

Summary
Untreated sewage discharges and limited agricultural manure management practices contribute to fecal pollution in rural Brazilian waterways. Few microbial source tracking studies have tested host-specific indicators in underdeveloped regions and have mostly focused on Bacteroidales. Sequencing of sewage, human feces, and animal feces with Illumina HiSeq revealed Prevotellaceae as the most abundant family in humans, with Lachnospiraceae and Ruminococcaceae also comprising a large proportion of the microbiome. These same families were also dominant in animals. Bacteroides, the most commonly utilized human-specific marker in the US, was present in very low abundance. We used oligotyping to identify Prevotella and Blautia sequences that can distinguish human fecal contamination. Thirty-five of 61 Blautia oligotypes and 13 of 108 Prevotella oligotypes in humans were specific to or highly abundant (i.e. host-preferred) compared to pig, dog, horse, and cow sources. Certain human Prevotella and Blautia oligotypes increased more than an order of magnitude along a polluted river transect in rural Brazil, but traditional fecal indicator levels followed a steady or even decreasing trend. While both Prevotella and Blautia oligotypes distinguished human and animal fecal pollution in Brazil surface waters, Blautia appears to contain more discriminatory and globally applicable markers for tracking sources of fecal pollution.
INTRODUCTION
Fecal pollution in surface water sources constitutes a significant public health threat worldwide. Waterborne illnesses, related to the inadequate provision of water and sanitation services, are responsible for four billion cases of diarrhea and 1.8 million deaths each year, mostly impacting children five years of age and younger living in the rural communities of developing countries (Esrey et al., 2001; WHO, 2007). The lack of sanitation and drinking water infrastructures within these rural communities creates a self-perpetuating cycle of waterborne diseases (Esrey et al., 2001; WHO, 2013b). In Brazil, these include diarrheal illnesses caused by a broad array of pathogens including Escherichia coli, Campylobacter, Giardia, Cryptosporidium, helminth, and enteric viruses (Guerrant et al., 1983; Franzolin et al., 2005; Moreno et al., 2010) to schistosomiasis, a systemic parasitic disease with a morbidity/mortality rate estimated at over 250,000 per year (WHO, 2013a). While there has been significant progress in controlling schistosomiasis through repeated mass administration of drugs, it is still the second leading cause of death from parasitic infections in Brazil (Blanton et al., 2011).
Data from legally-mandated fecal contamination surveillance in Brazil have shown that regions with the highest prevalence of diseases overlap with areas of low sanitation (Barreto et al., 2007). Although this relationship has been demonstrated, identifying human sources in waterways with fecal pollution is less certain. Untreated sewage discharges and limited agricultural manure management practices contribute to fecal pollution in Brazilian waterways (de Oliveira et al., 2012). Exposure to human feces is generally a greater health risk than exposure to animal feces (Soller et al., 2010); therefore, identifying the source contamination is important for risk assessment (USEPA, 2005; Field and Samadpour, 2007; Santo Domingo et al., 2007; Stewart et al., 2013). Traditional fecal indicator bacteria, such as E. coli and enterococci, are found in the feces of humans and animals alike, which limit their potential to accurately assess human health risk. Host specific alternative indicators may therefore be particularly useful in areas where there is ubiquitous fecal pollution in surface waters (Newton et al., 2011; Roslev and Bukh, 2011).
The majority of research to identify host-specific genetic markers (i.e., a specific sequence within the DNA of fecal bacteria) have focused on Bacteroidales (Bernhard and Field, 2000; Dick et al., 2005; Reischer et al., 2006; Kildare et al., 2007; Fremaux et al., 2009). Geographic variability and host-specificity can factor into the efficacy of markers in discrimination of host sources, (Bernhard and Field, 2000; Harwood et al., 2009; Shanks et al., 2013). Only a few studies have explored the applicability of these markers in underdeveloped regions (Jenkins et al., 2009; Ahmed et al., 2010; Reischer et al., 2013). One study in Kenya tested human- and cow-specific Bacteroidales assays and demonstrated that the cow-specific assay could detect 94% of samples, however, detection of human fecal pollution was much less, with only 65% of human fecal samples positive with one human-specific assay (Jenkins et al., 2009). Reischer and colleagues (2013) found cattle and ruminant Bacteroidales markers were widely reliable in a study spanned across16 countries on six continents; however, human Bacteroidales genetic markers were less prevalent and stable, suggesting more reliable markers were needed. Recent human microbiome studies demonstrate a difference in the microbial community across geography, age and time (De Filippo et al., 2010; Lozupone et al., 2012; Yatsunenko et al., 2012). Differences in cultures and diets may also cause variation in the human gut microbiota, which manifest in the relative abundances of Bacteroides versus Prevotella (De Filippo et al., 2010; Wu et al., 2011). Considering these findings, more research is needed to evaluate if the “human-specific” genetic markers developed in the US and EU are relevant in other regions of the world (Reischer et al., 2013). Further, a broader array of taxonomic groups other than Bacteroidales may need to be explored to identify robust indicators that can be applied globally for tracking human sources of fecal pollution.
With deep sequencing technologies becoming more commonplace, it is now possible to identify multiple microorganisms that provide a fecal signature within a sample (Newton et al., 2013) more reliably than a single marker (Unno et al., 2010; Dubinsky et al., 2012; Newton et al., 2013). Given the complexity and depth of massively parallel, high-throughput sequencing datasets, the use of sensitive methods that can distinguish closely related but distinct organisms is critical for accurate identification of markers associated with certain hosts. Oligotyping is a recently described method that facilitates high-resolution partitioning of the 16S rRNA gene amplicon data into “oligotypes” using Shannon entropy (Eren et al., 2013b). By recovering subtle nucleotide variation among reads oligotyping can distinguish organisms with more than 99% identity over the sequenced region (Eren et al., 2011; Eren et al., 2013b). Previous studies identified oligotypes within the genus Blautia that distinguish human and animal sources with remarkable accuracy (Eren et al., 2014).
In this study, we used Illumina HiSeq for ultra-deep sequencing of the bacterial communities associated with sewage, and fecal samples collected from humans and other animals including pig, dog, horse and cow feces. Processing and sequencing of sewage, and human and pig fecal samples is described elsewhere (Eren et al., 2014). Processing of dog, horse and cow fecal samples is described in Supplemental Text 1, along with other methods specific to this study. We applied an extremely stringent quality filtering on our sequencing reads to eliminate the vast majority of sequencing errors (Eren et al., 2013a) to avoid false positives in our findings. We explored the oligotypes that can discriminate hosts, and evaluated their environmental applicability by including our analyses the river water samples collected from the Jiquiriçá River in Jenipapo, Brazil and the Lucaia River in Salvador, Brazil. Ongoing work by our group is examining the correlation of human fecal contamination in the Jiquiriçá River with risk of Schistosoma mansoni infection. We contribute to this goal by describing the oligotype signature profile in human samples from Brazil and the imprint of this signature on contaminated surface water.
RESULTS & DISCUSSION
Microbial community structure of human fecal and sewage samples in Brazil
Brazilian human fecal samples and sewage shared common fecal taxa in similar proportions (Figure 1), but sewage also contained taxa that have been previously associated with pipe infrastructure and surface water samples (McLellan et al., 2010; VandeWalle et al., 2012; Shanks et al., 2013). Prevotella, within the family Prevotellaceae, was the most abundant genus, comprising >36% of the average Brazilian human fecal sample and >10% of the Brazilian sewage sample. Genera belonging to the families Lachnospiraceae (e.g., Blautia, Roseburia) and Ruminococceae (e.g., Ruminococcus, Oscillibacter, Faecalibacterium) also made up a large proportion of the sewage (20%) and human fecal (40%) samples when combined. The latter two families have been identified as dominant families in US human fecal and sewage samples (Turnbaugh et al., 2009; McLellan et al., 2010; McLellan et al., 2013). Notably abundant in US human fecal samples, but less prevalent in Brazil human fecal samples, are the Bacteroides, which make up ~11% of US human fecal samples and ~2% of US sewage (Dethlefsen et al., 2008; Turnbaugh et al., 2009; McLellan et al., 2010), but comprised <1% of the Brazil human and sewage samples.
Figure 1.

Microbial community populations of sewage and human fecal samples collected in Brazil. A 50 mL sewage sample was from Embasa (Empresa Baiana de Águas e Saneamento - The Bahian Water and Sanitation Company), a water treatment facility in Salvador, Brazil, on August 13, 2012. DNA extraction was carried out as described previously by our laboratory (McLellan et al. 2010). The human fecal samples (n=10) were collected as part of a 2009 schistosomiasis survey (Blanton et al., 2011). The V6 hypervariable regions of the 16S rRNA gene from community genomic DNA were amplified and sequenced using Illumina HiSeq as described previously (Eren et al., 2013a). Taxonomy was assigned to sequences using GAST (Huse et al., 2008) and taxonomic counts were normalized to the maximum number of sequences. Nearly 15 million bacterial sequence reads were generated (~1 million reads per sample). Sequence data is available in the National Center for Biotechnology Information (NCBI) Short Read Archive under the accession number SRP041262. Taxonomic counts were normalized to maximum number of sequences and abundance parameters were set from 1 to 100%. The relative abundance of the top 20 genera occurring in sewage (n=1) and humans (average of n=10) are presented.
Populations consuming carbohydrate-rich diets in Africa and South America have higher proportions of Prevotella their gut microbiomes, whereas Bacteroides tend to dominate the guts of individuals from Europe and North America who typically consume a higher protein diet (De Filippo et al., 2010; Wu et al., 2011; Yatsunenko et al., 2012). Our research further supports this geographical distinction, as the Brazilian human fecal and sewage samples had a higher abundance of Prevotella versus Bacteroides. In contrast to differences in the relative abundance of Bacteroides between the US and Brazil, the genus Blautia had a consistent signal in both populations. On average, Blautia makes up 1.5% of US and 1.3% of Brazilian human fecal samples and 0.9% of US sewage and 2.0% of Brazilian sewage (McLellan et al., 2010; Newton et al., 2011; Eren et al., 2014).
The most widely used markers for human fecal contamination target human-specific Bacteroides spp., which have been found to be widely applicable to tracking sewage in many areas including the US, Europe and Australia (Gourmelon et al., 2007; Reischer et al., 2007; Ahmed et al., 2009; Ahmed et al., 2010; Sauer et al., 2011), but a lower sensitivity was reported in a study that included transcontinental comparisons, suggesting that there may be targets in the human microbiome better suited for microbial source tracking (Reischer et al., 2013). We recently described an assay, termed Lachno2, for a Blautia spp. found in humans but absent in cow and chicken samples and have used it to demonstrate chronic sewage contamination in an US estuary (Newton et al., 2011). Given the rich array of sequences within Blautia that can identify host sources, this taxonomic group shows promise for further alternative indicator development (Eren et al., 2014). Genetic markers for human fecal pollution based on Lachnospiraceae, and more specifically Blautia, may prove more universally applicable than Bacteroides, given the higher proportion of this taxonomic group within the Brazilian fecal and sewage samples. Further, Prevotella may serve as a better target in regions such as rural Brazil, where the standard diet favors its dominance in the gut microbiome. Future studies could examine Prevotella population structure in humans across a broad geographic area to develop a human Prevotella targeted assay, either alone or in conjunction with a Blautia assay.
Comparison of Brazil human and animal fecal microbial communities
All animal samples contained 14 common fecal families, but in different relative proportions; genera within these fecal families varied among samples as well (Table 1). A comparison of the 14 most abundant fecal-associated families in Brazilian humans, animals, and sewage showed that Ruminococcaceae had the highest relative abundance, making up 13% to 30% of each sample, followed by Prevotellaceae and Lachnospiraceae (Table 1). These three families comprised >80% of the human sequence reads and >60% of the sewage and pig samples, but were less abundant in the cow, horse, and dog fecal samples. Enterobacteriaceae, a large family of facultative anaerobes that contain pathogenic members, made up >11% of the dog, horse and cow fecal samples, >5% of the sewage samples, but <2% of the human and pig fecal samples. Bacteroidaceae, the family containing the genus Bacteroides, comprised ~3% of the dog, horse and cow samples, but was considerably lower in the human and pig fecal samples. This particular suite of organisms, notably the top four fecal families, could be useful for tracking animal fecal contamination in the surface waters of Brazil. Studies have identified animal specific markers based on Bacteroides; however, further study is needed to test their applicability to other regions of the world (Dick et al., 2005; Fogarty and Voytek, 2005; Fremaux et al., 2009; Mieszkin et al., 2010). These assays target the V2 to V4 regions, whereas our study targeted the V6 region. Therefore additional full length sequencing and or deep sequencing of additional regions is needed to make direct comparisons.
Table 1.
Average taxonomic composition of fecal-associated bacteria in human fecal samples, sewage, and ten Brazilian animal fecal samples (three pigs, three dogs, two cows and two horses) collected from Jenipapo, Brazil, in August 2012. Data are based on the percentage of sequence reads associated with the taxonomic classification of these fourteen previously reported families of fecal bacterial (McLellan et al. 2010; Turnbaugh et al. 2009; Newton et al. 2013).
| Family | Embasa Sewage % | Average Human % | Average Pig % | Average Dog % | Average Horse % | Average Cow % |
|---|---|---|---|---|---|---|
| Ruminococcaceae | 18 | 25 | 30 | 13 | 23 | 18 |
| Prevotellaceae | 19 | 38 | 19 | 10 | 13 | 12 |
| Lachnospiraceae | 27 | 20 | 16 | 10 | 18 | 8.3 |
| Enterobacteriaceae | 5.4 | 2.0 | 1.4 | 36 | 11 | 20 |
| Porphyromonadaceae | 4.8 | 0.50 | 7.3 | 8.4 | 5.8 | 10 |
| Veillonellaceae | 7.9 | 4.4 | 11 | 3.2 | 3.8 | 2.7 |
| Rikenellaceae | 1.5 | 3.6 | 4.6 | 3.2 | 12 | 7.0 |
| Clostridiaceae | 1.3 | 2.3 | 1.7 | 3.5 | 4.1 | 8.1 |
| Erysipelotrichaceae | 4.4 | 2.7 | 3.9 | 3.5 | 2.0 | 2.1 |
| Bacteroidaceae | 4.9 | 0.30 | 0.30 | 3.8 | 3.1 | 3.4 |
| Lactobacillaceae | 0.70 | 0.10 | 1.5 | 3.8 | 1.7 | 4.2 |
| Bifidobacteriaceae | 4.9 | 1.4 | 0.30 | 0.90 | 1.0 | 0.60 |
| Fusobacteriaceae | 0.30 | 0 | 1.9 | 1.8 | 0.90 | 3.7 |
| Enterococcaceae | 0.10 | 0 | 0 | 0.10 | 0.10 | 0.10 |
Distribution of Blautia and Prevotella oligotypes among Brazil sewage, human and animal fecal microbial communities
We utilized oligotyping to investigate sequences that could distinguish human fecal pollution from animal sources. We identified 61 Blautia oligotypes in the ten human fecal samples, representing a total of 134,595 sequence reads (Table 2, Figure 2). Thirty-five oligotypes (79,469 sequence reads; 58%) were human-specific (found in all human samples and only human samples; n= 6), human-associated (found only in some human samples; n=26), or human-preferred (found at significantly higher abundance in humans than other sources; n=3) when compared to the animals in our study (pigs, dogs, horses, and cows). A total of 33 of the 35 human oligotypes were also present in the sewage sample from the Embasa wastewater treatment plant in Salvador, Brazil. The top three human-specific oligotypes comprised 11% of Blautia in humans and 6.2% of the sewage reads from the genus Blautia. Previous work in our laboratory has demonstrated that certain Blautia oligotypes are shared between US and Brazil sewage, suggesting some Blautia genetic markers may be universally applicable (Eren et al., 2014). Assays based on additional human-specific Blautia oligotypes could be used in conjunction with the Lachno2 assay to enhance specificity of human source identification.
Table 2.
Relative abundance of human-related Blautia and Prevotella oligotypes
| Sequence read abundance | % of reads in genus (from humans)a | % of reads in genus ( from sewage)b | |
|---|---|---|---|
| Blautia | |||
| Total Human Oligotypes (n=61) | 134,595 | 98%c | 97% c |
| Human-Specific, Preferred, or Associated Oligotypes (n=35) | 79,469 | 58% | 32% |
| Top three human-specific oligotypes | |||
| AACTCGGACCCCTAA | 9910 | 7.2% | 1.0% |
| AACTCGTCTACTTGA | 3945 | 2.9% | 2.3% |
| AGCTCACTCGTTTGA | 1151 | 0.8% | 2.9% |
| Prevotella | |||
| Total Human Oligotypes (n=108) | 3,792,564 | 99%c | 99%c |
| Human-Associated and Preferred Oligotypes (n=12) | 114,255 | 3.0% | 1.6% |
| Top three human-associated/preferred oligotypes | |||
| CCATACGTAGACTCTTCTG | 95,700 | 2.5% | 0.009% |
| CCATACGATGACAATTCCG | 4,635 | 0.12% | 0.91% |
| TCGGAAGATTGATACCGTG | 2,918 | 0.077% | 0.012% |
compared to total human Blautia or Prevotella sequence reads
compared to total sewage Blautia or Prevotella sequence reads
a small percentage of total reads from human and sewage were not used in oligotyping after quality filtering.
Figure 2.

Distribution of Blautia oligotypes for samples from Brazil sewage, human and animal fecal microbial communities. Oligotyping analysis (Eren et al., 2013b) was performed on 175,985 reads identified as Blautia from 27 samples using 15 nucleotide positions following the initial entropy analysis. Base locations of interest based on entropy values included: 1, 2, 14, 15, 16, 23, 24, 25, 31, 35, 36, 38, 40, 53, and 56. To reduce the noise, each oligotype was required to have a most abundant unique sequence with a minimum abundance of 100. Oligotypes that did not meet this criterion were removed from the analysis. The final number of quality-controlled oligotypes was 81, and they represented 169,295 reads (equivalent to 96.2% of all reads analyzed).
Although Prevotella was noted as the dominant organism in the Brazilian human fecal samples, the two most dominant Prevotella oligotype in humans were also dominant in animals (Table 2, Figure 3). We did not find any human-specific oligotypes in Prevotella, however, nine of the 108 Prevotella oligotypes identified in the ten human fecal samples were human-associated, and another three were human-preferred. Eleven of these oligotypes were represented in sewage. The top three human-preferred Prevotella oligotypes comprised 2.7% of all Prevotella in human samples and 1.0 % of sewage samples.
Figure 3.
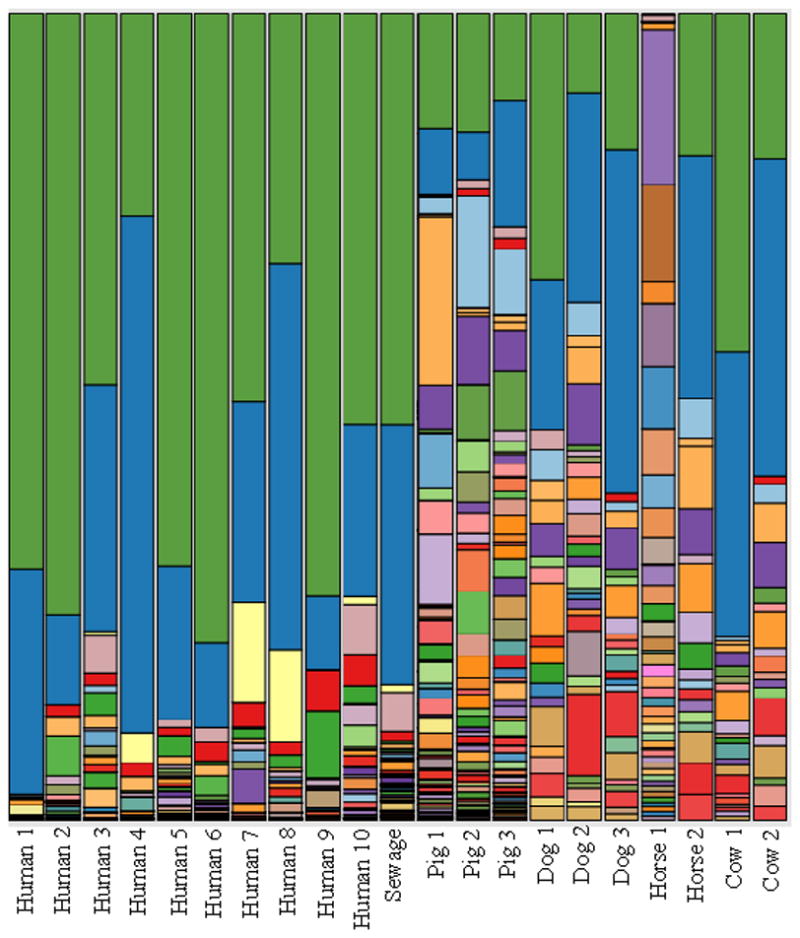
Distribution of Prevotella oligotypes for samples from Brazil sewage, human and animal fecal microbial communities. Oligotyping analysis was performed on 4,676,785 reads identified as Prevotella from 27 samples using 19 nucleotide positions following the initial entropy analysis. Base locations of interest included: 10, 13, 15, 17, 19, 20, 21, 23, 24, 25, 27, 34, 35, 37, 38, 40, 50, 54 and 55. To reduce the noise, each oligotype was required to have a most abundant unique sequence with a minimum abundance of 400. Oligotypes that did not meet this criterion were removed from the analysis. The final number of quality-controlled oligotypes was 188, and they represented 4,641,000 reads (equivalent to 99.2% of all Prevotella reads analyzed).
Although Prevotella was the most abundant genus in terms of total sequence reads, the majority of the sequence reads were not specific to the mammals surveyed. The relative abundance of one of the human- preferred Prevotella oligotypes an order of magnitude higher than the human Blautia oligotypes, however Prevotella had no strictly host-specific oligotypes. In contrast, more than half of the Blautia oligotypes distinguished humans from animals. These results suggest that Blautia oligotypes may be a more discriminatory fecal source marker, while Prevotella oligotypes may offer sensitivity for detection. It should be noted that our sequence reads are very short and that higher diversity may occur in other regions of the Prevotella 16S rRNA gene (Fogarty and Voytek, 2005; Haugland et al., 2010) that could allow for discrimination of Prevotella from humans and animals. The combination of these two fecal indicators could be very useful for simultaneously tracking total fecal pollution as well as the relative contribution from humans.
Tracking human fecal contamination in surface waters with oligotypes versus E. coli and Enterococcus spp. sequence abundance
The relative abundance of sequence reads identified as Prevotella, Blautia, E. coli and Enterococcus varied at sites along the Jiquiriçá River (Figure 4). Prevotella spp. and Blautia spp. both increased considerably at site 5 and steadily increased at subsequent sites downstream (sites 6 and 7), suggesting cumulative impacts of sewage input. In contrast to the increasing trend of our fecal signature organisms, traditional fecal indicator bacteria populations followed a steady or even decreasing trend. E. coli steadily decreased from sites 3 to 5, with only a slight increase at sites 6 and 7; and Enterococcus spp. steadily decreased with each downstream site. For this analysis, all sequence reads assigned to the genus Enterococcus were used since 98.5% of them were not assigned a species. Previous work in our lab has demonstrated that the V6 region may not resolve different species within this genus as >98% of presumptive enterococci isolates on mEI agar were assigned to the genus Enterococcus, but with no species assigned (Koskey et al., 2014).
Figure 4.

Human-specific Blautia and Prevotella oligotypes and HiSeq Illumina sequence reads identified as Prevotella spp., Blautia spp., and fecal indicator bacteria (E. coli and Enterococcus spp.) at five sites along the Jiquiriçá River in Jenipapo, Brazil. The water samples were collected on August 18, 2012 as part of a microbial source tracking and schistosomiasis survey (Ponce Terashima et al., unpublished). As with the sewage and fecal samples, sequence reads were generated using HiSeq Illumina sequencing, targeting the V6 hypervariable region of the 16S rRNA gene. Taxonomy was assigned to sequences using GAST, and taxonomic counts were normalized to the maximum number of sequences. Abundance parameters were set from 0 to 100%; sequence reads are reported on a log-scale. All classified and unclassified (species_NA) species from Prevotella, Blautia, and Enterococcus were counted. Sequences classified as E. coli and unclassifiable Enterobacteriaceae (non-distinguishable between Escherichia and Shigella) were added to E. coli counts. The top three human-specific Blautia and human-preferred Prevotella oligotypes are reported as sequence read counts.
The human Blautia and Prevotella oligotypes showed great facility in tracking the human fecal contamination along the Jiquiriçá River. Human-specific/preferred Blautia and Prevotella oligotypes were low or absent in sites 3 and 4, but appeared at sites 5–7 (Figure 4), consistent with the overall increase in the relative abundance of these two genera.
We also examined the presence of human-specific Blautia and Prevotella oligotypes in the Lucaia River, a historic river in Salvador, Brazil, near the Embasa treatment facility, that now generally appears to be a sewer ditch. This sample had a similar oligotype composition to the Embasa sewage and Jiquiriçá River sites 5–7, demonstrating the prevalence and traceability of Blautia and Prevotella oligotypes in both rural and urban environmental samples. Our cluster analyses illustrated that the human, Embasa sewage, downstream Jiquiriçá River and the Lucaia River samples are similar in Blautia oligotype composition, while the animal (three dogs, three pigs, two cows, and one horse) and upstream Jiquiriçá River samples (sites 3 and 4) are similar (Supplementary Figure S1). The Prevotella oligotypes also defined distinctions between the human, sewage (Embasa and the Lucaia River) and downstream Jiquiriçá River sites 5–7 and the animal samples and upstream Jiquiriçá River sites; however, the similarities were less pronounced than those of the Blautia oligotypes (Supplementary Figure S2). These findings further confirm the distinction of human/sewage Blautia oligotypes from animals and demonstrate our ability to track human fecal contamination in the environment.
Conclusions
We identified Prevotella as the dominant genus in Brazilian human fecal samples and second most abundant in Brazilian sewage. The genus Blautia was also consistently present in human and sewage samples. While both genera have demonstrated their applicability in distinguishing human and animal fecal pollution in Brazil surface waters, Blautia appears to contain more discriminatory and globally applicable markers for human sources. Blautia and Prevotella oligotypes may be a useful combination for identifying source of fecal pollution: Prevotella might function as a “general” indicator to assess total fecal pollution, while the host-specific/preferred sequences identified in Blautia and Prevotella would assess the amount of human influence. This is particularly important in rural regions of the world, where sewage handling and animal waste are both likely to impact waterways (Esrey et al., 2001). Overall, the notable differences in the taxonomic composition between Brazilian and US human fecal and sewage microbial communities reiterate the need to develop alternative indicators with global applicability for tracking of human fecal pollution in surface waters.
Supplementary Material
Acknowledgments
This work was supported by the National Oceanic and Atmospheric Administration, Great Lakes Oceans and Human Health Graduate Traineeship Grant to A.M.K. NA06OAR4310119 and through NIH funding on grant R01AI091829-01A1 to S.L.M. We also acknowledge the contributions of Hilary G. Morrison, Sharon L. Grim, Joseph H. Vineis and Mitchell L. Sogin at the Marine Biological Laboratory for sequencing and bioinformatics support.
References
- Ahmed W, Goonetilleke A, Powell D, Gardner T. Evaluation of multiple sewage-associated Bacteroides PCR markers for sewage pollution tracking. Water Res. 2009;43:4872–4877. doi: 10.1016/j.watres.2009.08.042. [DOI] [PubMed] [Google Scholar]
- Ahmed W, Yusuf R, Hasan I, Goonetilleke A, Gardner T. Quantitative PCR assay of sewage-associated Bacteroides markers to assess sewage pollution in an urban lake in Dhaka, Bangladesh. Can J Microbiol. 2010;56:838–845. doi: 10.1139/w10-070. [DOI] [PubMed] [Google Scholar]
- Barreto ML, Genser B, Strina A, Teixeira MG, Assis AMO, Rego RF, et al. Effect of city-wide sanitation programme on reduction in rate of childhood diarrhoea in northeast Brazil: assessment by two cohort studies. Lancet. 2007;370:1622–1628. doi: 10.1016/S0140-6736(07)61638-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhard AE, Field KG. A PCR assay to discriminate human and ruminant feces on the basis of host differences in Bacteroides-Prevotella genes encoding 16S rRNA. Appl Environ Microbiol. 2000;66:4571–4574. doi: 10.1128/aem.66.10.4571-4574.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanton RE, Blank WA, Costa JM, Carmo TM, Reis EA, Silva LK, et al. Schistosoma mansoni population structure and persistence after praziquantel treatment in two villages of Bahia, Brazil. International Journal for Parasitology. 2011;41:1093–1099. doi: 10.1016/j.ijpara.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, et al. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:14691–14696. doi: 10.1073/pnas.1005963107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Oliveira LK, Fleck JD, Comerlato J, Kluge M, Bergamaschi B, Fabres RB, et al. Enteric viruses in water samples from Brazilian dairy farms. Agricultural Water Management. 2012;111:34–39. [Google Scholar]
- Dethlefsen L, Huse S, Sogin ML, Relman DA. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 2008;6:e280. doi: 10.1371/journal.pbio.0060280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick LK, Bernhard AE, Brodeur TJ, Santo Domingo JW, Simpson JM, Walters SP, Field KG. Host distributions of uncultivated fecal Bacteroidales bacteria reveal genetic markers for fecal source identification. Appl Environ Microbiol. 2005;71:3184–3191. doi: 10.1128/AEM.71.6.3184-3191.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubinsky EA, Esmaili L, Hulls JR, Cao Y, Griffith JF, Andersen GL. Application of Phylogenetic Microarray Analysis to Discriminate Sources of Fecal Pollution. Environmental Science & Technology. 2012;46:4340–4347. doi: 10.1021/es2040366. [DOI] [PubMed] [Google Scholar]
- Eren AM, Vineis JH, Morrison HG, Sogin ML. A filtering method to generate high quality short reads using Illumina paired-end technology. PLoS One. 2013a:8. doi: 10.1371/journal.pone.0066643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eren AM, Zozaya M, Taylor CM, Dowd SE, Martin DH, Ferris MJ. Exploring the diversity of Gardnerella vaginalis in the genitourinary tract microbiota of monogamous couples through subtle nucleotide variation. PLoS One. 2011;6:e26732. doi: 10.1371/journal.pone.0026732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eren AM, Maignien L, Sul WJ, Murphy LG, Grim SL, Morrison HG, Sogin ML. Oligotyping: differentiating between closely related microbial taxa using 16S rRNA gene data. Methods in Ecology and Evolution. 2013b;4:1111–1119. doi: 10.1111/2041-210X.12114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eren AM, Sogin ML, Morrison HG, Vineis JH, Fisher JC, Newton RJ, McLellan SL. A single genus in the gut microbiome reflects host preference and specificity. Isme Journal. 2014 doi: 10.1038/ismej.2014.97. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esrey SA, Andersson I, Hillers A, Sawyer R. Closing the Loop: Ecological Sanitation for Food Security. 2001 [PubMed] [Google Scholar]
- Field KG, Samadpour M. Fecal source tracking, the indicator paradigm, and managing water quality. Water Res. 2007;41:3517–3538. doi: 10.1016/j.watres.2007.06.056. [DOI] [PubMed] [Google Scholar]
- Fogarty LR, Voytek MA. Comparison of bacteroides-prevotella 16S rRNA genetic markers for fecal samples from different animal species. Appl Environ Microbiol. 2005;71:5999–6007. doi: 10.1128/AEM.71.10.5999-6007.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzolin MR, Alves RCB, Keller R, Gomes TAT, Beutin L, Barreto ML, et al. Prevalence of diarrheagenic Escherichia coli in children with diarrhea in Salvador, Bahia, Brazil. Memorias Do Instituto Oswaldo Cruz. 2005;100:359–363. doi: 10.1590/s0074-02762005000400004. [DOI] [PubMed] [Google Scholar]
- Fremaux B, Gritzfeld J, Boa T, Yost CK. Evaluation of host-specific Bacteroidales 16S rRNA gene markers as a complementary tool for detecting fecal pollution in a prairie watershed. Water Research. 2009;43:4838–4849. doi: 10.1016/j.watres.2009.06.045. [DOI] [PubMed] [Google Scholar]
- Gourmelon M, Caprais MP, Segura R, Le Mennec C, Lozach S, Piriou JY, Rince A. Evaluation of two library-independent microbial source tracking methods to identify sources of fecal contamination in French estuaries. Appl Environ Microbiol. 2007;73:4857–4866. doi: 10.1128/AEM.03003-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrant RL, Kirchhoff LV, Shields DS, Nations MK, Leslie J, de Sousa MA, et al. Prospective study of diarrheal illnesses in northeastern Brazil: patterns of disease, nutritional impact, etiologies, and risk factors. J Infect Dis. 1983;148:986–997. doi: 10.1093/infdis/148.6.986. [DOI] [PubMed] [Google Scholar]
- Harwood VJ, Brownell M, Wang S, Lepo J, Ellender RD, Ajidahun A, et al. Validation and field testing of library-independent microbial source tracking methods in the Gulf of Mexico. Water Res. 2009;43:4812–4819. doi: 10.1016/j.watres.2009.06.029. [DOI] [PubMed] [Google Scholar]
- Haugland RA, Varma M, Sivaganesan M, Kelty C, Peed L, Shanks OC. Evaluation of genetic markers from the 16S rRNA gene V2 region for use in quantitative detection of selected Bacteroidales species and human fecal waste by qPCR. Systematic and Applied Microbiology. 2010;33:348–357. doi: 10.1016/j.syapm.2010.06.001. [DOI] [PubMed] [Google Scholar]
- Huse SM, Dethlefsen L, Huber JA, Welch DM, Relman DA, Sogin ML. Exploring microbial diversity and taxonomy using SSU rRNA hypervariable tag sequencing. PLoS Genet. 2008;4:e1000255. doi: 10.1371/journal.pgen.1000255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins MW, Tiwari S, Lorente M, Gichaba CM, Wuertz S. Identifying human and livestock sources of fecal contamination in Kenya with host-specific Bacteroidales assays. Water Res. 2009;43:4956–4966. doi: 10.1016/j.watres.2009.07.028. [DOI] [PubMed] [Google Scholar]
- Kildare BJ, Leutenegger CM, McSwain BS, Bambic DG, Rajal VB, Wuertz S. 16S rRNA-based assays for quantitative detection of universal, human-, cow-, and dog-specific fecal Bacteroidales: a Bayesian approach. Water Res. 2007;41:3701–3715. doi: 10.1016/j.watres.2007.06.037. [DOI] [PubMed] [Google Scholar]
- Koskey AM, Fisher JC, Traudt MF, Newton RJ, McLellan SL. Analysis of the gull fecal microbial community reveals the dominance of Catellicoccus marimammalium in relation to culturable Enterococci. Appl Environ Microbiol. 2014;80:757–765. doi: 10.1128/AEM.02414-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature. 2012;489:220–230. doi: 10.1038/nature11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLellan SL, Huse SM, Mueller-Spitz SR, Andreishcheva EN, Sogin ML. Diversity and population structure of sewage-derived microorganisms in wastewater treatment plant influent. Environ Microbiol. 2010;12:378–392. doi: 10.1111/j.1462-2920.2009.02075.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLellan SL, Newton RJ, Vandewalle JL, Shanks OC, Huse SM, Eren AM, Sogin ML. Sewage reflects the distribution of human faecal Lachnospiraceae. Environ Microbiol. 2013;15:2213–2227. doi: 10.1111/1462-2920.12092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mieszkin S, Yala JF, Joubrel R, Gourmelon M. Phylogenetic analysis of Bacteroidales 16S rRNA gene sequences from human and animal effluents and assessment of ruminant faecal pollution by real-time PCR. J Appl Microbiol. 2010;108:974–984. doi: 10.1111/j.1365-2672.2009.04499.x. [DOI] [PubMed] [Google Scholar]
- Moreno AC, Filho AF, do Gomes TA, Ramos ST, Montemor LP, Tavares VC, et al. Etiology of childhood diarrhea in the northeast of Brazil: significant emergent diarrheal pathogens. Diagn Microbiol Infect Dis. 2010;66:50–57. doi: 10.1016/j.diagmicrobio.2008.03.017. [DOI] [PubMed] [Google Scholar]
- Newton RJ, Vandewalle JL, Borchardt MA, Gorelick MH, McLellan SL. Lachnospiraceae and Bacteroidales alternative fecal indicators reveal chronic human sewage contamination in an urban harbor. Appl Environ Microbiol. 2011;77:6972–6981. doi: 10.1128/AEM.05480-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton RJ, Bootsma MJ, Morrison HG, Sogin ML, McLellan SL. A microbial signature approach to identify fecal pollution in the waters off an urbanized coast of Lake Michigan. Microbial Ecology. 2013;65:1011–1023. doi: 10.1007/s00248-013-0200-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reischer GH, Kasper DC, Steinborn R, Mach RL, Farnleitner AH. Quantitative PCR method for sensitive detection of ruminant fecal pollution in freshwater and evaluation of this method in alpine karstic regions. Appl Environ Microbiol. 2006;72:5610–5614. doi: 10.1128/AEM.00364-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reischer GH, Kasper DC, Steinborn R, Farnleitner AH, Mach RL. A quantitative real-time PCR assay for the highly sensitive and specific detection of human faecal influence in spring water from a large alpine catchment area. Lett Appl Microbiol. 2007;44:351–356. doi: 10.1111/j.1472-765X.2006.02094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reischer GH, Ebdon JE, Bauer JM, Schuster N, Ahmed W, Astrom J, et al. Performance characteristics of qPCR assays targeting human- and ruminant-associated bacteroidetes for microbial source tracking across sixteen countries on six continents. Environ Sci Technol. 2013;47:8548–8556. doi: 10.1021/es304367t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roslev P, Bukh AS. State of the art molecular markers for fecal pollution source tracking in water. Applied Microbiology and Biotechnology. 2011;89:1341–1355. doi: 10.1007/s00253-010-3080-7. [DOI] [PubMed] [Google Scholar]
- Santo Domingo JW, Bambic DG, Edge TA, Wuertz S. Quo vadis source tracking? Towards a strategic framework for environmental monitoring of fecal pollution. Water Res. 2007;41:3539–3552. doi: 10.1016/j.watres.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Sauer EP, VandeWalle JL, Bootsma MJ, McLellan SL. Detection of the human specific Bacteroides genetic marker provides evidence of widespread sewage contamination of stormwater in the urban environment. Water Research. 2011;45:4081–4091. doi: 10.1016/j.watres.2011.04.049. [DOI] [PubMed] [Google Scholar]
- Shanks OC, Newton RJ, Kelty CA, Huse SM, Sogin ML, McLellan SL. Comparison of the microbial community structures of untreated wastewaters from different geographic locales. Applied and Environmental Microbiology. 2013;79:2906–2913. doi: 10.1128/AEM.03448-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soller JA, Schoen ME, Bartrand T, Ravenscroft JE, Ashbolt NJ. Estimated human health risks from exposure to recreational waters impacted by human and non-human sources of faecal contamination. Water Res. 2010;44:4674–4691. doi: 10.1016/j.watres.2010.06.049. [DOI] [PubMed] [Google Scholar]
- Stewart JR, Boehm AB, Dubinsky EA, Fong TT, Goodwin KD, Griffith JF, et al. Recommendations following a multi-laboratory comparison of microbial source tracking methods. Water Research. 2013;47:6829–6838. doi: 10.1016/j.watres.2013.04.063. [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unno T, Jang J, Han D, Kim JH, Sadowsky MJ, Kim OS, et al. Use of Barcoded Pyrosequencing and Shared OTUs To Determine Sources of Fecal Bacteria in Watersheds. Environmental Science & Technology. 2010;44:7777–7782. doi: 10.1021/es101500z. [DOI] [PubMed] [Google Scholar]
- USEPA. Microbial Source Tracking Guide Document. Washington, DC: Office of Research and Development; 2005. [Google Scholar]
- VandeWalle JL, Goetz GW, Huse SM, Morrison HG, Sogin ML, Hoffmann RG, et al. Acinetobacter, Aeromonas and Trichococcus populations dominate the microbial community within urban sewer infrastructure. Environmental Microbiology. 2012;14:2538–2552. doi: 10.1111/j.1462-2920.2012.02757.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO. Combating waterborne disease at the household level. Geneva, Switzerland: 2007. [Google Scholar]
- WHO. Schistosomiasis Fact Sheet. World Health Organization; 2013a. http://www.who.int/mediacentre/factsheets/fs115/en/index.html. [Google Scholar]
- WHO. Progress on sanitation and drinking-water 2013 update. World Health Organization; 2013b. [Google Scholar]
- Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA, et al. Linking Long-Term Dietary Patterns with Gut Microbial Enterotypes. Science. 2011;334:105–108. doi: 10.1126/science.1208344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, et al. Human gut microbiome viewed across age and geography. Nature. 2012;486:222. doi: 10.1038/nature11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.