

Abstract
Intratumoral hypoxia and Hypoxia Inducible Factor-1α (HIF-1α)-dependent CD39/CD73 ecto-enzymes may govern the accumulation of tumor-protecting extracellular adenosine and signaling through the A2A adenosine receptors (A2AR) in tumor microenvironments (TME). Here, we explored the conceptually novel motivation to use supplemental oxygen as a treatment to inhibit the hypoxia/HIF-1α-CD39/CD73-driven accumulation of extracellular adenosine in the TME in order to weaken the tumor protection. We report that hyperoxic breathing (60% O2) decreased the TME hypoxia, as well as levels of HIF-1α and downstream target proteins of HIF-1α in the TME according to proteomics studies in mice. Importantly, oxygenation also down-regulated the expression of adenosine-generating ecto-enzymes and significantly lowered levels of tumor-protecting extracellular adenosine in the TME. Using supplemental oxygen as a tool in studies of the TME, we also identified FHL-1 as a potentially useful marker for the conversion of hypoxic into normoxic TME. Hyperoxic breathing resulted in the up-regulation of antigen-presenting MHC-class I molecules on tumor cells and in the better recognition and increased susceptibility to killing by tumor-reactive cytotoxic T cells. Therapeutic breathing of 60% oxygen resulted in the significant inhibition of growth of established B16.F10 melanoma tumors and prolonged survival of mice. Taken together, the data presented here provide proof-of principle for the therapeutic potential of systemic oxygenation to convert the hypoxic, adenosine-rich and tumor-protecting TME into a normoxic and extracellular adenosine-poor TME that, in turn, may facilitate tumor regression. We propose to explore the combination of supplemental oxygen with existing immunotherapies of cancer.
Keywords: Hypoxia, HIF-1α, Adenosine, CD73, Cancer, Immunology
Introduction
The intratumoral hypoxia and Hypoxia Inducible Factor-1α (HIF-1α)-dependent pathways are tumor-protecting [1] and were shown to up-regulate the tandem activity of ecto-enzymes CD39 (ecto-ATPase/ADPase) and CD73 (ecto-5'-nucleotidase) [2]. These enzymes represent the major source of extracellular adenosine in tissue microenvironments including the tumor microenvironment (TME) [3].
The Hypoxia-HIF-1α-CD39/CD73-mediated accumulation of adenosine results in signaling through the cAMP-elevating A2A and A2B (A2AR/A2BR) adenosine receptors (Fig. 1). Extracellular adenosine and A2AR/A2BR were shown to be critical and non-redundant in the protection of inflamed normal tissues [4-11] and in the protection of tumors from the anti-tumor immune response [3-5].
Fig. 1. Hyperoxia reduces intratumoral hypoxia and HIF-1α, down-regulates HIF-1α-target genes, and increases levels of HIF-1α regulating proteins including Four and a half LIM domains-1 (FHL-1).
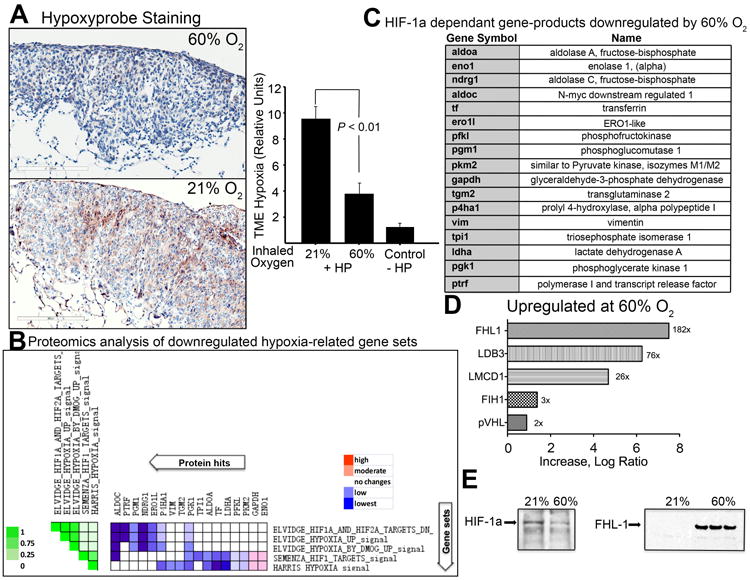
(A) Immunohistochemical staining of intratumoral hypoxia using the in vivo hypoxia marker, Hypoxyprobe. Positive Hypoxyprobe staining is shown in brown in the light micrograph. Shown is the average intensity of staining OD/mm2 (mean ± SEM, P < 0.01, n = 3). Mice bearing MCA205 pulmonary tumors breathing 60% oxygen for 3h were injected i.v. with Hypoxyprobe. (B) Expression of Hypoxia-HIF-1α dependent gene products in the TME as demonstrated by comparative whole cell proteomics analysis of known HIF-1α targets in the TME. The heat map shows the clustered genes in the leading edge subsets with the expression values represented as colors. The set-to-set graph (left) uses color intensity to show the overlap between subsets: the darker the color (on the left in green), the greater the overlap between the subsets. Leading edge analysis using GSEA software showed that five gene sets were highly correlated with the observed HIF-α regulated proteins. Only those protein sets that fall into < 0.25 false discovery rate (FDR) and were significant in the GSEA output are shown, as described previously (SI, Ref. 4-6). (C) The full names of hypoxia-regulated proteins identified by whole cell proteome analysis. (D) Proteomics analysis demonstrate HIF-1α regulating proteins (FIH, VHL, and FHL-1) are increased in tumors from mice breathing 60% oxygen. Tumor sections representing hypoxic regions (HIF-1αhigh) from mice breathing 21% O2 were compared to normoxic regions of the TME in mice breathing 60% oxygen. (E) Immunoblot assays from tumors of mice breathing 21% or 60% oxygen. Shown are representative samples of HIF-lα (left panel) and FHL-1 (right panel) from 5 independent assays.
There is a well-appreciated need in therapeutic treatments to i) weaken tumor hypoxia and/or ii) inhibit HIF-1α [1, 6-8], iii) inhibit CD39/CD73-mediated adenosine formation [9, 10], and iv) antagonize the A2AR/A2BR in the TME [3,4, 11-13]. In view of the importance of extracellular adenosine in tumor protection, one of the goals of this research was to test the hypothesis whether the early stages of Hypoxia→HIF-lα signaling govern the downstream molecular events of the hypoxia-adenosinergic pathway (i.e. TME hypoxia → HIF-α → CD39/CD73 → [Adenosine] → A2AR/A2BR → intracellular cAMP → PKA → cAMP Response Element (CRE) → CREB pathway) (Fig. S1). Recently, the concept of extracellular adenosine-mediated tumor protection has received additional strong support from the analysis of massive human data sets from more than 6000 triple negative and chemotherapy-resistant breast cancers. Resistance to chemotherapy and immunotherapy was shown to be due to an over-expression of extracellular adenosine-generating CD73, resulting in stronger immunosuppression by adenosine-A2AR/A2BR signaling in the TME [14].
Here, we aimed to address the need in novel approaches to decrease the tumor-protecting effects of tumor hypoxia. Indeed, it could be therapeutically valuable to decrease the intensity of tumor hypoxia-driven and CD73-mediated accumulation of extracellular adenosine in the TME by using already available and safe drugs or treatments. We reasoned that by inhibiting the upstream events of hypoxia-HIF-1α-governed signaling pathways, oxygenation may also inhibit the downstream CD39/CD73-[extracellular adenosine]high-A2AR/A2BR-mediated tumor protection in the TME (Fig. 1). This, in turn, may enable tumor rejection. The experimental observations described here validate these assumptions.
Methods
Animals
Female C57BL/6N (B6) mice, 8-12 weeks old, were purchased from Charles River Laboratories. These animals were housed in a specific pathogen-free environment according to the National Institute of Health guidelines. All animal experimentation conforms to protocols approved by the Institutional Animal Care and Use Committee.
Tumors
MCA205 fibrosarcoma is a 3-methylcholanthrene-induced tumor of B6 origin and B16-F10.P1 is a poorly immunogenic subclone of the spontaneously arising B16/BL6 melanoma (SI, Ref. 10-12). B16-F10.P1 is the parent strain of CL8-1 (transfected with MHC Class I) [4]. To establish tumors, B6 mice were injected s.c. (1×105) or i.v. (3×105) with tumor cells resuspended in 200 μl of HBSS. Mice with subcutaneous tumors were placed in either 21% or 60% oxygen after tumors became palpable (between day 7 and 10). Tumor size was measured in two dimensions using vernier calipers.
Hyperoxic breathing
Mice were placed in small animal care units with well-controlled gas composition to mimic protocols of supplemental oxygen delivery to humans [28]. Self-contained oxygen generators (Airsep) were used to ensure desired levels of oxygen were maintained inside each unit. Hypercapnic acidosis was avoided by replacing traditional mouse cage tops with aerated wire lids and by using Sodasorb (Grace&Co) (SI, Ref. 1, 2). CO2 levels inside the chamber never exceeded 0.4%, while hypercapnia typically occurs at levels higher than 2%. Levels of CO2 and O2 in the chambers were carefully monitored (see full details in supporting information).
Hypoxyprobe Analysis
Hypoxyprobe™-1 Plus Kit (Millipore) was used to detect hypoxia in tumors. Mice with established MCA205 tumors were placed in 21% or 60% oxygen for 3h then injected with 80 mg/kg of Hypoxyprobe. After 1.5h of labeling, lungs were snap frozen in n-hexane and 4-μm cryosections were prepared from 10-20 different cutting surfaces and immunohistochemistry was performed.
Immunohistochemistry
Immunohistochemistry was performed at Harvard Medical School Pathology Department at Brigham and Women's Hospital. Four μm thick acetone-fixed, O.C.T-embedded tissue sections were prepared. The slides were soaked in -20°C methanol-acetic acid for 2 min then air-dried for 20 min at room temperature. Slides were pre-treated with Peroxidase Block (DAKO). Primary anti-Hypoxyprobe (Millipore) was applied at a concentration of 1:100 at room temperature for 1h. Rabbit anti-rat immunoglobulin antibody was applied at a concentration of 1:750 in DAKO diluent for 1h. Slides were detected with anti-rabbit Envision+ kit (DAKO). Immunoperoxidase staining was developed using a DAB chromogen (DAKO) and counterstained with hematoxylin. Tumor hypoxia was annotated using SpectrumTM Plus and Aperio's ScanScope® slide scanners by the Pathology Department at Brigham and Women's Hospital. See SI for additional details of fluorescent immunostaining.
Whole cell proteomics
Mice bearing 7-day established MCA205 intradermal tumors were placed in either 21% or 60% oxygen. On day 21, tumors were excised and homogenized in protein extraction lysing buffer as recommended by manufacturer (Thermo Fisher Scientific). After denaturing and enzymatic digestion by trypsin, the 21% O2 and 60% O2 samples were labeled with differential stable-isotopic labeling using dimethylation reaction. A two-dimensional HPLC separation was performed by high/low pH reverse phase HPLC and a Q-Exactive Orbitrap mass spectrometer (Thermo Fisher Scientific) to acquire the MS and MS/MS spectra of isotopically labeled peptide signals. Leading edge analysis using GSEA software showed that five gene sets were highly correlated with the observed HIF-α regulated proteins. Only those protein sets that fall into < 0.25 false discovery rate (FDR) and were significant in the GSEA output are shown (SI, Ref 3-6). See SI for full details on proteomics and assays to pre-screen hypoxic versus normoxic TMEs.
Western Blot
Mice with established tumors were placed in either 21% or 60% oxygen for 10 days and tumors were snap frozen in liquid nitrogen and then homogenized in lysis buffer. After fractionation using SDS/PAGE followed by semi-dry transfer, samples were detected using mAb (1:500, Santa Cruz) against mouse HIF-1α, FLH-1, or VEGF. Levels of beta-actin were detected using anti-mouse beta-actin mAb (1:5,000, Sigma-Aldrich). See SI for additional details.
In vivo extracellular adenosine measurements
Mice with established MCA205 tumors were used to analyze levels of extracellular adenosine. Microdialysis probes (Bioanalytical Systems) were placed in the center, edge and in normal tissue next to tumors. Microdialysate (isotonic saline, 100 U of heparin, and 20 mM EHNA adenosine deaminase inhibitor) was perfused at 2 mL/min for 2.5h using an infusion pump (Braintree Scientific). Following the initial probe and an equilibration period, mice were placed in 21% or 60% oxygen for 3h and re-probed for levels of adenosine in the same tumor area. Adenosine levels were measured by reversed-phase liquid chromatography-tandem mass spectrometry using a triple quadrupole mass spectrometer with 13C10-adenosine as an internal standard (SI, Ref. 7-9). See SI for additional details.
RT-PCR
Mice with established MCA205 tumors were placed in either 21% or 60% oxygen for 72h. RT-PCR was performed using the RT2 SYBR green PCR master mix (Superarray) on an Applied Biosciences 7300 PCR platform. Primers were purchased from SABiosciences (Qiagen, Valencia, CA). House-keeping genes were included as controls.
Cytoxicity Assay
MCA205 cells were cultured in 21% oxygen or 60% oxygen for 4 days in vitro. Cells were collected by trypsinization and labeled with 100 μCi sodium chromate (Amersham Biosciences) per 2 × 106 cells for 1h at 37°C. In vitro activated T cells from the tumor draining lymph nodes of MCA205 inoculated mice were used as effector cells and seeded into 96-well round-bottomed plates at the indicated effector/target (E/T) ratios. 51Cr-labeled MCA-205 cells at 1 × 104 cells per well were used as target cells. After 4h incubation at 37°C, radioactivity released from lysed target cells was counted on a γ-counter. See SI for calculations of Percent Specific Lysis.
Flow cytometry
Solid or pulmonary tumors were homogenized and passed through a 70 μm strainer. Tumor cells and lymphocytes were recovered using 40% Percoll separation, incubated with monoclonal antibodies (1:50, BD Pharmingen and eBioscience) in FACS buffer (PBS + 0.5% BSA) and acquired on a FACSCalibur. Analysis was performed using CellQuest (BD Biosciences) and FlowJo (Tree Star) software.
Statistics
Significance of differences in survival was calculated using Log-rank (Mantel-Cox) Test and differences in tumor size was evaluated by Two-way ANOVA. Significance in differences in immunohistochemistry, extracellular adenosine levels, RNA, and flow cytometry was evaluated by the Student's T test.
Results
Our assays were designed to test whether breathing supplemental oxygen (60% O2) may inhibit the tumor hypoxia-HIF-1α driven biochemical pathways specifically the tumor-protecting CD39/CD73-dependent accumulation of extracellular adenosine ([Ado]high) in the TME. Sixty percent oxygen was selected since it has been used clinically with human patients for other indications as well as in our previous studies of lung inflammation in mice [28].
As expected, Fig. 1A demonstrates that continuous breathing of 60% oxygen reduced intratumoral levels of hypoxia in mice. This was reflected in the significant reduction in immunostaining by the molecular hypoxia marker Hypoxyprobe, which identifies deeply hypoxic areas with oxygen tension less than 1% [15]. Fig. S2 demonstrates that breathing 60% oxygen was also capable of decreasing the pathophysiological hypoxia not only in tumors, but also in inflamed tissue microenvironments. In parallel studies of mice with acute sepsis induced by cecal ligation and puncture (see supplementary methods), breathing 60% oxygen reduced hypoxia in surrounding tissues (Fig. S2).
Results of the studies shown in Fig. 1 confirmed that breathing 60% oxygen increased the local intratumoral oxygen tension by decreasing the number of deeply hypoxic areas (0% to 1% oxygen) in the TME. It is likely that 60% oxygen also oxygenated less hypoxic areas, but the use of Hypoxyprobe is limited by its inability to detect changes in hypoxia between 1% and 3% oxygen tension [15].
In order to investigate the oxygenation-associated changes in TME in more detail, we performed comparative whole cell proteomics analyses of the TME in mice breathing 21% (ambient) or 60% oxygen (Fig. 1 B and C). The expectation was that breathing 60% oxygen may lead to the degradation of HIF-1α by increasing the oxygen tension in the TME above 1% to 3%, which is required for the stabilization of HIF-l α. This was predicted to affect many proteins down-stream of HIF-1 α, including HIF-1α itself (Fig. 1E, left panel).
The results of the proteomics gene set enrichment analysis (GSEA) provide information on the enriched functions/pathways in a differential protein list. Data in Fig. 1 B-D and Fig. S3 revealed that breathing 60% oxygen reduced the protein expression of known HIF-1α targets and resulted in the down-regulation of hypoxia- and HIF-1α related gene sets. The list of HIF-1α-related proteins that were significantly down-regulated are shown in Fig. 1C. These data confirm and extend to the protein level the previous observations of other scientists [16-18] who have reported the oxygen-dependent manner of the expression of these gene sets on the mRNA level. The proteins listed in Fig. 1C are known to play important roles in many biological processes regulated by hypoxia (e.g. carbohydrate catabolic and metabolic processes, gluconeogenesis pathway, etc).
The observations and confirmations of expected changes in hypoxia-HIF-1α dependent events due to oxygenation of the TME from an unbiased proteomics screen also provided an important internal control. Indeed, since the parallel analyses of the TME confirmed that hyperoxic breathing did reduce the intratumoral levels of HIF-1α (Fig. 1E, left panel), it provided more confidence in the interpretations of subsequent findings of the effects of hyperoxia on the expression of other proteins, which were not yet identified as governed by hypoxia-HIF-1α signaling.
In order to further clarify differences in the proteome of the hypoxic versus normoxic TME and to identify proteins that may serve as molecular markers for normoxic TMEs, SDS PAGE separated proteins from different areas of tumors were evaluated by HIF-1α immunoblotting prior to proteomics analysis. In these experiments, tumor sections representing hypoxic regions (HIF-1α high) from mice breathing 21% O2 were compared to normoxic regions of the TME in mice breathing 60% oxygen (Fig. S4).
Interestingly, proteomics screen of the TME identified FHL-1 (Four and a half LIM domains-1), an inhibitor of HIF-1α signaling (19), to be dramatically upregulated in the normoxic regions of tumors from mice breathing 60% oxygen (Fig. 1D). The strong increase in FHL-1 protein levels observed in proteomics studies was subsequently confirmed by western blot analysis (Fig. 1E, right panel). In the proteomics analysis, FHL-1 was up-regulated significantly higher than any other gene product in the TME of mice breathing 60% oxygen (Fig. 1D), suggesting that FHL-1 could serve as a marker for more oxygenated TMEs. These observations have also identified FHL-1 as a candidate molecule, which might mediate the effects of 60% oxygen by preventing association of HIF-1a with the critical co-activator p300/CBP [19].
Interestingly, the proteomics screen of the TME also demonstrated the 60% oxygen-mediated increase of other HIF-1α regulating proteins such as Factor Inhibiting HIF-1 (FIH-1) [1] as well as von Hippel-Lindau (VHL) [20] (Fig. 1D). Taken together, these findings are in agreement with the biochemical assumptions that breathing 60% oxygen reverses tumor hypoxia and HIF-1α regulated pathways in the TME.
It was important to test whether the chaotic tissue hypoxia and vasculature in the TME would be affected by 60% oxygen to such an extent that it would be accompanied by a decrease in the levels of tumor-protecting extracellular adenosine. To this end, direct measurements of extracellular adenosine in the TME were performed using equilibrium microdialysis probes placed intratumorally in a growing tumor. Following an initial microdialysis probe collection, mice were placed in 21% or 60% oxygen for 3h and re-probed for levels of adenosine in the same tumor area. Subsequent quantitative determination by high performance liquid chromatography-tandem mass spectrometry (HPLC-MS) revealed that tumors from mice breathing 60% oxygen had significantly reduced levels of extracellular adenosine compared to those returned to ambient oxygen (Fig. 2 A). Importantly, tumor areas with initially high levels of extracullular adenosine were significantly reduced by supplemental oxygenation (Fig. S5).
Fig. 2. Hyperoxic breathing inhibits both upstream and downstream stages of the hypoxia-HIF-1α-CD39/CD73-A2 Adenosine receptor tumor-protecting pathway.
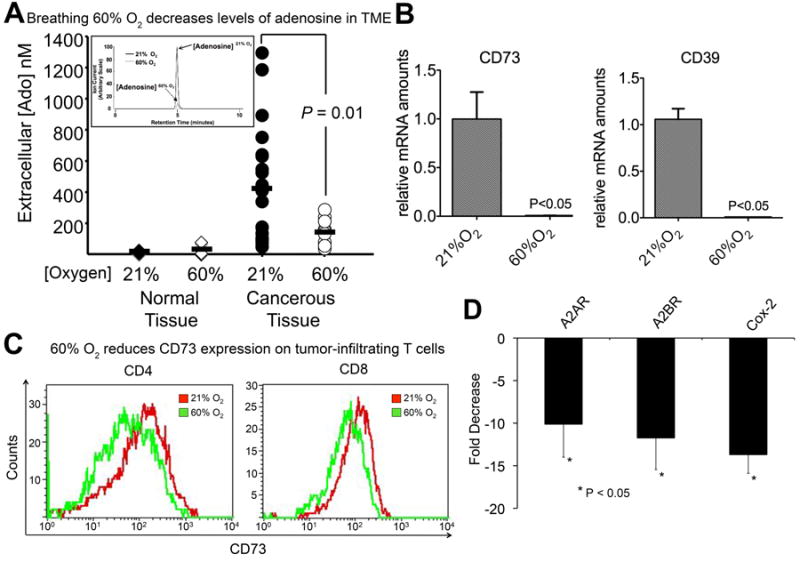
(A) Levels of extracellular adenosine in the TME following 60% oxygen breathing. Microdialysis probes were placed in MCA205 intradermal tumors. After initial probe of the TME, mice were placed in 21% or 60% oxygen for 3h and new probes were placed in the same tumor area. Adenosine levels were measured by reversed-phase liquid chromatography-tandem mass spectrometry using a triple quadrupole mass spectrometer with 13C10-adenosine as an internal standard (P = 0.01) as in (SI, Ref. 7-9). (B) RT-qPCR analysis of CD73 and CD39 in B16 s.c. tumors from mice breathing 21% or 60% O2 for 72 h (P < 0.05, n = 3). (C) Expression of CD73 on the surface of TME-infiltrating T cells. Lymphocytes were isolated from the TME of mice bearing MCA205 pulmonary tumors breathing 21% and 60% oxygen and the expression of CD73 was measured by flow cytometry (CD4, P < 0.02, n = 4; CD8, P < 0.02, n = 4). (D) Breathing 60% oxygen down-regulates immunosuppressive A2AR, A2BR, and COX-2. Mice bearing established MCA205 pulmonary tumors were treated with 21% or 60% oxygen for 72 h and total RNA was extracted and subjected to RT-qPCR analysis (P < 0.05, n = 3).
It was also observed that intratumoral levels of extracellular adenosine were significantly higher than in normal adjacent dermal tissue, confirming the TME is rich in extracellular adenosine. Moreover, the high levels of adenosine are distributed chaotically within different locations of the TME, which might be expected in a chaotically grown tumor (Fig. S5).
Further analysis of the TME demonstrated that breathing 60% oxygen was capable of reducing the expression of the adenosine-generating ecto-enzymes CD39 and CD73 (Fig. 2B). This provided a possible explanation for the decrease in extracellular adenosine levels in the TME after supplemental oxygenation. The down-regulation of CD73 by oxygenation in the TME was also observed during analysis of tumor-infiltrating CD8+ and CD4+ T cells (Fig. 2 C).
Interestingly, breathing 60% oxygen also resulted in the down-regulation of tumor-protecting [4] adenosine receptors A2AR, A2BR, and even of key enzymes of other anti-inflammatory pathways, e.g. Prostaglandin E Synthase-2 (COX-2) (Fig. 2 D). In support of these in vivo TME studies, COX-2 has been shown to be regulated by hypoxia and A2AR-mediated cAMP induction in vitro [21-22]. Since there is no A2A/A2B receptor reserve on T cells [23], the lower expression of A2AR/A2BR translates directly into reduced production of intracellular cyclic AMP. These data indicate that oxygenation decreases all stages of the hypoxia-driven and CD39/CD73-adenosine high-A2AR/A2BR-cAMP pathway.
The reduction in hypoxia and destabilization of HIF-1α in mice breathing 60% oxygen also affected other HIF-1α downstream targets, particularly those involved in the angiogenesis pathways. Fig. 3 A shows that hyperoxic breathing significantly reduced the levels of VEGF in the TME. This was also confirmed by RT-qPCR analysis of tumor tissue (Fig. S6). We then hypothesized that the reduction in VEGF levels would alter neovascularization of tumors in these mice. Indeed, immunohistochemical analyses of intratumoral PECAM-1 (CD31) demonstrated a slight, but statistically significant reduction in the expression of the angiogenic marker in mice breathing 60% oxygen (Fig. 3 B and C). In parallel assays, no change was detected in the expression of the VEGF receptor on tumor cells (Fig. 3 B).
Fig. 3. Hyperoxia affects HIF-1α downstream targets in angiogenesis pathways.
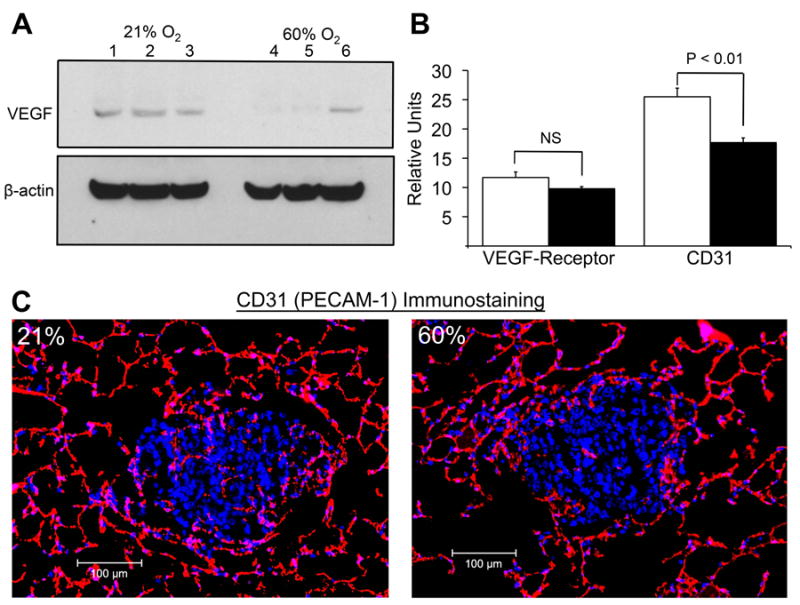
(A) Immunoblot analysis of VEGF in the TME. Mice with MCA205 pulmonary tumors were exposed to 21% or 60% oxygen for 72 h; shown are representative samples. (B)(C) Immunohistochemical staining of intratumoral CD31 in mice breathing 21% (left panel) and 60% oxygen (right panel) (20x magnification, P < 0.01, n = 3). Mice with established MCA205 pulmonary tumors were placed in either 21% or 60% oxygen for 72 h. Also shown is the effect of hyperoxic breathing on the levels of the VEGF Receptor (CD309) on tumor cells.
The ability of supplemental oxygenation to favorably change the TME by potentially weakening tumor escape mechanisms was confirmed in assays of several different tumor models and anatomical locations. The anti-tumor effects of oxygenation were reflected in the increased expression of antigen-presenting MHC-class I molecules on the surface of tumors in mice breathing 60% oxygen (Fig. 4 A), suggesting that oxygenation may increase the susceptibility to recognition and lethal hit delivery by CTL. Strong support for this interpretation is provided by observations that tumor cells identified to be more hypoxic (HPhi) in vivo had lower levels MHC-I class I expression (Fig. 4 B).
Fig. 4. Hyperoxic breathing increases tumor cell recognition and killing by antitumor T cells, enhances tumor regression, and improves survival of tumor-bearing mice.
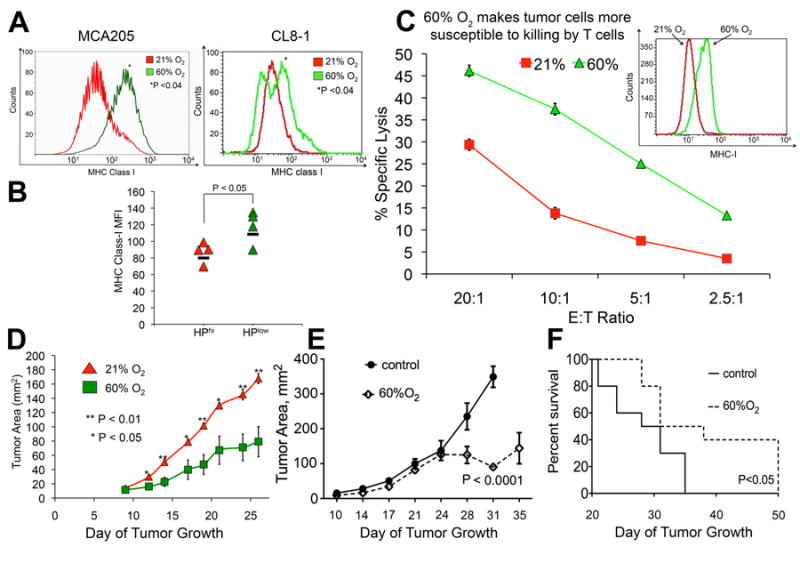
(A) Expression of antigen-presenting MHC class I molecules on the surface of MCA205 fibrosarcoma and B16 melanoma tumors (H2Kb transfected B16, CL8-1 line) Mean fluorescent intensity of MHC-I expression was measured by flow cytometry in mice breathing 60% or 21% oxygen (MCA205, P < 0.04, n = 3; CL8-1, P < 0.04, n = 4). (B) Tumor cells exposed to higher levels of hypoxia had lower surface expression of MHC class I (P < 0.05, n = 4). (C) Hyperoxia is increases lethal hit delivery by cytotoxic T lymphocytes. MCA205 cells cultured in 21% or 60% oxygen for 4 days in vitro demonstrated an increase in the expression of MHC-I as measured by flow cytometry (inset, P < 0.01). The cells were 51Cr-labeled and used in a cytotoxicity assay with culture-activated T cells from MCA205 tumor draining lymph nodes at different effector/target (E/T) ratios. After 4 h, radioactivity released from lysed target cells was counted on a γ-counter (*P < 0.01). (D) Tumor regression of B16 (H2Kb transfected B16, CL8-1 line) tumor-bearing mice following 60% oxygen breathing. After s.c. tumor establishment identified by the first appearance of palpable tumors, mice were placed in either 21% or 60% oxygen and tumor size was measured in two dimensions (*P <0.0.5, **P< 0.01, n = 5 mice). (E) Tumor regression of B16 melamona tumors following 60% oxygen breathing. Mice bearing established B16 s.c. melanomas were placed in either 21% or 60% oxygen and tumor size was measured in two dimensions (P < 0.0001, n = 10). (F) Prolonged survival in mice with established B16 tumors breathing 60% oxygen. Mice bearing established B16 s.c. melanomas were placed in either 21% or 60% oxygen. Tumor-bearing mice were euthanized when tumor reached 2 cm in one direction (P < 0.05, n = 10).
These data support the view that hypoxia within the TME may promote tumor evasion by making the tumor cells less recognizable. Supplemental oxygenation, therefore, may prevent tumor evasion and facilitate the recognition of tumors by T cells, weakening tumor protection. Since antigen-presenting MHC class I molecules are essential for the priming of anti-tumor T cells, we hypothesized that tumor cells exposed to 60% oxygen would be more susceptible to recognition and lysis by tumor-reactive T cells. Fig. 4 C demonstrates that tumor cells with hyperoxia-induced increase in MHC class-I expression were significantly more vulnerable to lethal hit delivery by cytotoxic T lymphocytes.
To examine whether the improved tumor destruction demonstrated in in vitro assays would translate to in vivo tumor-regression, we treated tumor-bearing mice with 60% oxygen. Fig. 4 D shows that breathing 60% oxygen results in significant tumor regression in mice bearing established subcutaneous B16 melanoma tumors (H2Kb transfected B16, CL8-1 line). An increase in the MHC class I expression on the poorly immunogenic parent strain (Fig. S7) may also have contributed to the observed tumor regression of B16 tumors following hyperoxic breathing (Fig. 4E). Moreover, breathing 60% oxygen resulted in the improved survival of B16 melanoma tumor-bearing mice (Fig. 4F). These data confirm the original assumption that oxygenation converts the TME from tumor-protecting to tumor-destructing.
Discussion
It has long been recognized that tumors are hypoxic, tumor hypoxia is a poor prognosis factor, and that established tumors are protected by hypoxia and HIF-1α-mediated biochemical pathways [1]. However, HIF-1 and HIF-2 as well as von Hippel-Lindau (VHL) may have different roles in carcinogenesis [1] [20]. Thus, the Hypoxia-driven and CD39/CD73-A2AR/A2BR-adenosinergic immunosuppression represents a major tumor-protecting mechanism (Fig. 5) [4, 24] [12, 13] [25].
Fig. 5. Hyperoxic breathing may prevent tumor hypoxia-dependent accumulation of tumor-protecting extracellular adenosine ([Ado]high) in tumor microenvironments.
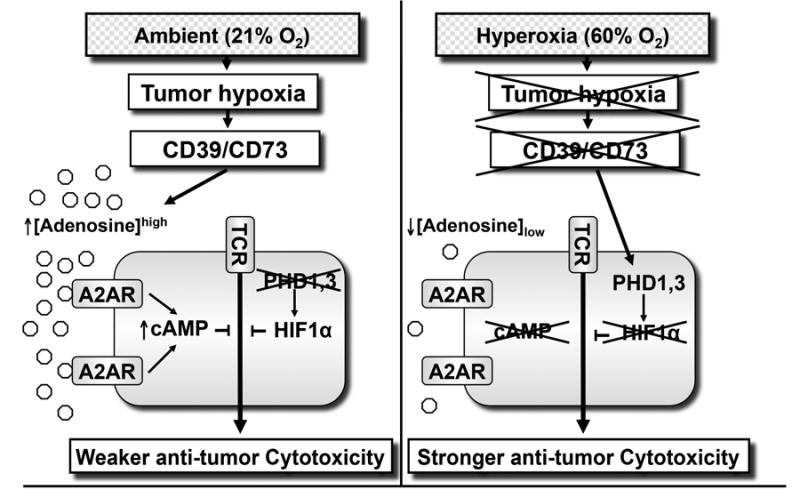
Left panel: Tumor hypoxia/HIF-1α-driven and CD39/CD73 ecto-enzyme-generated extracellular [Adenosine]high protects tumors by inhibiting T cells and NK cells through activation of intracellular cAMP-elevating A2A or A2B adenosine receptors (A2AR/A2BR). TME hypoxia may also protect tumors through the stabilization of HIF-1α. At tissue oxygen tension above ∼3%, oxygen-sensing prolyl hydroxylases (PHD) target HIF-1α for degradation, thereby preventing T cell inhibition. Right panel: Hyperoxic breathing 60% [Oxygen]high is expected to weaken the first upstream stage (TME hypoxia) of hypoxia-HIF-1α signaling and thereby inhibit the formation of extracellular [Adenosine]high. This, in turn, may weaken tumor protection and enable T-and NK cells to reject tumors.
However, while the potential benefits of anti-tme hypoxia treatments and hif-1α inhibitors are well recognized [1,6], there is still an acute medical need in such treatments in the immunotherapy of cancer.
This study demonstrates the previously un-appreciated property of oxygen to predictably and therapeutically recondition the TME by reducing hypoxia and levels of extracellular adenosine. Supplemental oxygenation converts the hypoxic and extracellular adenosine-rich TME into a less hypoxic and extracellular adenosine-poor TME. This, in turn, may result in less favorable conditions for cancerous cells, while simultaneously creating less hostile conditions for the anti-tumor immunity. Thus, by altering the hypoxia-adenosinergic landscape of the TME, hyperoxic breathing may create a more immunopermissive TME, subsequently increasing the vulnerability of tumors to tumor-reactive immune cells (Fig. 5).
These data offer molecular justification to use supplemental oxygen in therapies of cancer in a way that has not been done, and in combination with anti-tumor immunity. Indeed, our motivation to test 60% oxygen is conceptually different from other previous medical uses of oxygen. Here, we used oxygenation in order to weaken the immunosuppression in TME, while previous motivations to use supplemental oxygen were guided by the hypothesis that high oxygen would enhance reactive oxygen species (ROS) formation in the radiotherapy of cancer [26]; or that 70-100% oxygen alone may be directly toxic to tumors [27]. Therefore, the settings of tumor growth in pre-clinical studies were not designed to allow for the development of the anti-tumor immune response following the oxygen-mediated weakening of immunosuppression in the TME. These data provide mechanistic reasoning for breathing 60% oxygen as a treatment to weaken all upstream and down-stream stages of the tumor-protecting Hypoxia/HIF-1α-CD39/CD73- [Adenosine] High-A2AR/A2BR pathway (Fig. 5).
It was found that supplemental oxygen i) decreases hypoxia in inflamed tissues or in tumors (Fig. 1, S2); ii) decreases levels of extracellular adenosine and adenosine generating enzymes in the TME (Fig. 2); iv) reduces neovascularization (Fig. 3); and iii) increases the expression of tumor antigen-expressing molecules, thereby improving the recognition of tumors by tumor-reactive immune cells (Fig. 4). Importantly, the changes in the TME mediated by hyperoxia resulted in significantly delayed tumor growth in vivo and longer survival (Fig. 4).
It remains a likely possibility that the reduction in the levels of extracellular adenosine following oxygenation could be not only because of the decreased generation of adenosine by ecto-enzymes CD39/CD73, but also due to enhanced degradation of extracellular adenosine by adenosine degrading enzymes [29]. The experimental testing of the effects of oxygen on different mechanisms of adenosine metabolism and degradation may be complicated. Due to the critical necessity in the regulation of the levels of extracellular adenosine, there are many different enzymes and pathways involved in adenosine metabolism; which becomes increasingly complex when considering the shuttling of adenosine through many adenosine transporters and the very short half-life of extracellular adenosine in vivo [30].
It is important to point out the possible differential roles of supplemental oxygenation on normal, non-transformed cells during tumorigenesis and on hypoxia and HIF-1α in the TME during the growth and rejection of established tumors. Our data suggest that the destruction of tumors due the lower levels of immunosuppressive and tumor-protecting extracellular adenosine most likely takes place much earlier than the tumor-supporting effects of increased oxygenation of tumors due to e.g. HIF-1α dependent increase in EPO, VEGF and neovascularization [1] [7] [18].
As suggested by the data shown here, systemic oxygenation may indeed provide better oxygenation through existing blood vessels. While the increased oxygenation could potentially benefit some of the tumor cells, it simultaneously may unleash the anti-tumor cytotoxic cells from hypoxia and adenosine mediated inhibition. Thus, the overall outcome would be the death of even the more oxygenated, “healthier” tumor cells. It is also possible that by reducing HIF-1α signaling, breathing 60% oxygen may contribute to the normalization of tumor vasculature, further contributing to tumor oxygenation [31]. Future studies will further characterize the effects of breathing 60% oxygen on the time-scale of neovascularization as well as on already established blood vessels.
Since supplemental oxygen is already a widely used and safe medical treatment, these data may have accelerated clinical implications. We envision the combination of supplemental oxygen with existing immunotherapies of cancer such as cancer vaccines, adoptive cell transfer, and/or blockade of immunological negative regulators (e.g. CTLA-4/PD-1).
However, as an important exclusion criteria, 60% oxygen should not be given to cancer patients during episodes of simultaneous acute inflammation. This includes episodes of acute lung and liver inflammation, among others. Oxygenation may not only enable stronger anti-tumor activities by de-inhibited tumor reactive immune cells, but also accomplish stronger inflammatory damage of normal tissues by de-inhibited myeloid cells or T cells activated by other antigens. Earlier, we attracted attention to such possible caveats in using antagonists of A2AR, which act downstream of the Hypoxia→HIF-1α stages [35].
Indeed, the delivery of 60% oxygen should be considered with an understanding that while long-term treatment with 60% oxygen has been proven to be safe, our previous reports have shown that 60% oxygen exacerbates acute inflammatory lung injury [28] by inhibiting the Hypoxia-HIF-lα -[Adenosine]High-A2AR pathway. Thus, the weakening of this physiological tissue-protecting mechanism may increase inflammatory damage not only in cancerous tissues (Fig. 2), but also in normal tissues of vital organs (Fig. S2). This excessive collateral tissue damage was originally shown to be due to overactive neutrophils [28]. Similar effects on inflamed lungs were observed by others who implicated macrophages [32] and pulmonary invariant natural killer T (iNKT) cells in the effects of hyperoxia [33].
It will be interesting in future studies to compare in parallel the effects of oxygenation on mice with increased tumor burden versus mice with acute lung inflammation. Studies in mice with high numbers of lung tumors may directly clarify whether tumor-reactive T cells de-inhibited by supplemental oxygenation will not only inflict damage to cancerous tissue, but also collateral damage to normal tissues. The expectation is that in contrast to myeloid cells and due to their localized mechanism of lethal hit delivery [34], tumor-reactive T cells de-inhibited by 60% oxygen will still be selective for tumor-antigens. Thus, the tumor-reactive T cells will likely cause much less collateral damage when de-inhibited by oxygen compared to myeloid cells during episodes of acute inflammation [28][32][33].
While considering the issues of patient compliance in using 60% oxygen-delivering masks, it will be appealing to explore the re-purposing of hyperbaric chambers or hypoxic tents which are used by athletes, as a more comfortable option for oxygenation of cancer patients. Future investigations will determine whether other concentrations of oxygen, including hyperbaric oxygen, are capable of providing therapeutic benefit.
Taken together, these data highlight the potentially powerful benefits of supplemental oxygen as a co-adjuvant in combination with other existing cancer treatments in order to weaken the tumor hypoxia-HIF-1α-driven and CD39/CD73→A2AR/A2BR-mediated tumor protective pathway in the hypoxic TME.
Supplementary Material
Key Messages.
Oxygenation decreases levels of tumor protecting hypoxia
Oxygenation decreases levels of tumor protecting extracellular adenosine.
Oxygenation decreases expression of HIF-alpha dependent tumor-protecting proteins.
Oxygenation increases MHC class I expression and enables tumor regression.
Acknowledgments
This work was supported by the funding from Northeastern University and NIH grants (PI: M.S.) R01 CA-111985; R01 CA-112561; R21 AT 002788. We thank Dr. Richard Marsh, an expert in the field of energetics, kinematics and kinetics, for assistance with monitoring gas compositions in hyperoxia units.
Footnotes
Disclosure: The authors declare they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
Author Contributions: S.H., J.K., D.L., S.S, A.O. and M.S. performed and/or analyzed cancer immunology assays; S.R., J.L.K. and S.H. performed, enumerated and interpreted immunohistochemistry assays. E.P. and T.S. designed and used custom-made RNA arrays to scan lung tumor TME. D.S. and B.K. performed and analyzed all proteomics studies of tumor tissue. B.B, J.R. and E.K.J. performed and analyzed measurements of extracellular adenosine. M.S. designed the overall approach, directed research and wrote the manuscript with S.H. and J.K.
References
- 1.Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148:399–408. doi: 10.1016/j.cell.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Synnestvedt K, Furuta GT, Comerford KM, Louis N, Karhausen J, Eltzschig HK, Hansen KR, Thompson LF, Colgan SP. Ecto-5′-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J Clin Invest. 2002;110:993–1002. doi: 10.1172/JCI15337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eltzschig HK, Sitkovsky MV, Robson SC. Purinergic signaling during inflammation. N Engl J Med. 2013;368:1260. doi: 10.1056/NEJMc1300259. [DOI] [PubMed] [Google Scholar]
- 4.Ohta A, Gorelik E, Prasad SJ, Ronchese F, Lukashev D, Wong MK, Huang X, Caldwell S, Liu K, Smith P, Chen JF, Jackson EK, Apasov S, Abrams S, Sitkovsky M. A2A adenosine receptor protects tumors from antitumor T cells. Proc Natl Acad Sci USA. 2006;103 doi: 10.1073/pnas.0605251103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sitkovsky MV, Kjaergaard J, Lukashev D, Ohta A. Hypoxia-adenosinergic immunosuppression: tumor protection by T regulatory cells and cancerous tissue hypoxia. Clin Cancer Res. 2008;14:5947–5952. doi: 10.1158/1078-0432.CCR-08-0229. [DOI] [PubMed] [Google Scholar]
- 6.Dewhirst MW, Cao Y, Moeller B. Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat Rev Cancer. 2008;8:425–437. doi: 10.1038/nrc2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang H, Qian DZ, Tan YS, Lee K, Gao P, Ren YR, Rey S, Hammers H, Chang D, Pili R, Dang CV, Liu JO, Semenza GL. Digoxin and other cardiac glycosides inhibit HIF-1α synthesis and block tumor growth. Proc Natl Acad Sci USA. 2008;105:19579–19586. doi: 10.1073/pnas.0809763105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee K, Qian DZ, Rey S, Wei H, Liu JO, Semenza GL. Anthracycline chemotherapy inhibits HIF-1 transcriptional activity and tumor-induced mobilization of circulating angiogenic cells. Proc Natl Acad Sci USA. 2009;106:2353–2358. doi: 10.1073/pnas.0812801106. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 9.Feng L, Sun X, Csizmadia E, Han L, Bian S, Murakami T, Wang X, Robson SC, Wu Y. Vascular CD39/ENTPD1 directly promotes tumor cell growth by scavenging extracellular adenosine triphosphate. Neoplasia. 2011;13:206–216. doi: 10.1593/neo.101332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stagg J, Divisekera U, McLaughlin N, Sharkey J, Pommey S, Denoyer D, Dwyer KM, Smyth MJ. Anti-CD73 antibody therapy inhibits breast tumor growth and metastasis. Proc Natl Acad Sci U S A. 2010;107:1547–1552. doi: 10.1073/pnas.0908801107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lukashev D, Ohta A, Sitkovsky M. Hypoxia-dependent anti-inflammatory pathways in protection of cancerous tissues. Cancer Metastasis Rev. 2007;26:273–279. doi: 10.1007/s10555-007-9054-2. [DOI] [PubMed] [Google Scholar]
- 12.Waickman AT, Alme A, Senaldi L, Zarek PE, Horton M, Powell JD. Enhancement of tumor immunotherapy by deletion of the A2A adenosine receptor. Cancer Immunol Immunother. 2012;61:917–926. doi: 10.1007/s00262-011-1155-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jin D, Fan J, Wang L, Thompson LF, Liu A, Daniel BJ, Shin T, Curiel TJ, Zhang B. CD73 on tumor cells impairs antitumor T-cell responses: a novel mechanism of tumor-induced immune suppression. Cancer Res. 2010;70:2245–2255. doi: 10.1158/0008-5472.CAN-09-3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Loi S, Pommey S, Haibe-Kains B, Beavis PA, Darcy PK, Smyth MJ, Stagg J. CD73 promotes anthracycline resistance and poor prognosis in triple negative breast cancer. Proc Natl Acad Sci USA. 2013;110:11091–11096. doi: 10.1073/pnas.1222251110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Raleigh JA, Dewhirst MW, Thrall DE. Measuring Tumor Hypoxia. Semin Radiat Oncol. 1996;6:37–45. doi: 10.1053/SRAO0060037. [DOI] [PubMed] [Google Scholar]
- 16.Manalo DJ, Rowan A, Lavoie T, Natarajan L, Kelly BD, Ye SQ, Garcia JG, Semenza GL. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood. 2005;105:659–669. doi: 10.1182/blood-2004-07-2958. [DOI] [PubMed] [Google Scholar]
- 17.Elvidge GP, Glenny L, Appelhoff RJ, Ratcliffe PJ, Ragoussis J, Gleadle JM. Concordant regulation of gene expression by hypoxia and 2-oxoglutarate-dependent dioxygenase inhibition: the role of HIF-1α, HIF-2α, and other pathways. J Biol Chem. 2006;281:15215–15226. doi: 10.1074/jbc.M511408200. [DOI] [PubMed] [Google Scholar]
- 18.Semenza GL. Hypoxia-inducible factor 1 and cardiovascular disease. Annu Rev Physiol. 2014;76:39–56. doi: 10.1146/annurev-physiol-021113-170322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hubbi ME, Gilkes DM, Baek JH, Semenza GL. Four-and-a-half LIM domain proteins inhibit transactivation by hypoxia-inducible factor 1. J Biol Chem. 2012;287:6139–6149. doi: 10.1074/jbc.M111.278630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shen C, Kaelin WG., Jr The VHL/HIF axis in clear cell renal carcinoma. Semin Cancer Biol. 2013;23:18–25. doi: 10.1016/j.semcancer.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chida M, Voelkel NF. Effects of acute and chronic hypoxia on rat lung cyclooxygenase. Am J Physiol. 1996;270:L872–878. doi: 10.1152/ajplung.1996.270.5.L872. [DOI] [PubMed] [Google Scholar]
- 22.Klein T, Shephard P, Kleinert H, Komhoff M. Regulation of cyclooxygenase-2 expression by cyclic AMP. Biochim Biophys Acta. 2007;1773:1605–1618. doi: 10.1016/j.bbamcr.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 23.Armstrong JM, Chen JF, Schwarzschild MA, Apasov S, Smith PT, Caldwell C, Chen P, Figler H, Sullivan G, Fink S, Linden J, Sitkovsky M. Gene dose effect reveals no Gs-coupled A2A adenosine receptor reserve in murine T-lymphocytes: studies of cells from A2A-receptor-gene- deficient mice. Biochem J. 2001;354:123–130. doi: 10.1042/0264-6021:3540123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ohta A, Sitkovsky M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature. 2001;414:916–920. doi: 10.1038/414916a. [DOI] [PubMed] [Google Scholar]
- 25.Zhang B. CD73: a novel target for cancer immunotherapy. Cancer Res. 2010;70:6407–6411. doi: 10.1158/0008-5472.CAN-10-1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Busk M, Horsman MR. Relevance of hypoxia in radiation oncology: pathophysiology, tumor biology and implications for treatment. Q J Nucl Med Mol Imaging. 2013;57:219–234. [PubMed] [Google Scholar]
- 27.Margaretten NC, Witschi H. Effects of hyperoxia on growth characteristics of metastatic murine tumors in the lung. Cancer Res. 1988;48:2779–2783. [PubMed] [Google Scholar]
- 28.Thiel M, Chouker A, Ohta A, Jackson E, Caldwell C, Smith P, Lukashev D, Bittmann I, Sitkovsky MV. Oxygenation inhibits the physiological tissue-protecting mechanism and thereby exacerbates acute inflammatory lung injury. PLoS Biol. 2005;3:e174. doi: 10.1371/journal.pbio.0030174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ren J, Mi Z, Stewart NA, Jackson EK. Identification and quantification of 2′,3′-cAMP release by the kidney. J Pharmacol Exp Ther. 2009;328:855–865. doi: 10.1124/jpet.108.146712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Longhi MS, Robson SC, Bernstein SH, Serra S, Deaglio S. Biological functions of ecto-enzymes in regulating extracellular adenosine levels in neoplastic and inflammatory disease states. J Mol Med (Berl) 2013;91:165–172. doi: 10.1007/s00109-012-0991-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang Y, Goel S, Duda DG, Fukumura D, Jain RK. Vascular normalization as an emerging strategy to enhance cancer immunotherapy. Cancer Res. 2013;73:2943–2948. doi: 10.1158/0008-5472.CAN-12-4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aggarwal NR, D'Alessio FR, Eto Y, Chau E, Avalos C, Waickman AT, Garibaldi BT, Mock JR, Files DC, Sidhaye V, Polotsky VY, Powell J, Horton M, King LS. Macrophage A2A adenosinergic receptor modulates oxygen-induced augmentation of murine lung injury. Am J Respir Cell Mol Biol. 2013;48:635–646. doi: 10.1165/rcmb.2012-0351OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nowak-Machen M, Schmelzle M, Hanidziar D, Junger W, Exley M, Otterbein L, Wu Y, Csizmadia E, Doherty G, Sitkovsky M, Robson SC. Pulmonary natural killer T cells play an essential role in mediating hyperoxic acute lung injury. Am J Respir Cell Mol Biol. 2013;48:601–609. doi: 10.1165/rcmb.2012-0180OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sitkovsky MV, Paul WE. Immunology. Global or directed exocytosis? Nature. 1988;332:306–307. doi: 10.1038/332306a0. [DOI] [PubMed] [Google Scholar]
- 35.Ohta A, Sitkovsky M. Caveats in promising therapeutic targeting of the anti-inflammatory A2 adenosine receptors: The notes of caution. Nature Reviews Drug Discovery. 2006 jpet.108.146712 [pii] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.