

Abstract
Improving cellulolytic enzyme production by plant biomass degrading fungi holds great potential in reducing costs associated with production of next-generation biofuels generated from lignocellulose. How fungi sense cellulosic materials and respond by secreting enzymes has mainly been examined by assessing function of transcriptional regulators and via transcriptional profiling. Here, we obtained global proteomic and phosphoproteomic profiles of the plant biomass degrading filamentous fungus Neurospora crassa grown on different carbon sources, i.e. sucrose, no carbon, and cellulose, by performing isobaric tags for relative and absolute quantification (iTRAQ)-based LC–MS/MS analyses. A comparison between proteomes and transcriptomes under identical carbon conditions suggests that extensive post-transcriptional regulation occurs in N. crassa in response to exposure to cellulosic material. Several hundred amino acid residues with differential phosphorylation levels on crystalline cellulose (Avicel) or carbon-free medium vs sucrose medium were identified, including phosphorylation sites in a major transcriptional activator for cellulase genes, CLR1, as well as a cellobionic acid transporter, CBT1. Mutation of phosphorylation sites on CLR1 did not have a major effect on transactivation of cellulase production, while mutation of phosphorylation sites in CBT1 increased its transporting capacity. Our data provides rich information at both the protein and phosphorylation levels of the early cellular responses to carbon starvation and cellulosic induction and aids in a greater understanding of the underlying post-transcriptional regulatory mechanisms in filamentous fungi.
Keywords: Neurospora crassa, Proteome, Phosphoproteome, Cellulase, Carbon starvation, Plant biomass
1. Introduction
Plant biomass degradation is a common strategy used by many fungi to obtain nutrients. Saprotrophic fungi have the ability to sense the presence of plant cell wall material and to produce a rich repertoire of lignocellulolytic enzymes that assists in the conversion of complex plant biomass polysaccharides into simple sugars. Humans have exploited this capacity; plant cell wall degrading enzymes are widely used in industry for commercial food processing and paper production (Kuhad et al., 2011). Lignocellulosic biofuel production that utilizes these enzymes to decompose nonfood crops holds great promise as a sustainable energy solution, but remains economically prohibitive due to the high cost associated with commercial production of the enzyme mixtures. Further improvement of fungal hosts for higher enzyme production warrants a deeper understanding of transcriptional and translational regulatory networks that mediate production of these proteins.
A great deal of effort has been made to elucidate transcriptional responses to cellulosic materials in saprotrophic fungi such as Aspergillus species, Trichoderma reesei, and Neurospora crassa, which has led to the discovery of transcriptional activators involved in the expression of lignocellulolytic enzymes (Coradetti et al., 2012; Delmas et al., 2012; Ries et al., 2013, and reviewed in Tani et al., 2014). For example, XLR1/XlnR is critical for xylanase production in N. crassa and Fusarium graminearum (Brunner et al., 2007; Sun et al., 2012a) and for both hemicellulase and cellulase production in T. reesei and Aspergillus niger (Mach-Aigner et al., 2008; Stricker et al., 2008; van Peij et al., 1998). In N. crassa, CLR1 and CLR2 are essential for cellulase production, as is a clr-2 ortholog in Aspergillus nidulans (ClrB) and Aspergillus oryzae (ManR) (Coradetti et al., 2012; Ogawa et al., 2013). However, simple manipulation of the transcript level of an individual transcriptional activator to achieve high cellulolytic enzyme production in the absence of inducers derived from plant biomass has only been successful with a single-point-mutation in xyr-1 in T. reesei and via mis-expression of clr-2 in N. crassa (Coradetti et al., 2013; Derntl et al., 2013). These data indicate that additional proteins and multifaceted post-transcriptional functions are engaged in regulation/activation of these transcription factors.
Many industrial cellulase hyper-secreting fungi were generated by classical mutagenesis, and comparative genome sequencing studies have provided genome-wide insights into mutational changes (Le Crom et al., 2009; Liu et al., 2013b; Porciuncula Jde et al., 2013). Interestingly, many of these mutations are in genes encoding proteins involved in post-transcriptional processes, suggesting they play an important role in production and secretion of plant cell wall degrading enzymes. Studies in systems ranging from bacterial, yeast and to human cells have revealed only a modest correlation between mRNA levels and protein abundance, implying regulation by mRNA stability, translational efficiency, and protein degradation that affect final protein levels and activity (Schwanhausser et al., 2011; Taniguchi et al., 2010; Vogel et al., 2010; Vogel and Marcotte, 2012). In addition, post-translational modifications, especially phosphorylation, often regulate protein function, protein turnover, protein–protein interactions as well as intracellular signal transduction (Cohen, 2000; Manning et al., 2002).
Previous quantitative proteomics-based analyses of filamentous fungi grown on cellulosic materials were limited to the secretomes or a small fraction of cellular proteins (Adav et al., 2012; Chundawat et al., 2011; de Oliveira et al., 2011; Do Vale et al., 2012; Herpoel-Gimbert et al., 2008; Liu et al., 2013a; Phillips et al., 2011). Only a few studies have reported on the regulation of the cellulolytic response by phosphorylation. For example, the DNA binding function of T. reesei CRE1, involved in carbon catabolite repression, is regulated by phosphorylation (Cziferszky et al., 2002). Reversible phosphorylation of A. oryzae XlnR in response to D-xylose has also been reported (Noguchi et al., 2011). However, a systematic comparison of proteome and phosphoproteome of cellulolytic fungi grown on different carbon sources has not been performed. Such a study may provide a rich treasure trove of information that will help to improve our understanding of fungal cellular events associated with plant biomass degradation.
To achieve this goal, here we present a global view of changes in both protein abundance and phosphorylation events in N. crassa in response to sucrose or cellulose, vs no carbon source, using isobaric peptide tags for relative and absolute quantification (iTRAQ)-based LC–MS/MS analyses. The iTRAQ method is based on covalent labeling of isobaric tags onto the N-terminal and lysine residues. While the same peptides across experimental conditions labeled with different iTRAQ reagents are indistinguishable by mass, different masses will be generated in the tandem MS by releasing the reporter ions for the 4-plex iTRAQ method. Here we show that a comparison between protein abundance and mRNA measurements reveals extensive post-transcriptional regulation in the fungal response to cellulose. We subsequently tested functional importance of identified phosphorylation sites in the transcriptional regulator CLR1 and a cellobionic acid transporter, cbt-1 (NCU05853), by mutational analyses and functional assays. Our results indicate that in N. crassa, proteome remodeling and phosphorylation in response to the presence of plant material may play an important role in rewiring metabolism for efficient plant cell wall deconstruction and utilization.
2. Research material and methods
2.1. Cell culture and media shift experiments
Culture conditions for N. crassa wild type FGSC 2489 and media shift experiments were described previously (Coradetti et al., 2012). Briefly, conidia were inoculated into 100 mL of liquid Vogel’s minimal medium (VMM) (Vogel, 1956) with 2% (w/v) sucrose at 106 conidia/mL and grown at 25 °C in constant light and shaking (200 rpm) for 16 h. Cultures were centrifuged at 2000g for 10 min and washed in VMM without a carbon source, followed by growth in 100 mL fresh VMM with either 2% sucrose, 2% Avicel® PH-101 (Sigma Aldrich, MO), or with no carbon source added.
2.2. Avicel content analyses
The hemicellulose content in Avicel were estimated by measuring the abundance of xylose and mannose in the hydrolysate of Avicel. Avicel was hydrolyzed into monosaccharides using a two-step acid hydrolysis method from the National Renewable Energy Laboratory (http://www.nrel.gov/docs/gen/fy13/42618.pdf) with small modifications. In brief, 100 mg of Avicel was suspended in 1 mL of 72% (wt/wt) sulfuric acid and incubated at 30 °C for 30 min. Deionized water (28 mL) was added to dilute the sulfuric acid to 4% (wt/wt). The bottle was sealed and autoclaved for 1 h at 121 °C. The Avicel hydrolysate was allowed to cool before calcium carbonate was added to neutralize the pH to 5–6. The supernatant was further diluted 100 fold with water and subjected to high-performance anion exchange chromatography (HPAEC) analysis. HPAEC was performed on a ICS-3000 HPLC (Thermo Fisher) using a CarboPac PA20 analytical column (3 × 150 mm) and a CarboPac PA20 guard column (3 × 30 mm) at 30 °C. Following injection of 25 μL of diluted sample, elution was performed at 0.4 mL/min using 2 mM KOH as mobile phase.
The cellodextrin and xylodextrin content in the 2% Avicel medium were analyzed after the medium was autoclaved for 15 min. 25 μL of the supernatant was directly injected to and analyzed on the same HPLC equipped with a CarboPac PA200 analytical column (3 × 150 mm) and a CarboPac PA200 guard column (3 × 30 mm) at 30 °C. The elution was performed using 0.1 M NaOH as the mobile phase at 0.4 mL/min with following sodium acetate gradients, 0 mM for 1 min, increasing to 80 mM in 8 min, increasing to 300 mM in 1 min, keeping at 300 mM for 2 min, followed by re-equilibration at 0 mM for 3 min. Carbohydrates were detected using pulsed amperometric detection (PAD) and peaks were analyzed and quantified using the Chromeleon software package. Autoclaved Avicel contains 500 ± 149 nM glucose, 41 ± 16 nM cellobiose, 84 ± 35 nM cellotriose, 71 ± 11 nM cellotetraose, 74 ± 15 nM cellopentaose, and 20 ± 5 nM cellohexaose. Xylodextrin concentration is below 10 nM.
2.3. Protein sample preparation
Mycelia were harvested at 4 h after media shift by filtering the culture through a Buchner funnel and immediately freezing mycelia in liquid nitrogen. Frozen mycelia were ground into powder using freezing-milling method using a Spex Sample Prep 6770, followed by cell lysis and protein extraction. Proteins were isolated using TRIzol Reagent (Life Technologies) according to manufacturer’s instructions; no enrichment for soluble vs membrane proteins was applied. Briefly, for 100 mg of mycelia powder, 1 mL of TRIzol and 100 μL zirconia beads were added, and the mixture was homogenized. Two hundred μL of chloroform was added to each homogenate and vortexed. After centrifugation, RNA was removed by discarding the aqueous phase, and DNA were precipitated by adding 0.3 mL of 100% ethanol to the interphase-organic phase mixture and centrifuged. Proteins remaining in the phenol–ethanol supernatant were precipitated by adding 3 volumes of acetone. Precipitated proteins were washed with 0.3 M guanidine hydrochloride (in 95% ethanol) three times, followed by a final wash with 100% ethanol. Dried protein pellets were solubilized in 6 M guanidine hydrochloride (in 50 mM NH4HCO3, pH 7.4).
2.4. Trypsin digest and iTRAQ labeling of peptides
Proteins were reduced with 5 mM dithiothreitol for 1 h at 37 °C, and subsequently alkylated with 10 mM iodoacetamide. Samples were diluted 10-fold with 25 mM NH4HCO3 pH 8.0 and digested with Promega sequencing grade modified trypsin at 1:50 enzyme-to-substrate ratio. After 3 h of digestion at 37 °C, the samples were further diluted 2-fold with 25 mM NH4HCO3 and the same amount of trypsin was added to the samples, which were incubated at room temperature overnight (~16 h). The digested samples were then acidified with 10% trifluoroacetic acid (TFA) to pH~3. Tryptic peptides were desalted on SUPELCO, Discovery C18 SPE columns and dried using a SpeedVac (SC250 Express) (ThermoSavant).
Desalted peptides were labeled with 4-plex iTRAQ reagents (AB SCIEX) according to the manufacturer’s instructions. For 1 mg peptide, 10 units of labeling reagent were used. Peptides were dissolved in 300 μL of 0.5 M TEAB pH 8.5 solution and labeling reagent was added in 700 μL of ethanol. After 1 h of incubation at room temperature, 3 mL of 0.05% TFA was added before 30 min incubation to stop the reaction and to hydrolyze the unreacted iTRAQ reagents. Differentially labeled peptides were mixed and subsequently desalted on SUPELCO, Discovery C18 SPE columns.
To reduce the potential bias caused by different iTRAQ labels, two samples prepared under the identical carbon conditions, but from the two independent experiments, were labeled with different labels, i.e., the three samples from no-carbon, sucrose, and Avicel media in experiment 1 were labeled with 114, 115, and 116 iTRAQ tags respectively, and their counterparts from experiment 2 were labeled with 116, 114, and 115 iTRAQ tags, respectively (Fig. 1). Peptides from trypsin digest of all six samples were also mixed in equal amounts to create the reference sample, labeled with 117 iTRAQ tag, and used in the two independent LC–MS/MS runs. By normalizing each individual data point to the reference data in each iTRAQ experiment, all the data across the two iTRAQ experiments can be directly compared.
Fig. 1.

Experimental design for proteomic and phosphoproteomic quantification analysis. Three cultures of FGSC 2489 were prepared in VMM with 2% sucrose and grown for 16 h, then transferred to VMM with either sucrose, Avicel, or no carbon. After 4 h growth in the new media, the cultures were harvested; proteins were extracted and digested into peptides. Two independent iTRAQ experiments were performed with two biological replicates of each condition. A reference sample was made by pooling equal amount of peptides from all six samples to allow normalization across both iTRAQ experiments. In each iTRAQ experiment, this reference sample was labeled with the same iTRAQ reagent (117 tag). Samples from three medium conditions in each biological replicates were labeled with different iTRAQ reagents (114, 115 and 116 tag) and combined with the iTRAQ labeled reference sample prior to the fractionation using the pH 7.5 RPLC. The majority (~95%) of the 12 concatenated peptide fractions was subjected to phosphopeptide enrichment by immobilized metal affinity chromatography (IMAC), followed by LC–MS/MS analysis. The rest of the 12 peptide fractions from each iTRAQ experiment was analyzed by LC–MS/MS directly.
2.5. Phosphoproteome and proteome sample preparation
Approximately 2.6 mg of 4-plex iTRAQ peptides for each experiment were separated on a Reversed Phase XBridge C18 column (Waters) (250 mm × 4.6 mm column containing 5 μM particles and a 4.6 mm × 20 mm guard column) at flow rate 0.5 mL/min using an Agilent 1200 HPLC System equipped with a quaternary pump, degasser, diode array detector, peltier-cooled autosampler, and fraction collector (set at 4 °C). Samples loaded onto the C18 column were washed for 35 min in solvent A (10 mM triethylammonium bicarbonate (TEAB), pH 7.5) for equilibration before a 112 min LC gradient. The LC gradient started with a linear increase of solvent A to 10% of solvent B (10 mMTEAB, pH 7.5, 90% MeCN) in 6 min, then linearly increased to 30% B in 86 min, 10 min to 42.5% B, 5 min to 55% B and another 5 min to 100% solvent B. A total of 96 equal volume fractions were collected into a 96 well plate through the LC gradient. The high pH RP (pH 7.5) fractions were combined into 12 fractions using the concatenation strategy reported in our previous study (Wang et al., 2011). For proteome analysis, a ~5% aliquot of each concatenated fraction was dried down and re-suspended in 0.1% TFA to a peptide concentration of 0.10 μg/μL. The rest of the concatenated fractions (95%) were subjected to immobilized metal affinity chromatography (IMAC) for phosphopeptide enrichment. Briefly, magnetic Fe3+–NTA-agarose beads were prepared using the Ni–NTA-agarose beads following the protocols reported (Ficarro et al., 2009). Peptides were reconstituted in 500 μL IMAC binding/wash buffer (80% MeCN, 0.1% TFA) and incubated for 30 min with 125 μL of the 5% pre-conditioned bead suspension. After incubation, the beads were washed 4 times each with 500 μL of wash buffer. Phosphorylated peptides were eluted from the beads using 125.0 μL of 1:1 acetonitrile/2.5% ammonia in 2 mM phosphate buffer (pH 8). Samples were acidified to pH ~ 3.5 and concentrated to 5–10 μL and subsequently reconstituted to 30 μL with 0.1% TFA for LC–MS/MS analysis.
2.6. Mass-spectrometry based analysis
All peptide samples were analyzed using an automated homebuilt constant flow nano-LC system (Agilent) coupled to an LTQ Orbitrap Velos mass spectrometer (Thermo Fisher Scientific) (Nguyen et al., 2012). Electrospray emitters were custom made using either 360 μm o.d. (phophoproteomics) or 150 μm (globals) o.d. × 20 μmi.d. chemically etched fused silica. The nano LC system for phosphoproteomics analysis has an online 4-cm × 360 μm o.d. × 150 μmi.d. C18 SPE column (5-μmJupiter C18, Phenomenex, Torrence, CA) to desalt each phosphopeptide sample (20 μL), which is connected to a home-made 60-cm × 360 μm o.d. × 50 μm i.d. capillary column (3-μm Jupiter C18, Phenomenex, Torrence, CA). Mobile phase flow rate was 100 nL/min and consisted of 0.1 M acetic acid in water (A) and 0.1 M acetic acid in 70:30 (v/v) acetonitrile: water (B) with a gradient profile as follows (min:%B); 0:0, 5:10, 140:35, 160:60, 165:90, 170:90. For global proteome analysis, an on-line 4-cm × 360 μm o.d. × 150 μm i.d. SPE column packed with 3.6-μm Aeris Widepore XB-C18 and a 35-cm × 360 μm o.d. × 75 μm i.d. fused-silica capillary analytical column (3 μm Jupiter C18) were used. Mobile phases consisted of 0.1% formic acid in water (A) and 0.1% formic acid acetonitrile (B) operated at 300 nL/min with a gradient profile as follows (min:%B); 0:5, 2:8, 20:12, 75:35, 97:60, 100:85.
The LTQ Orbitrap Velos mass spectrometer was operated in the data-dependent mode acquiring higher-energy collisional dissociation (HCD) scans (R = 7500, 5 × 104 target ions) after each full MS scan (R = 30,000, 3 × 106 target ions) for the top ten most abundant ions within the mass range of 300–1800 m/z. A mass isolation width of 2.5 Thomson was used to isolate ions prior to HCD. All HCD scans used a normalized collision energy of 45 and a maximum inject time of 1000 ms. The dynamic exclusion time was set to 60 s and charge state screening was enabled to reject unassigned and singly charged ions.
2.7. Peptide identification and quantification
For peptide identification MS/MS spectra were searched against a decoy Neurospora protein sequence database using SEQUEST (Eng et al., 1994). Search parameters included: no enzyme specificity for proteome data and trypsin enzyme specificity for phosphoproteome data with a maximum of two missed cleavages, ±50 ppm precursor mass tolerance, ±0.05 Da product mass tolerance, and carbamidomethylation of cysteines and iTRAQ labeling of lysines and peptide N-termini as fixed modifications. Allowed variable modifications were oxidation of methionine residue for proteome data and phosphorylation of serine, threonine or tyrosine residues for phosphoproteome data. MSGF spectra probability value was also calculated for peptides identified from SEQUEST search. Measured mass accuracy and MSGF spectra probability were used to filter identified peptides to <0.4% false discovery rate (FDR) at spectrum level and <1% FDR at peptide level using the decoy approach. iTRAQ reporter ions were extracted using the MASIC software (Monroe et al., 2008) within 10 ppm mass tolerance of each expected iTRAQ reporter ion from each MS/MS spectrum. The median ratios of all non-phosphorylated peptides from proteome study were used to normalize the ratios of all phosphorylated peptides. The sum of the individual iTRAQ reporter ion values from all MS/MS spectra for a given peptide/protein was used for calculating their relative abundance across different conditions.
2.8. Data analysis
Individual data points in each 4-plex iTRAQ experiment were normalized using the data of the pooled sample as the reference. To correct any systematic error due to pipetting, data were further normalized by the median of iTRAQ reporter ion of the individual sample. Fold change of each protein or phosphopeptide was calculated by dividing the data points from the two different carbon source conditions and transformed into Log2 scale. Protein or phosphopeptide with valid data from both replicates were subjected for downstream analysis. Only data with changes exceeding 2-fold (Log2 change≥1 or Log2 ≤ –1) in both biological replicates were considered differential. To compare mRNA and protein abundance changes, transcripts in FPKM (fragments per kilobase of exon per million fragments mapped) (Coradetti et al., 2012) were averaged from three biological replicates and transformed into ratios using the data from two different carbon sources. Statistical analyses and data plotting were performed in R software (http://www.rproject.org/). Functional category enrichment analysis was carried out through the Munich Information Center for Protein Sequence (MIPS) database (http://mips.helmholtz-muenchen.de/proj/funcatDB/) (Ruepp et al., 2004).
2.9. N. crassa strain construction and enzyme activity assays
The clr-1 (NCU07705) constitutive over-expression mutant strains were constructed by transforming a Δclr-1; his-3 mat A mutant with linear fragments of pMF272 containing the WT and mutant clr-1 coding sequences inserted under regulation of the ccg-1 promoter as in (Coradetti et al., 2013). Transformants were selected for histidine prototrophy (Margolin et al., 1997). Homokaryotic strains were obtained by backcrossing transformants with FGSC11029 (Δclr-1 mat a).
N. crassa strains were grown inVMMplus 2% sucrose for 16 h and the cultures were shifted to VMMplus 2% Avicel as described previously (Coradetti et al., 2013). Five μL of culture supernatant was taken at 8, 24, 48, and 72 h after media shift and assayed with Azo-CarboxyMethyl Cellulose (Azo-CMC; Megazyme) in a 200 μL reaction. Enzyme activity was measured by quantification of the released short-chain dye-conjugated oligomers from the long-chain substrates with a UV spectrophotometer at 590 nm wavelength.
2.10. Homology modeling
Homology model of CBT1 (NCU05853) was built by Modeller v9.12 (Eswar et al., 2007) using a xylose transporter XylE structure; PDB code 4GBZ (Sun et al., 2012b) as the template. Protein structure figure was prepared with the molecular visualization software PyMol (http://www.pymol.org).
2.11. Saccharomyces cerevisiae strain construction
A 2μm yeast plasmid expressing cbt-1 was previously described (Galazka et al., 2010). In this plasmid, the open reading frame of cbt-1 was fused with a GFP sequence at C-terminus and flanked with the S. cerevisiae PGK1 promoter and CYC transcriptional terminator in the pRS426 backbone. Point mutations of cbt-1 were introduced with the QuikChange mutagenesis kit (Agilent Technologies) and confirmed by sequencing. S. cerevisiae D452-2 (MATa leu2 his3 ura3 can1) (Kurtzman, 1994) was used as the recipient strain for the expression of cbt-1 and its mutants. Transformants were selected for uracil prototrophy.
2.12. Cellobionic acid uptake assays
S. cerevisiae expressing either the empty vector, WT, or mutated cbt-1 were grown in liquid synthetic complete medium lacking uracil until its growth enters the late log phase. Cells were harvested and washed three times with the assay buffer (5 mM MES, 100 mM NaCl, pH 6.0) and re-suspended to final OD of 20. The fluorescence intensity of GFP fusions and the optical density of the cell populations were measured by scanning the cultures at wavelengths of 485 nm and 600 nm, respectively, using Spectramax M2 plate reader. Cellobionic acid stock was prepared in the same assay buffer with a concentration of 200 μM cellobionic acid. Transport reactions were initiated by mixing equal volumes of cell suspension and substrate stock. Pilot experiments using three time points (5, 10 and 30 min) indicated a linear uptake rate in WT; reactions with both WT and mutants were performed at 30 °C with continuous shaking for 10 min. Cells were pelleted by centrifuging at 20,800g at 4 °C for 5 min and 400 μL of supernatant from each sample was transferred to an HPLC vial containing 100 μL 0.5 M NaOH. The concentration of remaining cellobionic acid was measured on a ICS-3000 HPLC (Dionex Corporation) using a CarboPac PA200 Analytical Column (3 × 150 mm) and a CarboPac PA200 guard column (3 × 30 mm) at 30 °C. Following injection of 25 μL of diluted samples, isocratic elution was performed at 0.4 mL/min with amobile phase containing 0.1 M NaOH and 60 mM sodium acetate. Cellobionic acid was eluted at 5.6 min and detected with pulsed amperometric detector. Peaks were analyzed using the Chromeleon software package (Thermo Scientific).
2.13. Fluorescence microscopy
S. cerevisiae cells were prepared for microscopy as described in cellobionic acid uptake assays (above). Confocal microscopy was performed on a Leica SD6000 microscope equipped with a Yokogawa CSU-X1 spinning disk head and 488-nm lasers controlled by Metamorph software (Molecular Devices). All images were acquired using a 100 × 1.4 NA oil immersion objective and the same imaging setting parameters.
3. Results and discussion
3.1. Quantitative analysis of the proteome of N. crassa following exposure to different carbon sources
To analyze changes in protein abundance and protein phosphorylation in N. crassa in the early response to cellulosic material, we used wild type strain FGSC 2489 and compared the global proteome and phosphoproteome of cells following exposure to sucrose, Avicel (crystalline cellulose) or no-carbon medium (Fig. 1). Strains were grown for 16 h in Vogel’s minimal medium (VMM) with 2% sucrose before being washed and re-suspended in fresh Vogel’s MM containing either 2% sucrose, 2% Avicel, or lacking any carbon sources. Proteins were harvested 4 h after transfer; this time point was chosen because substantial transcriptional induction of cellulase genes occurs on Avicel vs carbon-free media (Coradetti et al., 2012), allowing comprehensive comparison of datasets obtained by different methods. It is likely that early signal transduction events mediated by phosphorylation occur during this period. To compare the relative abundance of proteins and occupancy of phosphorylated residues of samples from different conditions, we used an iTRAQ labeling and liquid chromatography tandem mass spectrometry (LC-MS/MS) approach (see Material and methods). Two biological replicates for each carbon condition were prepared from two independent experiments (Fig. 1).
Relative quantification of protein abundance between two different media, i.e, no carbon vs sucrose (NC vs Suc) and Avicel vs no carbon (Avi vs NC) conditions, was performed for each replicate. In the global proteome analysis, we identified 79749 and 76498 peptides from replicate 1 and replicate 2, respectively, and they mapped to 3891 and 3862 proteins. Of these proteins, 3459 were identified in both replicates, representing 35% of proteins predicted in the N. crassa genome (Table S1). Eight hundred and fifty-seven of the 3459 proteins (25%) are annotated as hypothetical proteins, whose biological function is unknown. By using TMHMM prediction method (http://www.cbs.dtu.dk/services/TMHMM-2.0/) (Krogh et al., 2001), 1706 N. crassa genes were predicted to encode integral membrane proteins with transmembrane helices existing after the first 60 amino acids, representing 17% of the genome. Although our protein sample preparation was not tailored specifically for membrane protein extraction, 416 (24%) of the predicted integral membrane proteins were detected (Table S1). The protein abundance changes between the two replicates were well correlated (rp = 0.63 with 95% CI: 0.61–0.65 and p < 0.01 for fold changes on NC vs Suc in Log2 scale, and rp = 0.65 with 95% CI: 0.63–0.67 and p < 0.01 for fold changes on Avi vs NC in Log2 scale) (Fig. 2A and B).
Fig. 2.

Protein quantification by mass spectrometry and functional category analysis of differentially abundant proteins. (A) Fold changes in protein abundance for no carbon (NC) vs sucrose (Suc) duplicates. (B) Fold changes in protein abundance for Avicel (Avi) vs no carbon (NC) duplicates. (A and B) Proteins with ≥2-fold changes were considered differentially abundant and labeled in orange for increased abundance and in blue for decreased abundance. Proteins with less than 2-fold changes were labeled in black. (C) Functional category (Ruepp et al., 2004) analysis of differentially abundant proteins. Pie charts present the percentages of the differentially abundant proteins. (D) Table shows enrichment of differentially abundant proteins in functional categories.
We focused on proteins that showed a ≥2-fold change in abundance in both replicates, which we refer to as “differentially abundant proteins”. Our relative quantification analysis revealed that 139 proteins increased and 31 proteins decreased in abundance on NC vs Suc (Fig. 2A and B). Functional category analysis (FunCat) (Ruepp et al., 2004) was performed on these differentially abundant proteins (Cutoff: P-value < 1e–03) (Fig. 2C). About one third of the differentially abundant proteins on NC vs Suc fell into the functional category of metabolism. Proteins with increased abundance in response to carbon starvation were over-represented in functional categories of amino acid metabolism, nitrogen, sulfur and selenium metabolism and lipid, fatty acid and isoprenoid metabolism. Additional functional categories overrepresented included glycolysis and gluconeogenesis, tricarboxylic-acid pathway, and oxidation of fatty acids. Genes encoding some of these proteins are induced at transcriptional level upon glucose starvation in N. crassa (Xie et al., 2004). Proteins showing decreased abundance in NC vs Suc were enriched in the functional categories of nucleotide-sugar metabolism and metabolism of vitamin, cofactors, and prosthetic group.
Of the 139 proteins that showed greater abundance on NC vs Suc, there were five predicted sugar transporters, a glucoamylase precursor (NCU01517), clock controlled protein CCG1 (NCU03753), NmrA family transcriptional regulator (NCU09169), an extracellular β-glucosidase (NCU08755) and cellodextrin transporter 1 (CDT1/NCU00801). Genes encoding the high affinity glucose transporter/NCU10021, CCG1 and the glucoamylase precursor were all previously reported to be glucose-repressible genes (Ebbole, 1998; McNally and Free, 1988; Xie et al., 2004). CDT1 has been experimentally verified as a high affinity cellodextrin transporter (Galazka et al., 2010) and may be involved in signaling during cellulolytic induction (Znameroski et al., 2014). An ortholog of the NmrA family of transcriptional regulators (Nmr1-3) from M. oryzae modulates carbon catabolite repression (CCR) (Fernandez et al., 2012). Whether an increase in the N. crassa NmrA family transcriptional regulator (NCU09169) protein abundance promotes CCR de-repression in N. crassa will be an interesting avenue to pursue in future studies.
In the Avi vs NC comparison, 37 proteins increased in abundance and 210 proteins decreased in abundance. Proteins with increased abundance were enriched in the functional categories of polysaccharide metabolism, and sugar, glucoside, polyol and carboxylate metabolism, C-compound and carbohydrate transport, and non-vesicular cellular import (Fig. 2C and D). These data are consistent with cellular responses to cellulolytic induction based on transcriptional profiling data (Coradetti et al., 2012; Znameroski et al., 2012). However, in the protein set showing decreased abundance on Avi vs NC, we observed an overrepresentation of proteins involved in transcription and protein synthesis. In particular, proteins predicted to function in DNA conformation modification, RNA synthesis, RNA processing, and translation initiation were overrepresented (Fig. 2C and D). These data suggest a general global repression in both transcription and translational events as a cellular response to cellulolytic induction, which were not revealed by the previous transcriptome analysis (Coradetti et al., 2012).
Of the 37 proteins showing increased abundance on Avi vs NC, 12 were carbohydrate-active-enzymes (CAZymes), including CBHI, CBHII, and EGII (GH6-3). Three of these proteins were intracellular enzymes (β-glucosidase/NCU00130, a β-xylosidase/NCU01900, and a β-mannosidase/NCU00890) (Table S1). These data are consistent with the notion that, in the early cellulolytic response, short oligo-saccharides transported into cytosol are further broken down by intracellular enzymes, which may play a role in modulating signal transduction responses to plant biomass components. In addition to the CAZymes, two cellodextrin transporters (CDT1/NCU00801 and CDT2/NCU08114), both of which may also be involved in early signaling events (Znameroski et al., 2014), one predicted MFS sugar transporter/NCU05853, a translocation protein SEC62/NCU06333, intracelluar proteins involved in xylose degradation (a xylose reductase/NCU08384, a xylitol dehydrogenase/NCU00891, and a-D-xylulose kinase/NCU11353) also increased in abundance under Avicel conditions compared to NC (Table S1), in addition to ten proteins of unknown function. Our previous transcriptional studies showed that induction of xylose utilization genes is correlated with exposure to Avicel (Coradetti et al., 2012), and we detected ~2.6% xylose and 1.2% mannose in Avicel hydrolysate (see Material and methods). The induction of xylose utilization genes is absent when cellobiose is used as an inducer (Znameroski et al., 2012). Thus, the detection of xylose utilization proteins in this study is likely due to residual xylan in Avicel.
The MFS sugar transporter/NCU05853 (cbt-1) transports aldonic acids, products from reactions of polysaccharide monooxygenases (PMO) on crystalline cellulose or cellobiose dehydrogenases (CDH) on cellodextrin; N. crassa is able to utilize aldonic acid as a carbon source (Li, X., unpublished results). Both PMO1 and PMO2 showed increased abundance in Avi vs NC conditions. Although a strain carrying a deletion of cbt-1 exhibits no growth defect on Avicel (Znameroski et al., 2012) (Li, X., unpublished results), the early increase in protein abundance of both CBT1 and PMO1/PMO2 implies a preferred strategy of degrading crystalline cellulose by N. crassa.
Proteins destined for secretion enter the endoplasmic reticulum (ER) by two routes, signal recognition particle (SRP)-dependent co-translational translocation and SRP-independent posttranslational translocation (Rapoport, 2007); SRP-independent post-translational translocation requires the Sec62/Sec63 complex. In the Avi vs NC comparison, we detected an increase of SEC62 (NCU06333), but a decrease in abundance of the DnaJ subunit, SEC63 (NCU00169). Additionally, several other proteins involved in protein sorting and translocation, including COPII coat assembly protein SEC16 (NCU03819), glucosidase II subunit beta (NCU05606), calnexin (NCU09265) and an additional DnaJ domain-containing protein (NCU02490) also decreased in abundance. As both calnexin and glucosidase II assist in proper folding of glycoproteins in the ER (D’Alessio et al., 1999; Song et al., 2001), and DnaJ/Hsp40 family proteins are required to prevent the unfolded protein response (UPR) (Fama et al., 2007), a decrease of these proteins suggests a reduced folding capacity of the ER. In many organisms, transcriptional activation of UPR target genes occurs via a process mediated by IRE1-dependent hac-1 mRNA splicing (reviewed in Walter and Ron, 2011). In fact, increased hac-1 splicing and transcriptional expression of UPR-associated genes such as BiP and PDI were observed when N. crassa was exposed to Avicel (Benz et al., 2014a). The cause and effect relationship between proteins involved in secretion and the UPR under cellulosic conditions in filamentous fungi warrants further investigation.
Among the differentially abundant proteins, NCU00801/CDT1, a cellodextrin transporter, is the only protein that showed an increase in abundance in both NC vs Suc and Avi vs NC comparisons. NCU07503, encoding a hypothetical protein, was the only one that decreased in abundance in both comparisons. Thirteen proteins showed increased abundance on NC vs Suc, but decreased abundance on Avi vs NC, and most of them are hypothetical proteins.
3.2. Proteome and transcriptome changes in response to different carbon sources
Using mRNA expression level as a proxy for the abundance and activities of the corresponding proteins has been common practice, however, large-scale surveys of both transcriptome and proteome in bacteria, yeast, and mammals showed only modest correlations between transcript and protein expression (reviewed in Maier et al., 2009; Schwanhausser et al., 2011; Vogel and Marcotte, 2012). We therefore compared protein abundance data obtained in this study with published mRNA transcript abundance data obtained under identical conditions (Coradetti et al., 2012) (Table S2). In a comparison between NC vs Suc conditions, the protein and mRNA expression changes of 3391 genes correlated with R2 of 0.43, indicating that ~43% of the variation in protein abundance can be explained by the change in mRNA abundance (Fig. 3A). Previous genome-wide measurements of both mRNAs and protein abundance by either absolute quantification or relative quantification showed a similar correlation, in range of r = 0.25–0.6 in bacteria, yeast or animals (de Godoy et al., 2008; Lundberg et al., 2010; Maier et al., 2009; Schwanhausser et al., 2011; Vogel and Marcotte, 2012). These data suggest that, in addition to the potential measurement noise, post-transcriptional, translational and protein degradation regulation contribute at least as much as transcription to protein abundance in N. crassa and mostly likely, in other filamentous fungi. However, mRNA changes on Avi vs NC correlated poorly with the protein changes (R2 = 0.07) (Fig. 3B), especially in proteins that showed reduced abundance. There was no correlation between mRNA levels and the 210 proteins showing reduced abundance on Avi vs NC (rp = 0.00) in contrast to the mRNA and protein levels of the 37 proteins that showed increased abundance (rp = 0.57) (Fig. 3B). As mentioned previously, this 210-protein set consists of many proteins functioning in RNA synthesis and protein synthesis (Fig. 2D), including eIF3a and eIF3c, which were verified as two essential translation initiation factors in N. crassa (Smith et al., 2013). Such a high discordance between mRNA and protein level has previously been observed in S. cerevisiae following responses to perturbation to different stimuli (Fournier et al., 2010; Lee et al., 2011; Vogel et al., 2011). These data suggest that exposure to Avicel may trigger a global reduction in transcription and translational capacities, but with targeted transcriptional and post-transcriptional regulation to support production of plant cell wall degrading enzymes. Further experiments with time series of measurements would be helpful in monitoring dynamics between transcriptional and post-transcriptional regulation during cellulase production.
Fig. 3.
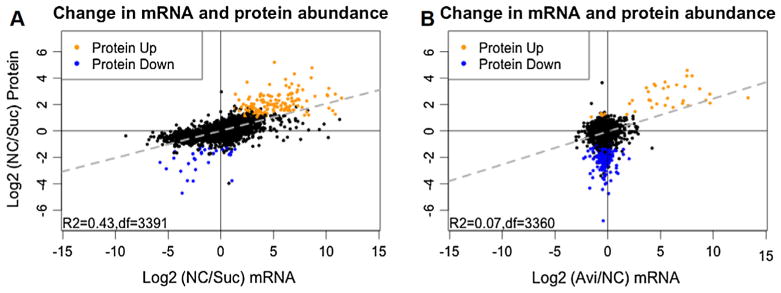
Correlation between mRNA level change and protein abundance change. Scatter plot of mRNA change vs protein abundance change of (A) 3391 genes in no carbon (NC) vs sucrose (Suc) comparison and (B) 3360 genes in Avicel (Avi) vs no carbon (NC) comparison, with ≥2-fold increase in protein abundance (orange) and ≥2-fold decrease in protein abundance (blue). Genes with protein abundance change less than 2 folds were labeled in black. Linear regression (dashed line in gray) shows that mRNA and protein changes on NC vs Suc correlate better than those on Avi vs NC (R2 = 0.43 vs R2 = 0.07).
3.3. Quantitative analysis of the phosphoproteome of N. crassa following exposure to different carbon sources
An analysis of the N. crassa phosphoproteome at early time-points may unveil signaling events associated with cellulolytic induction that may be difficult to detect at later stages of utilization. To uncover phosphorylation events associated with early cellular responses to different carbon sources, we performed phosphoproteome profiling on the same samples used for proteome experiments. We identified 8814 (from 2120 proteins) and 12216 phosphopeptides (from 2473 proteins) from replicate 1 and replicate 2, respectively, with 5882 of them overlapping and mapping to 1942 proteins (Table S3). The phosphopeptide abundance changes correlated well between the two replicates (rp = 0.65 with 95% CI: 0.61–0.64 for fold changes on NC vs Suc in Log2 scale, and rp = 0.66 with 95% CI: 0.64–0.67 for fold changes on Avi vs NC in Log2 scale) (Fig. S1A; Fig. 4A and B). Phosphopeptide abundance changes less than 2-fold were filtered and the remaining peptides were subjected to downstream analysis. Of these phosphopeptides, 295 (representing 143 proteins) increased in abundance and 291 (representing 186 proteins) decreased in abundance in the NC vs Suc comparison (Fig. S1A). There were 423 phosphopeptides (representing 257 proteins) with increased abundance on Avi vs NC and 999 phosphopeptides (representing 404 proteins) that showed decreased abundance (Fig. S1B).
Fig. 4.

Fold changes of phosphopeptides after normalization with protein abundance changes and functional category analysis of differential phosphorylated proteins. (A) Data from duplicate samples for fold changes in phosphopeptide abundance for no carbon (NC) vs sucrose (Suc) and (B) Avicel (Avi) vs no carbon (NC) were well correlated (rp = 0.47 and rp = 0.37 respectively). (A and B) Phosphopeptides with ≥2-fold changes were considered differentially phosphorylated and labeled in orange for increased abundance vs in blue for decreased abundance. Phosphopeptides with less than 2-fold changes were labeled in black. (C) FunCat (Ruepp et al., 2004) analysis of proteins with differential phosphorylation levels. Pie charts show percentages of phosphoproteins in each functional category. (D) Enrichment of phosphoproteins in particular functional categories.
Because the change in phosphopeptide abundance could be merely a result of changes in protein abundance, we normalized our phosphoproteome data with the proteome data. This step unavoidably filtered out some 835 and 953 low abundant phosphoproteins from the 2120 identified phosphpoproteins from replicate 1 and the 2473 identified phosphoproteins from replicate 2 respectively, due to their absence in the proteome data set, but presence in phosphoproteome data set, a result from the enrichment step for phosphopeptides. Such filtered-out phosphoproteins could play important roles in cellulolytic responses and included two transcription factors, one essential for induction of cellulases and cellulose deconstruction (CLR1) (Coradetti et al., 2012) and one essential for induction of hemicellulases and xylan deconstruction (XLR1) (Sun et al., 2012a). Four phosphopeptides from CLR1 showed Ser108 and Thr120 phosphorylation increased in abundance on Avi vs NC. One phosphopeptide from XLR1 showed Ser182 phosphorylation increased in abundance on NC vs Suc.
After correction for protein level changes, we filtered the data by setting ≥2-fold change as the cutoff, which resulted in 135 phosphopeptides from 79 proteins that showed increased abundance and 160 phosphopeptides from 108 proteins that showed decreased abundance on NC vs Suc. In the Avi vs NC comparison, 432 phosphopeptides from 243 proteins showed increased abundance, while 108 phosphopeptides from 77 proteins decreased (Fig. 4A and B; Table S4). Although most differentially phosphorylated proteins had only one phosphopeptide pass the 2-fold change criterion, some proteins were heavily phosphorylated in one condition vs the other. Many of these heavily phospho-modified proteins are hypothetical proteins with unknown functions (Table S4). Ten proteins were found to be highly phosphorylated at one or some regions but de-phosphorylated at other regions on NC vs. Suc. This protein set included orotidine-5′-phosphate decarboxylase, PYR4 (NCU03488) and eukaryotic peptide chain release factor ERF2 (NCU04790). Eighteen such proteins appeared in the comparison between Avi vs NC, and included the S/T protein kinase STK46 (NCU06638), an actin cytoskeleton-regulatory complex protein (NCU06171), a 60S ribosomal protein L13 (NCU05554), and a myosin type-2 heavy chain 2 (NCU00551).
Functional category analysis was performed for the four sets of differentially phosphorylated proteins. Proteins highly phosphorylated on NC vs Suc were slightly over-represented in the category of non-vesicular ER transport and cell fate. Proteins highly dephosphorylated under the same conditions were over-represented in the categories of phosphate metabolism and cAMP/cGMP mediated signal transduction (Fig. 4C and D). Proteins highly phosphorylated in response to Avicel were enriched in phosphate metabolism, cell cycle, transcriptional control, translation initiation, regulation of protein activity, cell growth/morphogenesis, cAMP/cGMP mediated signal transduction, cell aging, and actin cytoskeleton. Proteins showing a decrease in phosphorylation on Avicel vs NC were only enriched in RNA binding (Fig. 4D). Proteins with functional property of RNA binding appearing in both up and down datasets suggests that proteins in this functional category are targets of differential phosphorylation modification in response to Avicel.
When comparing Avi vs NC, we found that the Ser233 of NCU11187/signal recognizing particle (SRP)-receptor alpha subunit, and Ser16 of NCU00621/TORC1 growth control complex subunit Kog1 both showed hyper-phosphorylation. In S. cerervisiae, a homolog of NCU11187 is SRP101 and is involved in protein targeting to the ER (Ogg et al., 1998). In S. cerevisiae, Kog1 is an essential subunit of TOR complex 1 (TORC1). TORC1 plays an important role in mediating cell growth in response to nutrient availability and cell stress (Inoki et al., 2005; Loewith et al., 2002); an increase in cellulase production in N. crassa after inhibiting TOR pathway using rapamycin has been reported (Xiong Y. et al., submitted). Although both phosphoproteins are quite conserved in primary amino acid sequences with their homologs from other fungal species, the phosphorylated amino acids identified in this study were only conserved among filamentous fungi (Fig. S2).
3.4. Phosphorylation of CBT1 (NCU05853) regulates transporting capability
Unlike S. cerevisiae, N. crassa is able to utilize cellobionic acids released from cellulose degradation as a carbon source. In response to Avicel, CBT1 (NCU05853) showed increased mRNA and protein abundance levels. In addition, we detected an increase in phosphorylation at Ser524 in the comparison of NC vs Suc and at Thr525 in the comparison of Avi vs NC. CBT1 is a member of the major facilitator superfamily, containing 12 transmembrane helices and a cytoplasmic domain. Homologs of CBT1 can be found in many filamentous fungi, but the conservation of these two sites (Ser524 and Thr525) is limited to fungi that are closely related to N. crassa (Fig. S3). Nonetheless, protein structure modeling of CBT1 based on xylose transporter XylE structure (Sun et al., 2012b) revealed an intriguing localization of the two phosphorylated amino acids to the intracellular C-terminus of the transporter. The cytoplasmic domain is mainly composed of a 68-residue insertion (P219-K286) between TM-6 and TM-7, and a 62-residue C-terminal loop (E480-P541). A large number of acidic (24 glutamate or aspartate) and basic (18 lysine or arginine) residues are found on the cytoplasmic domain, indicating extensive ionic interactions. The phosphorylated Ser524 and Thr525 residues are located between two short intracellular helices at the C-terminus (Fig. 5A). Notably, a pair of arginine residues is adjacent to this double phosphorylation site. As one of predictable changes resulting from phosphorylation is an alteration in the charge of this motif, we tested through phosphosite amino acid substitution if a charge swap would turn the transporter to a more activated state. We generated mutated versions of CBT1 and compared their cellobionic acid transporting efficiency to WT. As N. crassa has redundant pathways to consume cellobionic acids, we expressed CBT1 in S. cerevisiae, which lacks cellobionic transport and utilization capacities. Four mutated cbt-1 alleles were created: (1) Ser524 and Thr525 to alanine (RRAA); (2) Ser524 and Thr525 to glutamate (RREE); (3) Arg522 and Arg523 to glutamates (EEST); and (4) Arg522, Arg523, Ser524, and Thr525 to glutamate (EEEE); only EEEE would change the net charge to −4. GFP fusions of the wild type and the mutated CBT1 transporters were expressed under control of the PDK1 promoter. WT and mutant CBT1 proteins, except EEST, localized to the plasma membrane as expected (Fig. 5B). Transformants bearing EEST-GFP did not express the fusion protein well and lacked proper localization, and were therefore excluded from downstream analysis. Expression levels of GFP fusion proteins of different CBT1 variants were assessed by measurements of both GFP fluorescence intensity and optical density of cell populations. Strains expressing mutated CBT1 showed similar ratios of fluorescence intensity to optical density as the strain expressing WT CBT1 (Fig. S4), indicating similar protein abundance for WT and mutant CBT1 transporters. Cellobionic acid uptake efficiency was measured by mixing an equal amount of cells with 100 μMof cellobionic acid and monitoring cellobionic acid remaining in the supernatant. The transporting efficiency was further normalized by fluorescence intensity. Within 10 min, the EEEE mutant significantly exceeded WT in reducing cellobionic acid in the solution (p = 9e–04, independent t-test), while both RREE and RRAA were not statistically significantly different from WT (Fig. 5C). These data are consistent with our hypothesis that a charge inversion caused by the phosphorylation at Ser524 and Thr525 may reform ionic interactions and which leads to a more active conformation. However, the impact of phosphorylation of CBT1 might not be fully replicated by amino acid substitution, and the physiological influence and the detailed mechanism merits further investigation, particularly differences in transporter function in N. crassa where transport is coupled with intracellular cellobionic acid metabolism vs S. cerevisiae strains that bear various cbt-1 alleles, but are only capable of transport.
Fig. 5.

Mutational analysis of Ser525 and Thr525 of the cellobionic acid transporter, CBT1 (NCU05853). (A) Homology model of CBT1 based on the crystal structure of a xylose transporter (Sun et al., 2012b) showing the location of the “RRST” motif. Transmembrane domains are colored in red, helices from the intracellular domain are in magenta and cyan, and loops in green. Side chains of the “RRST” motif are shown as sticks. (B) Ectopically expressed WT and mutated CBT1 proteins localize to the plasma membrane in S. cerevisiae cells. Scale bar = 4 μm. (C) Cellobionic acid transport capacity of S. cerevisiae strains bearing WT and CBT1 variants were compared by measuring reduction of cellobionic acid in the medium after 10 min. Error bars represent standard deviation and are not visible when smaller than the column outline thickness.
3.5. An essential function of CLR1 in cellulase production does not require phosphorylation at Ser108 and Thr120
CLR1 is a transcription factor essential for expression of clr-2 and subsequent cellulase gene expression (Coradetti et al., 2012). Mis-expression of clr-2 induces cellulase gene expression and secretion in the absence of inducers derived from plant biomass (Coradetti et al., 2013), but constitutive over-expression of clr-1 does not. These data suggest that post-translational modification after cellulolytic induction may be required for activation of CLR1. We therefore tested whether the identified CLR1 phosphopeptides Ser108 and Thr120, which showed higher abundance on Avi vs NC (Table S3), played a role in activation of CLR1. These two amino acids are highly conserved among homologs of CLR1 in many filamentous fungi, but less so among the Aspergilli (Fig. S3B). Although CLR2/ClrB function for cellulase gene expression/secretion is conserved in both N. crassa and A. nidulans, CLR1/ClrA function is not; deletion of clrA only mildly affected cellulase production in A. nidulans (Coradetti et al., 2012). We created three Δclr-1 N. crassa strains, each expressing a mutated version of CLR1 with V5 and histidine tags fused to the C-terminus and under the regulation of the ccg-1 promoter: (1) Pc1-STVH is wild type CLR1; (2) Pc1-AAVH has both Ser108 and Thr120 mutated into alanine (A); (3) Pc1-DEVH has Ser108 mutated into a aspartate (D) and Thr120 mutated into a glutamate (E). We first evaluated WT and strains carrying the constitutively expressed WT clr-1 and clr-1 mutant alleles for growth on Avicel. Although the Δclr-1 strain is unable to grow on Avicel (Coradetti et al., 2012), the two strains carrying mutant clr-1 alleles (Pc1-AAVH and Pc1-DEVH) grew indistinguishably from wild type FGSC 2489 or the Pc1-STVH (wild type clr-1) strains on Avicel (data not shown). Consistent with the growth phenotype on Avicel, no substantial difference in cellulase activity was observed among the Pc1-STVH, Pc1-AAVH, and Pc1-DEVH strains (Fig. 6); a Δclr-1 strain showed no enzyme activity. These data indicate that phosphorylation at Ser108 and Thr120 of CLR1 is dispensable for the essential conversion of CLR1 to an active state and subsequent induction of cellulase gene expression and activity. Phosphorylation of transcription activators has often been observed concomitantly upon variation in nutrient inductions; however, perturbation of the phosphorylation events do not always prevent activation of transcription. For example, phosphorylation of GAL4 at Ser837 was correlated with activity of GAL4, but substitution of the residue for alanine has no detectable effect on transcriptional activation (Sadowski et al., 1991). Although the phosphorylation sites identified in this study are not sufficient to activate CLR1, it is possible that phosphorylation of CLR1 is a consequence of activation or phosphorylation may be involved in modulation of CLR1 activity. However, assays to distinguish the role of phosphorylation in the modulation of CLR1 activity would require either in vitro assays or the construction of reporter strains that have higher sensitivity to discern modulation of activation.
Fig. 6.

Enzyme activity measurements of culture supernatants from N. crassa strains bearing CLR1 (NCU07705) variants. N. crassa WT strain (FGSC 2489), Δclr-1, and mutants constitutively expressing either a WT copy of clr-1 (Pc1-STVH) or mutated clr-1 alleles (Pc1-AAVH or Pc1-DEVH) were pre-grown on VMM with 2% sucrose for 16 h, and subsequently transferred to VMM with 2% Avicel. Culture supernatants were taken at 8, 24, and 48 h after medium shift and assayed for cellulase activity using Azo-CMcellulose as a substrate. Enzyme activity data are expressed as arbitrary unit (AU).
4. Conclusions
In this study, we evaluated the proteome and the phosphoproteome when N. crassa is exposed to sucrose, vs carbon starvation or Avicel. We chose these conditions to enable comparisons with RNA-seq data that focuses on both this time point and on these carbon sources. We identified a large number of proteins (~3460) within the N. crassa proteome, many of which had not previously been identified and are currently annotated as “hypothetical proteins” (~25% of the proteome). Of the 3459 proteins identified in both replicate samples, only a small proportion changed in abundance when N. crassa was switched from sucrose to either carbon starvation or Avicel. The differential proteome on Avicel vs. carbon starvation also revealed that a subset of CAZymes increased in abundance, presumably to initiate cellulose degradation and signaling. In this case, mRNA induction was mirrored by an elevation in protein abundance. In contrast, a great portion of proteins with reduced abundance on Avicel had uncorrelated mRNA changes, suggesting post-transcriptional regulation of protein abundance in response to exposure to different carbon sources.
In addition to protein abundance changes associated with carbon source, we also evaluated phosphorylation changes. This effort resulted in the identification of a large number of phosphopeptides and which mapped to 1942 proteins (Table S3). Interestingly, in contrast to the proteome dataset, a considerable number of identified phosphoproteins showed differences in phosphorylation between the three different carbon conditions, particularly in the Avi vs NC comparison. Our data revealed phosphorylation events on proteins that are filamentous fungal specific, and phosphorylation events on non-conserved residues of conserved proteins, both of which could be specific for cellulolytic responses. We tested the functional significance of phosphorylation sites in two proteins, one a transcriptional activator required for induction of the major regulator of cellulase gene expression, CLR2 (Coradetti et al., 2012) and the cellobionic acid transporter CBT1. Phosphosite mutations in CLR1 did not dramatically affect the ability of N. crassa to sense and respond to cellulose. However, phosphosite mutations in CBT1 increased initial transport capacity. These experiments were performed in S. cerevisiae, as this species has proven to be very useful in analysis of N. crassa membrane proteins that transport components of plant biomass, such as the cellodextrin transporters CDT1, CDT2 (Galazka et al., 2010) and a galacturonic acid transporter (GAT1) (Benz et al., 2014b). Our data suggest that phosphorylation of CBT1 in response to exposure of N. crassa to Avicel is a functionally important modulator of cellobionic acid transport. Our data on the proteomic and phosphoproteomic response of N. crassa associated with exposure to sucrose, Avicel and carbon starvation conditions provides a useful dataset for researchers working on various aspects of metabolism in filamentous fungi and during carbon starvation and cellulose utilization in particular.
Supplementary Material
Acknowledgments
This work was funded by Grants from the Energy Biosciences Institute to N.L.G. and to J.H.D.C. The authors also acknowledge the partial funding support from P41GM103493 (to R.D.S.) for the proteomics and phosphoproteomics analyses. Portions of this project were supported by the U.S. Department of Energy (DOE) Office of Biological and Environmental Research (OBER) Panomics program at Pacific Northwest National Laboratory (PNNL) and performed in the Environmental Molecular Sciences Laboratory (EMSL), a U.S. Department of Energy (DOE) OBER national scientific user facility located at PNNL in Richland, Washington. PNNL is a multi-program national laboratory operated by Battelle Memorial Institute for the DOE under Contract DE-AC05-76RL01830.
Appendix A. Supplementary material
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.fgb.2014.05.005.
References
- Adav SS, Chao LT, Sze SK. Quantitative secretomic analysis of Trichoderma reesei strains reveals enzymatic composition for lignocellulosic biomass degradation. Mol Cell Proteomics. 2012;11 (M111):012419. doi: 10.1074/mcp.M111.012419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benz JP, Chau BH, Zheng D, Bauer S, Glass NL, Somerville CR. A comparative systems analysis of polysaccharide-elicited responses in Neurospora crassa reveals carbon source-specific cellular adaptations. Mol Microbiol. 2014a;91:275–299. doi: 10.1111/mmi.12459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benz JP, Protzko RJ, Andrich JMS, Bauer S, Dueber JE, Somerville CR. Identification and characterization of a galacturonic acid transporter from Neurospora crassa and its application for Saccharomyces cerevisiae fermentation processes. Biotechnol Biofuels. 2014b;7:20. doi: 10.1186/1754-6834-7-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner K, Lichtenauer AM, Kratochwill K, Delic M, Mach RL. Xyr1 regulates xylanase but not cellulase formation in the head blight fungus Fusarium graminearum. Curr Genet. 2007;52:213–220. doi: 10.1007/s00294-007-0154-x. [DOI] [PubMed] [Google Scholar]
- Chundawat SP, Lipton MS, Purvine SO, Uppugundla N, Gao D, Balan V, Dale BE. Proteomics-based compositional analysis of complex cellulase-hemicellulase mixtures. J Proteome Res. 2011;10:4365–4372. doi: 10.1021/pr101234z. [DOI] [PubMed] [Google Scholar]
- Cohen P. The regulation of protein function by multisite phosphorylation – a 25 year update. Trends Biochem Sci. 2000;25:596–601. doi: 10.1016/s0968-0004(00)01712-6. [DOI] [PubMed] [Google Scholar]
- Coradetti ST, Craig JP, Xiong Y, Shock T, Tian C, Glass NL. Conserved and essential transcription factors for cellulase gene expression in ascomycete fungi. Proc Natl Acad Sci USA. 2012;109:7397–7402. doi: 10.1073/pnas.1200785109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coradetti ST, Xiong Y, Glass NL. Analysis of a conserved cellulase transcriptional regulator reveals inducer-independent production of cellulolytic enzymes in Neurospora crassa. Microbiologyopen. 2013;2:595–609. doi: 10.1002/mbo3.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cziferszky A, Mach RL, Kubicek CP. Phosphorylation positively regulates DNA binding of the carbon catabolite repressor Cre1 of Hypocrea jecorina (Trichoderma reesei) J Biol Chem. 2002;277:14688–14694. doi: 10.1074/jbc.M200744200. [DOI] [PubMed] [Google Scholar]
- D’Alessio C, Fernandez F, Trombetta ES, Parodi AJ. Genetic evidence for the heterodimeric structure of glucosidase II. The effect of disrupting the subunit-encoding genes on glycoprotein folding. J Biol Chem. 1999;274:25899–25905. doi: 10.1074/jbc.274.36.25899. [DOI] [PubMed] [Google Scholar]
- de Godoy LM, Olsen JV, Cox J, Nielsen ML, Hubner NC, Frohlich F, Walther TC, Mann M. Comprehensive mass-spectrometry-based proteome quantification of haploid versus diploid yeast. Nature. 2008;455:1251–1254. doi: 10.1038/nature07341. [DOI] [PubMed] [Google Scholar]
- de Oliveira JM, van Passel MW, Schaap PJ, de Graaff LH. Proteomic analysis of the secretory response of Aspergillus niger to D-maltose and D-xylose. PLoS ONE. 2011;6:e20865. doi: 10.1371/journal.pone.0020865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmas S, Pullan ST, Gaddipati S, Kokolski M, Malla S, Blythe MJ, Ibbett R, Campbell M, Liddell S, Aboobaker A, Tucker GA, Archer DB. Uncovering the genome-wide transcriptional responses of the filamentous fungus Aspergillus niger to lignocellulose using RNA sequencing. PLoS Genet. 2012;8:e1002875. doi: 10.1371/journal.pgen.1002875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derntl C, Gudynaite-Savitch L, Calixte S, White T, Mach RL, Mach-Aigner AR. Mutation of the Xylanase regulator 1 causes a glucose blind hydrolase expressing phenotype in industrially used Trichoderma strains. Biotechnol Biofuels. 2013;6:62. doi: 10.1186/1754-6834-6-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do Vale LH, Gomez-Mendoza DP, Kim MS, Pandey A, Ricart CA, Ximenes FFE, Sousa MV. Secretome analysis of the fungus Trichoderma harzianum grown on cellulose. Proteomics. 2012;12:2716–2728. doi: 10.1002/pmic.201200063. [DOI] [PubMed] [Google Scholar]
- Ebbole DJ. Carbon catabolite repression of gene expression and conidiation in Neurospora crassa. Fungal Genet Biol. 1998;25:15–21. doi: 10.1006/fgbi.1998.1088. [DOI] [PubMed] [Google Scholar]
- Eng JK, McCormack AL, Yates JR. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen MY, Pieper U, Sali A. Comparative protein structure modeling using MODELLER. Curr Protoc Protein Sci. 2007;Chapter 2(Unit 2):9. doi: 10.1002/0471140864.ps0209s50. [DOI] [PubMed] [Google Scholar]
- Fama MC, Raden D, Zacchi N, Lemos DR, Robinson AS, Silberstein S. The Saccharomyces cerevisiae YFR041C/ERJ5 gene encoding a type I membrane protein with a J domain is required to preserve the folding capacity of the endoplasmic reticulum. Mol Cell Res. 2007;1773:232–242. doi: 10.1016/j.bbamcr.2006.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez J, Wright JD, Hartline D, Quispe CF, Madayiputhiya N, Wilson RA. Principles of carbon catabolite repression in the rice blast fungus: Tps1, Nmr1-3, and a MATE-family pump regulate glucose metabolism during infection. PLoS Genet. 2012;8:e1002673. doi: 10.1371/journal.pgen.1002673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ficarro SB, Adelmant G, Tomar MN, Zhang Y, Cheng VJ, Marto JA. Magnetic bead processor for rapid evaluation and optimization of parameters for phosphopeptide enrichment. Anal Chem. 2009;81:4566–4575. doi: 10.1021/ac9004452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier ML, Paulson A, Pavelka N, Mosley AL, Gaudenz K, Bradford WD, Glynn E, Li H, Sardiu ME, Fleharty B, Seidel C, Florens L, Washburn MP. Delayed correlation of mRNA and protein expression in rapamycin-treated cells and a role for Ggc1 in cellular sensitivity to rapamycin. Mol Cell Proteomics. 2010;9:271–284. doi: 10.1074/mcp.M900415-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galazka JM, Tian C, Beeson WT, Martinez B, Glass NL, Cate JH. Cellodextrin transport in yeast for improved biofuel production. Science. 2010;330:84–86. doi: 10.1126/science.1192838. [DOI] [PubMed] [Google Scholar]
- Herpoel-Gimbert I, Margeot A, Dolla A, Jan G, Molle D, Lignon S, Mathis H, Sigoillot JC, Monot F, Asther M. Comparative secretome analyses of two Trichoderma reesei RUT-C30 and CL847 hypersecretory strains. Biotechnol Biofuels. 2008;1:18. doi: 10.1186/1754-6834-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Ouyang H, Li Y, Guan KL. Signaling by target of rapamycin proteins in cell growth control. Microbiol Mol Biol Rev. 2005;69:79–100. doi: 10.1128/MMBR.69.1.79-100.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- Kuhad RC, Gupta R, Singh A. Microbial cellulases and their industrial applications. Enzyme Res. 2011;2011:280696. doi: 10.4061/2011/280696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtzman CP. Molecular taxonomy of the yeasts. Yeast. 1994;10:1727–1740. doi: 10.1002/yea.320101306. [DOI] [PubMed] [Google Scholar]
- Le Crom S, Schackwitz W, Pennacchio L, Magnuson JK, Culley DE, Collett JR, Martin J, Druzhinina IS, Mathis H, Monot F, Seiboth B, Cherry B, Rey M, Berka R, Kubicek CP, Baker SE, Margeot A. Tracking the roots of cellulase hyperproduction by the fungus Trichoderma reesei using massively parallel DNA sequencing. Proc Natl Acad Sci USA. 2009;106:16151–16156. doi: 10.1073/pnas.0905848106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MV, Topper SE, Hubler SL, Hose J, Wenger CD, Coon JJ, Gasch AP. A dynamic model of proteome changes reveals new roles for transcript alteration in yeast. Mol Syst Biol. 2011;7:514. doi: 10.1038/msb.2011.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Li J, Zhao S, Zhang R, Wang M, Miao Y, Shen Y, Shen Q. Secretome diversity and quantitative analysis of cellulolytic Aspergillus fumigatus Z5 in the presence of different carbon sources. Biotechnol Biofuels. 2013a;6:149. doi: 10.1186/1754-6834-6-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Zhang L, Qin Y, Zou G, Li Z, Yan X, Wei X, Chen M, Chen L, Zheng K, Zhang J, Ma L, Li J, Liu R, Xu H, Bao X, Fang X, Wang L, Zhong Y, Liu W, Zheng H, Wang S, Wang C, Xun L, Zhao GP, Wang T, Zhou Z, Qu Y. Long-term strain improvements accumulate mutations in regulatory elements responsible for hyper-production of cellulolytic enzymes. Sci Rep. 2013b;3:1569. doi: 10.1038/srep01569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, Bonenfant D, Oppliger W, Jenoe P, Hall MN. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell. 2002;10:457–468. doi: 10.1016/s1097-2765(02)00636-6. [DOI] [PubMed] [Google Scholar]
- Lundberg E, Fagerberg L, Klevebring D, Matic I, Geiger T, Cox J, Algenas C, Lundeberg J, Mann M, Uhlen M. Defining the transcriptome and proteome in three functionally different human cell lines. Mol Syst Biol. 2010;6:450. doi: 10.1038/msb.2010.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mach-Aigner AR, Pucher ME, Steiger MG, Bauer GE, Preis SJ, Mach RL. Transcriptional regulation of xyr1, encoding the main regulator of the xylanolytic and cellulolytic enzyme system in Hypocrea jecorina. Appl Environ Microbiol. 2008;74:6554–6562. doi: 10.1128/AEM.01143-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier T, Guell M, Serrano L. Correlation of mRNA and protein in complex biological samples. FEBS Lett. 2009;583:3966–3973. doi: 10.1016/j.febslet.2009.10.036. [DOI] [PubMed] [Google Scholar]
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- Margolin BS, Freitag M, Selker EU. Improved plasmids for gene targeting at the his-3 locus of Neurospora crassa by electroporation. Fungal Genet Newslett. 1997;44:34–36. [Google Scholar]
- McNally MT, Free SJ. Isolation and characterization of a Neurospora glucose-repressible gene. Curr Genet. 1988;14:545–551. doi: 10.1007/BF00434079. [DOI] [PubMed] [Google Scholar]
- Monroe ME, Shaw JL, Daly DS, Adkins JN, Smith RD. MASIC: a software program for fast quantitation and flexible visualization of chromatographic profiles from detected LC-MS(/MS) features. Comput Biol Chem. 2008;32:215–217. doi: 10.1016/j.compbiolchem.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TH, Brechenmacher L, Aldrich JT, Clauss TR, Gritsenko MA, Hixson KK, Libault M, Tanaka K, Yang F, Yao Q, Pasa-Tolic L, Xu D, Nguyen HT, Stacey G. Quantitative phosphoproteomic analysis of soybean root hairs inoculated with Bradyrhizobium japonicum. Mol Cell Proteomics. 2012;11:1140–1155. doi: 10.1074/mcp.M112.018028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi Y, Tanaka H, Kanamaru K, Kato M, Kobayashi T. Xylose triggers reversible phosphorylation of XlnR, the fungal transcriptional activator of xylanolytic and cellulolytic genes in Aspergillus oryzae. Biosci Biotechnol Biochem. 2011;75:953–959. doi: 10.1271/bbb.100923. [DOI] [PubMed] [Google Scholar]
- Ogawa M, Kobayashi T, Koyama Y. ManR, a transcriptional regulator of the beta-mannan utilization system, controls the cellulose utilization system in Aspergillus oryzae. Biosci Biotechnol Biochem. 2013;77:426–429. doi: 10.1271/bbb.120795. [DOI] [PubMed] [Google Scholar]
- Ogg SC, Barz WP, Walter P. A functional GTPase domain, but not its transmembrane domain, is required for function of the SRP receptor beta-subunit. J Cell Biol. 1998;142:341–354. doi: 10.1083/jcb.142.2.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips CM, Iavarone AT, Marletta MA. Quantitative proteomic approach for cellulose degradation by Neurospora crassa. J Proteome Res. 2011;10:4177–4185. doi: 10.1021/pr200329b. [DOI] [PubMed] [Google Scholar]
- de Porciuncula JO, Furukawa T, Mori K, Shida Y, Hirakawa H, Tashiro K, Kuhara S, Nakagawa S, Morikawa Y, Ogasawara W. Single nucleotide polymorphism analysis of a Trichoderma reesei hyper-cellulolytic mutant developed in Japan. Biosci Biotechnol Biochem. 2013;77:534–543. doi: 10.1271/bbb.120794. [DOI] [PubMed] [Google Scholar]
- Rapoport TA. Protein translocation across the eukaryotic endoplasmic reticulum and bacterial plasma membranes. Nature. 2007;450:663–669. doi: 10.1038/nature06384. [DOI] [PubMed] [Google Scholar]
- Ries L, Pullan S, Delmas S, Malla S, Blythe M, Archer DB. Genome-wide transcriptional response of Trichoderma reesei to lignocellulose using RNA sequencing and comparison with Aspergillus niger. BMC Genomics. 2013;14:541. doi: 10.1186/1471-2164-14-541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruepp A, Zollner A, Maier D, Albermann K, Hani J, Mokrejs M, Tetko I, Guldener U, Mannhaupt G, Munsterkotter M, Mewes HW. The FunCat, a functional annotation scheme for systematic classification of proteins from whole genomes. Nucleic Acids Res. 2004;32:5539–5545. doi: 10.1093/nar/gkh894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadowski I, Niedbala D, Wood K, Ptashne M. GAL4 is phosphorylated as a consequence of transcriptional activation. Proc Natl Acad Sci USA. 1991;88:10510–10514. doi: 10.1073/pnas.88.23.10510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M. Global quantification of mammalian gene expression control. Nature. 2011;473:337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- Smith MD, Gu Y, Querol-Audi J, Vogan JM, Nitido A, Cate JH. Humanlike eukaryotic translation initiation factor 3 from Neurospora crassa. PLoS ONE. 2013;8:e78715. doi: 10.1371/journal.pone.0078715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Sata J, Saito A, Usui M, Azakami H, Kato A. Effects of calnexin deletion in Saccharomyces cerevisiae on the secretion of glycosylated lysozymes. J Biochem. 2001;130:757–764. doi: 10.1093/oxfordjournals.jbchem.a003046. [DOI] [PubMed] [Google Scholar]
- Stricker AR, Mach RL, de Graaff LH. Regulation of transcription of cellulases- and hemicellulases-encoding genes in Aspergillus niger and Hypocrea jecorina (Trichoderma reesei) Appl Microbiol Biotechnol. 2008;78:211–220. doi: 10.1007/s00253-007-1322-0. [DOI] [PubMed] [Google Scholar]
- Sun J, Tian C, Diamond S, Glass NL. Deciphering transcriptional regulatory mechanisms associated with hemicellulose degradation in Neurospora crassa. Eukaryot Cell. 2012a;11:482–493. doi: 10.1128/EC.05327-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Zeng X, Yan C, Sun X, Gong X, Rao Y, Yan N. Crystal structure of a bacterial homologue of glucose transporters GLUT1-4. Nature. 2012b;490:361–366. doi: 10.1038/nature11524. [DOI] [PubMed] [Google Scholar]
- Tani S, Kawaguchi T, Kobayashi T. Complex regulation of hydrolytic enzyme genes for cellulosic biomass degradation in filamentous fungi. Appl Microbiol Biotechnol. 2014 doi: 10.1007/s00253-014-5707-6. http://dx.doi.org/10.1007/s00253-014-5707-6. [DOI] [PubMed]
- Taniguchi Y, Choi PJ, Li GW, Chen H, Babu M, Hearn J, Emili A, Xie XS. Quantifying E. coli proteome and transcriptome with single-molecule sensitivity in single cells. Science. 2010;329:533–538. doi: 10.1126/science.1188308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Peij NN, Gielkens MM, de Vries RP, Visser J, de Graaff LH. The transcriptional activator XlnR regulates both xylanolytic and endoglucanase gene expression in Aspergillus niger. Appl Environ Microbiol. 1998;64:3615–3619. doi: 10.1128/aem.64.10.3615-3619.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel C, de Abreu RS, Ko D, Le SY, Shapiro BA, Burns SC, Sandhu D, Boutz DR, Marcotte EM, Penalva LO. Sequence signatures and mRNA concentration can explain two-thirds of protein abundance variation in a human cell line. Mol Syst Biol. 2010;6:400. doi: 10.1038/msb.2010.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel C, Marcotte EM. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat Rev Genet. 2012;13:227–232. doi: 10.1038/nrg3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel C, Silva GM, Marcotte EM. Protein expression regulation under oxidative stress. Mol Cell Proteomics. 2011;10 (M111):009217. doi: 10.1074/mcp.M111.009217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel H. A convenient growth medium for Neurospora (medium N) Microb Genet Bull. 1956;13:2–43. [Google Scholar]
- Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- Wang Y, Yang F, Gritsenko MA, Clauss T, Liu T, Shen Y, Monroe ME, Lopez-Ferrer D, Reno T, Moore RJ, Klemke RL, Camp DG, 2nd, Smith RD. Reversed-phase chromatography with multiple fraction concatenation strategy for proteome profiling of human MCF10A cells. Proteomics. 2011;11:2019–2026. doi: 10.1002/pmic.201000722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Wilkinson HH, Correa A, Lewis ZA, Bell-Pedersen D, Ebbole DJ. Transcriptional response to glucose starvation and functional analysis of a glucose transporter of Neurospora crassa. Fungal Genet Biol. 2004;41:1104–1119. doi: 10.1016/j.fgb.2004.08.009. [DOI] [PubMed] [Google Scholar]
- Znameroski EA, Coradetti ST, Roche CM, Tsai JC, Iavarone AT, Cate JH, Glass NL. Induction of lignocellulose-degrading enzymes in Neurospora crassa by cellodextrins. Proc Natl Acad Sci USA. 2012;109:6012–6017. doi: 10.1073/pnas.1118440109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Znameroski EA, Li X, Tsai JC, Galazka JM, Glass NL, Cate JH. Evidence for transceptor function of cellodextrin transporters in Neurospora crassa. J Biol Chem. 2014;289:2610–2619. doi: 10.1074/jbc.M113.533273. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.