

ABSTRACT
Cell signaling can control the dynamic balance between filamentous and monomeric actin by modulating actin regulatory proteins. One family of actin regulating proteins that controls actin dynamics comprises cyclase-associated proteins 1 and 2 (CAP1 and 2, respectively). However, cell signals that regulate CAPs remained unknown. We mapped phosphorylation sites on mouse CAP1 and found S307 and S309 to be regulatory sites. We further identified glycogen synthase kinase 3 as a kinase phosphorylating S309. The phosphomimetic mutant S307D/S309D lost binding to its partner cofilin and, when expressed in cells, caused accumulation of actin stress fibers similar to that in cells with reduced CAP expression. In contrast, the non-phosphorylatable S307A/S309A mutant showed drastically increased cofilin binding and reduced binding to actin. These results suggest that the phosphorylation serves to facilitate release of cofilin for a subsequent cycle of actin filament severing. Moreover, our results suggest that S307 and S309 function in tandem; neither the alterations in binding cofilin and/or actin, nor the defects in rescuing the phenotype of the enlarged cell size in CAP1 knockdown cells was observed in point mutants of either S307 or S309. In summary, we identify a novel regulatory mechanism of CAP1 through phosphorylation.
KEY WORDS: Actin dynamics, Actin cytoskeleton, Kinase, Cell signaling, Cell migration
INTRODUCTION
The actin cytoskeleton is essential for many fundamental cell functions such as morphogenesis, cytokinesis, endocytosis, and cell polarization and migration. The cytoskeleton undergoes dynamic changes by regulating the balance of monomeric actin (G-actin) and filamentous actin (F-actin), a process called actin dynamics. One of the types of actin dynamics is ‘treadmilling’ (Paavilainen et al., 2004; Pollard and Cooper, 2009), whereby actin filaments maintain a constant length but are in a state of dynamic equilibrium. During treadmilling, monomeric actin depolymerizes from the pointed end (or minus end) because it has a lower affinity, but polymerizes onto the barbed end (or plus end), which has a higher affinity. Energy to maintain this state comes from the hydrolysis of ATP. Treadmilling can be exploited by cells to cause rapid changes in dynamics. Actin depolymerizing factor (ADF) proteins, such as cofilin sever ‘aged’ actin filaments to create shorter fragments and, hence, increase the amount of filament pointed ends and consequent depolymerization of monomeric actin from these ends (Andrianantoandro and Pollard, 2006; Michelot et al., 2007). The monomers then undergo nucleotide exchange, i.e. replacing the bound ADP with ATP, and are recycled to the barbed end of the filament for the next round of actin polymerization (Pollard and Cooper, 2009). Repeated cycles of actin filament turnover drives actin cytoskeletal rearrangement, and cofilin plays a central role in driving actin filament turnover (DesMarais et al., 2005; Lappalainen and Drubin, 1997). The activity of cofilin is controlled by the reversible phosphorylation of S3 (Yang et al., 1998). Thus, by modulating phosphorylation at S3, cell signaling pathways can control the actin cytoskeleton in order to regulate cell motility and chemotaxis; although cofilin regulates actin dynamics by itself it is assisted by other proteins.
Members of the cyclase-associated protein (CAP) family are cytoskeletal proteins that interact with cofilin and actin to regulate actin dynamics (Hubberstey and Mottillo, 2002; Ono, 2013). Mutation or knockdown of CAPs leads to problems with the actin cytoskeleton, including aberrant cell morphology, polarity and motility (Bertling et al., 2004; Freeman and Field, 2000; Kawamukai et al., 1992; Vojtek et al., 1991; Zhang et al., 2013). CAP regulation of the actin cytoskeleton is widely conserved, and defects in S. cerevisiae depleted of the gene encoding CAP are rescued by expression of CAP gene homologues from even distant species (Kawamukai et al., 1992; Matviw et al., 1992; Zelicof et al., 1993; Zhou et al., 1998). CAPs regulate actin dynamics at multiple levels. They bind and sequester actin monomers to prevent them from polymerizing (Gerst et al., 1991; Gieselmann and Mann, 1992; Zelicof et al., 1996), which helps to maintain a sufficiently large and readily accessible G-actin pool that is essential for rapid actin cytoskeletal reorganization (Zhou et al., 2014). Studies over the past decade have revealed that several domains within CAPs bind to actin, and that CAPs also facilitate nucleotide exchange of ATP onto monomers (Hubberstey and Mottillo, 2002; Ono, 2013). CAP1 and CAP2 are the two mammalian CAP isoforms. Of the two, CAP1 has been studied more extensively and its function as a regulator of the actin cytoskeleton and cell migration has been solidly established (Bertling et al., 2004; Moriyama and Yahara, 2002; Zhang et al., 2013; Zhou et al., 2014). Our work here focuses on CAP1.
CAP homologues have three conserved structural domains, the N-terminal domain, the C-terminal domain and a proline-rich middle domain (Hubberstey and Mottillo, 2002; Ono, 2013). All three domains contribute to actin filament turnover through interactions with cofilin, and G- and F-actin (Ono, 2013). In mammals, the C-terminal domain binds and sequesters G-actin, and also catalyzes nucleotide exchange of ATP onto ADP-bound G-actin, whereas in yeast this function is further enhanced by the Wasp homology 2 (WH2) domain, which is located towards the C-terminus of the middle domain (Chaudhry et al., 2010; Makkonen et al., 2013). Nucleotide recharging on ADP–G-actin in complex with cofilin is a key rate-limiting step (Nishida, 1985) and CAPs relieve the inhibitory effect of cofilin on recharging. The N-terminal domain of CAP binds the cofilin–ADP–G-actin complex first for subsequent nucleotide exchange, and CAPs can also directly bind F-actin to promote its severing (Chaudhry et al., 2013; Normoyle and Brieher, 2012). Moreover, the proline-rich middle domain interacts with profilin (Bertling et al., 2007; Makkonen et al., 2013). Finally, CAPs were recently shown to assemble into a hexameric oligomer (Chaudhry et al., 2013). Regions within the N-terminal domain (Quintero-Monzon et al., 2009; Yusof et al., 2005; Yusof et al., 2006) and C-terminal domain (Dodatko et al., 2004; Zelicof et al., 1996) can mediate CAP assembly into oligomers. However, the regulatory signals that modulate CAPs, if there are any, remain unknown.
We report here that mouse CAP1 is a phosphorylatable protein, with at least two phosphorylatable regulatory (hereafter referred to as phospho-regulatory) sites. Phosphorylation at S307/S309 prevents the association with cofilin and expression of mutants that fail to undergo this phospho-regulation disrupts the actin cytoskeleton. We identify glycogen synthase kinase 3 (GSK3) as a kinase that phosphorylates residue S309 and suggest that S309 functions with S307 as a tandem phospho-regulatory site to control association with cofilin and actin. Thus, phosphorylation of S309 by GSK3 (and potentially other kinases) is part of a regulatory mechanism that might facilitate association and dissociation of CAP1 with partner proteins cofilin and actin, essential interactions to promote actin filament turnover.
RESULTS
Mapping of phosphorylation sites on CAP1
We first tested potential phosphorylation of CAP1 in vivo through metabolic labeling of cells with radiolabeled orthophosphate, as previously described (Woodring et al., 2004) followed by immunoprecipitation of CAP1 from cell lysates with an anti-CAP1 monoclonal antibody (Freeman and Field, 2000). Radioactivity was detected in CAP1, suggesting that CAP1 is, indeed, phosphorylatable (data not shown). We next mapped phosphorylation sites on mouse CAP1 by mass spectrometry assays. To do this, we transiently expressed 6×His-CAP1 in HEK293T cells (Zhang et al., 2013). 6×His-CAP1 in the lysate was precipitated by using Ni-NTA beads and the 6×His-CAP1 band was excised from a Coomassie-Blue-stained protein gel and subjected to mass spectrometry. As shown in Table 1, a total of nine phosphorylation sites were identified in three independent assays. Some of these sites, such as S36, S307, S309 and T314, were well-conserved among the mammalian CAP1 homologues. The NCBI ID numbers of the CAP homologues compared by us are as following: (1) S. pombe Cap1: NP_587817.1; (2) M. musculus CAP1: NP_031624.2; (3) H. sapiens CAP1: NP_001099000.1; (4) S. cerevisiae CAP (SRV2): NP_014261.1; (5) R. norvegicus CAP1: NP_071778.2; (6) C.elegans CAS-2: NP_491324.1; (7) D. melanogaster Capt: AAF51408.2; (8) A. thaliana CAP1: NP_195175.1; (9) M. musculus CAP2: NP_080332.1.
Table 1. Phosphorylation sites identified on mouse CAP1.
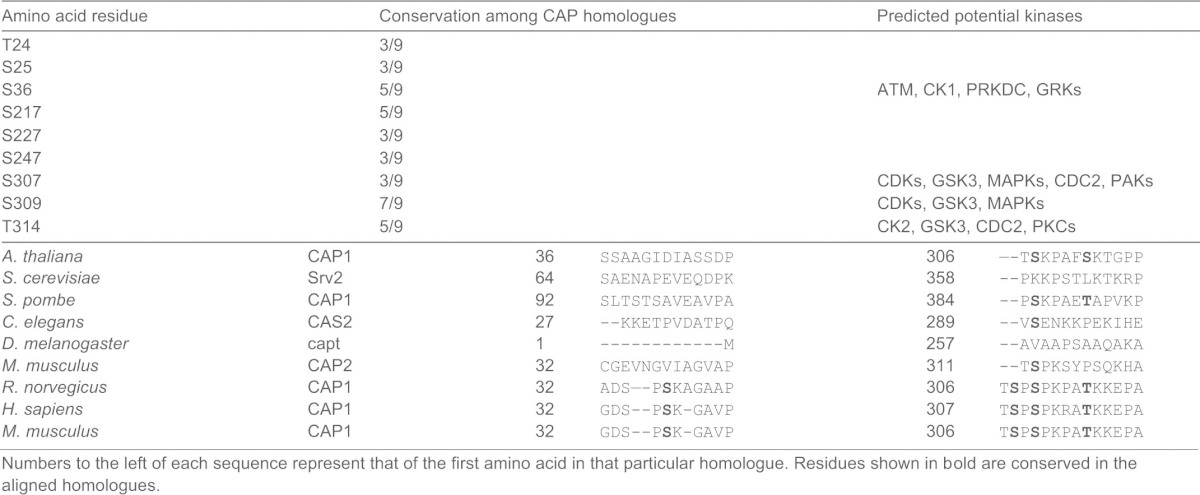
Numbers to the left of each sequence represent that of the first amino acid in that particular homologue. Residues shown in bold are conserved in the aligned homologues.
S36 and S307/S309 are phospho-regulatory sites
To assess the significance of each phosphorylation site, we generated CAP1 point mutants, expressed the mutants in NIH3T3 cells and then evaluated their effects on the actin cytoskeleton. The phosphorylation sites were mutated to either alanine (A), which mimics a non-phosphorylatable residue, or aspartic acid (D), which is phosphomimetic. Where two phosphorylation sites are adjacent to each other (T24/S25) or are separated by a single residue (S307/S309), we mutated both sites together. As a result, we generated a total of fourteen constructs that express CAP1 mutants fused to green fluorescent protein (GFP), as listed in supplementary material Table S1. These mutants were: T24A/S25A, T24D/S25D, S36A, S36D, S217A, S217D, S227A, S227D, S247A, S247D, S307A/S309A (hereafter referred to as AA), S307D/S309D (hereafter referred to as DD), T314A and T314D.
We next stably expressed the mutants in NIH3T3 cells and compared them with wild-type CAP1 (WTCAP1). Comparable expression levels of the exogenous proteins were verified by western blotting with a GFP antibody (Fig. 1A, shown only for WTCAP1 and S36A, S36D, AA and DD – the four mutants characterized further). Using phase-contrast microscopy, we found that expression of WTCAP1 caused increased peripheral ruffles (data not shown). In contrast, mutation of two phosphorylation sites, S36 and S307 and/or S309 caused significant, yet distinct, morphological changes. Many cells that expressed the S36D mutant were smaller, with multiple randomly positioned lamellipodia. The cells expressing the DD mutant were larger and had reduced peripheral ruffling. In contrast, AA-expressing cells – albeit also larger – still had well-developed lamellipodia. We also found that cells expressing T314A had a phenotype similar to that of AA-expressing cells. To quantify the changes, we measured the cell area of 50 cells from three fields; results from three independent experiments were analyzed using Student's t-test as shown in Fig. 1B. Consistently, S36D-expressing cells were significantly reduced in size, whereas AA-, DD- and T314-expressing cells were significantly increased in size when compared to cells expressing WTCAP1. We also scored cells with prominent peripheral ruffling (lamellipodia) and found that the DD- and S36A-expressing cells had the least number of cells with lamelipodia (Fig. 1C). These data suggest that CAP1 is regulated by phosphorylation within two regions, the first is near residue S36 and the second is within amino acid residues 307–314. The rest of the mutants did not cause any obvious morphological changes compared to WTCAP1, suggesting that their phosphorylation has a less significant role in the regulation of CAP1 function.
Fig. 1.

Expression of phospho mutants of S36 and S307/S309 in NIH3T3 fibroblasts caused morphological changes. (A) Expression of GFP-fusion WTCAP1 and mutants. Cell lysates from the NIH3T3 stable cells harboring an empty GFP vector, a plasmid expressing GFP-fusion WTCAP1 and the four mutants were resolved on 10% SDS-PAGE followed by western blotting using a GFP antibody. GAPDH was used as a loading control. (B) Effects of phospho mutants on cell size. The area of 50 cells per field were measured individually (using ImageJ software) from three fields for each experiment. Results from three independent experiments were analyzed by using Student's t-test, the error bars represent ±s.e.m; *P<0.05, **P<0.01. (C) Mutations S36A or S307D/S309D reduced the percentage of cells with lamellipodia (LP). Cells with lamellipodia were counted and the percentages were calculated. 100 cells per field were counted from three fields for each cell type, and results from three independent experiments were analyzed and shown as in B.
Expression of S36 or S307/S309 mutants in NIH3T3 cells results in changes of the actin cytoskeleton
We next stained cells with fluorescently tagged (Alexa-Fluor-488- or Alexa-Fluor-594-conjugated) phalloidin to visualize F-actin. When we examined cells cultured on Nunc chamber slides for wide-field fluorescence imaging, we found that control cells expressing the GFP vector rarely developed lamellipodia but had prominent stress fibers (Fig. 2A, upper left panel, cells indicated by arrows). Expression of WTCAP1 did yield lamellipodia (Fig. 2A, upper right panel, cells indicated by arrows), a characteristic of motile cells because these structures help establish cell polarity and are required for cell migration. Interestingly, many S36D-expressing cells had a disrupted actin cytoskeleton that appeared collapsed, some of them had lamellipodium-like protrusions on the ‘sides’ of otherwise spindle-shaped cells (Fig. 2A, middle right panel, arrows indicate cells with a contracted actin cytoskeleton and arrowheads indicate randomly positioned protrusions). Both the DD- and AA-expressing cells had enhanced stress fibers (Fig. 2A, bottom panels, cells indicated by arrows), and looked similar to the vector-expressing control cells. The stress fibers in DD- and AA-expressing cells are also similar to those observed in CAP1 knockdown fibroblasts and HeLa cells (Bertling et al., 2004; Zhang et al., 2013), presumably because of reduced depolymerization of actin filaments due to impaired CAP1 activity. However, AA-expressing cells had more lamellipodia than DD-expressing cells. We calculated the percentage of cells with prominent lamellipodia in a way similar to the one described for Fig. 1D, and found that S36D- and AA-expressing cells had similarly high percentages of cells with developed lamellipodia as WTCAP1-expressing cells (Fig. 2B). The changes in cell morphology and the actin cytoskeleton caused by the mutants are briefly summarized in supplementary material Table S1.
Fig. 2.

Actin cytoskeletal alterations caused by expression of CAP1 phospho mutants. (A) Wide-field fluorescence imaging shows alterations in lamellipodia and stress fibers in cells that express the mutants. The actin cytoskeleton was stained with fluorescently tagged phalloidin (Alexa-Fluor 594). Cells indicated by arrows show the typical changes to the actin cytoskeleton, as discussed for S36D- and S307/S309-mutant-expressing cells; arrowheads indicate randomly positioned protrusions in the S36D-expressing cells. (B) Percentage of cells with prominent lamellipodia. Fifty cells were counted for each cell type and the experiment was repeated for three times. The data were analyzed using Student's t-test and shown as mean ± s.d; **P <0.01). (C) Confocal fluorescence microscopy showing further details of the actin cytoskeletal alterations. F-actin was stained with fluorescently tagged phalloidin (Alexa-Fluor 488) and images were taken using a 60×lenses with a BD Pathway 855 imaging system (GFP is stably expressed at low levels and does not interfere with Alexa-Flour 488 in microscopy). Arrows indicate changes to the stress fibers of each cell type, as discussed; arrowheads indicate lamellipodia. (D) Transwell migration assays revealed reduced motility in both S36D and DD cells. The data were analyzed and are shown as in B; *P<0.05, ** P<0.01. The experiment was repeated three times with similar results.
We further examined the actin cytoskeleton by conducting confocal microscopy. In addition to enhanced lamellipodia, cells expressing WTCAP1 had remarkably reduced stress fibers (Fig. 2C, right top panel indicated by arrowheads and arrows). The mutants also affected stress fibers in some of the S36D-expressing cells where the stress fibers had a discontinuous structure (Fig. 2C, right middle panel, indicated by arrows), in contrast to the straight stress fibers seen in control cells as well as stress fibers typical seen in mammalian cells (not shown). We speculate that the abnormal stress fiber structure contributes to the contracted actin cytoskeleton in these cells. Also, we confirmed significantly enhanced stress fibers in DD- and AA-expressing cells that were also somewhat thicker than those in GFP-vector-expressing cells (Fig. 2C, bottom panels, indicated by arrows). Thick stress fibers are known to hinder cell migration (Tojkander et al., 2012). Stress fibers in AA-expressing cells were similar to those of DD-expressing cells; however, unlike DD-expressing cells, AA-expressing cells still developed lamellipodia (Fig.2C, bottom left panel, arrowheads).
The changes in lamellipodia and stress fibers of S36D- and DD-expressing cells suggest that expression of these mutants affects cell migration, such as observed previously in Akt1-knockout mouse embryonic fibroblasts (MEFs) cells (Zhou et al., 2006). Because changes to lamellipodia and stress fibers were most pronounced in S36D- and DD-expressing cells, we tested the motility of these cells in Transwell migration assays. As shown in Fig. 2D, expression of WTCAP1 stimulated cell migration when compared to cells expressing the GFP vector. In contrast, expression of either S36D or DD significantly reduced migration. These results suggest that the randomly oriented protrusions in S36D cells caused difficulty in establishing and maintaining cell polarity during directional cell migration. In addition, it is also possible that the discontinuous stress fibers hindered cell migration, because stress fibers generate and transduce tension force to pull up the trailing edge of the cell during migration. As for DD-expressing cells, they had the lowest rate of migration, probably because of defects in their stress fibers and cell polarity.
Effects of S307/S309 phosphorylation on association of CAP1 with cofilin and actin, and evidence that the two residues function in tandem
Changes to the actin cytoskeleton caused by the S307/S309 mutations suggest an impaired activity of CAP1, which led us to reason that phosphorylation on these sites may affect the association of CAP1 with cofilin or actin, two binding partners of CAP1. To avoid the complications of interpreting results caused by the presence of endogenous CAP1, we used CAP1-knockdown HeLa cells to re-express WTCAP1 or CAP1 mutants (Zhang et al., 2013). Plasmids expressing WTCAP1 or CAP1 mutants were modified by introducing mismatches to the small hairpin RNA (shRNA)-target sequence so that they were not recognized by shRNA used to stably knock down CAP1, but without changing the amino acid sequence. Stable re-expression of 6×His-tagged WTCAP1, S36A, S36D, AA or DD mutants was confirmed by western blotting and immunofluorescence (Fig. 3 and Fig. 5A). We tested cofilin binding in a GST–cofilin pull-down assay that we had developed previously (Zhang et al., 2013). Interestingly, we found striking differences between the DD and AA mutants in their capabilities to bind cofilin; the DD mutant virtually lost cofilin binding, whereas the AA mutant had drastically increased cofilin binding that is much higher than that of WTCAP1 (Figs 3A, quantified results shown in Fig. 3B). We also found that the S36A mutant, similar to the DD mutant, had reduced cofilin binding. It is noteworthy that NIH3T3 cells expressing the S36A or DD mutant had the lowest number of lamellipodia (Fig. 1D); these results suggest that cofilin binding is crucial for CAP1 activity in facilitating the formation of these F-actin-rich structures, and that cofilin binding is regulated by phosphorylation of S36 and S307/S309.
Fig. 3.

Altered binding of cofilin and actin for the S307/S309 mutants, and evidence that S307 and S309 function in tandem. (A) GST-cofilin pull-down revealed loss of cofilin binding of the DD mutant, whereas the AA mutant showed drastically increased cofilin binding. Equal amounts of GST-cofilin bound to glutathione beads (∼10–20 µg) were added to lysates containing ∼400 µg total protein harvested from CAP1 knockdown HeLa cells re-expressing WTCAP1 or the mutants. Tagged CAP1 co-precipitated with cofilin was detected with a 6×His antibody and GST-cofilin was detected with a cofilin antibody. (B) Quantification of results shown in A. Three independent experiments of GST–cofilin pull-down were measured by densitometry, and the results were analyzed and shown in the graph; error bars represent ±s.e.m.; *P<0.05, **P<0.01. (C) Actin co-precipitation assays reveal reduced actin binding for the AA mutant. Cell lysates prepared from CAP1-knockdown HeLa cells re-expressing WTCAP1 or the mutants were incubated with Ni-NTA beads, and the beads were precipitated and washed with lysis buffer containing 20 mM imidazole. Samples were resolved on SDS-PAGE, and detected with antibodies against actin and 6×His in western blotting. (D) Quantification of results shown in C. Three independent experiments were measured by densitometry, results were analyzed and are shown in the graph; error bars represent ±s.e.m.; **P<0.01. (E,F) A single S307A or S309 mutation did not mimic the drastic increase in cofilin binding, the experiments and data analysis were done in a similar manner as described for A and B. (G,H) A single S307A or S309A mutation did not mimic reduced actin binding. Experiments and data analysis were done as described for C and D; **P<0.01. In panels B, D, F and H, the y-axis represents the capability for cofilin and/or actin binding; the value for WT CAP1 was set at 1, and values for the mutants are relative to those of WT CAP1.
Fig. 5.

Effects of single and double mutations of CAP1 phosphorylaton sites on the subcellular localization of CAP1 and the rescue of the enlarged-cell-size phenotype in the CAP1-knockdown HeLa cells. (A) Confocal images showing cells re-expressing a CAP1 mutant (S309A, S309D, AA, DD, S36A or S36D) or WTCAP1. Cells were fixed with 3.7% formaldehyde and stained with mouse anti-Xpress followed by Alexa-Flour 594 for CAP1 and Alexa-Flour-488-conjugated phalloidin for F-actin. The arrows indicate peripheral localization of CAP1 mutants (AA and S309A). (B) Quantification of cell area (measured with ImageJ software) carried out similarly as done for that shown in Fig. 1C. Results from three independent experiments were analyzed using Student's t-test and plotted in the graph where the error bars represent ±s.e.m; **P<0.01.
We next examined actin binding in lysates of S36A-, AA- or DD-expressing HeLa cells by using Ni-NTA beads to precipitate 6×His-CAP1, followed by western blotting to detect co-precipitated actin (Zhang et al., 2013). We found that, although the DD mutation did not affect actin binding, the AA mutation had significantly reduced actin binding (Fig. 3C, quantified results shown in Fig. 3D). Thus phosphorylation can modulate actin binding as well.
Because AA and/or DD mutants harbor mutations at both S307 and S309, we constructed point mutations at either S307 or S309 to determine whether one mutation alone affected cofilin or actin binding. As shown in Fig. 3E and quantified in Fig. 3F, neither S307A nor S309A showed increased binding to cofilin observed in the AA mutants. Similar results were observed when we tested actin binding. As shown in Fig. 3G and quantified in Fig. 3H, although the AA mutant had reduced actin-binding capability, single mutants (S307A and S309A) both bound normally to actin. These results suggest that S307 and S309 function in tandem, and phospho-regulation at both residues collaboratively controls the interaction of CAP1 with cofilin and actin.
Although CAP1 binds both G- and F-actin, it has a preference for G-actin (Ono, 2013). We next tested whether this preference is changed in the S307/S309 mutants. We treated lysates from HeLa cells re-expressing WTCAP1 and these mutants with latrunculin A to depolymerize the F-actin. As shown in supplementary material Fig. S1, treatment with latrunculin A increased actin binding for both WTCAP1 and the AA and DD mutants in a similar fashion, and the AA mutant consistently had the least actin binding with or without the treatment. These results suggest that the S307/S309 mutation of CAP1 does not alter its binding preference for G-actin.
GSK3 phosphorylates S309 on CAP1
To aid in identifying kinase(s) for the S307/S309 site, we generated a phospho-specific antibody designed to detect phosphorylated S307 and S309, as described in Materials and Methods. Western blotting of transiently expressed GFP-fusion WTCAP1, and the AA and DD mutants show that the phospho-specific antibody recognized GFP-WTCAP1 and endogenous CAP1 but not the GFP-fusion AA or DD mutants (supplementary material Fig. S2A). We further characterized the antibody in peptide blocking assays to see whether it only recognizes doubly phosphorylated antigen. As shown in supplementary material Fig. S2B, the doubly phosphorylated peptide (S307p/S309p) completely blocked detection of the phospho signal on CAP1 in cells, whereas the S309-phosphorylated peptide (S309p) modestly blocked the signal, suggesting that the antibody also recognizes CAP1 phosphorylated at S309 alone, although it seems substantially less effective in blocking the signal. We detected phospho signals on CAP1 from all cells that we tested, including HEK293T, HeLa, NIH3T3 and the pancreatic cell line hTERT-HPNE (Lee et al., 2003) (Figs 4 and 6; no data shown for NIH3T3). These results support the notion that S307/S309 is a phospho-regulatory site across different cell (and tissue) types, although mass spectrometry mapping was conducted with the protein expressed in HEK293T cells.
Fig. 4.

GSK3 phosphorylates S309 on CAP1. (A) Treatment of HEK293T cells with GSK3 inhibitors LiCl, 6-BIO and SB216763 all reduced CAP1 phosphorylation at S307/S309. Cells were treated with the inhibitors for 14 hrs, and cell lysates were prepared for western blotting with both CAP1 and phospho-CAP1 antibodies. (B) Signals from three independent experiments were measured using densitometry and analyzed using Student's t-test and plotted in the graph; error bars represent ±s.e.m; *P<0.05, ** P<0.01. <@?show=[to]?>(C) Knockdown of GSK3β with lentiviral shRNA in HeLa cells reduced S307/S309 phosphorylation. HEK293FT cells were transfected using Lentiviral Packaging Mix together with the shRNA constructs. Supernatant medium was collected 48 hrs post transfection and used to infect HeLa cells. The cells were harvested 48 hrs post infection, and lysates were prepared and used in western blotting. (D) CAP1 co-precipitates with GSK3β. Lysates from CAP1-knockdown HeLa cells re-expressing 6xHis-CAP1 or a control vector were incubated with Ni-NTA beads to precipitate 6xHis-CAP1. The precipitates were resolved on SDS-PAGE and blotted with both 6xHis and GSK3β antibodies. (E) Kinase assays mapping S309 as the GSK3 site. Peptides harboring an alanine mutation at either S307 or S309 with a phosphorylated or dephosphorylated T314 (a.a. 305–316, QTSPSPKPATKK) were used as substrates in the kinase assays to test their phosphorylation by GSK3β; the GSK3 substrate GSM peptide (RRRPASVPPSPSLSRHSSHQRR; the serine residue shown in bold is phosphorylated) was used as a positive control. Samples were resolved on a 10–20% Tris-Tricine gradient gel (Bio-Rad) and signals were detected by autoradiography. Asterisks indicate quantified signals from the phosphorylated peptide, the arrowhead indicates partially degraded substrate. (F) Kinase assay results supporting T314 as the priming site for S309. A dephosphorylated T314 abolished the phosphorylation by GSK3; also both GSK3α and GSK3β phosphorylated S309. In E and F, numbers below the gels indicate the intensity of the signals in relation to the positive control.
Fig. 6.

Effects of GSK3 inhibition on the actin cytoskeleton and CAP1 localization. (A) Treatment with 6-BIO reduced S307/S309 phosphorylation of CAP1 in hTERT-HPNE cells. (B) GSK3 suppression caused accumulation of stress fibers, reduced cell polarity and loss of CAP1 enrichment at the leading edge of the cell. Control cells (top panels) had well-established cell polarity and leading edges (indicated by arrows), cells treated with 6-BIO for 5 hrs lost this polarity (bottom panels; cells indicated by arrows). Cells were stained with CAP1 antibody and visualized with an Alex-Fluor 594 goat anti-mouse secondary antibody in addition to phalliodin conjugated to Alexa-Fluor 488. CAP1 is enriched at the leading edge of the control cells, whereas the cytosolic areas immediately behind the leading edges had very weak CAP1 staining (indicated by arrowheads). In contrast, CAP1 had a diffuse localization across the cytosol in cells treated with 6-BIO, including the area mentioned above. (C) GSK3 suppression caused relocalization of both WTCAP1 and the AA mutant. CAP1 knockdown HeLa cells that re-express WTCAP1, and AA or DD mutants were treated with 6-BIO, and tagged CAP1 was stained with an X-press antibody followed by visualization using an Alexa-Fluor 594 goat anti-mouse secondary antibody. The arrows indicate peripheral localization of WTCAP1 and the AA mutant, and both of which were abolished by suppression of GSK3. (D) GSK3 inhibition led to reduced cofilin activity, including downregulation of cofilin as well as elevated cofilin phosphorylation at S3. HeLa cells were treated with 5 µM 6-BIO for 5 hrs, and cofilin and S3-phosphorylated cofilin were detected in western blotting.
By using the Net-Phos2.0 program to identify potential kinases that may phosphorylate CAP1, the Ser/Thr kinase GSK3 (Kim and Kimmel, 2000; Woodgett, 2001) was predicted to be one of the kinases to phosphorylate both S307 and S309 (Table 1). Because GSK3 is a regulator of cell polarity and migration (Kim and Kimmel, 2006; Sun et al., 2009; Wu et al., 2011), we further investigated GSK3. Three commonly used GSK3 inhibitors LiCl (Klein and Melton, 1996), 6-BIO (6-bromoindirubin-3′-oxime) (Meijer et al., 2003) and SB216763 (Coghlan et al., 2000) were used in western blots with the phospho-CAP1 antibody. Treatment of HEK293T cells with all three inhibitors reduced S307/S309 phosphorylation (Fig. 4A, quantified in Fig. 4B). The PI3 kinase inhibitor LY294002 caused a modest stimulation of the phosphorylation, consistent with a negative regulation of GSK3 through PI3K/Akt signaling (Clodfelder-Miller et al., 2005). We found that forskolin, an activator of protein kinase A (PKA), reduced the phosphorylation, which may result from indirect inhibition of GSK3 through activated PKA (Fang et al., 2000). We also tested several other inhibitors – including the ERK inhibitor U0126, the JNK inhibitor XVI and the ROCK inhibitor RKI-1447 – and none of them reduced phosphorylation at S307/S309 (supplementary material Fig. S3A, quantified in supplementary material Fig. S3B). Thus, the inhibitor data suggest that GSK3 is a kinase targeting S307/S309. We also directly tested several kinases that regulate the actin cytoskeleton and/or cell migration, including Akt, LIM domain kinase 1 and the serine/threonine-protein kinase PAK1 in kinase assays, using purified porcine CAP1 as a substrate; however, none of these kinases phosphorylated CAP1 (data not shown). We next silenced GSK3β in HeLa cells by using two previously validated lentiviral shRNA constructs (Yoeli-Lerner et al., 2009). As expected, the constructs efficiently knocked down GSK3β and also reduced phosphorylation of S307/S309 (Fig. 4C). We further found that GSK3 co-precipitates with CAP1 in HeLa cells (Fig. 4D) or with CAP1 purified from bacteria (data not shown), suggesting a physical interaction between CAP1 and GSK3. Taken together, these results strongly support the conclusion that GSK3 phosphorylates CAP1 at S307/S309.
To find out whether S307 or S309 is the GSK3 phosphorylation site, we performed in vitro kinase assays and using synthesized CAP1 peptides as substrates. GSK3 only phosphorylates substrates that are primed through prior phosphorylation of a serine or threonine residue located four or five residues after its actual phosphorylation site, i.e. [S/T]xxx[S(P)/T(P)] or [S/T]xxxx[S(P)/T(P)], where x can be any amino acid (Cole et al., 2004; Frame and Cohen, 2001). Because we also mapped T314 as a site of phosphorylation (Table 1), the combination of S309/T314 (separated by four residues), but not S307/T314 (separated by six residues), fits the motif for a GSK3 site (Cole et al., 2004). We, thus, predicted S309 as the GSK3 phosphorylation site, with T314 as its priming site. To test this we synthesized peptides on the basis of the mouse CAP1 sequence (QTSPSPKPATKK, amino acid residues 305–316) harboring S307A or S309A mutations together with a phosphorylated or dephosphorylated T314 (all indicated in bold). As shown in Fig. 4E, GSK3β phosphorylated the S307/S309/T314(P) peptide, but a single S309A mutation (S307/A309/T314(P)) was sufficient to abolish the phosphorylation. In contrast, an S307A mutation (A307/S309/T314P) had no effect on its phosphorylation by GSK3β. A peptide derived from the known GSK3 substrate glycogen synthase 1 from muscle (GYS1) (RRRPASVPPSPSLSRHSSHQRR; the serine residue shown in bold is phosphorylated) was used as a positive control (Ryves et al., 2002). Thus, we mapped S309 as the actual GSK3 site. We further demonstrated that T314 is the priming site for S309. Whereas S307/S309/T314(P) was phosphorylated by GSK3β, the same peptide harboring a non-phosphorylated T314 (S307/S309/T314) was not phosphorylated by GSK3β (Fig. 4F). Finally, we also demonstrate that GSK3α, the other GSK3 isoform, phosphorylated S309 as well (Fig. 4F). Although we found that GSK3 can regulate S307/S309, we note that other cell signals might also regulate this site because treating cells with phorbol 12-myristate 13-acetate (PMA) reduced phosphorylation at S307/S309. Interestingly, when we tested the association between CAP1 and cofilin, we found that the PMA treatment increased cofilin binding (supplementary material Fig. S4). These results further support our conclusion that dephosphorylated CAP1 has increased binding with cofilin.
Roles of S307 and S309 in CAP1 function and localization
Our biochemical assay results support the notion that S307 and S309 – a GSK3 site – function in tandem to regulate CAP1 function. To further assess the role for each amino acid residue that impacts on CAP1 function in cells, we expressed AA, DD, S309A or S309D mutants in CAP1-knockdown HeLa cells – which were depleted of at least 90% endogenous CAP1 (Zhang et al., 2013). As shown in Fig. 5A and quantified in Fig. 5B, expression of WTCAP1 rescued the phenotype of the enlarged cell size of the CAP1-depleted cells as previously observed by us (Zhang et al., 2013). In contrast, the AA and DD mutants failed to rescue the phenotype of the enlarged cell size (Fig. 5A,B), consistent with the phenotypes of NIH3T3 cells caused by these mutants that suggest impaired CAP1 activities. Interestingly, the S309A and S309D mutants were both able to rescue the phenotype of the enlarged cell size (Fig. 5A,B), consistent with normal actin and cofilin binding these S309 point mutants (Fig. 3E-H). In fact, mutants S309A and S309D had an even more pronounced effect on cell size as compared to the effect by WTCAP1, because large cells were not observed in cells rescued by these mutants. In contrast, in WTCAP1-rescued cells some large cells were still observed (data not shown). We also stained the transfected CAP1 with an X-press antibody and found that the AA mutant localized predominantly to the cell periphery (indicated by arrows in Fig. 5A) much more strongly than WTCAP1, whereas the DD mutant showed a diffuse localization across the cytosol (Fig. 5A). The S309A mutant also localized to the cell periphery (indicated by arrows), although not as strongly as the AA mutant. The S309D mutant had a diffuse localization similar to the DD mutant (Fig. 5A). These results suggest that phosphorylation of S309 affects CAP1 localization, whereas phosphorylation of both S307 and S309 affects localization as well as activity. We also tested the rescue capability of the S36A and S36D mutants, and found that both mutants were able to rescue the phenotype of the enlarged cell size of CAP1-depleted cells (supplementary material Fig. S5).
Inhibition of GSK3 leads to actin cytoskeletal changes and loss of peripheral CAP1 localization
We next tested effects of GSK3 inhibition on the actin cytoskeleton and CAP1 in hTERT-HPNE pancreatic cells, which have very well-established cell polarity. We first confirmed that GSK3 inhibition also reduced CAP1 phosphorylation in this cell line (Fig. 6A). In a wound-healing assay, cells treated with vehicle control (Fig. 6B, upper panels) showed well-established cell polarity (leading edge indicated by arrows). In contrast, cells treated with 6-BIO lost cell polarity (Fig. 6B, lower panels, indicated by arrows). LiCl had a similar effect (not shown). These cells also had significantly enhanced and well organized stress fibers compared to the control cells. Moreover, treatment with 6-BIO also abolished CAP1 enrichment at the leading edge of the cell (Fig. 6B). Instead, CAP1 showed a diffuse localization across the cytosol, including the cytosolic area immediately behind the leading edge, where virtually no CAP1 staining was observed in the control cells (Fig. 6B, upper panels, indicated by arrowheads). The stress fiber phenotype is consistent with those observed in NIH3T3 cells that expressed mutants DD and AA, neither of the mutants was able to be regulated through reversible phosphorylation. However, the localization of CAP1 in GSK3-suppressed cells – in which CAP1 is expected to be not phosphorylated at S309 – is not consistent with the cell peripheral localization of the S309A and AA mutants (Fig. 5A). Thus, an unknown localization mechanism may be involved in these cells, which is independent of phospho-regulation of CAP1 by GSK3. We tested this by inhibiting GSK3 with 6-BIO in CAP1-knockdown HeLa cells that re-expressed WTCAP1 and the AA or DD mutants, and found that treatment abolished the peripheral localization of both WTCAP1 and the AA mutant (Fig. 6C), supporting this is, indeed, the case. We next looked into potential effects suppression of GSK3 might have on cofilin, a binding partner of CAP1, and found that the treatment with the inhibitor reduced cofilin expression and also elevated levels of cofilin phosphorylated on S3, suggesting loss of cofilin activity (Fig. 6D). The effect of GSK3 inhibition on cofilin expression and phosphorylation are consistent with the phenotype of enhanced stress fibers and loss of cell polarity. Because CAP1 activity depends on cofilin (Moriyama and Yahara, 2002), the effect of GSK3 on cofilin may be epistatic to its effect on CAPs.
Increased phosphorylation of S307/S309 in cells with a static actin cytoskeleton
To assess the role that S307/S309 phosphorylation has in regard to CAP1 function within the actin cytoskeleton, we examined the possible correlation of phosphorylated S307/S309 and actin dynamics by comparing the phosphorylation in cells that undergo dynamic reorganization of the actin cytoskeleton with phosphorylation in cells that have a static actin cytoskeleton. For this, one batch of HEK293T cells was cultured in suspension, whereupon their reorganization of the actin cytoskeleton is substantially reduced. Another batch was cultured on fibronectin-coated plates, on which they undergo dynamic rearrangement of the actin cytoskeleton during early and rapid spreading stage. Interestingly, we found remarkably elevated S307/S309 phosphorylation in cells grown in suspension, or when cells were cultured for extended time periods (e.g. 48 hrs; the left lane labeled as 0 min; Fig. 7A) and had reached near full confluence. In contrast, phosphorylation is significantly reduced in cells cultured on fibronectin-coated plates during the spreading stage. We quantified both CAP1 and phospho-CAP1 signals, calculated the ratio of phospho-CAP1 to CAP1 (Fig. 7B), and found CAP1 was significantly reduced in cells during spreading stage. These results suggest that cells that undergo a dynamic reorganization of the actin cytoskeleton have reduced phosphorylation. We propose that CAP1 phosphorylated at S307/S309 is inactive, consistent with the observation that the phosphomimetic DD mutant binds poorly to cofilin and is located throughout the cytosol.
Fig. 7.
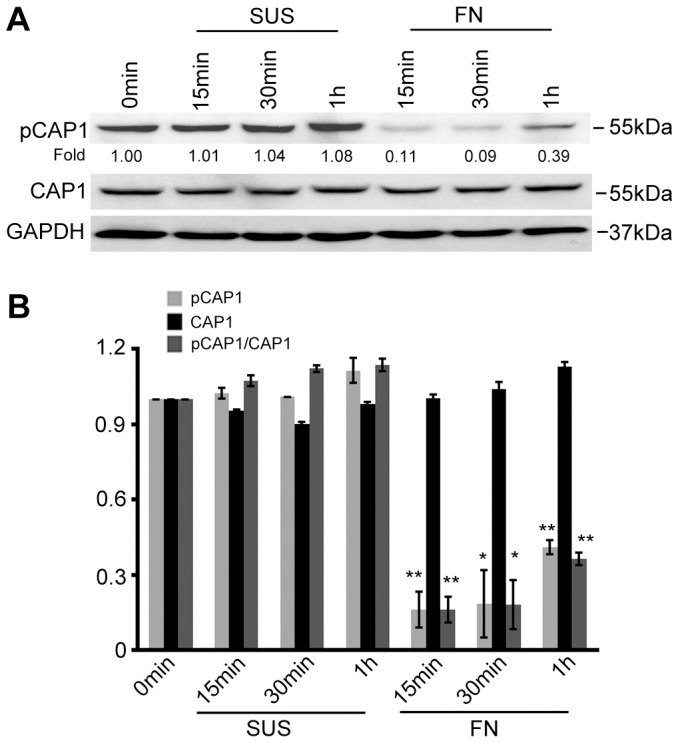
Phosphorylation at S307/S309 is reduced in cells undergoing dynamic actin cytoskeletal reorganization. (A) In HEK293T cells, CAP1 phosphorylation at S307/S309 was compared between cells cultured in suspension (SUS) and cells cultured on fibronectin-coated culture dishes (FN). Cells were harvested at the indicated time points and lysates were prepared for western blotting with antibodies against CAP1 and CAP1 phosphorylated at S307/S309 (pCAP1). GAPDH was used as a loading control. (B) Densitometric quantification of CAP1 and phospho-CAP1 signals from western blots of three independent experiments; ratios of phospho-CAP1 (pCAP1) versus CAP1 were calculated and plotted in the bar graph, error bars represent ±s.e.m. *P<0.05; **P<0.01.
DISCUSSION
We identify for the first time a phosphorylation-dependent regulatory mechanism of the versatile actin cytoskeletal protein CAP1. We show that regulation of both phosphorylation sites – at S307 and S309 – is important for CAP1 function as both play a role in the interaction of CAP1 with cofilin and actin. Single-point mutations at S307 or S309 did not alter binding to cofilin or actin; thus phosphorylation of S307 (by another kinase together with that of S309 collaboratively regulate CAP1 function. We found that GSK3 phosphorylates S309 by using inhibitors, shRNA and in-vitro kinase assays. GSK3 function has primarily been studied in its role in development and Wnt signaling (Wu and Pan, 2010). However, it also regulates cell polarity and migration, and GSK3 targets that modulate tubulin can mediate some of the relevant cell signals. The role for GSK3 in actin regulation is less established, although ARHGAP35 has been reported to be a GSK3 target (Jiang et al., 2008) and, thus, GSK3 might regulate the actin cytoskeleton through Rho.
The double mutation of S307D/S309D (mimicking a constitutively phosphorylated site) prevented cofilin binding, whereas the non-phosphorylatable double mutant S307A/S309A showed drastically increased cofilin binding, suggesting that phosphorylation serves as a regulatory mechanism to control association and/or dissociation of cofilin and CAP1. Releasing cofilin (and actin) from association with CAP1 following completion of nucleotide exchange on G-actin would allow continuous turnover of actin filaments by recycling cofilin. Although the DD mutant lost cofilin binding, it had normal actin binding. In contrast, although the AA mutant had dramatically increased cofilin binding, its actin binding was reduced. Actin binding is not necessary for yeast CAP to bind to cofilin and assist depolymerization or filament severing (Chaudhry et al., 2013), consistent with our results that the S36A and the DD mutants, which lost cofilin association, had the fewest lamellipodia. Of note, there are two actin binding sites on CAP1, the cofilin–ADP–G-actin binding site at the N-terminus and the G-actin binding/sequestering site at the C-terminus (Ono, 2013). The reduced actin binding in the AA mutant may reflect reduced binding of G-actin at the C-terminal domain, because binding to cofilin (which is in a complex with ADP–G-actin that initially binds to the N-terminal domain) is drastically increased. The observation that the DD mutant, which cannot bind to cofilin, also caused changes to lamellipodia that are very similar to those observed in CAP1-knockdown cells, suggested that phosphorylation inactivates CAP1. Inactivation of CAP1 by phosphorylation is also in line with the fact that GSK3 typically phosphorylates and inactivates its substrates (Woodgett, 2001). Our results suggest that the DD mutant is acting dominantly. This could be because of (1) the inability to bind the cofilin–ADP–G-actin to complete the nucleotide exchange or (2) the interference with CAPs itself because the functional form of CAP proteins appear to be a hexameric oligomer. These results support a universal role for mammalian CAP1 in balancing the ratio of actin filaments and monomers, a property that cells can use to modulate cell polarity and migration (Bertling et al., 2004; Zhang et al., 2013). Moreover, given the important function for CAP1 in controlling actin dynamics, it would not be surprising if multiple kinases phosphorylate S307 and/or S309 on CAP1 in order to allow other cellular signals to regulate this protein.
Our findings that the AA and DD mutants both caused changes to the actin cytoskeleton also suggest that phosphorylation does not merely inactivate CAP1 but, rather, is a step within a regulatory cycle, in which alternating phosphorylation and dephosphorylation serves to control cycles of association and dissociation of cofilin and actin. The remarkable difference of the subcellular localization of the AA and DD mutant also support this notion. In this model, phosphorylation at S307/S309 on CAP1 may facilitate the release of cofilin so it can be recycled for the next round of severing. Also, kinase(s) and phosphatase(s) are expected to work in concert to achieve the reversible phosphorylation of CAP1 to drive the continuous cycles of actin filament turnover under physiological conditions, although we have yet to identify any specific phosphatase for these residues.
An unexpected finding of this study is that phosphorylation at S307/S309 regulates cofilin binding because this site is located near the C-terminus of CAP1, which is distant from the minimal cofilin binding site that – in turn – is closer to the N-terminus. It thus remains unclear how phosphorylation at S307/S309 can regulate binding to cofilin. However, we note that the cofilin binding site has not been clearly defined. Additionally, mutations at the extreme N-terminal domain of the yeast CAP homologue (Srv2) can prevent localization and SH3 binding at the P2 site, which is near the equivalent of S307/S309 (Yu et al., 1999). Thus there are other CAP mutations distant from direct binding sites that can affect binding to partners. We suggest that phosphorylation can prevent cofilin binding indirectly.
Inhibition of GSK3 can cause loss of cell polarity as well as accumulation of stress fibers. We propose that GSK3 regulates CAP1 through at least two mechanisms. First, GSK3 (and potentially other kinases) phosphorylate CAP1 at S309 and promote CAP1 localization to the cytosol. Second, phosphorylation at S309 affects protein–protein interactions with actin and cofilin. Our data suggest that phosphorylation occurs not exclusively at S309 but is likely to require a ‘priming’ phosphorylation at T314, and requires at least another kinase to phosphorylate S307. The relative contribution of GSK3 in regulating CAP1 may vary in different cells compared to other kinases. The loss of this phospho-regulation by GSK3 inhibition is expected to disrupt CAP1 function and actin dynamics. Additionally GSK3 can also, as observed by us, affect actin dynamics through other mechanisms, such as cofilin phosphorylation and downregulation (Fig. 6). We show that reduced cofilin activity (including reduced expression) could be responsible for the unexpected CAP1 localization in GSK3-suppressed cells, overriding the direct effects of GSK3 on CAP1.
The results that compare the single point mutations of S309 and S307 with the AA and DD double mutations in biochemical and cell biological analyses support the notion that the two residues function in tandem. However, it remains to be determined whether the differences in binding cofilin and actin, as well as the subcellular localization of CAP1, are solely responsible for the cellular phenotype caused by these mutants. The cellular effects may involve additional mechanisms, such as effects on nucleotide exchange, of G-actin and CAP1 oligomerization, and studies are underway to unravel these potential scenarios. In addition to S307/S309, we also identified S36 as another phospho-regulatory site, although we have yet to identify the responsible kinase. Phospho-regulation may also be employed in fungal CAP, as we previously found that the N-terminal domain of CAP homologues from L. .edodes and S. pombe both interact with 14-3-3 protein homologues (Zhou et al., 2000), a family of chaperon proteins that bind phosphoproteins. We did not observe significant alterations in the morphology of cells that express CAP1 harboring mutations at the other phosphorylation sites that we found; however, we cannot exclude the possibility that they impact on other aspects of CAP1, such as subcellular localization. Some of these sites, such as S227 and S247 are within the middle domain, which is involved in CAP localization in budding yeast (Freeman et al., 1996).
In summary, our work reveals a phosphorylation-dependent regulatory mechanism of CAP1, by identifying a direct phosphorylation of CAP1 by GSK3 at S309, which, together with phosphorylation of S307, collaboratively regulates CAP1 function through association with cofilin and actin in order to control the actin cytoskeleton. These findings contribute mechanistic insights into how cell signaling controls the actin cytoskeleton through molecules that regulate actin dynamics.
MATERIALS AND METHODS
Miscellaneous reagents, cell culture, transfection and western blotting
Monoclonal antibodies against human CAP1 has been previously described (Freeman and Field, 2000). Mouse anti-GAPDH, goat anti-actin and rabbit anti-GSKβ were from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). Antibodies against GSK3α and phosphorylated cofilin were from Cell Signaling Technology Inc. (Danvers, MA). Anti-His antibody, Alexa-Fluor-488-conjugated phalloidin, Alexa-Fluor-594-conjugated phalloidin, Alexa-Fluor-488 goat anti-mouse IgG (H+L), fibronectin and platelet-derived growth factor (PDGF) were all from Life Technologies Inc. (Carlsbad, CA). Cofilin antibody was from the Cytoskeleton Inc. (Denver, CO). The phospho-specific polyclonal antibody against S307/S309 was developed by synthesis and injection of the peptides APKPQTS(P)PS(P)PKPA (amino acid residues 301–313) into rabbits followed by affinity-purification of the antibody (Genscript Inc., Piscataway, NJ). Lentiviral packaging mix was from Sigma Aldrich (St Louis, MO). PH797804, LY294002 and MEK inhibitor U0126 were from Sellekchem (Houston, TX). Forskolin and H-89 were from LC Laboratories (Woburn, MA). 6-BIO was from Biovision Inc. (Milpitas, CA) and SB216763 was from Cayman Chemical Co. (Ann Arbor, MI). JNK inhibitor XVI and ROCK inhibitor RKI-1447 were from EMD Millipore (Billerica, MA). Peptide substrates were synthesized by Neo-PeptideTM (Cambridge, MA). Fetal bovine serum (FBS) was from Hyclone Laboratories Inc. (Logan, UT). NIH3T3, HEK293T and HeLa cells were cultured in Dulbecco's modified Eagle's medium (DMEM), hTERT-HPNE cells were maintained in 75% DMEM supplemented with 25% medium M3 base. Cell transfection was conducted using FuGene 6 from Roche Diagnostics according to the manufacturer's protocol. Recombinant active GSK3α and GSK3β, as well as control GSK3 substrate peptide glycogen synthase 1 from muscle (GSM) [RRRPASVPPSPSLSRHSSHQRR; the serine residue shown in bold is phosphorylated] were from Millipore (Billerica, MA). Tissue culture dishes for immunofluorescence were from MatTek Corp. (Ashland, MA) and Transwell plates were from Corning Inc. (Corning, NY). Western blotting was conducted following standard procedures, and cell culture, transfection and preparation of cell lysates were conducted following procedures previously described (Zhou et al., 2003).
Mapping of phosphorylation sites on CAP1 by mass spectrometry
Metabolic labeling of cells with radiolabeled orthophosphate was done as previously described (Woodring et al., 2004). A plasmid expressing mouse CAP1 with a 6×His-tag at the N-terminus was transfected into HEK293T cells, and the expressed 6×His-tagged CAP1 was precipitated from the cell lysate with Ni-NTA beads as described (Zhang et al., 2013). The samples were resolved on SDS-PAGE followed by Coomassie Blue staining of the gel. The tagged CAP1 band was isolated, reduced with dithiothreitol (DTT) and trypsin-digested. Part of the recovered sample was used for protein identification, the remaining sample was loaded onto a TiO2 column for purification of the phosphorylated peptides (Cantin et al., 2007). The samples were subjected to a nanospray/Qstar-XL analysis and the data obtained was searched against the Swiss protein database. The mass spectrometry and data analysis were performed by the Proteomics Core Facility at the University of the Pennsylvania Perelman School of Medicine.
Construction of plasmids and establishment of NIH3T3 cells stably expressing the mutants
The plasmid that expresses a 6×His-tagged mouse CAP1 has been described previously (Zhang et al., 2013). To express the S36 and S307/S309 mutants in CAP1 knockdown HeLa cells, DNA fragments of mouse CAP1 mutants were amplified by using high-fidelity DNA polymerase and sub-cloning them into the pcDNA4 vector, following similar procedures as described for the WT CAP1 gene (Zhang et al., 2013). Point mutants of CAP1 yielding non-phosphorylatable (A) or phosphomimetic (D) amino acid site(s) were generated by using pEGFP-N1 vector and the primers listed in supplementary material Table S2. NIH3T3 cells stably expressing WTCAP1 or the point mutants were generated by transfection of cells, followed by selection with 1000 µg/ml neomycin for 2 weeks and pooling surviving cells.
Phase-contrast imaging and immunofluorescence
Phase-contrast imaging and immunofluorescence were conducted as described (Zhang et al., 2013). The actin cytoskeleton was stained with fluorescent (Alexa-Fluor-488- or Alexa-Fluor-594-conjugated) phalloidin. For immunofluorescence of CAP1, cells were blocked with 3% BSA for 1 hr after fixation and permeabilization, followed by incubation with a CAP1 monoclonal antibody diluted at 1∶200 (Freeman and Field, 2000) or an X-press antibody. Signals were visualized by incubation with an Alexa-Fluor-594 goat anti-mouse secondary antibody. The samples were observed and wide-field images were taken using a Zeiss fluorescence microscope as previously described (Zhou et al., 2003); wide-field fluorescence and confocal imaging was conducted as described previously (Zhang et al., 2013).
Measurement of cell area, percentage of cells with lamellipodia and Transwell migration assays
Areas of NIH3T3 and HeLa cells were measured by using ImageJ software and analyzed as described previously (Zhang et al., 2013). To quantify and compare the percentage of cells showing peripheral ruffling, cells with prominent peripheral ruffling were counted and the percentage was calculated. 100 cells per field were counted from three fields for each experiment, and results from three independent experiments were analyzed using Student's t-test and plotted in the graph where the error bars represent +s.e.m. Transwell assays were conducted as previously described (Zhou et al., 2006). Briefly, sub-confluent stable cells were serum-starved overnight and ∼3×104 cells were plated in triplicate into Transwell inserts with 8 µm pores placed in 12-well plates containing DMEM supplemented with 10 µg/ml PDGF. Cells were incubated for ∼16 hrs before they were stained, and migrated cells were counted.
Kinase assays
Kinase assays were conducted similarly as previously described (Zhou et al., 2006). 20 µg peptide substrate was mixed with 0.2 µg GSK3 (α or β) in a 25 µl reaction mixture containing 20 µM non-radiolabeled ATP in 1×kinase assay buffer (10 mM MgCl2, 40 mM HEPES pH 7.4). Reactions were initiated by the addition of 5 µCi γ-32P ATP; the samples were then incubated at 30°C for 30 min and reactions were terminated by adding 25 µl of 2×Tris-Tricine sample buffer (Bio-Rad). Samples were boiled and resolved on a 10–20% Tris-Tricine gradient gel (Bio-Rad), and signals were detected by autoradiography.
Supplementary Material
Acknowledgments
The authors thank Shoichiro Ono (Emory University, Atlanta, GA), Peter S. Klein (University of Pennsylvania, Philadelphia, PA), Pamela J. Woodring (The Salk Institute, La Jolla, CA) and Changhui Wang (University of Pennsylvania, Philadelphia, PA) for helpful discussions, Alex Toker (Harvard University, Cambridge, MA) for the gift of GSK3β shRNA constructs and Chao-Xing Yuan at the Penn Proteomics Core Facility for his excellent technical assistance.
Footnotes
Competing interests
The authors declare no competing interests.
Author contributions
G.-L.Z conceived, designed and supervised most of the project, and wrote the manuscript. G.-L.Z., H.Z., H.W. and P.G. conducted the experiments and analyzed the data. J.F. conceived and supervised the project at the early stage, and H.Z. organized the figures and tables.
Funding
Work was supported by the Arkansas Biosciences Institute and an Institutional Development Award (IDeA) from the NIGMS of the National Institutes of Health [grant number: P20GM103429-12] to G.-L.Z., who is also supported by a pilot grant from Arkansas Breast Cancer Research Program and the University of Arkansas for Medical Sciences Translational Research Institute [CSTA Grant Award number UL1TR000039]. J.F. is supported by the National Institutes of Health [grant number R01 GM48241]. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.156059/-/DC1
References
- Andrianantoandro E., Pollard T. D. (2006). Mechanism of actin filament turnover by severing and nucleation at different concentrations of ADF/cofilin. Mol. Cell 24, 13–23 10.1016/j.molcel.2006.08.006 [DOI] [PubMed] [Google Scholar]
- Bertling E., Hotulainen P., Mattila P. K., Matilainen T., Salminen M., Lappalainen P. (2004). Cyclase-associated protein 1 (CAP1) promotes cofilin-induced actin dynamics in mammalian nonmuscle cells. Mol. Biol. Cell 15, 2324–2334 10.1091/mbc.E04-01-0048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertling E., Quintero-Monzon O., Mattila P. K., Goode B. L., Lappalainen P. (2007). Mechanism and biological role of profilin-Srv2/CAP interaction. J. Cell Sci. 120, 1225–1234 10.1242/jcs.000158 [DOI] [PubMed] [Google Scholar]
- Cantin G. T., Shock T. R., Park S. K., Madhani H. D., Yates J. R., 3rd (2007). Optimizing TiO2-based phosphopeptide enrichment for automated multidimensional liquid chromatography coupled to tandem mass spectrometry. Anal. Chem. 79, 4666–4673 10.1021/ac0618730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhry F., Little K., Talarico L., Quintero-Monzon O., Goode B. L. (2010). A central role for the WH2 domain of Srv2/CAP in recharging actin monomers to drive actin turnover in vitro and in vivo. Cytoskeleton 67, 120–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhry F., Breitsprecher D., Little K., Sharov G., Sokolova O., Goode B. L. (2013). Srv2/cyclase-associated protein forms hexameric shurikens that directly catalyze actin filament severing by cofilin. Mol. Biol. Cell 24, 31–41 10.1091/mbc.E12-08-0589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clodfelder-Miller B., De Sarno P., Zmijewska A. A., Song L., Jope R. S. (2005). Physiological and pathological changes in glucose regulate brain Akt and glycogen synthase kinase-3. J. Biol. Chem. 280, 39723–39731 10.1074/jbc.M508824200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coghlan M. P., Culbert A. A., Cross D. A., Corcoran S. L., Yates J. W., Pearce N. J., Rausch O. L., Murphy G. J., Carter P. S., Roxbee Cox L. et al. (2000). Selective small molecule inhibitors of glycogen synthase kinase-3 modulate glycogen metabolism and gene transcription. Chem. Biol. 7, 793–803 10.1016/S1074-5521(00)00025-9 [DOI] [PubMed] [Google Scholar]
- Cole A. R., Knebel A., Morrice N. A., Robertson L. A., Irving A. J., Connolly C. N., Sutherland C. (2004). GSK-3 phosphorylation of the Alzheimer epitope within collapsin response mediator proteins regulates axon elongation in primary neurons. J. Biol. Chem. 279, 50176–50180 10.1074/jbc.C400412200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DesMarais V., Ghosh M., Eddy R., Condeelis J. (2005). Cofilin takes the lead. J. Cell Sci. 118, 19–26 10.1242/jcs.01631 [DOI] [PubMed] [Google Scholar]
- Dodatko T., Fedorov A. A., Grynberg M., Patskovsky Y., Rozwarski D. A., Jaroszewski L., Aronoff-Spencer E., Kondraskina E., Irving T., Godzik A. et al. (2004). Crystal structure of the actin binding domain of the cyclase-associated protein. Biochemistry 43, 10628–10641 10.1021/bi049071r [DOI] [PubMed] [Google Scholar]
- Fang X., Yu S. X., Lu Y., Bast R. C., Jr, Woodgett J. R., Mills G. B. (2000). Phosphorylation and inactivation of glycogen synthase kinase 3 by protein kinase A. Proc. Natl. Acad. Sci. USA 97, 11960–11965 10.1073/pnas.220413597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frame S., Cohen P. (2001). GSK3 takes centre stage more than 20 years after its discovery. Biochem. J. 359, 1–16 10.1042/0264-6021:3590001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman N. L., Field J. (2000). Mammalian homolog of the yeast cyclase associated protein, CAP/Srv2p, regulates actin filament assembly. Cell Motil. Cytoskeleton 45, 106–120 [DOI] [PubMed] [Google Scholar]
- Freeman N. L., Lila T., Mintzer K. A., Chen Z., Pahk A. J., Ren R., Drubin D. G., Field J. (1996). A conserved proline-rich region of the Saccharomyces cerevisiae cyclase-associated protein binds SH3 domains and modulates cytoskeletal localization. Mol. Cell. Biol. 16, 548–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerst J. E., Ferguson K., Vojtek A., Wigler M., Field J. (1991). CAP is a bifunctional component of the Saccharomyces cerevisiae adenylyl cyclase complex. Mol. Cell. Biol. 11, 1248–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gieselmann R., Mann K. (1992). ASP-56, a new actin sequestering protein from pig platelets with homology to CAP, an adenylate cyclase-associated protein from yeast. FEBS Lett. 298, 149–153 10.1016/0014-5793(92)80043-G [DOI] [PubMed] [Google Scholar]
- Hubberstey A. V., Mottillo E. P. (2002). Cyclase-associated proteins: CAPacity for linking signal transduction and actin polymerization. FASEB J. 16, 487–499 10.1096/fj.01-0659rev [DOI] [PubMed] [Google Scholar]
- Jiang W., Betson M., Mulloy R., Foster R., Lévay M., Ligeti E., Settleman J. (2008). p190A RhoGAP is a glycogen synthase kinase-3-beta substrate required for polarized cell migration. J. Biol. Chem. 283, 20978–20988 10.1074/jbc.M802588200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamukai M., Gerst J., Field J., Riggs M., Rodgers L., Wigler M., Young D. (1992). Genetic and biochemical analysis of the adenylyl cyclase-associated protein, cap, in Schizosaccharomyces pombe. Mol. Biol. Cell 3, 167–180 10.1091/mbc.3.2.167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim L., Kimmel A. R. (2000). GSK3, a master switch regulating cell-fate specification and tumorigenesis. Curr. Opin. Genet. Dev. 10, 508–514 10.1016/S0959-437X(00)00120-9 [DOI] [PubMed] [Google Scholar]
- Kim L., Kimmel A. R. (2006). GSK3 at the edge: regulation of developmental specification and cell polarization. Curr. Drug Targets 7, 1411–1419 10.2174/1389450110607011411 [DOI] [PubMed] [Google Scholar]
- Klein P. S., Melton D. A. (1996). A molecular mechanism for the effect of lithium on development. Proc. Natl. Acad. Sci. USA 93, 8455–8459 10.1073/pnas.93.16.8455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappalainen P., Drubin D. G. (1997). Cofilin promotes rapid actin filament turnover in vivo. Nature 388, 78–82 10.1038/40418 [DOI] [PubMed] [Google Scholar]
- Lee K. M., Nguyen C., Ulrich A. B., Pour P. M., Ouellette M. M. (2003). Immortalization with telomerase of the Nestin-positive cells of the human pancreas. Biochem. Biophys. Res. Commun. 301, 1038–1044 10.1016/S0006-291X(03)00086-X [DOI] [PubMed] [Google Scholar]
- Makkonen M., Bertling E., Chebotareva N. A., Baum J., Lappalainen P. (2013). Mammalian and malaria parasite cyclase-associated proteins catalyze nucleotide exchange on G-actin through a conserved mechanism. J. Biol. Chem. 288, 984–994 10.1074/jbc.M112.435719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matviw H., Yu G., Young D. (1992). Identification of a human cDNA encoding a protein that is structurally and functionally related to the yeast adenylyl cyclase-associated CAP proteins. Mol. Cell. Biol. 12, 5033–5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer L., Skaltsounis A. L., Magiatis P., Polychronopoulos P., Knockaert M., Leost M., Ryan X. P., Vonica C. A., Brivanlou A., Dajani R. et al. (2003). GSK-3-selective inhibitors derived from Tyrian purple indirubins. Chem. Biol. 10, 1255–1266 10.1016/j.chembiol.2003.11.010 [DOI] [PubMed] [Google Scholar]
- Michelot A., Berro J., Guérin C., Boujemaa-Paterski R., Staiger C. J., Martiel J. L., Blanchoin L. (2007). Actin-filament stochastic dynamics mediated by ADF/cofilin. Curr. Biol. 17, 825–833 10.1016/j.cub.2007.04.037 [DOI] [PubMed] [Google Scholar]
- Moriyama K., Yahara I. (2002). Human CAP1 is a key factor in the recycling of cofilin and actin for rapid actin turnover. J. Cell Sci. 115, 1591–1601. [DOI] [PubMed] [Google Scholar]
- Nishida E. (1985). Opposite effects of cofilin and profilin from porcine brain on rate of exchange of actin-bound adenosine 5′-triphosphate. Biochemistry 24, 1160–1164 10.1021/bi00326a015 [DOI] [PubMed] [Google Scholar]
- Normoyle K. P., Brieher W. M. (2012). Cyclase-associated protein (CAP) acts directly on F-actin to accelerate cofilin-mediated actin severing across the range of physiological pH. J. Biol. Chem. 287, 35722–35732 10.1074/jbc.M112.396051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono S. (2013). The role of cyclase-associated protein in regulating actin filament dynamics – more than a monomer-sequestration factor. J. Cell Sci. 126, 3249–3258 10.1242/jcs.128231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paavilainen V. O., Bertling E., Falck S., Lappalainen P. (2004). Regulation of cytoskeletal dynamics by actin-monomer-binding proteins. Trends Cell Biol. 14, 386–394 10.1016/j.tcb.2004.05.002 [DOI] [PubMed] [Google Scholar]
- Pollard T. D., Cooper J. A. (2009). Actin, a central player in cell shape and movement. Science 326, 1208–1212 10.1126/science.1175862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintero-Monzon O., Jonasson E. M., Bertling E., Talarico L., Chaudhry F., Sihvo M., Lappalainen P., Goode B. L. (2009). Reconstitution and dissection of the 600-kDa Srv2/CAP complex: roles for oligomerization and cofilin-actin binding in driving actin turnover. J. Biol. Chem. 284, 10923–10934 10.1074/jbc.M808760200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryves W. J., Dajani R., Pearl L., Harwood A. J. (2002). Glycogen synthase kinase-3 inhibition by lithium and beryllium suggests the presence of two magnesium binding sites. Biochem. Biophys. Res. Commun. 290, 967–972 10.1006/bbrc.2001.6305 [DOI] [PubMed] [Google Scholar]
- Sun T., Rodriguez M., Kim L. (2009). Glycogen synthase kinase 3 in the world of cell migration. Dev. Growth Differ. 51, 735–742 10.1111/j.1440-169X.2009.01141.x [DOI] [PubMed] [Google Scholar]
- Tojkander S., Gateva G., Lappalainen P. (2012). Actin stress fibers—assembly, dynamics and biological roles. J. Cell Sci. 125, 1855–1864 10.1242/jcs.098087 [DOI] [PubMed] [Google Scholar]
- Vojtek A., Haarer B., Field J., Gerst J., Pollard T. D., Brown S., Wigler M. (1991). Evidence for a functional link between profilin and CAP in the yeast S. cerevisiae. Cell 66, 497–505 10.1016/0092-8674(81)90013-1 [DOI] [PubMed] [Google Scholar]
- Woodgett J. R. (2001). Judging a protein by more than its name: GSK-3. Sci. STKE 2001, re12. [DOI] [PubMed] [Google Scholar]
- Woodring P. J., Meisenhelder J., Johnson S. A., Zhou G. L., Field J., Shah K., Bladt F., Pawson T., Niki M., Pandolfi P. P. et al. (2004). c-Abl phosphorylates Dok1 to promote filopodia during cell spreading. J. Cell Biol. 165, 493–503 10.1083/jcb.200312171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D., Pan W. (2010). GSK3: a multifaceted kinase in Wnt signaling. Trends Biochem. Sci. 35, 161–168 10.1016/j.tibs.2009.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X., Shen Q. T., Oristian D. S., Lu C. P., Zheng Q., Wang H. W., Fuchs E. (2011). Skin stem cells orchestrate directional migration by regulating microtubule-ACF7 connections through GSK3β. Cell 144, 341–352 10.1016/j.cell.2010.12.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang N., Higuchi O., Ohashi K., Nagata K., Wada A., Kangawa K., Nishida E., Mizuno K. (1998). Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature 393, 809–812 10.1038/31735 [DOI] [PubMed] [Google Scholar]
- Yoeli-Lerner M., Chin Y. R., Hansen C. K., Toker A. (2009). Akt/protein kinase b and glycogen synthase kinase-3beta signaling pathway regulates cell migration through the NFAT1 transcription factor. Mol. Cancer Res. 7, 425–432 10.1158/1541-7786.MCR-08-0342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J., Wang C., Palmieri S. J., Haarer B. K., Field J. (1999). A cytoskeletal localizing domain in the cyclase-associated protein, CAP/Srv2p, regulates access to a distant SH3-binding site. J. Biol. Chem. 274, 19985–19991 10.1074/jbc.274.28.19985 [DOI] [PubMed] [Google Scholar]
- Yusof A. M., Hu N. J., Wlodawer A., Hofmann A. (2005). Structural evidence for variable oligomerization of the N-terminal domain of cyclase-associated protein (CAP). Proteins 58, 255–262 10.1002/prot.20314 [DOI] [PubMed] [Google Scholar]
- Yusof A. M., Jaenicke E., Pedersen J. S., Noegel A. A., Schleicher M., Hofmann A. (2006). Mechanism of oligomerisation of cyclase-associated protein from Dictyostelium discoideum in solution. J. Mol. Biol. 362, 1072–1081 10.1016/j.jmb.2006.08.008 [DOI] [PubMed] [Google Scholar]
- Zelicof A., Gatica J., Gerst J. E. (1993). Molecular cloning and characterization of a rat homolog of CAP, the adenylyl cyclase-associated protein from Saccharomyces cerevisiae. J. Biol. Chem. 268, 13448–13453. [PubMed] [Google Scholar]
- Zelicof A., Protopopov V., David D., Lin X. Y., Lustgarten V., Gerst J. E. (1996). Two separate functions are encoded by the carboxyl-terminal domains of the yeast cyclase-associated protein and its mammalian homologs. Dimerization and actin binding. J. Biol. Chem. 271, 18243–18252 10.1074/jbc.271.30.18243 [DOI] [PubMed] [Google Scholar]
- Zhang H., Ghai P., Wu H., Wang C., Field J., Zhou G. L. (2013). Mammalian adenylyl cyclase-associated protein 1 (CAP1) regulates cofilin function, the actin cytoskeleton, and cell adhesion. J. Biol. Chem. 288, 20966–20977 10.1074/jbc.M113.484535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G. L., Miyazaki Y., Nakagawa T., Tanaka K., Shishido K., Matsuda H., Kawamukai M. (1998). Identification of a CAP (adenylyl-cyclase-associated protein) homologous gene in Lentinus edodes and its functional complementation of yeast CAP mutants. Microbiology 144, 1085–1093 10.1099/00221287-144-4-1085 [DOI] [PubMed] [Google Scholar]
- Zhou G. L., Yamamoto T., Ozoe F., Yano D., Tanaka K., Matsuda H., Kawamukai M. (2000). Identification of a 14-3-3 protein from Lentinus edodes that interacts with CAP (adenylyl cyclase-associated protein), and conservation of this interaction in fission yeast. Biosci. Biotechnol. Biochem. 64, 149–159 10.1271/bbb.64.149 [DOI] [PubMed] [Google Scholar]
- Zhou G. L., Zhuo Y., King C. C., Fryer B. H., Bokoch G. M., Field J. (2003). Akt phosphorylation of serine 21 on Pak1 modulates Nck binding and cell migration. Mol. Cell. Biol. 23, 8058–8069 10.1128/MCB.23.22.8058-8069.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G. L., Tucker D. F., Bae S. S., Bhatheja K., Birnbaum M. J., Field J. (2006). Opposing roles for Akt1 and Akt2 in Rac/Pak signaling and cell migration. J. Biol. Chem. 281, 36443–36453 10.1074/jbc.M600788200 [DOI] [PubMed] [Google Scholar]
- Zhou G. L., Zhang H., Field J. (2014). Mammalian CAP (Cyclase-associated protein) in the world of cell migration: Roles in actin filament dynamics and beyond. Cell Adhes. Migr. 8, 55–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.