

Abstract
Although it has been previously demonstrated that an electrical current can be used to control biofilm growth on metal surfaces, the literature results are conflicting and there is no accepted mechanism of action. One of the suggested mechanisms is the production of hydrogen peroxide (H2O2) on metal surfaces. However, there are literature studies in which H2O2 could not be detected in the bulk solution. This is most likely because H2O2 was produced at a low concentration near the surface and could not be detected in the bulk solution. The goals of this research were (1) to develop a well-controlled system to explain the mechanism of action of the bioelectrochemical effect on 316L stainless steel (SS) surfaces and (2) to test whether the produced H2O2 can reduce cell growth on metal surfaces. It was found that H2O2 was produced near 316L SS surfaces when a negative potential was applied. The H2O2 concentration increased towards the surface, while the dissolved oxygen decreased when the SS surface was polarized to −600 mVAg/AgCl. When polarized and non-polarized surfaces with identical Pseudomonas aeruginosa PAO1 biofilms were continuously fed with air-saturated growth medium, the polarized surfaces showed minimal biofilm growth while there was significant biofilm growth on the non-polarized surfaces. Although there was no detectable H2O2 in the bulk solution, it was found that the surface concentration of H2O2 was able to prevent biofilm growth.
Keywords: biofilm, bioelectrochemical, biofilm control, hydrogen peroxide, cathode
Introduction
Although electrical current has been previously demonstrated to control biofilm growth on metal surfaces, the literature results are conflicting and there is no single accepted mechanism of action (Del Pozo et al. 2008). Some of the mechanisms proposed in the literature are (1) electrochemical generation of oxidants on the surface (Costerton et al. 1994), (2) contraction or expansion of biofilm structure (Stoodley et al. 1997), (3) electrolytic generation of oxygen during water electrolysis (Stewart et al. 1999), and (4) electrostatic and electrophoretic repulsive forces (Hong et al. 2008). However, none of these mechanisms is widely accepted because of conflicting results in the literature and the impossibility of comparing the different electrochemical systems. This may be partly due to the oversimplification that the magnitude of the electrical current itself determines the effect on biofilm growth. Because the research group responsible for the study reported here works with bioelectrochemical systems, information on current, potential, and internal resistances is needed to characterize the system, similar to microbial fuel cells (Logan et al. 2006). Thus, the term ‘bioelectrochemical effect’ is used to describe the electrochemical control of biofilm growth on metal surfaces.
To control biofilm growth using the bioelectrochemical effect, the minimum electrochemical system that can be used is a two-electrode cell in which a current or potential is applied to the metal surface with biofilm against another electrode. Shirtliff et al. (2005) used an annular reactor with a stainless steel surface to evaluate the bioelectrochemical effect. They applied a 370-μA cm−2 current and found that the applied current did not have any bactericidal effect. Similarly, Hong et al. (2008) used a flow chamber and applied a 15- μA cm−2 current and found that the applied current caused detachment of the biofilm on the indium tin oxide surface. They hypothesized that this was due to electrostatic and electrophoretic repulsive forces. Similar observations were made by van der Borden et al. (2004), who applied a current between 2.86 and 4.76 μA cm−2. Liu et al. (1997) inserted two electrodes into agar in a plate and found a zone where biofilm growth was inhibited. They found that this was due to hydrogen peroxide (H2O2) production. On the other hand, Del Pozo et al. (2009) applied a current between 1.626 × 103 and 1.626 × 106 μA cm−2 and did not detect H2O2; instead, they concluded that pH was the cause of decreased biofilm growth.
Other researchers have controlled the potential of surfaces to prevent cell growth. For example, Busalmen and de Sanchez (2001) poised a gold surface at negative potentials and observed a reduction in biofilm growth. They hypothesized that this was a result of electrostatic repulsion and pH changes caused by oxygen reduction. Similarly, Matsunaga and Lim (2000) poised nylon fishnets coated with conductive paint at negative potentials and observed biofilm detachment. Toxic substances such as H2O2, oxidizing radicals, and chlorine molecules were not detected. Since the work reported here focused on the bioelectrochemical effect only, the literature related to the combined use of antibiotics and electrical current known as the ‘bioelectric effect’ is not discussed (Del Pozo et al. 2008). The use of combined antibiotics and electrical current could involve different mechanisms than those discussed in this paper.
It is important to note that the literature studies discussed above used different electrochemical cells with different geometries and operated using either a controlled potential-potentiostatic system or a controlled current-galvanostatic system to achieve the bioelectrochemical effect. Regardless of the type of system, a specified current is required to understand the rate of reaction and a specified potential is required to assess the thermodynamic state of the system. To have reproducible or comparable data, both the potential (galvanostatic) and current (potentiostatic) data must be presented. Additionally, the potential of the electrode must be defined against a well-known reference electrode. Therefore, it is critical to use a well-defined electrochemical system to understand the mechanisms of action of the bioelectrochemical effect.
One suggested mechanism of action of the bioelectrochemical effect is H2O2 generation. However, this has never been investigated near metal surfaces (Davis et al. 1994). According to Equation (1), oxygen is reduced to water on metal surfaces (Bard et al. 1985; Freguia et al. 2007; De Schamphelaire et al. 2010; Qiao et al. 2010). However, at the same time, the partial reduction of oxygen leads to H2O2 production on metal surfaces (Equation (2)) (Bard and Faulkner 2001).
| (1) |
| (2) |
Although the standard reduction potential of H2O2 is +85 mVAg/AgCl, it is common to observe the onset of H2O2 production at negative polarization potentials because of large activation overpotentials (Vetter 1967). In bioelectrochemical systems where oxygen is the only cathodic reactant, cathodic current will most likely result in the production of H2O2.
By controlling the potential of the electrode, it is possible to generate H2O2 on 316L stainless steel (SS) surfaces. To the best of the authors’ knowledge, such generation of H2O2 on SS surfaces has not been experimentally demonstrated to be a mechanism of the bioelectrochemical effect. The goals of this research were (1) to develop a well-controlled system to explain the mechanism of action of the bioelectrochemical effect on SS surfaces and (2) to test whether electrochemically generated H2O2 can reduce biofilm growth on metal surfaces. A flat plate flow cell was designed to monitor in situ biofilm formation. Previously developed H2O2 microelectrodes were modified to measure H2O2 on SS surfaces. In addition, dissolved oxygen (DO) microelectrodes were used to measure the variation of DO near the SS surface. Since H2O2 production was only found near the SS surface, its biocidal efficacy on Pseudomonas aeruginosa PAO1 biofilms was tested. P. aeruginosa PAO1 biofilms were grown on SS surfaces and biofilm growth was quantified on polarized and non-polarized surfaces.
Materials and methods
Preparation of growth medium and inoculum
P. aeruginosa PAO1 tagged with green fluorescent protein was obtained from Dr Michael Franklin, Center for Biofilm Engineering, Montana State University. A defined medium with the following composition was used: Na2HPO4, 1830 mg l−1 (J.T. Baker®); KH2PO4, 1350 mg l−1 (J.T. Baker®); MgSO4 · 7H2O, 10 mg l−1 (J.T. Baker®); yeast extract, 10 mg l−1 (Bacto®); glucose, 1000 mg l−1 (Fisher®); (NH4)2SO4, 100 mg l−1 (J.T. Baker®). The medium was autoclaved at 121°C for 15 min l−1 of solution. The ammonium sulfate and yeast extract were autoclaved separately and then added to the growth medium. The glucose was filter-sterilized. After the medium was allowed to cool to room temperature, a trace elements solution (1 ml l−1) was filtered and added to it. The trace elements solution was prepared by adding MnCl2 · 4H2O, 357 mg l−1; CuCl2 · 2H2O, 228 mg l−1; CoCl2 · 2H2O, 317 mg l−1; (NH4)Mo7O4 ·H2O, 231 mg l−1; Na2B407 · 10H2O, 127 mg l−1; ZnCl2, 363 mg l−1; FeCl3, 2160 mg l−1; and CaCl2, 3700 mg l−1. Carbenicillin (150 μg ml−1 of USP grade disodium salt, Teknova) was added to the medium. A 1-ml vial of frozen (−85°C) P. aeruginosa PAO1 stock was thawed at room temperature. The stock was then used to inoculate 100 ml of the sterile defined medium, which was incubated on a shaker at 150 rpm at 30°C for 24 h. The optical density (OD) was measured at 600 nm and adjusted to 0.05 before the biofilm reactor was inoculated. An OD of 0.05 was chosen as optimal for obtaining a single layer of cells during initial attachment and quantifiable cell counts on the SS surface subsequently. The biofilm reactor was inoculated with 10 ml of inoculum using a sterile syringe.
Preparing the biofilm reactor
The experimental setup is shown in Figure 1. A custom-made flat plate flow cell was used to grow biofilms on SS surfaces. The reactor was designed in such a way that the SS working electrode and the graphite counter electrode (Glassmate grade GM-10, Poco Graphite, Inc., Decatur, TX, USA) were separated by a cation exchange membrane (Membranes International Inc., CMI-7000, 6 × 8 cm). The membrane isolated the reactions at the counter electrode from the SS working electrode. The reactor bottom was made of microscope cover glass (Thermo Scientific® Gold Seal Cover Glasses, Rectangles 48 × 60 mm, #1), which served as a viewing window for epifluorescence microscopy. In this way, the cells on the SS surface were imaged directly. Five autoclavable rubber separators were used between each two layers to prevent leakage. A graphite (1 × 2 cm) counter electrode and a custom-made Ag/AgCl reference electrode (Lewandowski and Beyenal 2007) were used in the upper compartment. Two SS coupons (1.18 × 1.18 cm) were used in the lower cells. The first SS coupon was the control surface (non-polarized) and the second SS coupon was polarized against the Ag/AgCl reference electrode. Platinum wires (CA Fine Wire Company, Pure TC Grade, MO, #M159850) were used to connect the SS coupons to external cables, which were connected to a custom-built potentiostat (Renslow et al. 2011). The connection resistances were always less than 5Ω.
Figure 1.
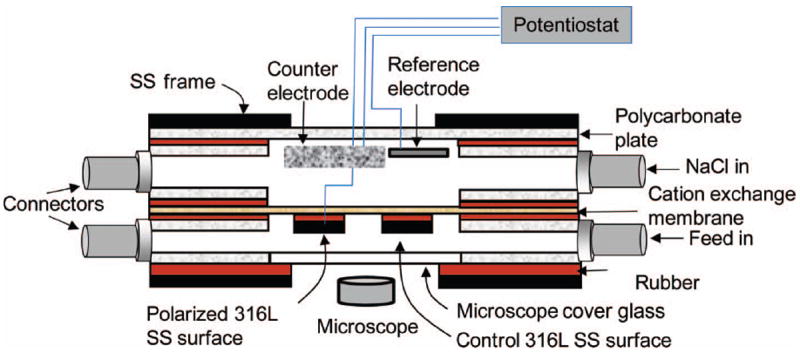
Schematic configuration of experimental setup and flat plate flow cell. The figure is not drawn to scale
Operating the biofilm reactor
The assembled flat plate flow cell (Figure 1) and tubing were autoclaved at 121°C for 15 min. After cooling to room temperature, the 316L SS working electrode and the counter and reference electrodes were connected to a custom-built potentiostat (Renslow et al. 2011). A two-channel peristaltic pump (Masterflex® L/S® Cole-Parmer Instrument Company) was used at a flow rate of 0.4 ml min−1 for the feed and 3M NaCl solution. The flow cell was inoculated aseptically from its inoculation port with 10 ml of inoculum. After 2 h of initial attachment (no flow), the flow was started and planktonic cells were washed out of the flow cell. Before polarization, it was verified that there were no bubbles in the reactor. For biofilm experiments, the polarization potential was set to −600 mVAg/AgCl.
Biofilm imaging and analysis
During operation, the flat plate flow cell was placed on an inverted epifluorescence microscope (Nikon® Eclipse Ti-S inverted microscope); a Nikon® DS-Qi1Mc camera was mounted on the microscope for imaging. A CFI Plan Fluor ELWD 40 × objective (N.A. 0.60, W.D. 3.7-2.7 mm) was used to image the cells. Every 8 h, at least 20 images were taken to obtain statistically representative data (Ica et al. 2012). The surface coverage was calculated from these images using NIS® software (Laboratory Imaging s.r.o., Praha, CZ). Surface coverage is the ratio of the area of biomass to the total area of the image. The higher the surface coverage, the higher the coverage of the surface by biomass.
Scanning electron microscopy
After treatment for 40 h, both the polarized and the non-polarized 316L SS surfaces were removed aseptically from the flat plate flow cell. The SS coupons were fixed overnight with 2.5% glutaraldehyde and 2%. paraformaldehyde in 0.1 M phosphate buffer followed by rinsing with 0.1 M phosphate buffer (3 × 10 min each). The SS coupons were then dehydrated gradually using 30%, 50%, 70%, and 95% alcohol (10 min each) and 100% alcohol (3 × 10 min each). Hexamethyldisilizane (HMDS) was used for overnight drying. The SS coupons were sputter-coated with gold prior to FEISEM (FEI 200F) imaging.
Microelectrode construction
The H2O2 and oxygen microelectrodes were constructed following the procedures given in Lewandowski and Beyenal (2007) except that the H2O2 microelectrode was modified so that it could operate on polarized surfaces. Briefly, an etched platinum wire was encased in a glass capillary which was pulled at a high temperature to seal the wire. The platinum tip was then exposed by grinding away the sealed tip using a diamond grinding wheel (Narishige®, model EG-4). The exposed tip was 10–15 μm in diameter. Platinum was electrochemically deposited onto the exposed platinum tip, which resulted in a porous platinum ball that increased the effective surface area and, consequently, the sensitivity of the microelectrode. A 5% cellulose acetate (Sigma-Aldrich®) membrane was then deposited on the platinum ball. The final tip diameter was ~20 μm. The H2O2 microelectrode was placed in a glass outer casing with the tip exposed to the solution. The glass junction was sealed with an agar (1.812-g l−1 R-2A Agar, Fluka®) salt bridge containing 1 mM sodium sulfate (Fisher®) and the outer case was filled with saturated KCl and Ag/AgCl reference filling solution (SP135-500, Fisher®). A Ag/AgCl reference electrode was inserted into the glass outer casing. The procedures given in the literature were followed to construct the dissolved oxygen microelectrode (Lewandowski and Beyenal 2007). The final tip diameter of the DO microelectrode was ~15 μm.
Operation of microelectrodes
The H2O2 and oxygen microelectrodes are amperometric sensors. A Keithley® 6517A Electrometer/High Resistance Meter was used as both the voltage source and a picoammeter. The H2O2 microelectrode was polarized to +600 mVAg/AgCl and the DO microelectrode was polarized to −800 mVAg/AgCl. The H2O2 microelectrode was calibrated in solutions of known H2O2 concentration ranging from 0 μm to 150 μm. The calibration curve was always linear in this concentration range. The oxygen microelectrode was calibrated in a solution with zero oxygen concentration and in air-saturated sterile growth medium. Each microelectrode was calibrated immediately before use and the calibrations were also verified after the measurements.
Reactor used for microelectrode measurements
Since the reactor shown in Figure 1 did not have a port for microelectrode measurements, a flat plate reactor with recycle was used (Nguyen et al. 2012). The SS coupons were placed at the bottom of this reactor so that it was possible to measure DO or H2O2 profiles on the SS surface. For control experiments, a clean SS coupon was used. For experiments with biofilms, P. aeruginosa PAO1 cells were allowed to attach to a SS surface using the procedure described earlier. After initial attachment, the growth medium was replaced several times to remove suspended cells. Air-saturated growth medium was continuously fed into the reactor. Microelectrode measurements with biofilm were conducted in a biosafety level II hood.
Microelectrode measurements
To determine the polarization potential at which H2O2 was produced at the SS surface, the H2O2 microelectrode was positioned approximately 100 μm from the SS surface and the potential was scanned from +200 mVAg/AgCl to −650mVAg/AgCl. The H2O2 concentration was monitored as a function of the potential of the SS. A distance of 100 μm from the SS surface was maintained to reduce the likelihood of breaking the microelectrode tip. The current values during the potential sweep were collected automatically by the potentiostat.
To measure depth profiles of H2O2 or DO, the tip of the microelectrode was positioned approximately 800 μm above the SS surface using micromanipulators (Narishige®). The microelectrode tip was then stepped down in 5-μm increments using custom microprofiler software until the tip was 100 μm from the SS surface. Concentrations were measured at each step. The measurement of profiles was stopped 100 μm from the SS surface to ensure that the tip of the microelectrode was not broken for repeated measurements. Each measurement was repeated at least three times in total. The locations of the microelectrode tip and the SS surface were monitored using a stereomicroscope (Zeiss® Stemi 2000 stereomicroscope). A Gamry® Series G™ 300 potentiostat (Gamry Instruments, Warminster, PA, USA) was used to polarize the SS coupon to −600 mVAg/AgCl during depth profile measurements. Subsequently, depth profiles were taken in the same position under non-polarized and polarized conditions by controlling the potentiostat. A graphite rod counter electrode (10 cm in length and 0.5 cm in diameter) and a Ag/AgCl reference electrode were used. Note that the current was allowed to stabilize before each depth profile was measured.
To find the SS surface, the tip of the microelectrode had to be broken by touching the surface, which did not allow replicate measurements with the same microelectrode. Therefore, depth profile measurements were stopped before the microelectrode tip touched the surface, except during the last measurement, when the tip was broken to determine the location of the SS surface. It was found that the profiles near the SS surface were always linear.
Results and discussion
The microelectrode measurements were conducted twice (two replicates) and repeated at least three times at each location. The biofilm experiments were conducted in replicates and repeated at three different times. The results were all statistically similar and led to identical conclusions. Selected representative measurements are shown here.
Quantifying H2O2 generation and oxygen consumption at the 316L SS surfaces
Figure 2 shows the variation in H2O2 concentration with potential at a position 100 μm from the SS surface. During the potential scan between +200 mVAg/AgCl and −650 mVAg/AgCl, the H2O2 concentration reached a transient concentration of 5.9 μm. The onset of detectable H2O2 at the microelectrode tip was at ~ −400 mVAg/AgCl, although it was expected to correspond with the onset of cathodic current at −200 mVAg/AgCl. The delay in detection of H2O2 at the microelectrode tip can be attributed to a combination of diffusion, catalytic decomposition of H2O2, and further reduction of H2O2 to water. According to Vetter (1967), oxygen reduction at metal electrodes proceeds initially with Equation (2). At more negative potentials, H2O2 is further reduced to water, which results in the complete reduction of oxygen to water as shown in Equation (1) (Vetter 1967). On iron electrodes, the transition from Equation (2) to Equation (1) occurs between −400 mVAg/AgCl and −800 mVAg/AgCl (Vetter 1967). Beyond −800 mVAg/AgCl, Equation (1) dominates and H2O2 production is minimal. Figure 2 shows the onset of the transition at −400 mVAg/AgCl and its continuation through −650 mVAg/AgCl. The observation of a potential range for H2O2 production provides a key point for the bioelectrochemical effect. The production of H2O2 cannot be understood by considering current values, alone. The limitation of reporting only the current values could explain the conflicting evidence on the role of H2O2 in inhibiting biofilm growth (Costerton et al. 1994; Busalmen and de Sanchez 2001; Shirtliff et al. 2005; Hong et al. 2008). At sufficiently negative potentials, H2O2 could go undetected by methods sampling the bulk solution.
Figure 2.
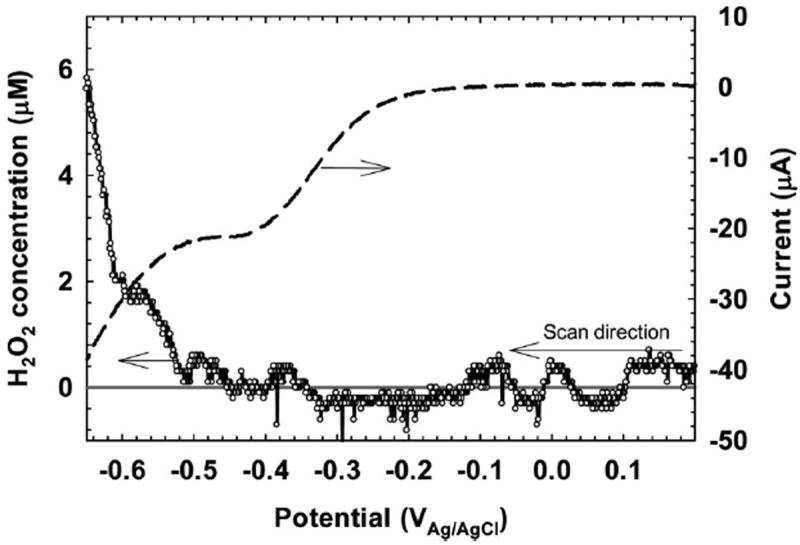
Transient H2O2 concentration variation with potential 100 μm from the SS surface. The black solid line represents the zero concentration line. The dashed line is the current measured during the potential sweep.
Figure 3A shows the H2O2 profiles that were measured above the SS surface, from the bulk to within 100 μm of the surface. The H2O2 concentration was 15.5 μm near the SS surface at a potential of −600 mVAg/AgCl. When the SS was not polarized, there was no detectable H2O2. However, Figure 3A shows that H2O2 was not detectable at a distance of 500 μm from the SS surface while it was polarized to −600 mVAg/AgCl. Considering that H2O2 degrades in water and that the flow cell was operated in continuous flow mode, H2O2 was unlikely to be found in the bulk solution. This result highlights the limitation of H2O2 measurements using bulk sampling methods in this type of system. However, it also emphasizes the suitability of the system for preventing biofilm growth near the surface. Based on the results presented here, it is expected that cells attached on the SS surface are exposed to H2O2 concentrations of at least 15.5 μm. The concentration of H2O2 could be enough to prevent monolayer cell coverage (~ 1 μm thick) from growing into a mature biofilm.
Figure 3.
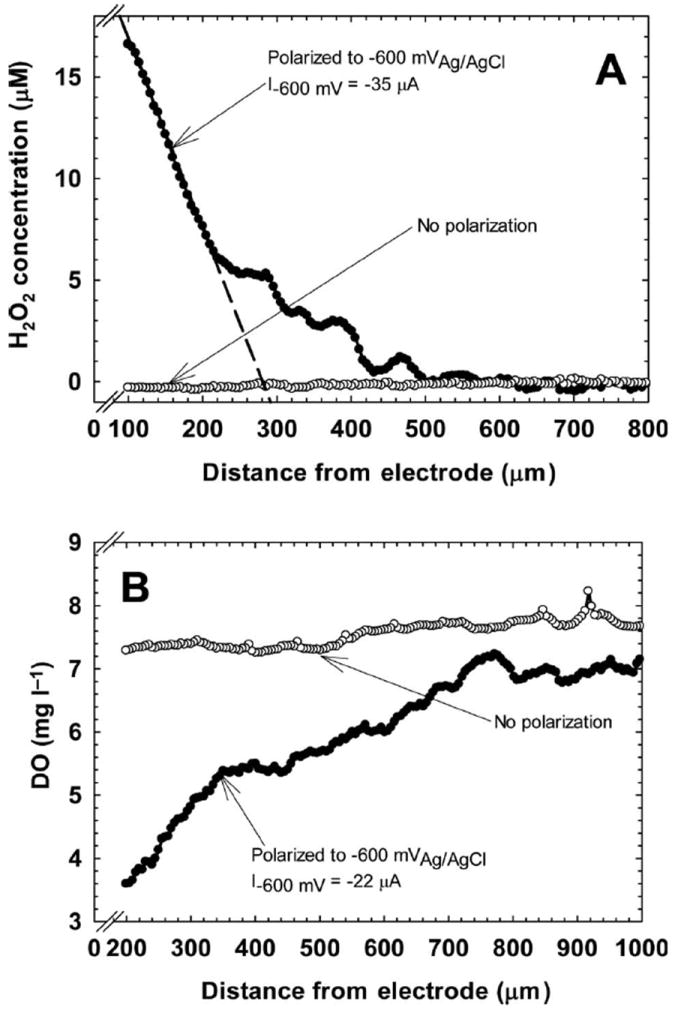
(A) H2O2 profiles above both non-polarized SS surfaces (○) and polarized SS surfaces (●). The black dashed line is fitted to points between 100 and 215 μm (R2 = 0.997). (B) Oxygen profiles above both non-polarized SS surfaces (○) and polarized SS surfaces (●). The current density was 25.14 μA cm−2.
Figure 3B shows the oxygen profiles that were measured above the SS, from the bulk to within 100 μm of the surface. The oxygen concentration decreased to ~ 3.5 mg l−1 near the SS surface from a bulk concentration of ~ 7.0 mg l−1 when the SS was polarized to −600 mVAg/AgCl. When the SS was not polarized, there was a slight decrease in oxygen concentration, suggesting that cells were utilizing oxygen to respire. The consumption of oxygen while a cathodic current was measured confirms oxygen reduction as the source of H2O2. The reduced DO at the SS surface also shows that the production of H2O2 at the surface drives the environment from an oxic to a suboxic condition. Thus, if H2O2 exposure is the primary mechanism of action of the bioelectrochemical effect, a simultaneous decrease in oxygen may also play a role. Since one of the proposed mechanisms of action, as described in the literature, is pH changes caused by the passing of current, pH variation also was quantified in the current study; there were no detectable pH changes near the SS surface (results not shown). This was expected, since the measured current was too low for the detection of significant pH changes at this buffer strength (Babauta et al. 2011).
One of the major limitations of this method of controlling biofilm growth is the requirement that oxygen be present at the SS surface. If a thick biofilm initially covered the SS surface, the DO concentration would be expected to be zero near the bottom of the biofilm, which would prevent H2O2 production (Renslow et al. 2011). Perhaps this, too, is a reason for the conflicting data in the literature. The method could be optimized for thicker biofilms by increasing the delivery of oxygen to the surface, but the added energy input would still make this method less efficient for thicker biofilms. Since the biofilm in the current study was a single layer of cells, there was oxygen available on the surface. Using these thin biofilms, it was possible to assess the effect of H2O2 production on biofilm growth.
Biofilm formation on the non-polarized and polarized SS coupons
To assess the effect of the H2O2 generated by applying a potential of −600 mVAg/AgCl, a monolayer of cells was exposed for 40 h, as shown in Figure 4. The initial cell surface coverage was ~ 1.9 ± 0.5% for both nonpolarized and polarized SS surfaces. There was no statistical difference in initial surface coverage by biofilms (P = 0.808). The surface coverage increased on the non-polarized SS surface to 99 ± 0.6% after 40 h, whereas the surface coverage remained almost constant for the polarized SS surface. The differences between the non-polarized and polarized SS surfaces were statistically significant (P ≤ 0.001). It was clear that uncontrolled cell growth on the non-polarized SS surfaces resulted in large biofilm clusters. In contrast, dim patches of light intensity behind a sparsely populated SS surface characterized the polarized SS surface. No small or large biofilm clusters were observable, signifying that biofilm growth was delayed. In a similar experiment, initially attached cells were treated with 50 μm H2O2 in the bulk solution and the surface coverage was monitored. Nearly identical results were observed for externally added H2O2 as for electrochemically produced H2O2 (data not shown). The addition of H2O2 to the bulk solution resulted in delaying biofilm growth. Interestingly, this suggests that the decrease in DO during oxygen reduction at the SS surface played only a minor role in delaying biofilm growth.
Figure 4.
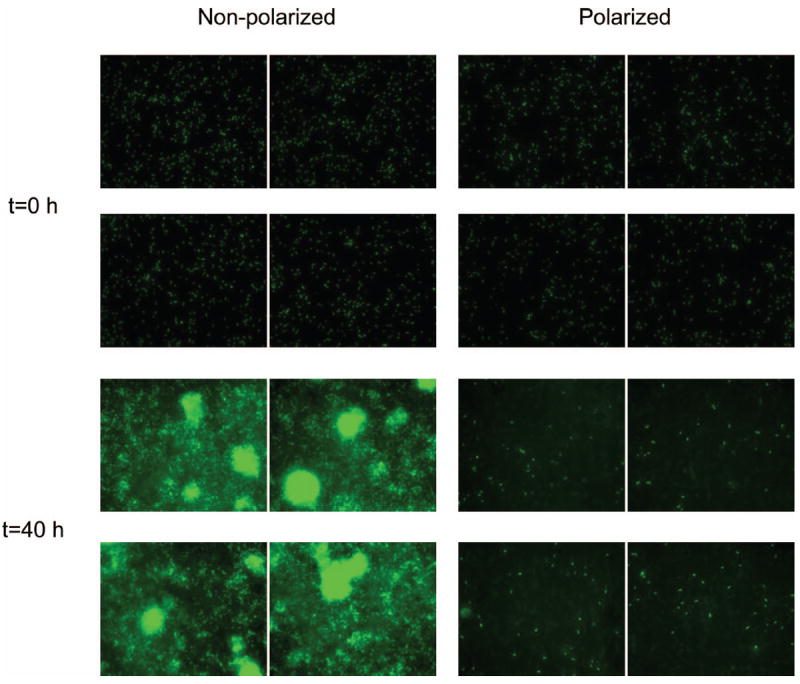
Images of P. aeruginosa PAO1 on non-polarized and polarized surfaces at t = 0 and t = 40 h (four images shown for each condition). The initial surface coverages were identical (1.9 ± 0.5). However, the final surface coverages were statistically different (P ≤ 0.001). The image size is 204.8 μm × 163.83 μm.
Figure 5A plots this time series and shows the variation with time of surface coverage of the SS surface. The non-polarized SS surface showed an exponential increase in surface coverage similar to the exponential growth measured by optical density. The polarized SS surface showed no measurable increase in surface coverage over time, which demonstrates the effectiveness of polarization in controlling biofilm growth. Over the length of the 40-h exposure, the current reached a steady state value of ~60 μA. Thus, it is thought that the monolayer of cells was exposed to at least 15.5 μm H2O2 under the polarized condition and a subsequent decrease in DO. Although seemingly small, these H2O2 concentrations have been shown to cause DNA damage (Linley et al. 2012).
Figure 5.
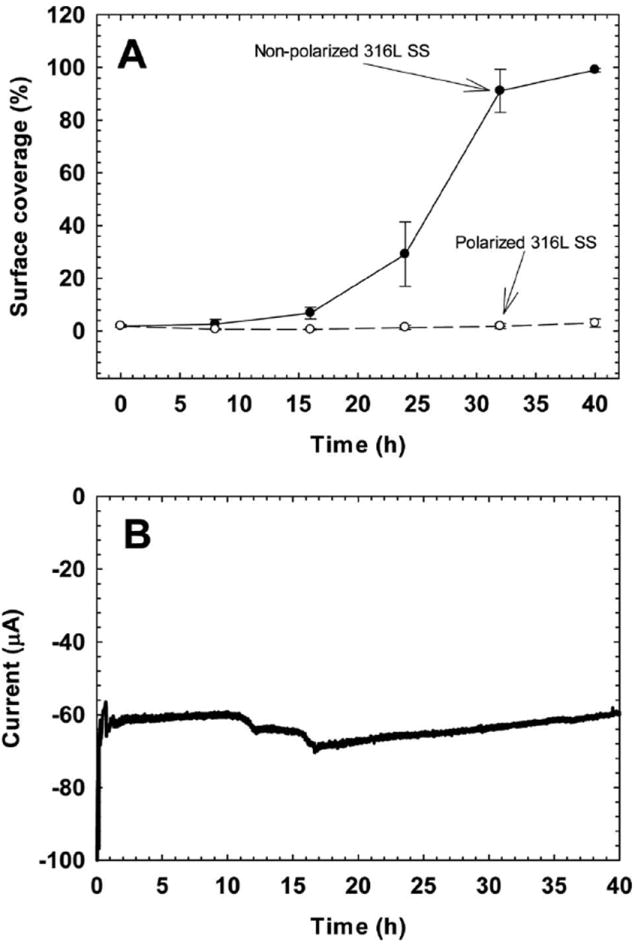
(A) The variation in surface coverage of polarized (○) and non-polarized (●) 316L SS surfaces over time. (B) The variation in current over time for the polarized 316L SS.
SEM analysis of biofilm growth on SS surfaces
To confirm the images presented in Figure 4, SEM images were taken of the SS coupons after fluorescent imaging. The SEM images in Figure 6 show that the non-polarized surface was covered with cells (Figure 6A), while fewer cells colonized the polarized surface (Figure 6B). Higher magnification of individual cells on the non-polarized SS surface showed intact cell walls (Figure 6C). On the polarized SS surface, membrane-compromised cells surrounded by scattered deposits of material were observed (Figure 6D). It has been shown that cell walls and cell membranes of P. aeruginosa 19142 are deformed when treated with H2O2 (DeQueiroz and Day 2007). This is because H2O2 targets thiol groups of enzymes on the cell wall (Denyer and Stewart 1998). Intracellular contents are lost and, as a result, cell volume decreases (Linley et al. 2012). Similar images were also obtained by Diao et al. (2004) when cells were treated with H2O2. The scattered deposits observed in Figure 6D were identified as cell debris. These two observations provide further evidence that the electrochemical production of H2O2 at the SS surface was the cause of the delayed biofilm growth.
Figure 6.
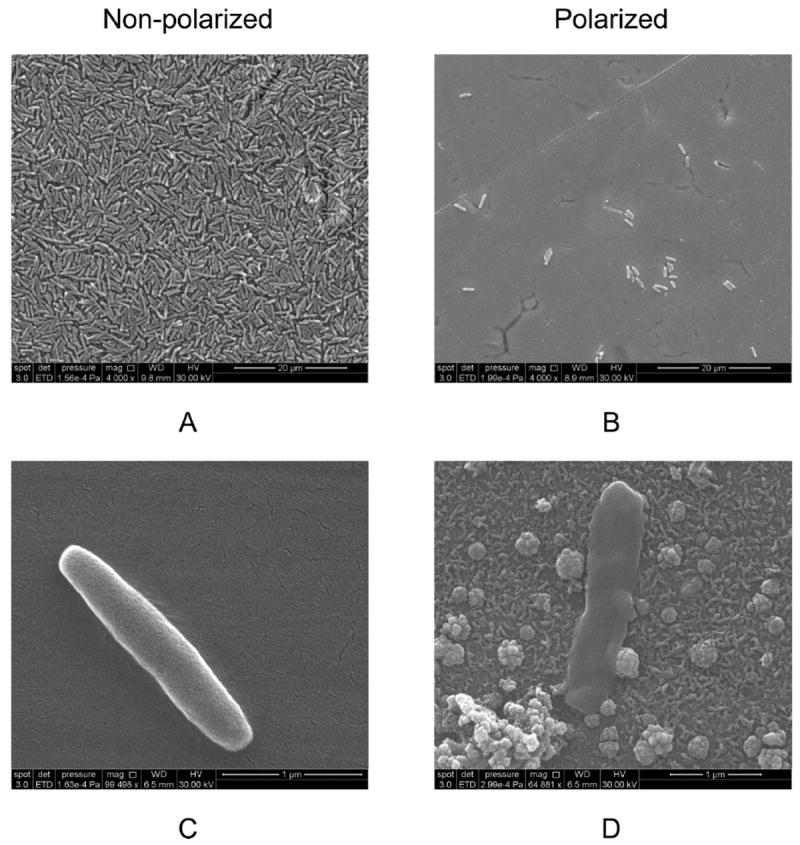
SEM images of cells on non-polarized (A and C) and polarized coupons (B and D).
Figures 4 and Figure 6B show that despite the 40-h polarization treatment, some cells were able to survive and persist on the polarized SS surface. This could be the result of heterogeneous current distribution on the SS surface, increased catalase production by these surviving cells, or a combination of both. Since these cells expressed GFP, they were most likely alive as demonstrated in the literature (Nancharaiah et al. 2005). Interestingly, catalase actively degrading H2O2 could be another reason for H2O2 not being detected in the bulk solution. This possibly explains why some of the previously published studies found H2O2, while others did not. Regardless of the mechanism that these cells used to survive, the bioelectrochemical effect did not kill all the cells on the polarized SS surfaces observed in this study. Thus, it is important to note that, although H2O2 has a biocidal effect on cells, the bioelectrochemical effect as a whole should be considered biostatic rather than biocidal.
Practical implications
In this research, it was demonstrated that H2O2 is produced on SS surfaces and that it can be used to prevent biofilm growth. However, for H2O2 production, the biofilm must be thin and allow oxygen penetration to the bottom. H2O2 cannot be produced when there is no DO on the SS surface. Thus, the thickness of the biofilm could be a critical factor when controlling biofilm growth electrochemically. Perhaps the best method of preventing cell growth on SS surfaces is to keep the potential of the surface at −600 mVAg/AgCl as soon as the process starts. Continuous H2O2 production may delay biofilm growth but may not eliminate long-term biofilm growth. Fewer cells might colonize but be able to survive and persist. Although a catalase-positive bacterium that can actively decompose H2O2 was used in this study, electrochemically produced H2O2 showed the biocidal effect. This technique could be more effective on catalase-negative bacteria. The presence of catalase could be critical in some systems if the biofilm producing catalase decomposes the produced H2O2 to oxygen. This could potentially prevent the biocidal effect of H2O2, as demonstrated by others (Liu et al. 1998; Stewart et al. 2000).
Conclusions
In this work, H2O2 production and dissolved oxygen consumption were quantified on polarized 316L SS surfaces using microelectrodes. The produced H2O2 was used to control P. aeruginosa PAO1 biofilm growth on the SS surfaces. The following conclusions have been reached: (1) H2O2 is produced at potentials below −400 mVAg/AgCl on SS surfaces. (2) When the SS surface is polarized to −600 mVAg/AgCl, the H2O2 concentration increases towards the surface, while the dissolved oxygen concentration decreases. There was no H2O2 production or dissolved oxygen consumption on non-polarized clean surfaces. (3) When polarized and non-polarized surfaces with identical biofilms continued to be fed with fresh medium, polarization prevented biofilm growth, while there was significant biofilm growth on non-polarized surfaces. (4) There was cell debris deposited on the polarized surfaces, while there was no cell debris on non-polarized surfaces, indicating the biocidal effect of H2O2. (5) Overall, H2O2 produced on a SS surface can be used to control biofilm growth. However, this requires a continuous flux of dissolved oxygen to the surface.
Acknowledgments
This work is supported by NSF-CAREER award #0954186 and the US Office of Naval Research (ONR), award #N00014-09-1-0090. The authors would like to thank Dr Michael Franklin for providing the P. aeruginosa PAO1 strain and Alice Dohnalkova for discussions about SEM images. The authors are also grateful to the Franceschi Microscopy and Imaging Center of Washington State University for the use of their facilities and staff assistance.
References
- Babauta JT, Nguyen HD, Beyenal H. Redox and pH microenvironments within Shewanella oneidensis MR-1 biofilms reveal an electron transfer mechanism. Environ Sci Technol. 2011;45:6654–6660. doi: 10.1021/es200865u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard AJ, Faulkner LR, editors. Electrochemical methods: fundamentals and applications. New York (NY): John Wiley & Sons, Inc; 2001. [Google Scholar]
- Bard AJ, Parsons R, Jordan J. Standard potentials in aqueous solutions. New York (NY): Marcel Dekker, Inc; 1985. p. 833. [Google Scholar]
- Busalmen JP, de Sanchez SR. Adhesion of Pseudomonas fluorescens (ATCC 17552) to nonpolarized and polarized thin films of gold. Appl Environ Microbiol. 2001;67:3188–3194. doi: 10.1128/AEM.67.7.3188-3194.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costerton JW, Ellis B, Lam K, Johnson F, Khoury AE. Mechanism of electrical enhancement of efficacy of antibiotics in killing biofilm bacteria. Antimicrob Agents Chemother. 1994;38:2803–2809. doi: 10.1128/aac.38.12.2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis CP, Shirtliff ME, Trieff NM, Hoskins SL, Warren MM. Quantification, qualification, and microbial killing efficiencies of antimicrobial chlorine-based substances produced by iontophoresis. Antimicrob Agents Chemother. 1994;38:2768–2774. doi: 10.1128/aac.38.12.2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Schamphelaire L, Boeckx P, Verstraete W. Evaluation of biocathodes in freshwater and brackish sediment microbial fuel cells. Appl Microbiol Biotechnol. 2010;87:1675–1687. doi: 10.1007/s00253-010-2645-9. [DOI] [PubMed] [Google Scholar]
- Del Pozo JL, Rouse MS, Patel R. Bioelectric effect and bacterial biofilms. A systematic review. Intl J Artif Organs. 2008;31:786–795. doi: 10.1177/039139880803100906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Pozo JL, Rouse MS, Mandrekar JN, Steckelberg JM, Patel R. The electricidal effect: reduction of Staphylococcus and Pseudomonas biofilms by prolonged exposure to low-intensity electrical current. Antimicrob Agents Chemother. 2009;53:41–45. doi: 10.1128/AAC.00680-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denyer SP, Stewart GSAB. Mechanisms of action of disinfectants. Int Biodeterior Biodegr. 1998;41:261–268. [Google Scholar]
- DeQueiroz GA, Day DF. Antimicrobial activity and effectiveness of a combination of sodium hypochlorite and hydrogen peroxide in killing and removing Pseudomonas aeruginosa biofilms from surfaces. J Appl Microbiol. 2007;103:794–802. doi: 10.1111/j.1365-2672.2007.03299.x. [DOI] [PubMed] [Google Scholar]
- Diao M, Li XY, Gu JD, Shi HC, Xie ZM. Electron microscopic investigation of the bactericidal action of electrochemical disinfection in comparison with chlorination, ozonation and Fenton reaction. Process Biochem. 2004;39:1421–1426. [Google Scholar]
- Freguia S, Rabaey K, Yuan Z, Keller J. Non-catalyzed cathodic oxygen reduction at graphite granules in microbial fuel cells. Electrochim Acta. 2007;53:598–603. [Google Scholar]
- Hong SH, Jeong J, Shim S, Kang H, Kwon S, Ahn KH, Yoon J. Effect of electric currents on bacterial detachment and inactivation. Biotechnol Bioeng. 2008;100:379–386. doi: 10.1002/bit.21760. [DOI] [PubMed] [Google Scholar]
- Ica T, Caner V, Istanbullu O, Nguyen DH, Ahmed B, Call DR, Beyenal H. Characterization of mono- and mixed-culture Campylobacter jejuni biofilms. Appl Environ Microbiol. 2012;78:1033–1038. doi: 10.1128/AEM.07364-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewandowski Z, Beyenal H. Fundamentals of biofilm research. Boca Raton (FL): CRC Press; 2007. p. 452. [Google Scholar]
- Linley E, Denyer SP, McDonnell D, Simons C, Maillard JY. Use of hydrogen peroxide as a biocide: new consideration of its mechanisms of biocidal action. J Antimicrob Chemother. 2012;67:1589–1596. doi: 10.1093/jac/dks129. [DOI] [PubMed] [Google Scholar]
- Liu WK, Brown MRW, Elliott TSJ. Mechanisms of the bactericidal activity of low amperage electric current (DC) J Antimicrob Chemother. 1997;39:687–695. doi: 10.1093/jac/39.6.687. [DOI] [PubMed] [Google Scholar]
- Liu XF, Roe F, Jesaitis A, Lewandowski Z. Resistance of biofilms to the catalase inhibitor 3-amino-1,2,4-triazole. Biotechnol Bioeng. 1998;59:156–162. [PubMed] [Google Scholar]
- Logan BE, Hamelers B, Rozendal RA, Schrorder U, Keller J, Freguia S, Aelterman P, Verstraete W, Rabaey K. Microbial fuel cells: methodology and technology. Environ Sci Technol. 2006;40:5181–5192. doi: 10.1021/es0605016. [DOI] [PubMed] [Google Scholar]
- Matsunaga T, Lim TK. Electrochemical prevention of biofouling. Electrochem. 2000;68:847–852. [Google Scholar]
- Nancharaiah YV, Venugopalan VP, Wuertz S, Wilderer PA, Hausner M. Compatibility of the green fluorescent protein and a general nucleic acid stain for quantitative description of a Pseudomonas putida biofilm. J Microbiol Meth. 2005;60:179–187. doi: 10.1016/j.mimet.2004.09.016. [DOI] [PubMed] [Google Scholar]
- Nguyen HD, Cao B, Mishra B, Boyanou MI, Kemner KM, Fredrickson JK, Beyenal H. Microscale geochemical gradients in Hanford 300 Area sediment biofilms and influence of uranium. Water Res. 2012;46:227–234. doi: 10.1016/j.watres.2011.10.054. [DOI] [PubMed] [Google Scholar]
- Qiao Y, Bao SJ, Li CM. Electrocatalysis in microbial fuel cells-from electrode material to direct electrochemistry. Energy Environ Sci. 2010;3:544–553. [Google Scholar]
- Renslow R, Donovan C, Shim M, Babauta J, Nannapaneni S, Schenk J, Beyenal H. Oxygen reduction kinetics on graphite cathodes in sediment microbial fuel cells. Phys Chem Chem Physics. 2011;13:21573–21584. doi: 10.1039/c1cp23200b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirtliff ME, Bargmeyer A, Camper AK. Assessment of the ability of the bioelectric effect to eliminate mixed-species biofilms. Appl Environ Microbiol. 2005;71:6379–6382. doi: 10.1128/AEM.71.10.6379-6382.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart PS, Wattanakaroon W, Goodrum L, Fortun SM, McLeod BR. Electrolytic generation of oxygen partially explains electrical enhancement of tobramycin efficacy against Pseudomonas aeruginosa biofilm. Anti-microb Agents Chemother. 1999;43:292–296. doi: 10.1128/aac.43.2.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart PS, Roe F, Rayner J, Elkins JG, Lewandowski Z, Ochsner UA, Hassett DJ. Effect of catalase on hydrogen peroxide penetration into Pseudomonas aeruginosa biofilms. Appl Environ Microbiol. 2000;66:836–838. doi: 10.1128/aem.66.2.836-838.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoodley P, deBeer D, LappinScott HM. Influence of electric fields and pH on biofilm structure as related to the bioelectric effect. Antimicrob Agents Chemother. 1997;41:1876–1879. doi: 10.1128/aac.41.9.1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Borden AJ, van der Werf H, van der Mei HC, Busscher HJ. Electric current-induced detachment of Staphylococcus epidermidis biofilms from surgical stainless steel. Appl Environ Microbiol. 2004;70:6871–6874. doi: 10.1128/AEM.70.11.6871-6874.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetter KJ. Electrochemical kinetics: theoretical and experimental aspects. New York (NY): Academic Press; 1967. p. 789. [Google Scholar]