

Abstract
Background
Conventional karyotyping (550 bands resolution) is able to identify chromosomal aberrations >5-10 Mb, which represent a known cause of intellectual disability/developmental delay (ID/DD) and/or multiple congenital anomalies (MCA). Array-Comparative Genomic Hybridization (array-CGH) has increased the diagnostic yield of 15-20%.
Results
In a cohort of 700 ID/DD cases with or without MCA, including 15 prenatal diagnoses, we identified a subgroup of seven patients with a normal karyotype and a large complex rearrangement detected by array-CGH (at least 6, and up to 18 Mb). FISH analysis could be performed on six cases and showed that rearrangements were translocation derivatives, indistinguishable from a normal karyotype as they involved a similar band pattern and size. Five were inherited from a parent with a balanced translocation, whereas two were apparently de novo. Genes spanning the rearrangements could be associated with some phenotypic features in three cases (case 3: DOCK8; case 4: GATA3, AKR1C4; case 6: AS/PWS deletion, CHRNA7), and in two, likely disease genes were present (case 5: NR2F2, TP63, IGF1R; case 7: CDON). Three of our cases were prenatal diagnoses with an apparently normal karyotype.
Conclusions
Large complex rearrangements of up to 18 Mb, involving chromosomal regions with similar size and band appearance may be overlooked by conventional karyotyping. Array-CGH allows a precise chromosomal diagnosis and recurrence risk definition, further confirming this analysis as a first tier approach to clarify molecular bases of ID/DD and/or MCA. In prenatal tests, array-CGH is confirmed as an important tool to avoid false negative results due to karyotype intrinsic limit of detection.
Keywords: GTG-banding, Array-CGH, Unbalanced derivative chromosomes, CNV, Genomic rearrangement, Intellectual disability
Background
GTG-banding karyotype is a standard procedure in the diagnosis of patients with unexplained intellectual disability/developmental delay (ID/DD), autism spectrum disorders (ASD), and multiple congenital anomalies (MCA) [1]. The limit for the detection of genomic rearrangements is estimated above 5–10 Mb at the 500–550 band level, at least in regions where the band pattern is distinctive [2]. Karyotyping is also widely used in prenatal testing even if its resolution is lower due to a more compact chromatin structure.
The detection of submicroscopic rearrangements by array-CGH has increased the diagnostic yield of patients with ID/DD and/or MCA of 15-20% [3–5], due to array-CGH higher resolution vs. karyotyping (50–100 kb on a 60 K Agilent platform) [3,6]. Indeed, since its introduction, array-CGH analysis has evidenced that karyotyping can also miss large (>7 Mb) and very large (>10 Mb) rearrangements [2]. An estimate of the number of these overlooked rearrangements is still unknown. These are likely to be mainly derivative chromosomes with deletions and duplications, involving chromosomal regions with a similar banding pattern and size. These anomalies can result from parental balanced translocations that malsegregate at meiosis. Given that ~0.5% of the general population is estimated to be carrier of a balanced rearrangement [7], derivative chromosomes apparently normal at karyotype may be more common than expected. Whereas balanced translocation are associated with infertility and recurrent miscarriages [8], the clinical consequences of derivative chromosomes can be lethal or lead to complex severe phenotypes.
This study includes seven patients presenting with complex developmental anomalies, an apparently normal karyotype and an unexpected large (>6 Mb) chromosomal rearrangement detected by array-CGH.
Results
In our survey of 700 patients with ID/DD and/or MCA analyzed by array-CGH as pre or postnatal test from 2008 to 2013, we identified 156 pathogenic or likely pathogenic rearrangements (manuscript in preparation). Seven cases from this cohort showed at least two large subtelomeric rearrangements - a deletion and a duplication spanning from 4.4 to 18 Mb - compatible with a derivative chromosome (Figures 1 and 2, and Table 1). In each case, one of the two genomic rearrangement was at least 6 Mb, although karyotype was reported normal. We confirmed the presence of a derivative chromosome by FISH in six subjects, and demonstrated it was inherited from a parent with a balanced translocation in five cases (Figure 2). In the remaining two, the rearrangement was apparently de novo (Table 1). The band pattern and sizing of the exchanged genomic region was very similar in all analyzed cases (Figure 1). The balanced chromosomal anomaly in the parent was also undetectable by karyotyping in four out of the five transmitted cases (Cases 1–5, Figure 1A). Rearrangements were always associated with complex developmental defects, summarized in Table 2 and described below.
Figure 1.
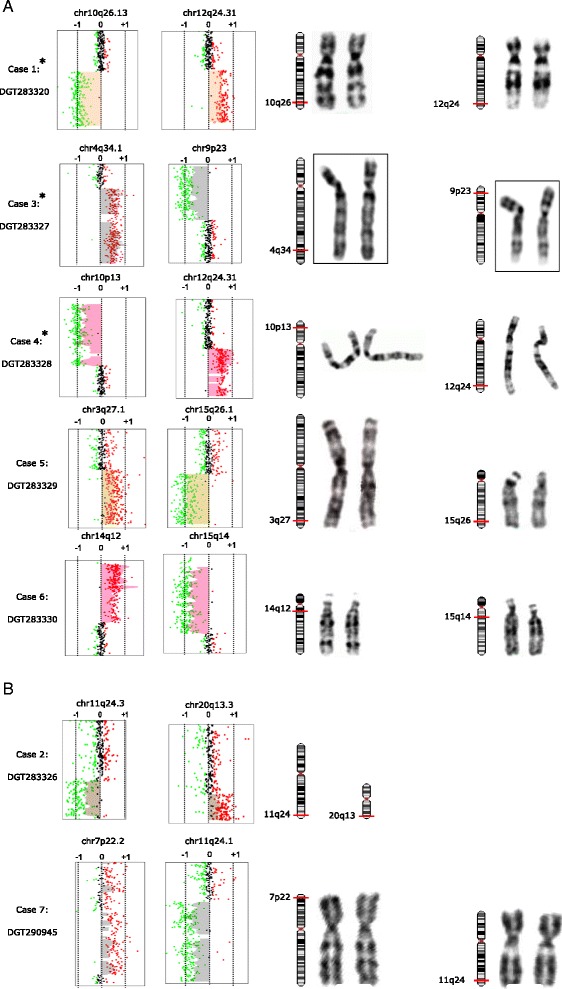
Array-CGH and karyotyping in cases with inherited and de novo chromosomal rearrangements. Panel A. Array-CGH analysis resulting in telomeric rearrangements on two different chromosomes (left). On the X-axis, the log ratio is reported (log2 intensity of [Cy5 fluorochrome/Cy3 fluorochrome)]. Expected values are from −0.7 to −1 for a deletion (green dots), 0 for normal (black dots), and +0.5 to +1 for a duplication (red dots). On the right side, GTG-banding of the chromosomes involved in the structural rearrangements. In cases marked by an asterisk, the chromosomes on the right are from the parent carrying the cryptic translocation, because patient karyotype image was not available. Breakpoints are indicated by red bars on chromosomal ideograms. In panel B, array-CGH analysis displays telomeric rearrangements in probands with de novo rearrangements.
Figure 2.
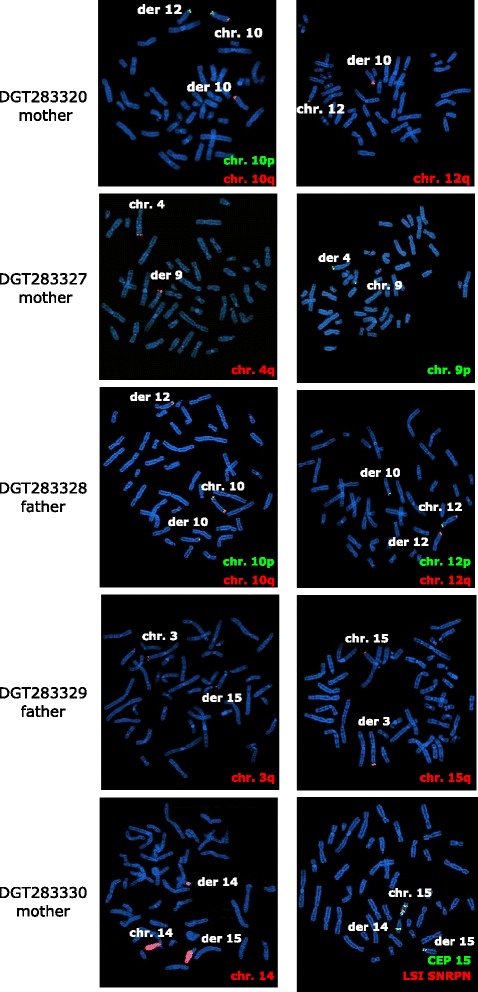
FISH analysis of the parents carrying balanced translocation. FISH probes hybridize on the normal homologues as well as on derivative chromosomes. Probes used in these analysis were the following: 3q SpectrumOrange (05 J04-003, Vysis TelVysion probe, Abbot, Illinois, USA), 4q SpectrumOrange (05 J04-004, Vysis TelVysion probe), 10p SpectrumGreen (05 J03-010, Vysis TelVysion probe), 10q SpectrumOrange (05 J04-010, Vysis TelVysion probe), 12p SpectrumGreen (05 J03-012, Vysis TelVysion probes), 12q SpectrumOrange (05 J04-012, Vysis TelVysion probe),15q SpectrumOrange (05 J04-015, Vysis TelVysion probe), painting 14 (LPP14R, CYTOCELL, Cambridge, UK), Prader-Willi/Angelman Region probe -LSI SNRP Spectrum orange/CEP15 (D15Z1) SpectrumAqua/LSI PML SpectrumGreen (05 J26-027, Vysis, Abbot).
Table 1.
Summary of the seven patients with large rearrangement missed by karyotype
| Patient code (Decipher) | Gender | Array-CGH (GRCh37/hg19) | Minimal region (Mb) | de novo /inherited | Cell source for karyotype | |
|---|---|---|---|---|---|---|
| 1 | DGT283320 | M | arr 10q26.13q26.3(124,500,982-135,404,471)x1, 12q24.31q24.33(125,178,836-133,819,092)x3 | 10.9: 8.6 | maternal translocation | Amniocytes |
| 2 | DGT283326 | F | arr 11q24.3q25(128,728,456-134,868,407)x1, 20q13.3(58,442,781-62,893,189)x3, | 6: 4.4 | de novo | Periph. Blood |
| 3 | DGT283327 | M | arr 4q34.1q35.2(172,930,618-190,896,674)x3, 9p24.3p23(271,257-12,907,826)x1, 14q21.1(43,881,311-44,623,069)x3 | 18: 12.6: 0.74 | maternal translocation | Periph. Blood |
| 4 | DGT283328 | M | arr 10p15.3p13(148,206-12,211,671)x1, 12q24.31q24.33(121,572,578-133,767,986)x3 | 12: 12.2 | paternal translocation | Periph. Blood |
| 5 | DGT283329 | M | arr 3q27.1q29(184,428,168-197,840,339)x3, 15q26.1q26.3( 90,857,664-102,383,473)x1 | 13: 11.5 | paternal translocation | Amniocytes |
| 6 | DGT283330 | M | arr 2p12p11.2(82,510,808-84,804,525)x1, 14q11.2q12(20,472,548-31,139,579)x3, 15q11.1q14(20,102,541-35,758,169)x1 | 2.2: 10.7: 15.6 | maternal translocation | Periph. Blood |
| 7 | DGT290945 | F | arr 7p22.3p22.2 (92.532-4.176.031)x3, 7p22.3p22.2 (7.044.310-15.709.683)x3, 11q24.1q25(122.467.330-134.868.407)x1 | 4: 8.6: 12.4 | de novo | Chorionic villi |
Table 2.
Genes involved in the rearrangements and their associated phenotypes
| Case | Age | Structural anomaly | Genes involved | OMIM associated genes a | Likely pathogenic genes (haploinsufficiency) | Patient’s features corresponding to the pathogenic genes |
|---|---|---|---|---|---|---|
| 1 | del 10q26.13q26.3 | 76 | 3 (AR) | - | ||
| 4 yrs | dup 12q24.31q24.33 | 54 | 1 (ADdn) | - | ||
| 3 (AR) | ||||||
| 2 | 10 yrs | del 11q24.3q25 | 29 | 1 (ADdn) | - | |
| 4 (AR) | ||||||
| dup 20q13.3 | 90 | 7 (ADh) | - | |||
| 3 | 6 yrs | del 9p24.3p23 | 29 | 1 (ADdn) | SLC1A1 (susceptibility to schizophrenia and psychotic disorder) | Patient too young to verify symptoms |
| 4 (AR) | SMARCA2 (Nicolaides-Baraitser synd.; dominant negative) | Intellectual disability, delayed speech, psychomotor development stooped posture and seizures | ||||
| 1 (AD) | DOCK8 (Intellectual disability) | Intellectual disability | ||||
| dup 4q34.1q35.2 | 53 | 9 (AR) | CCDC111 (susceptibility to high myopia) | Patient too young to verify symptoms | ||
| dup 14q21.1 | Gene desert | - | ||||
| 4 | 15 yrs | del 10p15.3p13 | 73 | 3 (ADh) | GATA3 (hypoparathyroidism, sensorineural deafness, and renal insufficiency) | Congenital hypoparathyroidism, deafness and renal disease |
| 1 (AR) | AKR1C4 (46XY sex reversal) | Cryptorchidism, hypospadia | ||||
| DHTKD1 (Charcot-Marie-Tooth type 2Q) | Patient too young to verify symptoms | |||||
| dup 12q24.31q24.33 | 108 | 1 (ADh) | P2RX2 (hearing loss) | May duplication affect hearing ability? | ||
| 1 (ADdn) | ||||||
| 9 (AR) | ||||||
| 5 | TOP | del 15q26.1q26.3 | 30 | 4 (ADh) | CHD2 (Childhood onset encephalopathy) | - |
| NR2F2 (Heart defects) | Ventricular septal defect | |||||
| 7 (AR) | IGF1R (Growth retardation) | IntraUterine Growth Retardation Heart malformation? | ||||
| dup 3q27.1q29 | 114 | 4 (ADh) | MEF2A (Coronary artery disease) | May duplication cause limb anomalies? | ||
| 1 (AD?) | TP63 (Heterodactily, ectodermal dysplasia, cleft lip palate syndrome 3) | |||||
| 9 (AR) | ||||||
| 6 | 1 yr | del 2p12p11.2 | 1 | 1 (AR) | ||
| del 15q11.1q14 | 60 | 5 (AR) | NPIA1 (Spastic paraplegia) | Patient too young to verify symptoms | ||
| MKRN3 (Precocious puberty) | Patient too young to verify symptoms | |||||
| MAGEL2 (Prader-Willy like, imprinted) | Maternally inherited deletion | |||||
| 7 (ADh) | NDN, SNRP, UBE3A (Angelman/Prader-Willy, imprinted) | Angelman features (maternally inherited deletion) | ||||
| CHRNA7 (15q13.3 syndrome) | Myoclonic seizures | |||||
| dup 14q11.2q12 | 60 | 4 ADh | ANG (Amyotrophic lateral sclerosis) | Patient too young to verify symptoms | ||
| 1 (ADgain) | CHD8 (Autism) | Duplication may be associated with psychomotor developmental delay | ||||
| 2 (AD?) | MYO6 (Atrial septal defect) | Pervious foramen ovale | ||||
| 6 (AR) | FOXG1 (Rett-like syndrome) | Duplication may be associated with neurocognitive impairment | ||||
| del 11q24.1q25 | 58 | 3 (ADh) | SCN3B (Brugada syndrome) | - | ||
| 7 | TOP | 1 (AD?) | CDON (Holoprosencephaly type 11) | Brain malformation | ||
| 10 (AR) | KIRREL3 (Intellectual disability) | - | ||||
| dup 7p22.3p22.2 | 42 | 4 (AR) | - |
Notes: aAR: autosomal recessive; ADdn: autosomal dominant, dominant negative effect; ADh: autosomal dominant, haploinsufficiency; AD?: autosomal dominant, unknown effect; ADgain: autosomal dominant with gain of function. TOP: Termination of Pregnancy.
Case 1. DGT283320
The proband was a 3-year-old boy, fifth child of non-consanguineous parents. He was born by Cesarean section at 39 weeks of gestation after an uneventful pregnancy. Neonatal weight was 2,740 g (3rd percentile [9]), length 47 cm (3rd percentile), and Occipital Frontal Circumference (OFC) 34.5 cm (50th percentile). Following birth, he needed immediate ventilation support (APGAR: 5/9). Since birth, he has shown severe hypotonia and feeding difficulties. Extensive metabolic workup was normal (levels of plasmatic and urinary amino acids, urinary organic acids profile, beta-N-acetyl-glucosamine, cerebroside beta-galactoside, arylsulfatase and chitotriosidade activity, lactic dehydrogenases, creatine kinase (CK) and serum creatinine). Isoelectrophoretic analysis ruled out main congenital disorders of glycosylation. Cerebral MRI detected hypoplastic corpus callosum, without other malformations. Neurological examination at 9 months revealed severe axial hypotonia, poor eye-hand coordination, and exotropia; he showed only mild facial dysmorphisms, namely a triangular face and thin upper lip (Figure 3A).
Figure 3.
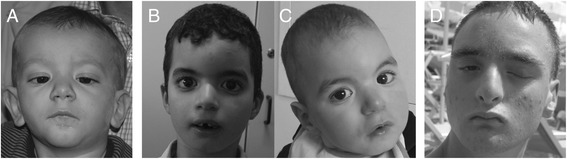
Dysmorphological features of cases 1, 3 and 4. A. Subject 1 at 18 months, shows mild facial dysmorphisms (triangular face, epicanthus, thin upper lip). B. Subject 3 (proband) at 6 yrs., dolico-trigonocephaly, thick eyebrows, mild synophris, mid-face hypoplasia. C. Younger brother of subject 3 at 2 yrs. showing similar dysmorphisms. D. Subject 4 at 14 yrs., showing bristly hair, left palpebral ptosis, broad nasal root, small ears.
The prenatal karyotype was normal (46,XY). Postnatal array-CGH analysis on lymphocytes detected a 10.9 Mb deletion at 10q26.13q26.3 and an 8.6 Mb duplication at 12q24.31q24.33 (Figure 1A, Tables 1 and 2). FISH analysis with probes specific for the subtelomeric regions of the long arms of chromosome 10 and 12 revealed a reciprocal balanced translocation in the mother (Figure 2). The balanced rearrangement in the mother was not evident upon GTG-banding analysis (Figure 1A, right).
Case 2. DGT283326
Female born from non-consanguineous healthy parents, after an uneventful pregnancy. Psychomotor delay and failure to thrive were reported. Delayed speech development was observed (first words at 4 yrs). At 7 yrs, clinical examination showed stature and weight below the 3rd centile, facial dysmorphisms (long, deeply grooved philtrum, thin upper lips), supernumerary nipples, pectus excavatum, hypertrichosis. The patient showed moderate intellectual delay.
Patient postnatal karyotyping, performed on blood, was reported normal (46,XX) (GTG-banding images were not available). Array-CGH analysis on the DNA extracted from proband whole blood showed a 6 Mb deletion at chromosome 11q24.3q25, and a 4.4 Mb duplication at 20q13.3 (Figure 1B, Tables 1 and 2). FISH analysis on the parents, using probes for the subtelomeric regions of the long arms of chromosome 11 and 20 was normal, suggesting a de novo event.
Case 3. DGT283327
The parents of this male proband were first cousins. He was, initially referred for DD at the age of 18 months. Family history was negative. He was born at full-term by Caesarean section after an uneventful pregnancy, with normal auxometric parameters. After birth, he received oxygen supplementation for mild respiratory distress (APGAR score 7/9). Cardiac ultrasound disclosed a modest ventricular septal defect that spontaneously resolved in a few months. Bilateral cryptorchidism and a metopic ridge with OFC at the 25th centile were also reported. Brain MRI detected a corpus callosum hypoplasia and several high-intensity lesions in the bilateral periventricular white matter T2-weighted imaging, due to perinatal hypoxia.
Methylation test for Prader-Willi syndrome and metabolic workup was normal. The patient was re-evaluated at 5 yrs during hospitalization for generalized seizures that occurred with fever. ElectroEncephaloGraphy (EEG) showed multifocal epileptic activities, and the patient was under valproic-acid therapy. He had a normal OFC (50 cm- 25th centile), dolico-trigonocephaly, thick eyebrows, mild synophrys, mid-face hypoplasia, retrognathia, and flat feet (Figure 3B). He started walking independently at the age of 4.5 yrs, but never developed any language skill. His younger brother presenting a similar phenotype (developmental delay, dolico-trigonocephaly, synophris, Figure 3C).
Patient postnatal karyotyping, performed on blood was reported as normal (46,XY) (GTG-banding images were not available). The subsequent array-CGH analysis on the DNA extracted from proband whole blood showed three rearrangements: an 18 Mb duplication at 4q34.1q35.2, a 12.6 Mb deletion at 9p24.3p23 and a 742 kb duplication at 14q21.1 (Figure 1A, Tables 1 and 2). The latter encompassed a gene desert region.
FISH analysis on the parents, using probes for the subtelomeric regions of the long arms of chromosome 4 and 9 revealed a reciprocal balanced translocation in the mother (Figure 2). The balanced rearrangement in the mother was not evident upon GTG-banding analysis (Figure 1A, right).
Case 4. DGT283328
The patient was the second son of unrelated parents. Family history was negative for ID/DD and/or MCA, and the pregnancy was uneventful. A low weight, left palpebral ptosis, hypospadias, and bilateral cryptorchidism were noted at birth. Further investigations disclosed right chorioretinal coloboma, bilateral mixed hearing loss, one sacral hemivertebra and bilateral bladder-ureteric reflux associated with right kidney dysplasia. The patient presented intermittent hypocalcaemia due to congenital hyperparathyroidism. A tethered spinal cord was diagnosed and surgically corrected at the age of 4 yrs. The patient was referred for genetic evaluation at the age of 13 yrs, during hospitalization for seizures. Brain MRI detected left hippocampus hypoplasia. He displayed profound intellectual disability (he was not able to walk unsupported and language was absent) and microcephaly; moreover he showed striking dysmorphisms, including bristly hair, left palpebral ptosis, broad nasal root, and a small ears (Figure 3D).
Patient postnatal karyotyping, performed on blood, was reported as normal (46,XY) (GTG-banding images were not available). The subsequent array-CGH analysis on the DNA extracted from proband whole blood showed a 12 Mb deletion at 10p15.3p13 and a 12.2 Mb duplication at 12q24.31q24.33 (Figure 1A, Tables 1 and 2). FISH analysis on the parents confirmed the presence of a derivative chromosome between chromosomes 10 and 12, with a balanced translocation inherited from the father (Figure 2). The balanced rearrangement in the father was not evident at GTG-banding analysis (Figure 1A, right).
Case 5. DGT283329
A pregnant woman was referred for very low levels of maternal serum PAPP-A, and multiple miscarriages. Pregnancy was interrupted at 21 weeks of gestation for severe Intrauterine Growth Restriction (IUGR) (<5th centile) and multiple developmental defects. Postmortem pathologic evaluation confirmed IUGR, and revealed midline fused eyebrows, marked hypognathia, nucal edema, short limbs, thymus hypoplasia, ventricular septal defect, and pulmonary and cerebellar hypoplasia. A diagnosis of Cornelia de Lange syndrome was suggested.
Prenatal karyotyping on amniocytes (46,XY) (Figure 1A, right), molecular analysis of NIPBL and SMC1, hypomethylation H19 and uniparental disomy of chromosome 7 were normal.
Array-CGH analysis performed on DNA extracted from autoptical fetal tissue showed a 13 Mb duplication at 3q27.1q29 and a 11.5 Mb deletion at 15q26.1q26.3 (Figure 1A, Tables 1 and 2). FISH analysis on the parents revealed a balanced translocation inherited from the father (Figure 2).
Case 6. DGT283330
First male child of non-consanguineous parents. Twin pregnancy with intrauterine death of one fetus at 6 weeks of gestation. Prenatal first trimester biochemical screening was normal. He was born by Caesarean section at 37 weeks of gestation. At birth, weight was 2,180 g (10th percentile), length 42.2 cm (<3rd percentile), OFC 34 cm (75th percentile), and APGAR score 4/8. He showed hypotelorism, bulbous nose, long and smooth philtrum and mild micrognathia. At 7 days, the patient presented with seizures. Right multifocal spike and wave anomalies, along with left occipital-temporal anomalies were detected by EEG, requiring multi-pharmacological treatment. Cerebral ultrasound and MRI revealed hypoplastic corpus callosum. Renal ultrasound showed bilateral pyelectasis and hydronephrosis, and echocardiogram revealed a patent foramen ovale. Cortical auditory evoked potentials and visually evoked potentials gave normal results. Neurological follow-up at 11 months of age revealed DD. The child experienced recurrent myoclonic seizures during the first year of life, and needed multidrug antiepileptic treatment to clinically control seizures. Despite the pharmacological therapy, multifocal spike and wave anomalies persisted at follow-up EEG.
Patient postnatal karyotyping was normal (46,XY) (Figure 1A, right). Array-CGH analysis showed three rearrangements: a 2.2 Mb deletion at 2p12p11.2, a 10.7 duplication at 14q11.2q12 and a 15.6 Mb deletion at 15q26.1q26.3 (Figure 1A, Tables 1 and 2). FISH analysis on the parents revealed a balanced translocation in the mother (Figure 2).
Case 7. DGT290945
A woman underwent pregnancy termination after ultrasonography diagnosis of fetal developmental defects. Fetal autopsy revealed a female fetus with IUGR, nuchal and occipital edema, flat nose, low-set ears, hypertelorism, thymus hypoplasia, preductal aortic arch and left heart hypoplasia, lung hypoplasia, corpus callosum agenesis, thoracic hemivertebrae, and short limbs.
Prenatal karyotyping on chorionic villi was normal (46,XX) (Figure 1B, right). Array-CGH analysis performed on DNA extracted from autoptic tissue from the fetus showed a ~13 Mb duplication at 7p22.3p22.2 and a 12.4 Mb deletion at 11q24.1q25 (Figure 1B, Tables 1 and 2). FISH analysis revealed the translocation was apparently de novo.
Discussion
In our cohort of ID/DD and/or MCA cases, routinely analyzed by array-CGH in pre or postnatal tests, we identified seven cases with derivative chromosomes missed by karyotyping, even if involving at least one region above 6 Mb. Large rearrangements detected by array-CGH missed by karyotyping have been previously reported [2], but our data suggest they are more common than expected, accounting for ∼ 4.5% of pathogenic array-CGH anomalies in our cohort (7/156). Moreover, three of our cases were prenatal diagnoses, out of a total of 15. Although these numbers are limited, it is important to note the utility of array-CGH in the presence of ultrasound anomalies, even with an apparently normal karyotype [10].
In five of the seven cases, we detected a balanced translocation in an healthy parent. In two, the derivative chromosome was apparently de novo, suggesting a germinal mosaic translocation in one parent. The translocation was also not detectable upon karyotyping in four out of the five cases, and had to be confirmed using telomeric FISH. Comparison of the two translocated chromosomes showed that the band pattern and sizing were highly similar. The rearrangements were therefore not detected using standard karyotyping due to the intrinsic technical limits of this analysis.
All cases had complex congenital anomalies and, in most, disease associated genes or disease candidate genes could be tracked in the deleted/duplicated segment. However, even if these rearrangements were large, the number of causative genes was always limited from one to four; in two patients no disease gene was presently annotated in the deletion/duplication. This suggests a minority of genes in our rearrangements were dose sensitive, and/or the pathogenicity of deleted/duplicated genes remains to be discovered.
Neurodevelopmental anomalies in patient 3 could be associated with the deletion encompassing DOCK8 (autosomal dominant mental retardation); SMARCA2, causing the autosomal dominant Nicolaides-Baraitser syndrome, may also have a role in the pathology, although reported mutations in this gene act as dominant negative [11,12]. In patient 4, the phenotype could be explained by the deletion of two genes: GATA3 (hypoparathyroidism, sensorineural deafness, and renal insufficiency) and AKR1C4 (46,XY sex reversal) [13,14]. Patient 6 was an Angelman syndrome phenocopy with atypical seizures. Indeed, he carried a large deletion spanning the AS/PWS critical region (maternally inherited) and the CHRNA7 gene related to 15q13.3 deletion syndrome, associated with intellectual disability and epilepsy. He also carried a 10.7 Mb duplication with possible involvement in the phenotype of the duplicated CHD8, implicated in autism [15], and FOXG1 (infantile growth retardation and epilepsy) [16–19].
In two additional patients, genes within the rearrangement were suggestive: in case 5, a duplication of TP63 may explain limb anomalies. Indeed, TP63 mutations cause at least five different types of ectodermal dysplasia syndromes, a non-syndromic split-hand/foot malformation (SHFM4) and non-syndromic cleft lip [20–23]; however its duplication has never been reported in a particular pathology. In the same patient, ventricular septal defects may be due to the NR2F2 deletion, a gene recently associated with non-syndromic atrioventricular septal defects (AVSDs) [24]. Finally, growth retardation may be caused by IGF1R haploinsufficiency. This gene has an important role in fetal growth and skeletal development, and Insulin-like Growth Factors (IGFs) have also been demonstrated to be involved in limb morphogenesis [25].
In case 7, brain malformations may be associated with CDON deletion, which is mutated in holoprosencephaly type 11 [26].
Conclusions
Large rearrangements above 6 Mb may remain undetected by karyotyping analysis, but can be an important cause of ID/DD and/or MCA, and be troublesome events in prenatal tests.
Methods
Patients
From 2008 to 2013, we assembled a cohort of over 700 ID/DD and/or MCA patients from the Pediatric Genetics Unit and the Medical Genetics Unit of the “Città della Salute e della Scienza” University Hospital (Torino, Italy). Fifteen cases were prenatal diagnoses. Seven patients presented a large rearrangement between 4.4 to 18 Mb, detected by array-CGH, but not revealed by karyotyping.
Karyotyping and array-CGH
Karyotyping on GTG-banded chromosomes from patients was performed on chorionic villi (1 patient), amniocytes (2 patients), and cultured lymphocytes (4 patients) according to standard protocols. Array-CGH was performed using a 60 K whole-genome oligonucleotide microarray (2.1 kb average probe spacing; 1.8 kb in Refseq genes) following the manufacturer’s protocol (Agilent Technologies, Santa Clara, California, USA). Slides were scanned using a G2565BA scanner, and analyzed using Agilent CGH Analytics software ver. 4.0.81 (Agilent Technologies Inc.) with the statistical algorithm ADM-2 and a sensitivity threshold of 6.0. Significant copy-number changes were identified by at least three consecutive aberrant probes. Reference human genomic DNA was GRCh37/hg19.
FISH analysis
Fluorescence in Situ Hybridization (FISH) analysis was performed on metaphase chromosomes to confirm array-CGH data. Commercial probes were selected for each patient and FISH was performed following the manufacturer’s protocol (Figure 2 legend). Slides were observed using a fluorescence microscope (Nikon, Eclipse 50i) and analyzed with Genikon software. Paternity was tested in de novo cases by microsatellite segregation.
Consent
Written informed consent was obtained from the patient’s guardian/parent/next of kin for the publication of this report and any accompanying images.
Acknowledgements
We are grateful to all family members who contributed to the study. This work was funded by the Associazione E.E. Rulfo per la Genetica Medica and MURST60% (to A. Brusco).
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
EDG and AB conception and design. EDG, ES, GDA, FF, AC, CM, EG, SC, FT, PP, MG, VA performed cytogenetic and molecular analyses; EB, EFB, VGN, GG, MG, GR, GM, MCS, EG, GBF collected patients, and performed clinical analyses; GB performed fetal autopsy and histopathology; EDG, EB, EFB, EG, GBF, AB analyzed data and wrote the manuscript; all authors read and approved final version.
Contributor Information
Eleonora Di Gregorio, Email: ele.digregorio@gmail.com.
Elisa Savin, Email: esavin@cittadellasalute.to.it.
Elisa Biamino, Email: biamino_elisa@yahoo.it.
Elga Fabia Belligni, Email: elga.belligni@yahoo.it.
Valeria Giorgia Naretto, Email: valeriagiorgia.naretto@gmail.com.
Gaetana D’Alessandro, Email: gdalessandro@cittadellasalute.to.it.
Giorgia Gai, Email: gai.giorgia@gmail.com.
Franco Fiocchi, Email: ffiocchi@cittadellasalute.to.it.
Alessandro Calcia, Email: alecalcia@gmail.com.
Cecilia Mancini, Email: cecilia.mancini3@gmail.com.
Elisa Giorgio, Email: elisa.giorgio84@libero.it.
Simona Cavalieri, Email: scavalieri@gmail.com.
Flavia Talarico, Email: ftalarico@cittadellasalute.to.it.
Patrizia Pappi, Email: patriziapappi@gmail.com.
Marina Gandione, Email: marina.gandione@unito.it.
Monica Grosso, Email: mgrosso@cittadellasalute.to.it.
Valentina Asnaghi, Email: vasnaghi@cittadellasalute.to.it.
Gabriella Restagno, Email: grestagno@hotmail.com.
Giorgia Mandrile, Email: giorgia.mandrile@gmail.com.
Giovanni Botta, Email: gbotta@cittadellasalute.to.it.
Margherita Cirillo Silengo, Email: margherita.cirillosilengo@unito.it.
Enrico Grosso, Email: egrosso@cittadellasalute.to.it.
Giovanni Battista Ferrero, Email: giovannibattista.ferrero@unito.it.
Alfredo Brusco, Email: alfredo.brusco@unito.it.
References
- 1.Vissers LE, de Vries BB, Veltman JA. Genomic microarrays in mental retardation: from copy number variation to gene, from research to diagnosis. J Med Genet. 2010;47(5):289–297. doi: 10.1136/jmg.2009.072942. [DOI] [PubMed] [Google Scholar]
- 2.Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, Church DM, Crolla JA, Eichler EE, Epstein CJ, Faucett WA, Feuk L, Friedman JM, Hamosh A, Jackson L, Kaminsky EB, Kok K, Krantz ID, Kuhn RM, Lee C, Ostell JM, Rosenberg C, Scherer SW, Spinner NB, Stavropoulos DJ, Tepperberg JH, Thorland EC, Vermeesch JR, Waggoner DJ, Watson MS, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86:749–764. doi: 10.1016/j.ajhg.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shaffer LG, Bejjani BA, Torchia B, Kirkpatrick S, Coppinger J, Ballif BC. The identification of microdeletion syndromes and other chromosome abnormalities: Cytogenetic methods of the past, new technologies for the future. Am J Med Genet C. 2007;145C(4):335–345. doi: 10.1002/ajmg.c.30152. [DOI] [PubMed] [Google Scholar]
- 4.Pickering DL, Eudy JD, Olney AH, Dave BJ, Golden D, Stevens J, Sanger WG. Array-based comparative genomic hybridization analysis of 1176 consecutive clinical genetics investigations. Genet Med. 2008;10(4):262–266. doi: 10.1097/GIM.0b013e31816b64ad. [DOI] [PubMed] [Google Scholar]
- 5.Lu X, Shaw CA, Patel A, Li J, Cooper ML, Wells WR, Sullivan CM, Sahoo T, Yatsenko SA, Bacino CA, Stankiewicz P, Ou Z, Chinault AC, Beaudet AL, Lupski JR, Cheung SW, Ward PA. Clinical implementation of chromosomal microarray analysis: summary of 2513 postnatal cases. PLoS One. 2007;2:e327. doi: 10.1371/journal.pone.0000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheung SW, Shaw CA, Yu W, Li J, Ou Z, Patel A, Yatsenko SA, Cooper ML, Furman P, Stankiewicz P, Lupski JR, Chinault AC, Beaudet AL. Development and validation of a CGH microarray for clinical cytogenetic diagnosis. Genetics in medicine : official journal of the American College of Medical Genetics. 2005;7:422–432. doi: 10.1097/01.GIM.0000170992.63691.32. [DOI] [PubMed] [Google Scholar]
- 7.Jacobs PA, Browne C, Gregson N, Joyce C, White H. Estimates of the frequency of chromosome abnormalities detectable in unselected newborns using moderate levels of banding. J Med Genet. 1992;29(2):103–108. doi: 10.1136/jmg.29.2.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Madan K. Balanced complex chromosome rearrangements: reproductive aspects. A review. Am J Med Genet A. 2012;158A(4):947–963. doi: 10.1002/ajmg.a.35220. [DOI] [PubMed] [Google Scholar]
- 9.Bertino E, Gilli G, Occhi L, Giuliani F, Di Nicola P, Spada E, Fabris C. Postnatal growth of preterm infants: which reference charts? Minerva Pediatr. 2010;62(3 Suppl 1):71–74. [PubMed] [Google Scholar]
- 10.D'Amours G, Kibar Z, Mathonnet G, Fetni R, Tihy F, Desilets V, Nizard S, Michaud JL, Lemyre E. Whole-genome array CGH identifies pathogenic copy number variations in fetuses with major malformations and a normal karyotype. Clin Genet. 2012;81(2):128–141. doi: 10.1111/j.1399-0004.2011.01687.x. [DOI] [PubMed] [Google Scholar]
- 11.Van Houdt JK, Nowakowska BA, Sousa SB, van Schaik BD, Seuntjens E, Avonce N, Sifrim A, Abdul-Rahman OA, van den Boogaard MJ, Bottani A, Castori M, Cormier-Daire V, Deardorff MA, Filges I, Fryer A, Fryns JP, Gana S, Garavelli L, Gillessen-Kaesbach G, Hall BD, Horn D, Huylebroeck D, Klapecki J, Krajewska-Walasek M, Kuechler A, Lines MA, Maas S, Macdermot KD, McKee S, Magee A, et al. Heterozygous missense mutations in SMARCA2 cause Nicolaides-Baraitser syndrome. Nat Genet. 2012;44:445–449. doi: 10.1038/ng.1105. [DOI] [PubMed] [Google Scholar]
- 12.Griggs BL, Ladd S, Saul RA, DuPont BR, Srivastava AK. Dedicator of cytokinesis 8 is disrupted in two patients with mental retardation and developmental disabilities. Genomics. 2008;91(2):195–202. doi: 10.1016/j.ygeno.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Esch H, Groenen P, Nesbit MA, Schuffenhauer S, Lichtner P, Vanderlinden G, Harding B, Beetz R, Bilous RW, Holdaway I, Shaw NJ, Fryns JP, Van de Ven W, Thakker RV, Devriendt K. GATA3 haplo-insufficiency causes human HDR syndrome. Nature. 2000;406:419–422. doi: 10.1038/35019088. [DOI] [PubMed] [Google Scholar]
- 14.Fluck CE, Meyer-Boni M, Pandey AV, Kempna P, Miller WL, Schoenle EJ, Biason-Lauber A. Why boys will be boys: two pathways of fetal testicular androgen biosynthesis are needed for male sexual differentiation. Am J Hum Genet. 2011;89(2):201–218. doi: 10.1016/j.ajhg.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blumenthal I, Ragavendran A, Erdin S, Klei L, Sugathan A, Guide JR, Manavalan P, Zhou JQ, Wheeler VC, Levin JZ, Ernst C, Roeder K, Devlin B, Gusella JF, Talkowski ME. Transcriptional consequences of 16p11.2 deletion and duplication in mouse cortex and multiplex autism families. Am J Hum Genet. 2014;94:870–883. doi: 10.1016/j.ajhg.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Falace A, Vanni N, Mallamaci A, Striano P, Zara F. Do regulatory regions matter in FOXG1 duplications? Eur J Hum Genet. 2013;21(4):365–366. doi: 10.1038/ejhg.2012.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brunetti-Pierri N, Paciorkowski AR, Ciccone R, Della Mina E, Bonaglia MC, Borgatti R, Schaaf CP, Sutton VR, Xia Z, Jelluma N, Ruivenkamp C, Bertrand M, de Ravel TJ, Jayakar P, Belli S, Rocchetti K, Pantaleoni C, D'Arrigo S, Hughes J, Cheung SW, Zuffardi O, Stankiewicz P. Duplications of FOXG1 in 14q12 are associated with developmental epilepsy, mental retardation, and severe speech impairment. Eur J Hum Genet. 2011;19:102–107. doi: 10.1038/ejhg.2010.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Striano P, Paravidino R, Sicca F, Chiurazzi P, Gimelli S, Coppola A, Robbiano A, Traverso M, Pintaudi M, Giovannini S, Operto F, Vigliano P, Granata T, Coppola G, Romeo A, Specchio N, Giordano L, Osborne LR, Gimelli G, Minetti C, Zara F. West syndrome associated with 14q12 duplications harboring FOXG1. Neurology. 2011;76:1600–1602. doi: 10.1212/WNL.0b013e3182194bbf. [DOI] [PubMed] [Google Scholar]
- 19.Yeung A, Bruno D, Scheffer IE, Carranza D, Burgess T, Slater HR, Amor DJ. 4.45 Mb microduplication in chromosome band 14q12 including FOXG1 in a girl with refractory epilepsy and intellectual impairment. Eur J Med Genet. 2009;52(6):440–442. doi: 10.1016/j.ejmg.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 20.McGrath JA, Duijf PH, Doetsch V, Irvine AD, de Waal R, Vanmolkot KR, Wessagowit V, Kelly A, Atherton DJ, Griffiths WA, Orlow SJ, van Haeringen A, Ausems MG, Yang A, McKeon F, Bamshad MA, Brunner HG, Hamel BC, van Bokhoven H. Hay-Wells syndrome is caused by heterozygous missense mutations in the SAM domain of p63. Hum Mol Genet. 2001;10:221–229. doi: 10.1093/hmg/10.3.221. [DOI] [PubMed] [Google Scholar]
- 21.Kantaputra PN, Hamada T, Kumchai T, McGrath JA. Heterozygous mutation in the SAM domain of p63 underlies Rapp-Hodgkin ectodermal dysplasia. J Dent Res. 2003;82(6):433–437. doi: 10.1177/154405910308200606. [DOI] [PubMed] [Google Scholar]
- 22.van Bokhoven H, Hamel BC, Bamshad M, Sangiorgi E, Gurrieri F, Duijf PH, Vanmolkot KR, van Beusekom E, van Beersum SE, Celli J, Merkx GF, Tenconi R, Fryns JP, Verloes A, Newbury-Ecob RA, Raas-Rotschild A, Majewski F, Beemer FA, Janecke A, Chitayat D, Crisponi G, Kayserili H, Yates JR, Neri G, Brunner HG. p63 Gene mutations in eec syndrome, limb-mammary syndrome, and isolated split hand-split foot malformation suggest a genotype-phenotype correlation. Am J Hum Genet. 2001;69:481–492. doi: 10.1086/323123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leoyklang P, Siriwan P, Shotelersuk V. A mutation of the p63 gene in non-syndromic cleft lip. J Med Genet. 2006;43(6):e28. doi: 10.1136/jmg.2005.036442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Al Turki S, Manickaraj AK, Mercer CL, Gerety SS, Hitz MP, Lindsay S, D'Alessandro LC, Swaminathan GJ, Bentham J, Arndt AK, Low J, Breckpot J, Gewillig M, Thienpont B, Abdul-Khaliq H, Harnack C, Hoff K, Kramer HH, Schubert S, Siebert R, Toka O, Cosgrove C, Watkins H, Lucassen AM, O'Kelly IM, Salmon AP, Bu'lock FA, Granados-Riveron J, Setchfield K, Thornborough C, et al. Rare variants in NR2F2 cause congenital heart defects in humans. Am J Hum Genet. 2014;94:574–585. doi: 10.1016/j.ajhg.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Agrogiannis GD, Sifakis S, Patsouris ES, Konstantinidou AE. Insulin-like growth factors in embryonic and fetal growth and skeletal development (Review) Mol Med Rep. 2014;10(2):579–584. doi: 10.3892/mmr.2014.2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bae GU, Domene S, Roessler E, Schachter K, Kang JS, Muenke M, Krauss RS. Mutations in CDON, encoding a hedgehog receptor, result in holoprosencephaly and defective interactions with other hedgehog receptors. Am J Hum Genet. 2011;89(2):231–240. doi: 10.1016/j.ajhg.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]