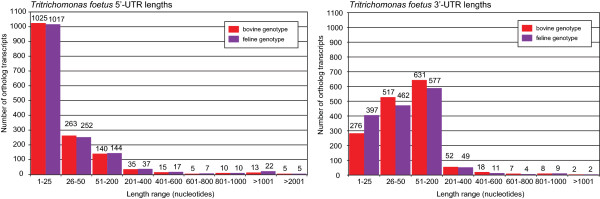
Length comparisons of untranslated regions (UTR) between the bovine and feline T. foetus . The number of transcripts from the 1,511 full-length bovine and feline orthologous transcripts with lengths (in nucleotides) of the 5′UTR (left) and 3′UTR (right) falling within the defined ranges.
To identify any patterns of correlation between the 3′UTR and 5′UTR length and the normalized expression count (RPKM), the 1,511 orthologs were plotted in a scatter plot (Figure 7). A non-linear model of regression was used to calculate a weighted R2 which takes into account the uneven variance between points across the graph, ensuring that all points contributed equally. In general, R2 < 0.01, indicated no correlation between the transcript expression and the length of the UTRs.
Figure 7.

Correlation between the length of UTRs and the normalized transcript expression (RPKM) for 1,511 shared ortholog. A non-linear, weighted sum of least square regression model was fitted to plots of the Log10 3′UTR and 5′UTR lengths against the Log10 of normalized expression counts (RPKM) of the respective transcript for the bovine and feline T. foetus genotypes. Regression values (R2) are quote on the individual plots. A-B. 3′UTR and 5′UTR plots respectively, for the feline T. foetus genotype. C-D. 3′UTR and 5′UTR plots respectively, for the bovine T. foetus genotypes.
Using the UTRscan algorithm [35] to search the UTRsite database [36] for known UTR regulatory motifs, a list of putative motifs were obtained for the two T. foetus genotypes. Overall, 14 different motifs were annotated in the UTR regions of 1,511 full-length orthologs between the two genotypes (Figure 8).
Figure 8.
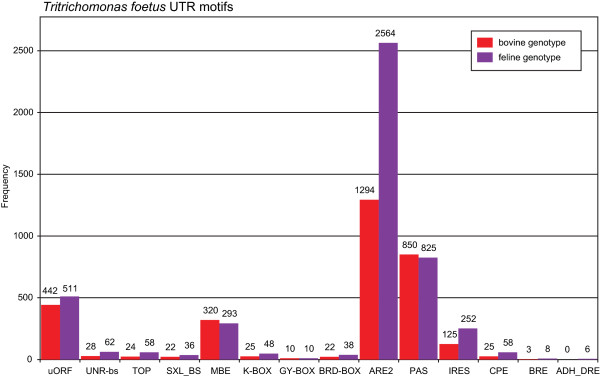
Frequency of regulatory motifs within the untranslated regions (UTR) of full-length orthologous transcripts. Frequency of hits to UTR motifs obtained through UTRscan searches of 1,511 full-length orthologous bovine and feline transcripts against the UTRsite of regulatory motifs. Actual number of hits is presented above each bar. Abbreviations: uORF; upstream open reading frame, UNR-bs; UNR binding site, TOP; terminal oligopyrimidine tract, SXL_BS; SXL bind site, MBE; Musashi binding element, K-Box; K-Box, GY-Box; GY-Box, BRD-Box; BRD-Box, ARE2; AU-rich element, PAS; polyadenylation signal, IRES; internal ribosome entry site, CPE; cytoplasmic polyadenylation element, BRE; Bruno responsive element, ADH_DRE; alcohol dehydrogenase down-regulation control element.
All motif patterns in the UTRs were common to both genotypes except for an alcohol dehydrogenase element associated with the 3′UTR of the feline genotype (ADH_DRE) (Figure 8). The most common motif in both genotypes was annotated to the AU-rich class-2 element (ARE2). This amounted to 28.2% and 30.6% of the number of unique hits in the bovine and feline genotypes, respectively. Polyadenylation signals (PAS) were found in 37.2% and 30.5% of the bovine and feline unique hits obtained.
Discovery of new proteases and protease inhibitors
Being a strict extracellular parasite, the ability of T. foetus parasites to attach to host cells is an essential prerequisite for the initiation and maintenance of infection [37]. During infection, the bovine host mounts a humoral defence against T. foetus, however, it is not necessarily sufficient to clear the infection [38]. The secretion of cysteine proteases (CPs) is thought to be an important facet of T. foetus virulence. CPs have been demonstrated to cleave and inactivate host protective antibodies, enabling the parasite to remain within the host [39]. To date, there have been 21 CPs identified in the bovine genotype, while only 8 CPs have been identified within the feline genotype [4, 21, 40]. Blast annotation identified a total of 665 and 623 hits to known proteases in the bovine and feline T. foetus transcriptomes, respectively. Both transcriptomes were also found to contain 11 hits for protease inhibitors. A total of 389 and 346 CPs with a corresponding 3 and 2 CP inhibitors were identified as belonging to the bovine and feline T. foetus respectively.
The full list of proteases was used in a search against the MEROPS peptidase database enabling the further identification of true proteases [41]. Of the initial list obtained through NCBI blast, only 483 and 445 bovine and feline T. foetus transcripts received hits from the MEROPS database. Of these hits, 243 bovine transcripts produced hits to a single protease active site, compared to 253 feline transcripts with unique hits. Some sequences obtained hits to multiple active site domains. A total number of 539 bovine and 498 feline active sites were obtained when the results were collapsed to show only unique types of protease active sites per transcript (Table 2). The largest group detected was the cysteine proteases active site architecture, amounting to 52.8% in bovine T. foetus and 50% in feline T. foetus of the total hits obtained. No hits were obtained for glutamic or asparagine proteases in either parasite transcriptomes. Similarly, the same pipeline carried out on Trichomonas vaginalis coding genes based on the draft genome produced 475 annotated “protease/peptidase/proteinase” related genes [42]. This comparably corresponded to 221 confirmed putative proteases possessing cysteine-specific active sites in T. vaginalis found in the MEROPS database (data not shown).
Table 2.
Frequency of protease active site present within the bovine and feline transcriptome
| Functional type | BovineT. foetus | FelineT. foetus |
|---|---|---|
| Cysteine | 285 | 249 |
| Serine | 99 | 95 |
| Glutamic | 0 | 0 |
| Metallo | 88 | 88 |
| Threonine | 22 | 22 |
| Aspartic | 9 | 9 |
| Inhibitors | 35 | 34 |
| Asparagine | 0 | 0 |
| Mixed | 0 | 0 |
| Unknown | 1 | 1 |
| Overall | 539 | 498 |
Two protease active sites were unique to either one of the parasite genotypes. In the bovine T. foetus, transcript Bc12_comp23753_c0_seq1 produced a hit to the serine active site S51 of the PC clan (MEROPS accession: MER001335) which was not present in the feline T. foetus protease list. Conversely, the feline transcript G10_comp5459_c0_seq1 produced a unique hit to the metalloprotease active site M20X of the MH clan (MEROPS Accession: MER001266) which was unmatched in the bovine T. foetus protease list.
Raw reads were mapped back onto the putative protease and counts were normalized using RPKM revealing 148 bovine and 113 feline T. foetus proteases being expressed at an RPKM of 500 or greater. This RPKM threshold was chosen to signify high expression of proteases. Of the highly expressed proteases, 42.3% of bovine and 39.9% of feline transcripts contained CP active sites, with the MEROPS C01A protease family represented in 65.08% and 40.47% of bovine and feline highly expressed CPs, respectively (Table 3, Additional file 1: Table S1 and Additional file 2: Table S2). The putative T. foetus CP sequences were aligned to previously published CP sequences to identify known CPs within our list [4, 40, 43]. Of the 20 bovine and 8 feline published CP sequences, 15 known bovine CPs were among the highly expressed proteases (RPKM >500), whereas CP7 and CP8 were the only known feline CPs with an RPKM above 500 in the feline transcriptome. Interestingly, while CP8 was the most transcribed protease in the bovine genotype, CP7 was found to be highly transcribed in the feline genotype.
Table 3.
Type distribution of highly expressed protease in bovine and feline T. foetus
| Protease type | BovineT. foetus | FelineT. foetus |
|---|---|---|
| Cysteine | 63 | 42 |
| Metallo | 34 | 26 |
| Serine | 26 | 16 |
| Threonine | 21 | 19 |
| Aspartic | 4 | 4 |
| Total | 148 | 113 |
Of the 11 inhibitors initially found in the BlastX transcriptome annotation, 9 feline T. foetus transcripts obtained a positive hit to an inhibitor active site from the MEROPS database, compared to only 8 bovine T. foetus transcripts with known inhibitor active site hits. Using BlastN for pairwise alignments of the bovine and feline inhibitor sequences, all bovine sequences align to a feline sequence with approx. 99% identity (Table 4). One putative feline inhibitor sequence (G10_comp9648_c0_seq1) did not align to any bovine sequences and produced a match to the MEROPS I04 protease family (MEROPS accession: MER003981).
Table 4.
Summary of aligned protease inhibitors and their predicted MEROPS family
| Feline transcript | Bovine transcript | Alignment | Identity | Predicted protease family | MEROPS accession number |
|---|---|---|---|---|---|
| G10_comp7804_c0_seq1 | Bc12_comp9941_c0_seq1 | 333/334 | 99% | I25B | MER018186 |
| G10_comp2876_c0_seq1 | Bc12_comp7451_c0_seq2 | 936/943 | 99% | I04 | MER018805, MER018695, MER023786 (bovine only) |
| G10_comp7790_c0_seq1 | Bc12_comp7451_c0_seq1 | 1139/1145 | 99% | I04 | MER018805, MER016306 (feline only), MER018807 (feline only), MER027490 (feline only), MER018695 (bovine only) |
| G10_comp7405_c0_seq1 | Bc12_comp9915_c0_seq1 | 293/297 | 99% | I25B | MER018937, MER018172 (bovine only) |
| G10_comp3687_c0_seq1 | Bc12_comp3242_c0_seq1 | 311/314 | 99% | I25B | MER018186 |
| G10_comp18864_c0_seq1 | Bc12_comp5569_c0_seq1 | 394/401 | 98% | I04 | MER018805 (feline only), MER003223 (feline only), MER018696 (bovine only), MER016306 (bovine only) |
| G10_comp17054_c0_seq1 | Bc12_comp4109_c0_seq1 | 899/903 | 99% | I04 | MER018698, MER023885 (bovine only) |
| G10_comp12847_c0_seq1 | Bc12_comp13520_c0_seq1 | 339/342 | 99% | I25B | MER181466, MER166026 |
Analysis of sequence divergence
Pairwise codon-alignments of the 1,511 full-length ortholog transcripts shared between the bovine and feline T. foetus, revealed that only 1,050 transcript pairs (69.5%) were sufficiently divergent to allow for Ka/Ks calculation. The aligned pairs of orthologs showed an average substitution of 10.6 nt ranging from a minimum of 4 nt to a maximum of 167 nt substitutions. While strong sequence conservation (Ka/Ks: <0.1) was detected in 80.41% of the coding orthologs analysed (Figure 9), weak purifying selection, that is, a low rate of protein change denoted by a Ka/Ks ratio within the 0.5 – 1.0 range was demonstrated by 14 orthologs pairs. A single ortholog pair had a Ka/Ks ratio of approximately 1, signifying neutral selection (no selective pressure), while only one other pair showed gene divergence with a Ka/Ks ratio greater than 1.
Figure 9.
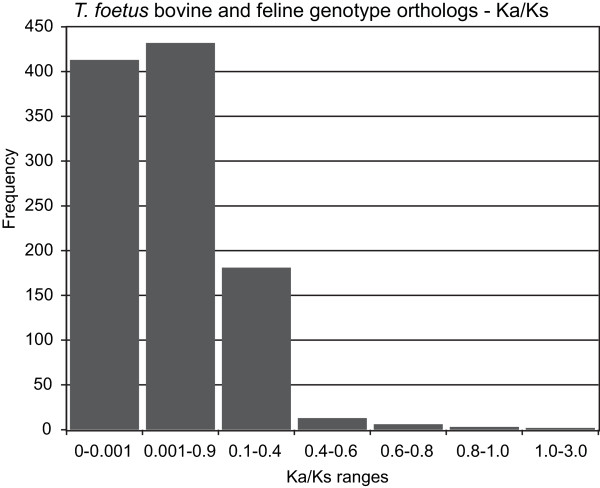
Frequency of Ka/Ks values for full-length orthologous transcripts between the bovine and feline genotype. Frequency of orthologous transcript pairs producing Ka/Ks values within various ranges.
Comparison of Gene Ontology (GO) terms between highly conserved, less-conserved and the divergent group of orthologs showed that “binding activity” is over-represented in all 3 groups. This functional category is represented by 66.5%, 50%, and 100% of orthologs belonging to the high purifying, weak purifying and the divergent ortholog set, respectively. Within the high purifying group (Ka/Ks <0.5), 211 sequences were specifically involved in ATP binding activity, while 13.5% of transcripts were metabolically active transcripts. The divergent group of orthologs presented hits to an unknown protein containing a Myb-like binding domain (GO: 0003677, GO:0003682).
Other GO categories that were obtained included “Transport activity” and “Translational activity” which were represented by 20% and 10% orthologs from the weak purifying subset, respectively.
Druggability
To explore in silico drug targeting pipelines between T. foetus genotypes, only the 1,511 predicted full-length orthologs were used for identification of potential drug target, with an added level of search stringency aimed at identifying non-host targets. Of the 1,511 ortholog pairs of transcripts blasted to their respective host proteome, 123 bovine T. foetus and 105 feline T. foetus transcripts were found to be unique to the parasite (i.e. not found in their respective hosts). Approximately half of the parasite-only transcripts obtained (bovine: 48/105; feline: 59/123) produced positive results to one or more druggable domains. From the feline BlastX results to druggable domains, 49.1% (416/846) of the domains adhere to the Lipinsky rule of 5 for small molecule binding. This number was lowered to 43% (184/424) in the bovine druggable transcripts (Additional file 3: Table S3 and Additional file 4: Table S4).
Discussion
In this study, we characterized draft transcriptomes of two genotypes of Tritrichomonas foetus; a trichomonad of veterinary significance, secondary in prominence only to the human Trichomonas vaginalis. In the absence of a genome or sufficient background proteomics, a de novo RNA-seq approach was used as an economical and high-throughput cell-wide gene discovery technique. Currently, only a small expressed sequence library of the bovine T. foetus is available in the public domain [40]. Here, existing expressed sequence data of the bovine T. foetus is augmented and we leverage the field by providing the first comprehensive expressed sequence library of a feline T. foetus genotype. Gene discovery via RNA sequencing projects provide an accurate representation of transcriptionally active regions of the genome, thus proving an accurate starting point for the unearthing of undiscovered genes [44, 45]. With the expression blueprints of the bovine and feline T. foetus genotypes, the first cell-wide comparison of shared genes was undertaken and an in silico novel drug target analysis was explored. A draft genome has previously been published for the related human pathogen Trichomonas vaginalis [42] . However, phylogenetically, T. vaginalis and T. foetus are significantly divergent, impeding extension of molecular information between the two parasites. Hence the sequence library we have constructed is currently the best depiction of the T. foetus genome to date and will reinforce the platform for future experimental studies on T. foetus cell biology and host-parasite interactions.
In general, the two T. foetus transcriptomes are comparable in size with a near identical GC content of approximately 35%. While this is equivalent to the GC content of T. vaginalis, it is lower than the previously published 41.9% for bovine T. foetus, presumably owing to small library-size bias of the latter [40, 42] . Given the very distinct fundamental host requirements and the vastly different host niches occupied by these two genotypes, we were interested to identify to what degree this would be reflected in their respective gene profiles. We found no biologically significant differences in the distribution of functional annotations between the two T. foetus transcriptomes, despite the slight variances in culture conditions of the two genotypes in this study. This suggests that the two genotypes possess remarkably similar basal functioning. Resemblances in functional capacity in transcriptome-wide studies of other protozoan groups are not uncommon [46, 47]. To our knowledge, this has not been documented in flagellate protozoans extending over a diverse host range such as T. foetus. Limited hints to host-origin were observed as approximately half of the transcripts were flagged as homologs between the genotypes, with 30% being orthologs shared from a common ancestor. As an alternative method of comparing the two T. foetus genotypes, the 100 most highly transcribed genes from each transcriptome were identified through counts of uniquely-mapped sequencing reads. Unsurprisingly, functional categories of highly expressed T. foetus genes included nutrition-related genes, transcription factors and oxygen scavenging genes, with over 50% of the sequences being identified as shared orthologs of the two genotypes. Although minor loss of detail is imminent, discarding multi-mapped reads for expression counts has shown overall reliability in depiction of highly expressed profiles from RNA-seq data [45, 48]. Of the comparable highly transcribed orthologs, the most notable was the 8-fold difference in actin expression between the two isolates. Actin is associated with a myriad of functions including whole cell and intracellular vacuole movement that contribute to parasite virulence [49–52]. Changes in cell morphology and increased interaction with host cells is associated with differential expression of actin in T. vaginalis [53]. While potential culture artefacts cannot be ruled out in the current study, the discrepancy of actin expression between the genotypes is worthy of further characterization to better understand T. foetus virulence.
Within their respective host niches, the bovine and feline T. foetus genotypes are exposed to extreme environmental constraints that place genes under selective pressure as an adaptive mechanism. These responses are seen in sequences as a ratio of synonymous to non-synonymous substitutions (Ka/Ks), which relates to the ratio of silent mutations to amino-acid changing mutations likely to alter protein functionality [54, 55]. Positive selective change was apparent in two pairs of shared T. foetus orthologs both, producing hits to a Myb DNA-binding domain containing protein. As one of the largest families of transcription factors, Myb domain-containing proteins act to regulate the transcription of genes that control and implement important biological processes such as growth, encystation and virulence [43, 56, 57]. Strong divergence of T. foetus transcription factors could imply unique adjustments of gene expression between the two genotypes according to their hosts. A total of 445 and 461 bovine and feline Myb-like proteins, respectively, were annotated in the transcriptomes, suggesting an important role for these transcription factors. In the related T. vaginalis, Myb-like nuclear proteins act to regulate transcription of a gene family that encode surface cytoadhesives such as the AP65 protein essential for the parasite adherence to host cells [58–61]. Taken together, the near identical transcriptomes and diversification of certain transcription factors suggest that transcription and post-transcription regulation form a major aspect of phenotypic host-specificity in T. foetus. It would seem that perhaps the pressure imposed by the differing hosts/niche environments has not, as of yet, been sufficient to drive molecular diversification at the amino acid level between these two genotypes. Alternatively, these may not be evident in the absence of specific cues for the host. The Myb-transcription factors present as ideal candidates for initial investigations into the extreme versatility of T. foetus that allows it to adapt rapidly to new, extreme environments presented by their non-original host.
Untranslated regions (UTRs) flanking either end of mRNA coding regions contain inherent information, such as motif sequences, that govern and regulate the downstream translation of a protein [32, 33]. Under varying environmental conditions, UTRs have the capacity to permit instantaneous phenotypic changes within the parasite to permit rapid responses to biological and environmental cues [32–34]. Here, we mined the untranslated regions (UTRs) for translation regulatory features that may be acting in the bovine and feline T. foetus genotypes. One such feature is the length of the UTRs which has been associated with mRNA stability and translational efficiency [62]. A compilation of UTR lengths from UTR databases indicates that across groups of living organisms, 3′UTR are longer than 5′UTRs [63]. Indeed the mean length of T. foetus UTRs in this study follows the same length bias. The validity of comparing UTR length to determine organism complexity, expression levels and breadth has generated mixed results [64–67]. In the closely related T. vaginalis, the length of the glycolytic glyceraldehydes-2-phosphate dehydrogenase 3′UTR did not show correlation to expression of the enzyme [65]. A more tangible manner of UTR translational regulation is mediated through binding of small mRNA-binding protein to cis-elements in the UTR region of the target protein [34]. Annotation of 5′ and 3′ UTRs to known patterns in this study provide a glimpse of putative regulatory motifs at play in T. foetus. Common motifs such as up-stream open reading frames (uORF) [68], internal ribosomal entry sites (IRES) [69] and AU-rich class 2 elements (ARE2) [70] have been identified here in the UTRs of both T. foetus genotypes. Functionality of several of the motifs found in T. foetus have been described in protozoan and in fact, in Plasmodium, uORF presents an interesting case as it regulates a virulence-associated trait according to host physiological factors [71]. The unique feline T. foetus genotype motif; ADH-DRE, has not been described in protozoans and is related to the down-regulation of the alcohol dehydrogenase gene [72, 73]. Nevertheless, finding common motif matches between T. foetus and other organisms in public databases confirms that the conserved nature of motif patterns extend to T. foetus. Here, we attempted to by-pass the inherent transcription level limitation of RNA-seq to provide an overview of putative translation-related mechanisms in T. foetus. Bioinformatics tools, however, are currently relatively un-reliable in determining true functional regulatory motifs [74]. Experimental characterisation of these motifs in T. foetus is required to elucidate regulatory relationships between regulators and the target in these parasites.
Proteases are expressed by all organisms, playing a critical role in protein catabolism. In parasites, however, these enzymes have evolved specialized functions and are directly involved in numerous host-parasite interactions. Proteases, also known as peptidases, can be classified into seven functional categories based on the principal residue occupying the active site: Aspartic, Cysteine, Glutamic, Serine, Threonine, Metallo and Mixed [41, 75, 76]. Each of these can be further divided into clans and families. Trichomonad parasites also release soluble proteases in vitro and in vivo; the best studied of these being the cysteine proteases (CP). Proteases released into the host milieu, as well as those on the surface of parasites, are important virulence factors involved in host-cell adherence, evasion of host immunity and host cell cytotoxicity [23–25, 39]. Approximately half of the proteases found in the transcriptomes in this study contained cysteine active sites and these were over-represented within a subset of highly expressed proteases. In bovine T. foetus, the most dominant CP family expressed was a papain type CP of the CA clan (C01A); a large family of CPs involved in facilitating infection in protozoa (reviewed in: [22]). This family is slightly less represented in the highest expressed proteases of the feline T. foetus. Regulation of the type of CP secreted by parasites has important implications for the extent of infection in the host. In the related T. vaginalis, secreted CP fraction has been demonstrated to promote host-cell apoptosis. Host-cell specificity of CPs, however, is apparent when incubation of T. vaginalis CP30 with non-host bovine cells fails to induce the same level of cellular destruction compared to the effect of bovine T. foetus CP8 on bovine cells [77, 78]. Here, we confirm that CP8 is the most transcribed CP in the bovine T. foetus as reported by Huang et al. [40]. To the contrary, CP7 was found to be more transcribed than CP8 in feline T. foetus suggesting that the increased expression of CP7 in feline genotype is a host-specific adapted virulence trait. CPs are capable of inducing varying levels of cellular destruction depending on cell source and type. The difference in the major type of CP expressed between the genotypes may explain the slightly varied pathology described in experimental cross-infection of the hosts but the significance of this has yet to be elucidated [10, 16] . Our analysis revealed that the bovine transcriptome contained more hits to proteases than the feline T. foetus transcriptome which could be due to the fractionally higher sequence reads obtained for the bovine T. foetus. The significance of this finding will require full genome sequencing and analysis.
The collection of expressed sequences from the bovine and feline T. foetus genotypes made available in this study presents an opportunity for low-cost in silico mining of novel drug targets worthy of experimental follow-up. With mounting reports of drug resistance and toxic host side-effects, the treatment of choice for human and feline trichomonad infections remains the 5-nitromidazoles drugs [12, 13, 79]. To date, significant, yet non-model protozoan species have been overlooked for in silico mining for druggable targets. Computational pipelines for drug-target discovery have been limited to the few high-profile protozoans with a sequenced genome or focused on identifying druggable features at the host-parasite interface [52, 80, 81]. Here we explore drug target identification for a unique case of a protozoan species with a broad, distantly-related host-range. Novel drug targets for experimental follow-up have to be compatible and non-toxic for the host-species undergoing treatment. In our analysis we intentionally only included shared, full-length T. foetus sequences to gain insights as to how the presence of endogenous host proteins could affect strategies for drug target identification of the same parasite species. By excluding similar host genes, a maximum of 5% of the druggable parasite-unique genes remained, the majority of which were not common between the two genotypes. While the list we generated from our analysis is purposed to be more illustrative rather than definitive; the findings stress the importance of taking different hosts into account as a part of target prioritization in more generalist parasites like T. foetus.
Conclusions
The expressed genes of the bovine and feline Tritrichomonas foetus genotypes offer insights into the breadth of both the T. foetus coding and non-coding genomes. This parasite represents an interesting biological model as it represents a unique case of a protozoan expanding its parasitic foothold across distantly related mammalian hosts. Despite extreme environmental conditions found between bovine urogenital tract and the feline digestive tract inhabited by the two genotypes, they possess near identical functional category distribution of expressed genes with no indication of molecular-level divergence. This reinforces the fact that taxonomically, the bovine and feline T. foetus represent two genotypes displaying intra-specific variation. Host-specific adaptation strategies appear to be focused on post-transcription regulation influenced by environmental cues within the two host niches. In this manner, expression patterns of virulence genes may differ in accordance to their host. Although RNA-seq has provided insights into expression patterns, proteomics studies need to be carried out to examine the full extent of these patterns between the genotypes. Nevertheless, the role of transcriptional and post-transcriptional regulation in T. foetus warrants consideration to guide further research since studies on an environmental-dependent gene in one genotype will not necessarily be representative of the other genotype. Hence, host genes and biology have to be taken into account, particularly in the design of new drug strategies. While in silico methods offer an ideal starting point for novel drug target identification, here we highlight the importance of taking both genotypes and their hosts into account to avoid downstream mis-identification of common drug targets. Taken as a whole, the cell-wide gene library of the bovine and feline T. foetus generated in this study is a useful platform to guide trichomonad research.
Methods
Cultures
Two Tritrichomonas foetus genotypes were used for this study; a genotype isolated from a bovine host and a feline genotype originating from a feline host. The bovine genotype; Tritrichomonas foetus BP-4: Beltsville (ATCC® 30003™, the American Type Culture Collection, Manassas, USA). The feline genotype; Tritrichomonas foetus Sydney-G10/I (cryopreserved in the culture collection at the Faculty of Veterinary Science, The University of Sydney) [15]. Both genotypes were axenically maintained at 37°C by 48 hour passages in a trypticase, yeast extract and maltose (TYM)-medium at pH 7.2. The media was supplemented with 10% (v/v) heat-inactivated lamb serum. To ensure adequate growth, media used for the bovine genotype was further supplemented with 0.05% (w/v) bacterial agar. A mixture of PenStrep and Fungizone was added at a final concentration of 100 μg/ml to both media to safeguard against biological contaminant growth.
Transcriptome
Sample preparation and RNA isolation
Bovine and feline T. foetus cells at the mid-exponential phase in culture were collected and 1 × 107 cells were pelleted at 3220 × g for 5 min. Cells were resuspended in 600 μl RTL buffer, according to the RNeasy Micro kit (Qiagen) protocol and homogenisation was carried out in a FastPrep® - 24 Instrument (MP Biomedicals, USA) for 30 seconds at 4 m/s. An in-column DNAase (Sigma-Aldrich) treatment step was carried out with an incubation time of 15 minutes at room temperature. RNA was eluted in 30 μl of sterile water and assessed both qualitatively and quantitatively using a 2100 Bioanalyzer (Agilent Technologies, Inc). Samples were then transferred into RNAstable® tubes (Biometrica) and preserved by drying in a Savant SpeedVac concentrator connected to a vapour trap for 1 hour, in accordance with the manufacturer’s instructions. Paired-end RNA sequencing on Illumina HiSeq2000 was performed at Macrogen (Seoul, Korea).
Transcriptome sequence analysis and assembly
Raw RNA sequenced reads were subjected to quality control analysis using FastQC (Babraham Bioinformatics). Sequenced reads were mapped onto a small indexed library of published T. foetus coding genes using a combination of bowtie (version bowtie/2.1.0) and TopHat (version tophat/2.0.8) and visualized using IGV (version igv/2.3.3-4G) [40, 82, 83]. A de novo approach using default parameters in Trinity, according to [27], was adopted for assembly of left and right reads belonging to each genotype individually, resulting in two libraries representing the feline and bovine expressed genome.
Ortholog prediction
To obtain a list of coding homologue pairs shared between the two T. foetus genotypes, a reciprocal blast was performed using the Galaxy platform with the assembled feline and bovine T. foetus transcriptomes [84–86]. Putative paralogues were removed by blasting the homolog pairs against the SwissProt database with an e-value cut-off of < 1 × 106. Top blast hits for each transcript pairs were collected and filtered using bash scripting to retrieve 7547 pairs of transcripts showing identical hits to the same protein, which were subsequently utilized as ortholog pairs.
The full-lengther Next algorithm [30] was implemented on the list of 7547 ortholog pairs using the invertebrate database and default parameters to identify full-length transcripts. Based on the criterion that both transcripts from each pair were full length (i.e. contained both a start and stop codon), 1151 pairs of transcripts were selected for further analysis. For each pair of full-length orthologues, bash scripting was used to isolate all coding regions, 5′UTR and 3′UTRs were extracted for further analysis.
Functional annotation and identification of highly expressed genes
The entire assembled transcriptomes and the two lists of 7547 orthologues were annotated through local BlastX searches against the NCBI non-redundant (nr) database abiding to a cut off e-value of 1 × 103 [28]. Gene Ontology (GO) level annotation of the assembled sequences were retrieved using default setting in Blast2GO using the results of the local NCBI BlastX [31]. Combined graphs were generated for each analysis with a level 3 cut-off and a minimum sequence threshold of 100 per category.
To identify the top 100 highly expressed genes between the bovine and feline T. foetus, a bash script was written to create .gtf files for each transcriptome. The assembled transcriptomes were indexed using Bowtie (version bowtie/2.1.0) and raw sequenced reads were mapped back onto the assembled transcriptomes using default TopHat (version tophat/2.0.8) settings [82, 83]. Qualimap compute-counts (version qualimap/0.7.1) [87] were subsequently used with the ‘uniquely-mapped-reads’ algorithm to count the number of raw reads that successfully mapped back onto each assembled transcriptome. Counts were normalized to transcript length using RPKM (reads per kilobase per million of reads). BlastX results of transcripts with the top 100 RPKM were extracted from the whole transcriptome blast against the NCBI nr database blast for comparison.
UTR extraction and annotation
Upon identification of the 1,511 full-length orthologous transcripts, the 5′ and 3′ UTRs were isolated based on identification of the start and stop codon predicted by the full-lengther Next algorithm [30]. Scripting enabled calculation of the lengths of the UTRs and comparative graphs were created. Scatter plots to compare the length of the 3′UTR and 5′UTRs to the normalized transcript expression counts (RPKM) were created and the sum of least square, straight line regression model was adopted in GraphPad Prime 6 (California, USA). A non-linear weighted R2 (weighted by 1/Y^2) was chosen to minimize the sum of the squares of the relative distance of the points from the line. A local version of Patsearch [88] was implemented with the UTRscan algorithm [35] to search for known motif patterns from UTRsite [36]. The UTR regions were extracted and only motifs annotated to within these regions were isolated.
Discovery of new proteases and protease inhibitors
The BlastX annotations of both the feline and the bovine transcriptomes were used to search for the synonymous terms; “protease”, “peptidase” and “proteinase”. Positive search hits to any of the terms were extracted creating two genotype-specific lists of predicated proteases. The lists were further mined for the term “inhibitor”, and all positive matches were removed and used to create separate lists of putative protease inhibitors from the bovine and feline T. foetus. Tritrichomonas foetus transcript IDs were used to retrieve corresponding nucleotide sequences from their respective assembled transcriptome ending with 665 bovine and 623 feline putative protease transcripts. These transcripts were submitted to the available online batch blasting tool on MEROPS peptidase database to search for similarities to known protease active sites [41, 75]. Similarly, the protease inhibitor sequences were subjected to the online blast search against the MEROPS inhibitor database. The resulting feline and bovine inhibitor nucleotide sequences were compared by pairwise alignment using a 2-sequence BlastN [28]. Tritrichomonas foetus transcripts with a positive hit against a MEROPS entry were considered a putative protease. As a comparative control for the proteases, identical searches were implemented on a list of 59,672 annotated Trichomonas vaginalis coding genes downloaded from TrichDB [89].
In order to identify which putative sequences in the two transcriptomes match to published T. foetus cysteine protease (CP) sequences, a 2-sequence BlastN pairwise nucleotide alignment between the MEROPS-confirmed bovine proteases and published bovine CP sequences from Huang et al. [40] and Šlapeta et al. [4] were carried out. Similarly, the BlastN was carried out between the MEROP-confirmed feline sequences and published feline CP sequences from Šlapeta et al. [4]. Qualimap compute-counts with the proportional algorithm was used to count the number of raw sequencing reads that mapped back onto the putative proteases [87]. Resultant expression count values were normalized to transcript length using RPKM and all transcripts with normalized expression values of 500 or greater were selected for comparison between the two genotypes.
Analysis of sequence divergence
Pairs of orthologous coding sequences were translated into protein sequences using a local version of TranslatorX and pairwise alignments of each pair were generated using ClustralW2 through an array bash scripting [90]. Pairwise proteins alignments were translated into codon alignments using Pal2Nal (v14) and the perl parseFastaIntoAXT.pl script which is distributed with kaks-calculator converted the resulting alignment file into the required format [91, 92]. The codon alignments were subsequently used to calculate substitution rates for non-synonymous (Ka) and synonymous (Ks) sites using a 14-model averaging method implemented in KaKs_Calculator2.0 [92].
Druggability
A host database of bovine protein sequences (the full official gene set v2 protein sequences) was retrieved from BovineGenome.org and the complete peptide database for the domestic cat was retrieved from ENSEMBL (release 6.2.74) [93]. The list of 1,511 ortholog pairs belonging to the bovine and feline T. foetus were queried against their respective host protein databases using BlastX (e-value 0.0001) [28]. Parasite transcripts that did not produce a common Blast hit with their respective host were extracted and queried in a further BlastX against known druggable domains retrieved from the ChEMBL’s DrugEBility database (e-value 0.0001) [94]. Positive hits were matched with domain information to identify which transcripts contain domains that satisfy Lipinski ‘rule of 5’ of druggability [95].
Availability of supporting datasets
The assembled transcriptome libraries supporting the results of this article are available in the LabArchives repository [96], [http://dx.doi.org/10.6070/H4GH9FWD]. All raw sequence read data has been submitted to the sequence read data (SRA) repository under the BioProject accession PRJNA246668 [http://www.ncbi.nlm.nih.gov/bioproject/PRJNA246668].
The additional tables (Additional file 1: Tables S1, Additional file 2: Table S2, Additional file 3: Table S3 and Additional file 4: Table S4) supporting the results of this article are included within the article.
Electronic supplementary material
Additional file 1: Highly expressed proteases of the bovine T. foetus genotype. Two excel documents showing assembled bovine transcript ID (from this study) that presents with a hit to the MEROPs database, in order of the highest estimation of protease expression (RPKM) within the bovine genotype. Transcript length, specific hits to MEROPS proteases family and MEROPS accession numbers are presented. Transcript sequences that successfully produce pairwise alignment to published bovine cysteine proteases are also shown. (XLSX 20 KB)
Additional file 2: Highly expressed proteases of the feline T. foetus genotype. Two excel documents showing assembled feline transcript ID (from this study) that presents with a hit to the MEROPs database, in order of the highest estimation of protease expression (RPKM) within the feline genotype. Transcript length, specific hits to MEROPS proteases family and MEROPS accession numbers are presented. Transcript sequences that successfully produce pairwise alignment to published feline cysteine proteases are also shown. (XLSX 41 KB)
Additional file 3: Non-host, putative bovine T. foetus genotype druggable domains. Bovine T. foetus non-host (i.e. unique to the parasite) transcripts which presented a positive hit to known druggable domains are presented, along with the EMBL DrugGAbility px number (SCOP domain), the associated pdb code and information regarding the druggable domain found. Transcript IDs are repeated in the table as more than one domain could associated within each transcript. Adherence to the Lipinski rule of 5 for druggablity is presented as “Y” for true or “N” for false. BlastX annotation and length of the assembled transcript is also presented. (XLSX 32 KB)
Additional file 4: Non-host, putative feline T. foetus genotype druggable domains. Feline T. foetus non-host (i.e. unique to the parasite) transcripts which presented a positive hit to known druggable domains are presented, along with the EMBL DrugGAbility px number (SCOP domain), the associated pdb code and information regarding the druggable domain found. Transcript IDs are repeated in the table as more than one domain could be associated within each transcript. Adherence to the Lipinski rule of 5 for druggablity is presented as “Y” for true or “N” for false. BlastX annotation and length of the assembled transcript is also presented. (XLSX 51 KB)
Acknowledgements
VMA is supported by the International Postgraduate Research Scholarship (IPRS) and an Australian Postgraduate Award (APA) tenable at The University of Sydney. VMA was a recipient of the Australian Society for Parasitology (ASP) Network Researcher Exchange, Training and Travel Award. This study was funded in part by the Dorothy Minchin Bequest (Faculty of Veterinary Science, University of Sydney), the School of Science and Health Research Seed Grant (University of Western Sydney) and the Spanish Ministry of Economy and Competitiveness (grant number BIO2012-40244) and FP7 STATegra (grant number 306000). We thank Dr. Pedro Seoane Zonjic and Dr. Giorgio Grillo for their active discussions and support with optimization of the full-lengther-next and UTRscan algorithms used in this study, respectively.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
VMA designed the study, carried out all laboratory work, bioinformatics, statistics and data analysis, result interpretations and drafted the manuscript; RL provided bioinformatics guidance, carried out the local blast analysis and scripts; DO’M contributed to preliminary bioinformatics analysis; CS contributed material and insights into the proteases; AC supervised the bioinformatics analysis and provided resources for the bioinformatics analysis; JŠ conceived and coordinated the study; all authors edited and commented on the paper. All authors read, approved and contributed to the final manuscript.
Contributor Information
Victoria Morin-Adeline, Email: victoria.morin-adeline@sydney.edu.au.
Rodrigo Lomas, Email: rodrigolomas@gmail.com.
Denis O’Meally, Email: omeally@gmail.com.
Colin Stack, Email: c.stack@uws.edu.au.
Ana Conesa, Email: aconesa@cipf.es.
Jan Šlapeta, Email: jan.slapeta@sydney.edu.au.
References
- 1.Cepicka I, Hampl V, Kulda J. Critical taxonomic revision of parabasalids with description of one new genus and three new species. Protist. 2010;161(3):400–433. doi: 10.1016/j.protis.2009.11.005. [DOI] [PubMed] [Google Scholar]
- 2.Tachezy J, Tachezy R, Hampl V, Šedinová M, Vaňáčová S, Vrlík M, Van Ranst M, Flegr J, Kulda J. Cattle pathogen Tritrichomonas foetus (Riedmüller, 1928) and pig commensal Tritrichomonas suis (Gruby & Delafond, 1843) belong to the same species. J Eukaryot Microbiol. 2002;49(2):154–163. doi: 10.1111/j.1550-7408.2002.tb00360.x. [DOI] [PubMed] [Google Scholar]
- 3.Mattos A, Solé-Cava AM, De Carli G, Benchimol M. Fine structure and isozymic characterization of trichomonadid protozoa. Parasitol Res. 1997;83(3):290–295. doi: 10.1007/s004360050249. [DOI] [PubMed] [Google Scholar]
- 4.Šlapeta J, Müller N, Stack CM, Walker G, Lew-Tabor A, Tachezy J, Frey CF. Comparative analysis of Tritrichomonas foetus (Riedmuller, 1928) cat genotype, T. Foetus (Riedmuller, 1928) cattle genotype and Tritrichomonas suis (Davaine, 1875) at 10 DNA loci. Int J Parasitol. 2012;42(13–14):1143–1149. doi: 10.1016/j.ijpara.2012.10.004. [DOI] [PubMed] [Google Scholar]
- 5.Mardones FO, Perez AM, Martinez A, Carpenter TE. Risk factors associated with Tritrichomonas foetus infection in beef herds in the province of Buenos Aires, Argentina. Vet Parasitol. 2008;153(3–4):231–237. doi: 10.1016/j.vetpar.2008.01.038. [DOI] [PubMed] [Google Scholar]
- 6.Parsonson IM, Clark BL, Dufty JH. Early pathogenesis and pathology of Tritrichomonas foetus infection in virgin heifers. J Comp Pathol. 1976;86(1):59–66. doi: 10.1016/0021-9975(76)90028-1. [DOI] [PubMed] [Google Scholar]
- 7.Rae DO, Crews JE, Greiner EC, Donovan GA. Epidemiology of Tritrichomonas foetus in beef bull populations in Florida. Theriogenology. 2004;61(4):605–618. doi: 10.1016/S0093-691X(03)00236-X. [DOI] [PubMed] [Google Scholar]
- 8.Rhyan JC, Stackhouse LL, Quinn WJ. Fetal and placental lesions in bovine abortion due to Tritrichomonas foetus. Vet Pathol. 1988;25(5):350–355. doi: 10.1177/030098588802500503. [DOI] [PubMed] [Google Scholar]
- 9.Gookin JL, Breitschwerdt EB, Ley MG, Gager RB, Benrud JG. Diarrhea associated with trichomonosis in cats. J Am Vet Med Assoc. 1999;215(10):1450–1454. [PubMed] [Google Scholar]
- 10.Stockdale HD, Dillon AR, Newton JC, Bird RC, Bondurant RH, Deinnocentes P, Barney S, Bulter J, Land T, Spencer JA, Lindsay DS, Blagburn BL. Experimental infection of cats (felis catus) with Tritrichomonas foetus isolated from cattle. Vet Parasitol. 2008;154(1–2):156–161. doi: 10.1016/j.vetpar.2008.02.024. [DOI] [PubMed] [Google Scholar]
- 11.Yaeger MJ, Gookin JL. Histologic features associated with Tritrichomonas foetus-induced colitis in domestic cats. Vet Pathol. 2005;42(6):797–804. doi: 10.1354/vp.42-6-797. [DOI] [PubMed] [Google Scholar]
- 12.Cudmore SL, Delgaty KL, Hayward-McClelland SF, Petrin DP, Garber GE. Treatment of infections caused by metronidazole-resistant Trichomonas vaginalis. Clin Microbiol Rev. 2004;17(4):783. doi: 10.1128/CMR.17.4.783-793.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gookin JL, Stauffer SH, Dybas D, Cannon DH. Documentation of in vivo and in vitro aerobic resistance of feline Tritrichomonas foetus isolates to ronidazole. J Vet Intern Med. 2010;24(4):1003–1007. doi: 10.1111/j.1939-1676.2010.0534.x. [DOI] [PubMed] [Google Scholar]
- 14.Yao C. Diagnosis of Tritrichomonas foetus-infected bulls, an ultimate approach to eradicate bovine trichomoniasis in US cattle? J Med Microbiol. 2013;62:1–9. doi: 10.1099/jmm.0.047365-0. [DOI] [PubMed] [Google Scholar]
- 15.Šlapeta J, Craig S, McDonell D, Emery D. Tritrichomonas foetus from domestic cats and cattle are genetically distinct. Exp Parasitol. 2010;126(2):209–213. doi: 10.1016/j.exppara.2010.04.024. [DOI] [PubMed] [Google Scholar]
- 16.Stockdale H, Rodning S, Givens M, Carpenter D, Lenz S, Spencer J, Dykstra C, Lindsay D, Blagburn B. Experimental infection of cattle with a feline isolate of Tritrichomonas foetus. J Parasitol. 2007;93(6):1429–1434. doi: 10.1645/GE-1305.1. [DOI] [PubMed] [Google Scholar]
- 17.Bernasconi CH, Bodmer M, Doherr MG, Janett F, Thomann A, Spycher C, Iten C, Hentrich B, Gottstein B, Müller N, Frey CF. Tritrichomonas foetus: prevalence study in naturally mating bulls in Switzerland. Vet Parasitol. 2014;200(3–4):289–294. doi: 10.1016/j.vetpar.2013.12.029. [DOI] [PubMed] [Google Scholar]
- 18.Mendoza-Ibarra JA, Pedraza-Díaz S, García-Peña FJ, Rojo-Montejo S, Ruiz-Santa-Quiteria JA, San Miguel-Ibáñez E, Navarro-Lozano V, Ortega-Mora LM, Osoro K, Collantes-Fernandez E. High prevalence of Tritrichomonas foetus infection in Asturiana de la Montana beef cattle kept in extensive conditions in northern Spain. Veter J. 2012;193(1):146–151. doi: 10.1016/j.tvjl.2011.09.020. [DOI] [PubMed] [Google Scholar]
- 19.Paris JK, Wills S, Balzer HJ, Shaw DJ, Gunn-Moore DA. Enteropathogen co-infection in UK cats with diarrhoea. BMC Vet Res. 2014;10:13. doi: 10.1186/1746-6148-10-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reinmann K, Müller N, Kuhnert P, Campero CM, Leitsch D, Hess M, Henning K, Fort M, Müller J, Gottstein B, Frey CF. Tritrichomonas foetus isolates from cats and cattle show minor genetic differences in unrelated loci ITS-2 and EF-1 alpha. Vet Parasitol. 2012;185(2–4):138–144. doi: 10.1016/j.vetpar.2011.09.032. [DOI] [PubMed] [Google Scholar]
- 21.Sun ZC, Stack C, Šlapeta J. Sequence differences in the diagnostic region of the cysteine protease 8 gene of Tritrichomonas foetus parasites of cats and cattle. Vet Parasitol. 2012;186(3–4):445–449. doi: 10.1016/j.vetpar.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 22.Atkinson HJ, Babbitt PC, Sajid M. The global cysteine peptidase landscape in parasites. Trends Parasitol. 2009;25(12):573–581. doi: 10.1016/j.pt.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burgess DE, Knoblock KF, Daugherty T, Robertson NP. Cytotoxic and hemolytic effects of Tritrichomonas foetus on mammalian cells. Infect Immun. 1990;58(11):3627–3632. doi: 10.1128/iai.58.11.3627-3632.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lucas JJ, Hayes GR, Kalsi HK, Gilbert RO, Choe Y, Craik CS, Singh BN. Characterization of a cysteine protease from Tritrichomonas foetus that induces host-cell apoptosis. Arch Biochem Biophys. 2008;477(2):239–243. doi: 10.1016/j.abb.2008.05.018. [DOI] [PubMed] [Google Scholar]
- 25.Thomford JW, Talbot JA, Ikeda JS, Corbeil LB. Characterization of extracellular proteinases of Tritrichomonas foetus. J Parasitol. 1996;82(1):112–117. doi: 10.2307/3284125. [DOI] [PubMed] [Google Scholar]
- 26.Fenchel T. Cosmopolitan microbes and their ‘cryptic’ species. Aquat Microb Ecol. 2005;41(1):49–54. doi: 10.3354/ame041049. [DOI] [Google Scholar]
- 27.Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29(7):644–U130. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 29.Bairoch A, Apweiler R. The SWISS-PROT protein sequence data bank and its new supplement TREMBL. Nucleic Acids Res. 1996;24(1):21–25. doi: 10.1093/nar/24.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lara AJ, Pérez-Trabado G, Villalobos DP, Díaz-Moreno S, Cantón FR, Claros MG. A web tool to discover full-length sequences - full-lengther. In: Corchado E, Corchado JM, Abraham A, editors. Innovations in Hybrid Intelligent Systems. Berlin Heidelberg: Springer-Verlag; 2007. pp. 361–368. [Google Scholar]
- 31.Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21(18):3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 32.Kleijn M, Scheper GC, Voorma HO, Thomas AAM. Regulation of translation initiation factors by signal transduction. Eur J Biochem. 1998;253(3):531–544. doi: 10.1046/j.1432-1327.1998.2530531.x. [DOI] [PubMed] [Google Scholar]
- 33.McCarthy JEG, Kollmus H. Cytoplasmic mRNA-protein interactions in eukaryotic gene expression. Trends Biochem Sci. 1995;20(5):191–197. doi: 10.1016/S0968-0004(00)89006-4. [DOI] [PubMed] [Google Scholar]
- 34.Pesole G, Grillo G, Larizza A, Liuni S. The untranslated regions of eukaryotic mRNAs: structure, function, evolution and bioinformatic tools for their analysis. Brief Bioinform. 2000;1(3):236–249. doi: 10.1093/bib/1.3.236. [DOI] [PubMed] [Google Scholar]
- 35.Pesole G, Liuni S. Internet resources for the functional analysis of 5 ′ and 3 ′ untranslated regions of eukaryotic mRNAs. Trends Genet. 1999;15(9):378–378. doi: 10.1016/S0168-9525(99)01795-3. [DOI] [PubMed] [Google Scholar]
- 36.Pesole G, Liuni S, Grillo G, Licciulli F, Mignone F, Gissi C, Saccone C. UTRdb and UTRsite: specialized databases of sequences and functional elements of 5 ′ and 3 ′ untranslated regions of eukaryotic mRNAs. Update 2002. Nucleic Acids Res. 2002;30(1):335–340. doi: 10.1093/nar/30.1.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Singh BN, Lucas JJ, Beach DH, Shin ST, Gilbert RO. Adhesion of Tritrichomonas foetus to bovine vaginal epithelial cells. Infect Immun. 1999;67(8):3847–3854. doi: 10.1128/iai.67.8.3847-3854.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Corbeil LB, Anderson ML, Corbeil RR, Eddow JM, BonDurant RH. Female reproductive tract immunity in bovine trichomoniasis. Am J Reprod Immunol. 1998;39(3):189–198. doi: 10.1111/j.1600-0897.1998.tb00353.x. [DOI] [PubMed] [Google Scholar]
- 39.Bastida-Corcuera F, Butler JE, Heyermann H, Thomford JW, Corbeil LB. Tritrichomonas foetus extracellular cysteine proteinase cleavage of bovine IgG2 allotypes. J Parasitol. 2000;86(2):328–332. doi: 10.1645/0022-3395(2000)086[0328:TFECPC]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 40.Huang K-Y, Shin J-W, Huang P-J, Ku F-M, Lin W-C, Lin R, Hsu W-M, Tang P. Functional profiling of the Tritrichomonas foetus transcriptome and proteome. Mol Biochem Parasitol. 2013;187(1):60–71. doi: 10.1016/j.molbiopara.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 41.Rawlings ND, Waller M, Barrett AJ, Bateman A. MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2014;42(D1):D503–D509. doi: 10.1093/nar/gkt953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carlton JM, Hirt RP, Silva JC, Delcher AL, Schatz M, Zhao Q, Wortman JR, Bidwell SL, Alsmark UCM, Besteiro S, Sicheritz-Ponten T, Noel CJ, Dacks JB, Foster PG, Simillion C, Van de Peer Y, Miranda-Saavedra D, Barton GJ, Westrop GD, Müller S, Dessi D, Fiori PL, Ren Q, Paulsen I, Zhang H, Bastida-Corcuera FD, Simoes-Barbosa A, Brown MT, Hayes RD, Mukherjee M, et al. Draft genome sequence of the sexually transmitted pathogen Trichomonas vaginalis. Science. 2007;315(5809):207–212. doi: 10.1126/science.1132894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sun CH, Palm D, McArthur AG, Svard SG, Gillin FD. A novel Myb-related protein involved in transcriptional activation of encystation genes in Giardia lamblia. Mol Microbiol. 2002;46(4):971–984. doi: 10.1046/j.1365-2958.2002.03233.x. [DOI] [PubMed] [Google Scholar]
- 44.Nookaew I, Papini M, Pornputtapong N, Scalcinati G, Fagerberg L, Uhlen M, Nielsen J. A comprehensive comparison of RNA-Seq-based transcriptome analysis from reads to differential gene expression and cross-comparison with microarrays: a case study in Saccharomyces cerevisiae. Nucleic Acids Res. 2012;40(20):10084–10097. doi: 10.1093/nar/gks804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008;18(9):1509–1517. doi: 10.1101/gr.079558.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Novaes J, Rangel L, Ferro M, Abe RY, Manha APS, de Mello JCM, Varuzza L, Durham AM, Madeira A, Gruber A. A comparative transcriptome analysis reveals expression profiles conserved across three Eimeria spp. of domestic fowl and associated with multiple developmental stages. Int J Parasitol. 2012;42(1):39–48. doi: 10.1016/j.ijpara.2011.10.008. [DOI] [PubMed] [Google Scholar]
- 47.Reid AJ, Vermont SJ, Cotton JA, Harris D, Hill-Cawthorne GA, Könen-Waisman S, Latham SM, Mourier T, Norton R, Quail MA, Sanders M, Shanmugam D, Sohal A, Wasmuth JD, Brunk B, Grigg ME, Howard JC, Parkinson J, Roos DS, Trees AJ, Berriman M, Pain A, Wastling JM. Comparative genomics of the apicomplexan parasites Toxoplasma gondii and Neospora caninum: coccidia differing in host range and transmission strategy. Plos Pathog. 2012;8(3):e1002567. doi: 10.1371/journal.ppat.1002567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Treangen TJ, Salzberg SL. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat Rev Genet. 2012;13(1):36–46. doi: 10.1038/nrg3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hill KL, Catlett NL, Weisman LS. Actin and myosin function in directed vacuole movement during cell division in Saccharomyces cerevisiae. J Cell Biol. 1996;135(6):1535–1549. doi: 10.1083/jcb.135.6.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Skillman KM, Diraviyam K, Khan A, Tang K, Sept D, Sibley LD. Evolutionarily divergent, unstable filamentous actin is essential for gliding motility in apicomplexan parasites. Plos Pathog. 2011;7(10):e1002280. doi: 10.1371/journal.ppat.1002280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bradley PJ, Sibley LD. Rhoptries: an arsenal of secreted virulence factors. Curr Opin Microbiol. 2007;10(6):582–587. doi: 10.1016/j.mib.2007.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ferreira RS, Guido RVC, Andricopulo AD, Oliva G. In silico screening strategies for novel inhibitors of parasitic diseases. Expert Opin Drug Discov. 2011;6(5):481–489. doi: 10.1517/17460441.2011.563297. [DOI] [PubMed] [Google Scholar]
- 53.Bricheux G, Brugerolle G. Molecular cloning of actin genes in Trichomonas vaginalis and phylogeny inferred from actin sequences. FEMS Microbiol Lett. 1997;153(1):205–213. doi: 10.1111/j.1574-6968.1997.tb10483.x. [DOI] [PubMed] [Google Scholar]
- 54.Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980;16(2):111–120. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- 55.Yang ZH, Bielawski JP. Statistical methods for detecting molecular adaptation. Trends Ecol Evol. 2000;15(12):496–503. doi: 10.1016/S0169-5347(00)01994-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rushton JJ, Davis LM, Lei WL, Mo XM, Leutz A, Ness SA. Distinct changes in gene expression induced by A-Myb, B-Myb and c-Myb proteins. Oncogene. 2003;22(2):308–313. doi: 10.1038/sj.onc.1206131. [DOI] [PubMed] [Google Scholar]
- 57.Gissot M, Briquet S, Refour P, Boschet C, Vaquero C. PfMyb1, a Plasmodium falciparum transcription factor, is required for intra-erythrocytic growth and controls key genes for cell cycle regulation. J Mol Biol. 2005;346(1):29–42. doi: 10.1016/j.jmb.2004.11.045. [DOI] [PubMed] [Google Scholar]
- 58.Alderete JF, Obrien JL, Arroyo R, Engbring JA, Musatovova O, Lopez O, Lauriano C, Nguyen J. Cloning and molecular characterization of 2 genes encoding adhesion proteins involved in Trichomonas vaginalis cytoadherence. Mol Microbiol. 1995;17(1):69–83. doi: 10.1111/j.1365-2958.1995.mmi_17010069.x. [DOI] [PubMed] [Google Scholar]
- 59.Garcia AF, Chang TH, Benchimol M, Klumpp DJ, Lehker MW, Alderete JF. Iron and contact with host cells induce expression of adhesins on surface of Trichomonas vaginalis. Mol Microbiol. 2003;47(5):1207–1224. doi: 10.1046/j.1365-2958.2003.03366.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ong SJ, Hsu HM, Liu HW, Chu CH, Tai JH. Multifarious transcriptional regulation of adhesion protein gene ap65-1 by a novel Myb1 protein in the protozoan parasite Trichomonas vaginalis. Eukaryot Cell. 2006;5(2):391–399. doi: 10.1128/EC.5.2.391-399.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tsai CD, Liu HW, Tai JH. Characterization of an iron-responsive promoter in the protozoan pathogen Trichomonas vaginalis. J Biol Chem. 2002;277(7):5153–5162. doi: 10.1074/jbc.M110234200. [DOI] [PubMed] [Google Scholar]
- 62.Tanguay RL, Gallie DR. Translational efficiency is regulated by the length of the 3′ untranslated region. Mol Cell Biol. 1996;16(1):146–156. doi: 10.1128/mcb.16.1.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mazumder B, Seshadri V, Fox PL. Translational control by the 3 ′-UTR: the ends specify the means. Trends Biochem Sci. 2003;28(2):91–98. doi: 10.1016/S0968-0004(03)00002-1. [DOI] [PubMed] [Google Scholar]
- 64.Rao YS, Wang ZF, Chai XW, Nie QH, Zhang XQ. Relationship between 5 ′ UTR length and gene expression pattern in chicken. Genetica. 2013;141(7–9):311–318. doi: 10.1007/s10709-013-9730-9. [DOI] [PubMed] [Google Scholar]
- 65.ter Kuile BH, Sallés FJ. The length of the combined 3 ′ untranslated region and poly (A) tail does not control rates of glyceraldehyde-3-phosphate dehydrogenase mRNA translation in three species of parasitic protists. J Bacteriol. 2000;182(12):3587–3589. doi: 10.1128/JB.182.12.3587-3589.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Maicas E, Shago M, Friesen JD. Translation of the Saccharomyces cerevisiae tcm1 gene in the absence of a 5′-untranslated leader. Nucleic Acids Res. 1990;18(19):5823–5828. doi: 10.1093/nar/18.19.5823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hughes MJG, Andrews DW. A single nucleotide is a sufficient 5′ untranslated region for translation in an eukaryotic in vitro system. FEBS Lett. 1997;414(1):19–22. doi: 10.1016/S0014-5793(97)00965-4. [DOI] [PubMed] [Google Scholar]
- 68.Morris DR, Geballe AP. Upstream open reading frames as regulators of mRNA translation. Mol Cell Biol. 2000;20(23):8635–8642. doi: 10.1128/MCB.20.23.8635-8642.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hellen CUT, Sarnow P. Internal ribosome entry sites in eukaryotic mRNA molecules. Genes Dev. 2001;15(13):1593–1612. doi: 10.1101/gad.891101. [DOI] [PubMed] [Google Scholar]
- 70.Barreau C, Paillard L, Osborne HB. AU-rich elements and associated factors: are there unifying principles? Nucleic Acids Res. 2005;33(22):7138–7150. doi: 10.1093/nar/gki1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Amulic B, Salanti A, Lavstsen T, Nielsen MA, Deitsch KW. An upstream open reading frame controls translation of var2csa, a gene implicated in placental malaria. PLoS Pathog. 2009;5(1):e1000256. doi: 10.1371/journal.ppat.1000256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Parsch J, Stephan W, Tanda S. A highly conserved sequence in the 3 ′-untranslated region of the Drosophila Adh gene plays a functional role in Adh expression. Genetics. 1999;151(2):667–674. doi: 10.1093/genetics/151.2.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Parsch J, Russell JA, Beerman I, Hartl DL, Stephan W. Deletion of a conserved regulatory element in the Drosophila Adh gene leads to increased alcohol dehydrogenase activity but also delays development. Genetics. 2000;156(1):219–227. doi: 10.1093/genetics/156.1.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bryant CH, Kemp GJL, Cvijovic M, Slelpi A first step towards learning which uORFs regulate gene expression. J Integr Bioinform. 2006;3(2):31. [Google Scholar]
- 75.Rawlings ND, Barrett AJ, Bateman A. MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2012;40(D1):D343–D350. doi: 10.1093/nar/gkr987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rawlings ND, Barrett AJ, Bateman A. MEROPS: the peptidase database. Nucleic Acids Res. 2010;38:D227–D233. doi: 10.1093/nar/gkp971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sommer U, Costello CE, Hayes GR, Beach DH, Gilbert RO, Lucas JJ, Singh BN. Identification of Trichomonas vaginalis cysteine proteases that induce apoptosis in human vaginal epithelial cells. J Biol Chem. 2005;280(25):23853–23860. doi: 10.1074/jbc.M501752200. [DOI] [PubMed] [Google Scholar]
- 78.Singh BN, Hayes GR, Lucas JJ, Beach DH, Gilbert RO. In vitro cytopathic effects of a cysteine protease of Tritrichomonas foetus on cultured bovine uterine epithelial cells. Am J Vet Res. 2005;66(7):1181–1186. doi: 10.2460/ajvr.2005.66.1181. [DOI] [PubMed] [Google Scholar]
- 79.LeVine DN, Gookin JL, Papich MG, Davidson GS. Twice-daily dosing of RDZ no longer recommended for treatment of intestinal Tritrichomonas foetus infection. J Feline Med Surg. 2014;16(2):198–198. doi: 10.1177/1098612X13506430. [DOI] [PubMed] [Google Scholar]
- 80.Brown JR, Magid-Slav M, Sanseau P, Rajpal DK. Computational biology approaches for selecting host-pathogen drug targets. Drug Discov Today. 2011;16(5–6):229–236. doi: 10.1016/j.drudis.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 81.Sateriale A, Bessoff K, Sarkar IN, Huston CD. Drug repurposing: mining protozoan proteomes for targets of known bioactive compounds. J Am Med Inform Assoc. 2014;21(2):238–244. doi: 10.1136/amiajnl-2013-001700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14(4):R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Blankenberg D, Von Kuster G, Coraor N, Ananda G, Lazarus R, Mangan M, Nekrutenko A, Taylor J. Galaxy: a web-based genome analysis tool for experimentalists. Curr Protoc Mol Biol. 2010;89:19.10.1–19.10.21. doi: 10.1002/0471142727.mb1910s89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Giardine B, Riemer C, Hardison RC, Burhans R, Elnitski L, Shah P, Zhang Y, Blankenberg D, Albert I, Taylor J, Miller W, Kent WJ, Nekrutenko A. Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 2005;15(10):1451–1455. doi: 10.1101/gr.4086505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Goecks J, Nekrutenko A, Taylor J, Galaxy Team Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010;11(8):R86. doi: 10.1186/gb-2010-11-8-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.García-Alcalde F, Okonechnikov K, Carbonell J, Cruz LM, Götz S, Tarazona S, Dopazo J, Meyer TF, Conesa A. Qualimap: evaluating next-generation sequencing alignment data. Bioinformatics. 2012;28(20):2678–2679. doi: 10.1093/bioinformatics/bts503. [DOI] [PubMed] [Google Scholar]
- 88.Grillo G, Licciulli F, Liuni S, Sbisa E, Pesole G. PatSearch: a program for the detection of patterns and structural motifs in nucleotide sequences. Nucleic Acids Res. 2003;31(13):3608–3612. doi: 10.1093/nar/gkg548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Aurrecoechea C, Brestelli J, Brunk BP, Carlton JM, Dommer J, Fischer S, Gajria B, Gao X, Gingle A, Grant G, Harb OS, Heiges M, Innamorato F, Iodice J, Kissinger JC, Kraemer E, Li W, Miller JA, Morrison HG, Nayak V, Pennington C, Pinney DF, Roos DS, Ross C, Stoeckert CJ, Jr, Sullivan S, Treatman C, Wang H. GiardiaDB and TrichDB: integrated genomic resources for the eukaryotic protist pathogens Giardia lamblia and Trichomonas vaginalis. Nucleic Acids Res. 2009;37:D526–D530. doi: 10.1093/nar/gkn631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Abascal F, Zardoya R, Telford MJ. TranslatorX: multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 2010;38:W7–W13. doi: 10.1093/nar/gkq291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Suyama M, Torrents D, Bork P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006;34:W609–W612. doi: 10.1093/nar/gkl315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang D, Zhang Y, Zhang Z, Zhu J, Yu J. KaKs_calculator 2.0: a toolkit incorporating gamma-series methods and sliding window strategies. Genomics Proteomics Bioinformatics. 2010;8(1):77–80. doi: 10.1016/S1672-0229(10)60008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Reese JT, Childers CP, Sundaram JP, Dickens CM, Childs KL, Vile DC, Elsik CG. Bovine Genome Database: supporting community annotation and analysis of the Bos Taurus genome. BMC Genomics. 2010;11:645. doi: 10.1186/1471-2164-11-645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gaulton A, Bellis LJ, Bento AP, Chambers J, Davies M, Hersey A, Light Y, McGlinchey S, Michalovich D, Al-Lazikani B, Overington JP. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012;40(D1):D1100–D1107. doi: 10.1093/nar/gkr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2012;64(1-3):3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 96.Morin-Adeline V, Lomas R, O’Meally D, Stack C, Conesa A, Šlapeta J: Bovine and felineTritrichomonas foetusassembled transcriptomes. LabArchives;http://dx.doi.org/10.6070/H4GH9FWD [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Highly expressed proteases of the bovine T. foetus genotype. Two excel documents showing assembled bovine transcript ID (from this study) that presents with a hit to the MEROPs database, in order of the highest estimation of protease expression (RPKM) within the bovine genotype. Transcript length, specific hits to MEROPS proteases family and MEROPS accession numbers are presented. Transcript sequences that successfully produce pairwise alignment to published bovine cysteine proteases are also shown. (XLSX 20 KB)
Additional file 2: Highly expressed proteases of the feline T. foetus genotype. Two excel documents showing assembled feline transcript ID (from this study) that presents with a hit to the MEROPs database, in order of the highest estimation of protease expression (RPKM) within the feline genotype. Transcript length, specific hits to MEROPS proteases family and MEROPS accession numbers are presented. Transcript sequences that successfully produce pairwise alignment to published feline cysteine proteases are also shown. (XLSX 41 KB)
Additional file 3: Non-host, putative bovine T. foetus genotype druggable domains. Bovine T. foetus non-host (i.e. unique to the parasite) transcripts which presented a positive hit to known druggable domains are presented, along with the EMBL DrugGAbility px number (SCOP domain), the associated pdb code and information regarding the druggable domain found. Transcript IDs are repeated in the table as more than one domain could associated within each transcript. Adherence to the Lipinski rule of 5 for druggablity is presented as “Y” for true or “N” for false. BlastX annotation and length of the assembled transcript is also presented. (XLSX 32 KB)
Additional file 4: Non-host, putative feline T. foetus genotype druggable domains. Feline T. foetus non-host (i.e. unique to the parasite) transcripts which presented a positive hit to known druggable domains are presented, along with the EMBL DrugGAbility px number (SCOP domain), the associated pdb code and information regarding the druggable domain found. Transcript IDs are repeated in the table as more than one domain could be associated within each transcript. Adherence to the Lipinski rule of 5 for druggablity is presented as “Y” for true or “N” for false. BlastX annotation and length of the assembled transcript is also presented. (XLSX 51 KB)