

Abstract
Hepatocellular carcinoma (HCC) is increasing in prevalence and is one of the most common cancers in the world. Chief amongst the risks of attaining HCC are hepatitis B and C infection, aflatoxin B1 ingestion, alcoholism and obesity. The later has been shown to promote non alcoholic fatty liver disease, which can lead to the inflammatory form non alcoholic steatohepatitis (NASH). NASH is a complex metabolic disorder that can impact greatly on hepatic function. The mechanisms by which NASH promotes HCC are only beginning to be characterized. Here in this review, we give an overview of the recent novel mechanisms published that have been associated with NASH and subsequent HCC progression. We will focus our discussion on inflammation and gut derived inflammation and how they contribute to NASH driven HCC.
Keywords: Nonalcoholic steatohepatitis, Hepatocellular carcinoma, Inflammation, Microbiome, Bile acids
Core tip: Non alcoholic steatohepatitis (NASH) is a metabolic inflammatory disease the can often advance to liver cancer. Previously, it was assumed that obesity, hepatocyte cellular death and insulin resistance were the dominant drivers of NASH progression to hepatocellular carcinoma. Herein, we discuss the latest concepts concerning the gut microbiome and bile acids, which have now been shown to have a role in promoting hepatic inflammation, and subsequent liver tumor growth.
INTRODUCTION
Hepatocellular carcinoma (HCC) is now the fifth cancer of greatest incidence worldwide. Hepatitis B and C leading to cirrhosis are the dominant risk factors and the most common causes of HCC[1]. But in recent years studies have found that non-alcoholic steatohepatitis (NASH) can promote liver fibrosis, end-stage liver failure, cirrhosis, and ultimately progression to HCC. NASH is a clinical and pathological syndrome that is not associated with increased alcoholic intake, but has histological features similar to alcoholic hepatitis, with prominent fatty deposition and fat storage in the liver parenchymal cells, that can promote inflammation and necrosis[2,3]. Day et al[4] proposed an initial theory for the pathogenesis of NASH, known as the “two-hit hypothesis”. Here it was suggested that the “first hit” of hepatic triglyceride accumulation or steatosis, increased the vulnerability of the liver to the “second hit” of injury due changes in inflammatory cytokines and/or adipokines, mitochrondrial dysfunction and elevated oxidative stress, that can together promote steatohepatitis and fibrosis. However, data from many sources has now attributed other “hits” that include insulin resistance and the metabolic syndrome. These include changes in serum cytokines, which have been extensively reviewed elsewhere[5-10], and more recently and of relevance to this review, inflammation, altered gut microflora and bile acids that can contribute to the generation of NASH and HCC.
NASH: A COMPLEX BIOLOGICAL ENTITY
Numerous clinical studies have shown strong links between NASH and consequent cirrhosis, which naturally increases the risk of progression to HCC[11-13]. Studies have also shown that HCC is now a major cause of mortality in NASH patients[14]. Importantly, work has illustrated in comparison to diabetes and hepatitis C virus that non alcoholic fatty liver disease (NAFLD)/NASH is a growing underlying etiological risk for HCC[15]. Significantly, research has shown that HCC is now occurring more frequently from non-cirrhotic NASH[16,17] (and reviewed in[18]). Taken together, these data suggest that NASH in the presence of cirrhosis or non-cirrhosis, can promote HCC through diverse pathways. However, an underlying theme that is now emerging from the literature is the role of inflammation in cancer and in particular HCC. Moreover, these pathways have activity in the presence or absence of cirrhosis and we believe that unbridled inflammation is one of the principle factors that can drive the progression from NASH to HCC, and enhance HCC growth. The targeting of these pathways offers a potential avenue of therapeutically restricting HCC growth. Thus, in this review we will focus on clarifying some of the principal and novel inflammatory mechanisms that have been recently described for promoting HCC in the presence of NASH.
FACTORS LINKING INFLAMMATION TO NASH AND HCC PROGRESSION
Cytokines
Cytokines represent a family of small bioactive proteins and peptides that have signaling qualities in mediating intercellular communication signals, cellular interactions, growth and differentiation in cells. Therefore, in disease states when imbalances in cytokine levels occur they have important effects in promoting aberrant signaling, and in particular modulating inflammatory responses[19-21]. Studies have shown that cytokines such as tumor necrosis factor (TNF)-α, leptin, adiponectin and interleukin-6 (IL-6) occupy important roles in hepatic pathology. Thus, we will now examine their relationship and role in progression from NASH to HCC.
TNF-α
Studies have shown that TNF-α has an important role in liver cancer progression and growth[22]. The binding of TNF-α to TNFR1 receptor activates the transcriptional regulator nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB)[23]. NF-κB consists of five different family members: RelA (p65), RelB, c-Rel, p50/p105, and p100/p52 that are localized to the cytoplasm by inhibitors of κB (IκB)[24]. In response to proinflammatory stimuli, the IκB kinase complex of IKKα and IKKβ subunits and the regulatory protein, NF-κB essential modulator (NEMO) or inhibitor of NF-κB kinase subunit gamma (IKKγ), phosphorylates IκB, leading to its degradation and the subsequent movement of NF-κB into the cell nucleus[25]. Genetic removal of components of this pathway, have illustrated the importance of NF-κB in liver inflammation and HCC.
In mice, the ablation of IKKβ from hepatocytes to foster NF-κB inactivation, and subsequent treatment with the mutagen diethylnitrosamine (DEN), resulted in a greater incidence of HCC[26]. To explain this phenotype it was found that on exposure to excess TNF-α, prolonged c-Jun N-terminal kinase (JNK) activation was stimulated, and resulted in increased hepatocyte apoptosis with compensatory hepatocyte proliferation. These events facilitated the accumulation of genetic errors to enhance HCC growth. Accordingly, increased JNK activity can also promote insulin resistance as JNK-/- mice have improved insulin sensitivity and less hepatic inflammation and fibrosis[27,28]. Alternatively, the removal of IKKβ from hepatocytes and Kupffer cells reduced the number and size of HCCs, after DEN treatment[26]. Mechanistically, it was found that the proliferation of hepatocytes after exposure to DEN, were dependent on the IKKβ induced production of the hepatic mitogens TNF-α, IL-6 and HGF from Kupffer cells.
Further studies showed that the deletion of NEMO from hepatocytes, which modulates the phosphorylation and degradation of IκBs in the TNF-α pathway, spontaneously promoted HCC development. It was found that NEMO absence completely blocked NF-κB activation and sensitized the NEMO-null hepatocytes to lipopolysaccharide (LPS) treatment, suggesting the involvement of the innate immune response and microbiome in HCC development[29].
To convey the importance of obesity in driving steatosis and inflammation, it has been shown that TNF-α and IL-6 are important in obesity driven HCC. Here liver cancer was promoted by DEN treatment of IL-6 or TNFR1 null mice and subsequent high-fat feeding. It was observed that compared to wild-type controls that IL-6 or TNFR1 null mice had significantly reduced steatosis, HCC number and size[30]. Collectively, these studies show the key role of the TNF-α pathway in hepatic inflammation, and suggest that imbalances in this pathway are important in the transition from NASH to HCC.
IL-6
IL-6 has been shown to have defined and important roles in HCC pathology. IL-6 is largely secreted by inflammatory cells and can bind to IL-6 receptors on hepatocytes and liver non-parenchymal cells to promote the binding of the signal-transducing receptor gp130 to the IL-6R complex, to activate Janus Kinase1 (JAK1). Subsequent activation and phosphorylation of the transcription factor signal transducer and activator of transcription-3 (STAT3) factor, promotes a transcriptional program to prevent apoptosis and initiate cell growth and differentiation. Studies have shown that phosphorylated STAT3 is expressed in up to 60% of human HCCs[31], and that increased IL-6 protein levels is associated with NASH as compared to steatosis in patients[32]. Moreover, IL-6 has been shown to be upregulated in two established models murine models of NAFLD[33], and mice overexpressing IL-6 and soluble IL-6R, have more extensive liver cancer than wild-type mice[34]. In agreement with the concept that IL-6 is a promoter of HCC, it has been shown that male IL-6 deficient mice generate less HCC than wild-type males after DEN treatment[35]. Finally, it has been demonstrated that IL-6 serum levels are increased in the majority of hepatic acute and chronic events[36]. Thus, these data suggest that IL-6 is associated with NASH and is a promoter of HCC formation and growth.
Leptin
Leptin (Ob) is a multifunctional adipocytokine secreted by fat cells and has a variety of biological effects mediated by its binding to a specific leptin receptor (ObR). It plays an important role in hepatic stellate cell (HSC) activation and hepatic fibrosis. It was found that the inhibition of endogenous leptin activity can retard HSC function and have powerful anti-fibrotic effects[37]. Moreover, leptin can stimulate tissue inhibitor of metalloproteinase 1 (TIMP-1) production via the JAK/STAT pathway to promote fibrogenesis[38,39]. The injection of leptin into carbon tetrachloride-treated mice increased the synthesis and secretion of pro-fibrotic genes in the liver including procollagen and TGFβ1. Leclercq et al[40] utilised leptin-deficient mice and found that the injection of leptin increased TGF-β1 levels and completely restored fibrosis. These data suggest that leptin plays an important role in liver fibrosis.
The relationship of leptin with NASH is less well determined. In patients Chittur et al[41] found that serum leptin levels were higher in NASH patients than in the normal group. Studies from another group, failed to find an association between leptin and NASH[42]. Leptin can regulate liver cancer development by promoting tumor cell proliferation and angiogenesis. Leptin acts on endothelial cells to promote tube formation and migration, while limiting leptin impairs angiogenesis[43]. In the presence of vascular endothelial growth factor, leptin-mediated neovascularization in the liver increased in line with NASH progression, indicating a pro-angiogenic role[44]. In HCC patients both leptin and ObR are expressed at higher levels in livers. Interestingly, poorly differentiated HCCs have greater blood vessel density and ObR expression, again suggesting an angiogenic role[45]. Furthermore, leptin has been shown to promote HCC proliferation, migration, and invasiveness through activation of the JAK/STAT pathway[46]. Taken together, these observations suggest that elevated serum leptin levels through increased adipose tissue mass, may promote progression by enhancing hepatic fibrogenesis, angiogenesis, and cancer cell division and behavior. Thus, it is plausible that increased leptin levels could promote progression from NASH to HCC. However, as studies in humans have not revealed a clear correlation it is possible that leptin is associated only with a subset of HCCs that originate in the background of NASH.
Adiponectin
Adiponectin is an anti-inflammatory cytokine produced by the adipose tissue. It has roles in regulating glucose and fatty acid metabolism, with decreased plasma concentrations correlating to increased BMI, insulin resistance, type 2 diabetes and atherosclerosis. Adiponectin circulates in the serum in different molecular forms: a low molecular weight trimer, a middle molecular weight hexamer, and high molecular weight multimers, that is considered to be the most biologically active. There are three known APN receptors: AdipoR1, AdipoR2 and T-cadherin that have distinct affinities for the various circulating forms of APN[47].
Given the different adiponectin forms and receptors, adiponectin has a plethora of activities. In the liver adiponectin activates AMPK to reduce hepatic gluconeogenesis, stimulate fatty acid oxidation, and limit hepatic de novo lipogenesis through inhibition of sterol-regulatory element binding protein-1c (SREBP-1c), a dominant regulator of triglyceride and fatty acid synthesis[48,49]. Adiponectin can also activate peroxisome proliferator-activated receptor α (PPARα) to promote fatty acid oxidation. Importantly, in the context of liver diseases adiponectin can limit inflammation by inhibiting the NF-κB activation to suppress TNF-α release[50,51]. Adiponectin can also further suppress macrophage function and the proliferation and migration of vascular smooth muscle cells[52].
In NASH, hypoadiponectinemia is an early feature, and it has been shown that low serum adiponectin levels are associated with increased hepatic steatosis and with necroinflammation[53]. Likewise, adiponectin null mice have more steatosis and fibrosis after high fat feeding, and develop more fibrosis on carbon tetrachloride treatment[54-56]. Moreover, adiponectin has strong hepatoprotective properties and can diminish steatosis and hepatic damage, in endotoxin and alcohol injury models by limiting hepatic production of TNF-α[57,58].
Nevertheless, studies have showed that in cirrhotic and HCC patients that impaired hepatic function is associated with increases in serum adiponectin levels[59-61]. It has been recently shown that in advanced NASH there is a significant correlation between increased serum bile acids, circulating adiponectin and reduced liver fat[62]. It is probable that similar mechanisms are operating in HCC patients. It remains open whether adiponectin can influence HCC once developed. In HCC cell lines adiponectin increased JNK activation and subsequent apoptosis in tumors, and promoted increased AMPK phosphorylation and the inhibition of the mammalian target of rapamycin (mTOR) phosphorylation to limit tumor growth in nude mice[63]. In a separate study, it was shown that adiponectin treatment decreased HCC tumor growth and macrophage infiltration, and secondary lung metastasis[64]. In summary, these data suggest that lower adiponectin levels associated with obesity are a driver of NASH and HCC formation.
EMERGING INFLAMMATORY PATHWAYS FOR PROMOTING NASH HCC DEVELOPMENT
The above illustrate that NASH driven HCC is associated with a robust inflammatory response. For some time the standard model assumed that the increased adipose mass associated with insulin resistance enhanced liver injury. Additionally, evidence also suggested that the accumulation of hepatic cellular fat could promote the release of reactive oxygen species to interfere with cellular functions such as cellular respiration to cause the release of toxic lipids species, to result in hepatocyte dysfunction and apoptosis (as reviewed by us[65]). However, recent studies now include another layer of factors that are responsible for contributing and perhaps enhancing the effect of inflammation and damage to the liver. Specifically, research has found that diet affects the constitution of the gut bacteria (microbiome) to subsequently influence inflammation. Furthermore, it has been demonstrated that the microbiome can modulate bile acids levels, which are now known to have both systemic and hepatic specific signaling. Together, these findings suggest a novel pathogenic pathway between the gut and liver driven by dietary changes that has the potential to generate hepatic inflammation and ultimately influence HCC.
Microbiome and NASH
Studies have linked NASH with dysbiosis of the gut. In NASH patients Wigg et al[66] observed small intestinal bacterial overgrowth and increased TNF-α levels. Similarly, in another study NASH patients had greater bacterial overgrowth, elevated expression of Toll-like receptor (TLR) 4 and increased levels of serum IL-8[67]. Commensurate with these changes qPCR for the major gut bacteria species has shown that NASH patients have less gut Gram-negative Bacteroidetes, than that observed in patients with simple steatosis[68]. Likewise, in NASH patients an increase in the abundance of alcohol producing bacteria has been observed, suggesting that these strains may be involved in NASH pathogenesis[69].
In view of these findings, interventional studies have been undertaken in rodents and patients. Treatment of mice fed the methionine-choline-deficient diet with the probiotic VSL#3 reduced liver fibrosis but had no effect on inflammation and steatosis[70]. However, in a genetic model, treatment of ApoE knock-out mice with VSL#3 had a more pronounced effect and reversed insulin resistance and steatohepatitis[71]. In choline-deficient/L-amino acid define (CDAA) fed rats a butyrate-producing probiotic reduced hepatic lipid deposition and significantly improved insulin resistance, serum endotoxin levels, and hepatic inflammatory indexes[72]. In patients there has been limited studies undertaken. A small Chinese study of 20 NASH patients has shown that probiotics can reduce liver fat and AST levels[73]. Similarly, in an Italian study of 66 NASH patients, the 33 that received probiotics had reduced TNF-α, C-reactive protein, AST, HOMO-IR, serum endotoxin levels, steatosis and NASH activity[74]. Together, these data suggest a role for the microbiome in mediating NASH and that the correction of gut dysbiosis to a more healthy phenotype can possibly be used as a therapy to limit NASH progression.
How does the microbiota induce hepatic inflammation and metabolism?
Numerous studies over the past decade have shed light on the role of the microbiota in modulating hepatic metabolism and inflammation. A focus point has been lipopolysaccharide (LPS), a large molecule present on and released by gram negative bacteria. LPS binds to the cluster of differentiation 14/TLR2/lymphocyte antigen 96 (CD14/TLR4/MD2) receptor complex on immune cells, which represents part of the innate immune system to promote a pro-inflammatory immune response. It has been shown that the consumption of a high fat diet can loosen the intestinal tight junctions, leading to the increased delivery of bacterial products like LPS via the portal vein to the liver. Moreover, as constituents of this signaling cascade have links with regulating metabolism, data has shown that LPS can also regulate insulin resistance. For example, obese ob/ob mice have intestinal overgrowth, increased intestinal permeability, more TNF-α, hepatic stellate cell activation and an enhanced LPS inflammatory response[75]. The treatment of ob/ob mice or HFD mice with antibiotics to alter the gut microbiota reduced glucose intolerance, body weight gain, fat mass development, lowered inflammation and oxidative stress. Moreover, the authors showed that the absence of CD14 in ob/ob CD14-/- mutant mice mimicked the metabolic and inflammatory effects of antibiotics, suggesting a key role for LPS[76]. In a separate study, leptin whose levels are induced by obesity has been shown to upregulate CD14, to promote hyper responsiveness to LPS and enhance progression to NASH[77]. These studies show a critical role for the TLR4 signaling complex in promoting inflammation and NASH.
Another arm of the innate immune system are the inflammasomes, which are a signaling complex consisting of caspase 1, apoptosis-associated speck-like protein containing (ASC) and nucleotide-binding oligomerization domain receptors (NOD-like receptors: NLRs). The complex can be stimulated by TLRs via LPS, but can also be activated by other bacterial ligands, depending on the NLR family member. A principle downstream action of the complex is to activate the caspase-1 cascade, leading to the production of pro-inflammatory cytokines IL-1β and IL-18. In an elegant study by Henao-Mejia et al[78], they showed that inflammasome-deficient mice (Casp1-/-, Asc-/-, Nlrp3-/- or IL-18-/-), fed a methionine and choline-deficient (MCD) diet had increased severity of NASH and decreased glucose tolerance. Critically, the authors showed that co-habitation of these mice with wild-type could result in the transfer of the NASH phenotype to the wild-type animals. Furthermore, they showed that the microbiota from these animals mediated the inflammatory response through TLR4, TLR9 and TNF-α. These data show that inflammatory cross-talk between the gut and liver has an important role in protecting the organism against metabolic disease, and that changes in the microbiome or that alternatively the decreased responsiveness of the innate immune system can lead to metabolic diseases such as diabetes and NASH.
Evidence of microbiota links to NASH driven HCC
The above evidence suggests that the microbiota can promote NASH a risk factor for HCC development. To test if the microbiota can influence HCC promotion, the group of Schwabe in a refined study, initiated tumorigenesis with DEN and followed with subsequent carbon tetrachloride treatment to promote fibrosis driven HCC[79]. They used this experimental protocol on TLR4 null mice and found that TLR4 absence limited HCC growth. Antibiotic treatment of wild-type mice subjected to the DEN/CCl4 treatment reduced tumor growth, suggesting that the microbiota and LPS through TLR4 promoted HCC. Further, the authors identified that the mitogen epiregulin, a ligand for the epidermal growth factor receptor (EGFR), was expressed by activated stellate cells, suggesting a mechanism by which activated HSCs could drive tumor cell proliferation. Moreover, they observed that the microbiota and TLR4 supported the expression of survival signals to promote tumor growth.
The gut microbiota apart from stimulating an immune response has other important functions. Specifically, they catalyse the generation of secondary bile acids such as deoxycholic acid (DCA), which is known to induce DNA damage[80]. Further to this concept, Yoshimoto et al[81] found that DCA can promote the activation of a senescence-associated secretory phenotype in HSCs, reflected by the secretion of IL-1β. They observed that the absence of IL-1β limited obesity-induced HCC development and similarly that antibiotic treatment could alleviate HCC development. Furthermore, the lowering of DCA or feeding of DCA to HFD-fed mice, respectfully limited or enhanced HCC growth. These data suggest in sum that bacterial metabolites can instigate hepatic inflammation either directly or indirectly via the generation of metabolites such as DCA that can enhance HCC growth and progression. These observations suggest a potential role for bile acids in NASH HCC progression.
Bile acids and NASH HCC
Primary bile acids are derived from cholesterol, the most abundant being chenodeoxycholic acid (CDCA) and cholic acid (CA). They are secreted into the intestine where modification by intestinal bacteria leads, as explained above, to the generation of the secondary bile acids, such as DCA and lithocholic acid (LCA). To maintain bile acids at physiological levels they are efficiently reabsorbed in the ileum and transported by the enterohepatic circulation back to the liver. For many years it was considered that bile acids were mere detergents, important for absorbing, transporting, and distributing dietary lipids, vitamins and steroids. However, it is now appreciated that they represent a distinct class of hormones that bind to highly specific receptors, the best described being the nuclear hormone receptor farnesoid X receptor (FXR) and the G-protein-coupled cell surface receptor TGR5.
FXR regulates bile acid synthesis by inhibiting the transcription of cholesterol 7α-hydroxylase (CYP7A1), the rate-limiting enzyme in the conversion of cholesterol to bile acids. FXR can also repress the SREBP-1c transcription to reduce triglyceride synthesis, and promote the β-oxidation of fatty acids through augmented PPARα signalling[82]. In agreement FXR null mice have elevated serum triglycerides and cholesterol and are prone to develop steatohepatitis[83]. Moreover, FXR agonists can antagonize NF-κB activity and limit hepatic inflammation in vivo[84]. In this light, studies have illustrated the potential of FXR agonists to treat NASH. In the mouse MCD model, treatment with WAY-362450, reduced liver injury and inflammation[85]. In a recent phase II clinical trial a study was undertaken to evaluate the effects of Obeticholic acid (OCA; INT-747, 6α-ethyl-chenodeoxycholic acid) on insulin sensitivity in patients with nonalcoholic fatty liver disease and type 2 diabetes mellitus. It was found that within 6 wk OCA increased insulin sensitivity and reduced the markers of liver inflammation treatment and fibrosis[86]. Given FXR’s important role in liver function, it has also been shown that FXR null mice can spontaneously develop liver tumors as they age, and treatment with CA further potentiated DEN-initiated liver cancer. Mechanistically, it was found that the null mice have increased levels of the proinflammatory cytokine IL-1β, activation of the Wnt/β-catenin pathway activation, and target gene c-myc[87,88].
TGR5 is expressed in the gall bladder, cholangiocytes, ileum, colon, brown and white adipose tissue and to a lesser extent in skeletal muscle, liver and immune cells[89]. TGR5 activation in muscle and brown adipose increases the intracellular secondary messenger cyclic adenosine monophosphate (cAMP), which in turn increases transcription of the Type II iodothyronine deiodinase gene (Dio2) and the prerequisite protein Type II iodothyronine deiodinase (D2), which converts thyroxine (T4) to triiodothyronine (T3). The net effect is augmented energy and oxygen consumption. TGR5 promotes hormone secretion in the gut, of intestinal glucagon-like peptide-1 (GLP-1), which in turn stimulates insulin release. The concept that TGR5 is involved in energy modulation, is supported by TRG5 null mice which accumulate fat faster than wild-type. Moreover, treatment of wild-type mice with the TGR5 specific agonist INT-777 increases brown adipose tissue D2 and energy metabolism, and improves obesity and steatosis[90]. In the liver, TGR5 is expressed in Kupffer cells, sinusoidal endothelial cells and cholangiocytes. In keeping with this expression pattern, TGR5 inhibits liver inflammation by suppressing NF-κB in macrophages and is protective, as TGR5 null mice have more liver damage after bile-duct ligation[91-93]. In sum, these observations show that TGR5 has been shown to repress hepatic inflammation, but as yet no definitive study has linked it with HCC progression in vivo. In sum, these data illustrate that elevated bile acids or inhibited bile acid signaling could be a deciding step in the progression from NASH to HCC.
CONCLUSION
Taken together we have illustrated here the critical importance of adipokines, inflammatory factors, the generation of inflammation through gut dysbiosis and bile acids in supporting and enhancing NASH driven liver tumor growth. Obviously, inflammation from other sources such as the adipose and liver generated injury due to steatosis and cholesterol are also consequential factors in NASH and HCC[65]. Jointly, these observations show that cross-talk between the fat and liver and gut and liver can contribute to NASH HCC. We illustrate these concepts in Figure 1.
Figure 1.
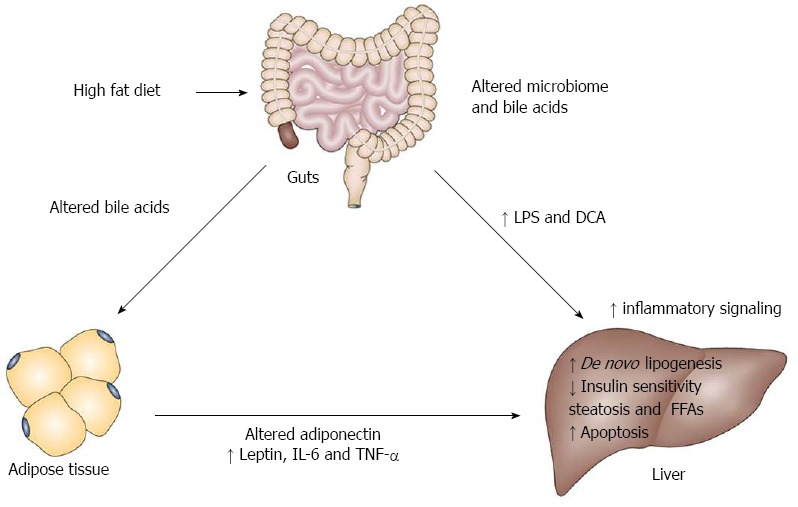
Model illustrating the role of inflammation in non alcoholic steatohepatitis driven hepatocellular carcinoma. The current literature supports the concept that hepatic inflammation can in part come from dietary changes that promote an altered microbiome to release inflammatory factors such as lipopolysaccharide (LPS) and the inflammatory bile acid deoxycholic acid (DCA). They impact on the liver to activate the innate immune system and a senescence - associated secretory phenotype in hepatic stellate cells. Bile acids can also modulate hepatic inflammation through farnesoid X receptor and TGR5 receptors. The increased adipose mass can produce less adiponectin and more leptin, interleukin-6 (IL-6) and tumor necrosis factor (TNF)-α that can further impact on the liver. The build up of fat and elevated FFAs, induce hepatocyte apoptosis, further amplifying the inflammatory effects. The net effect of the systemic and hepatic inflammation is to support neoplastic growth in the liver. It remains to be determined in appropriate mouse models whether bile acids can influence adipocyte function, non alcoholic steatohepatitis and hepatocellular carcinoma progression.
In light of the fact that NASH originates from being overweight and obese, the correction of the microbiome to a lean phenotype and utilization of bile acid agonists are attractive future therapeutic options to limit hepatic inflammation and NASH progression, and possible HCC formation. Nevertheless, it must be mentioned that few of these studies have demonstrated a genetic mechanism through which NASH HCC tumor initiation is influenced. Therefore, this suggests that in the context of NASH driven HCC there are clearly genetic and epigenetic factors that can promote the initiation of HCC. A review of the literature reveals that research into the association of NASH HCC with alterations in microRNA, methylation, chromatin remodeling and chromosomal changes is only in its infancy. Thus, even through the suppression of inflammation is an attractive target for limiting and restraining HCC growth, conceptually further research is urgently needed to elucidate the identity of genetic and epigentic factors that can initiate HCC in the presence of NASH.
Footnotes
Supported by Robert W Storr Bequest to the Sydney Medical Foundation University of Sydney (LH), Cancer Council NSW grant 1069733 (LH) and the West Translational Cancer Research Centre Partner Program funded by the Cancer Institute, NSW
P- Reviewer: Gong Y, Hashimoto N, Peck-Radosavljevic M S- Editor: Ma YJ L- Editor: A E- Editor: Zhang DN
References
- 1.Cabibbo G, Craxì A. Epidemiology, risk factors and surveillance of hepatocellular carcinoma. Eur Rev Med Pharmacol Sci. 2010;14:352–355. [PubMed] [Google Scholar]
- 2.Brunt EM. Pathology of nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol. 2010;7:195–203. doi: 10.1038/nrgastro.2010.21. [DOI] [PubMed] [Google Scholar]
- 3.Schuppan D, Schattenberg JM. Non-alcoholic steatohepatitis: pathogenesis and novel therapeutic approaches. J Gastroenterol Hepatol. 2013;28 Suppl 1:68–76. doi: 10.1111/jgh.12212. [DOI] [PubMed] [Google Scholar]
- 4.Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology. 1998;114:842–845. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 5.Gastaldelli A, Harrison S, Belfort-Aguiar R, Hardies J, Balas B, Schenker S, Cusi K. Pioglitazone in the treatment of NASH: the role of adiponectin. Aliment Pharmacol Ther. 2010;32:769–775. doi: 10.1111/j.1365-2036.2010.04405.x. [DOI] [PubMed] [Google Scholar]
- 6.Kugelmas M, Hill DB, Vivian B, Marsano L, McClain CJ. Cytokines and NASH: a pilot study of the effects of lifestyle modification and vitamin E. Hepatology. 2003;38:413–419. doi: 10.1053/jhep.2003.50316. [DOI] [PubMed] [Google Scholar]
- 7.Koek GH, Liedorp PR, Bast A. The role of oxidative stress in non-alcoholic steatohepatitis. Clin Chim Acta. 2011;412:1297–1305. doi: 10.1016/j.cca.2011.04.013. [DOI] [PubMed] [Google Scholar]
- 8.Loria P, Carulli L, Bertolotti M, Lonardo A. Endocrine and liver interaction: the role of endocrine pathways in NASH. Nat Rev Gastroenterol Hepatol. 2009;6:236–247. doi: 10.1038/nrgastro.2009.33. [DOI] [PubMed] [Google Scholar]
- 9.Michelotti GA, Machado MV, Diehl AM. NAFLD, NASH and liver cancer. Nat Rev Gastroenterol Hepatol. 2013;10:656–665. doi: 10.1038/nrgastro.2013.183. [DOI] [PubMed] [Google Scholar]
- 10.Yu J, Shen J, Sun TT, Zhang X, Wong N. Obesity, insulin resistance, NASH and hepatocellular carcinoma. Semin Cancer Biol. 2013;23:483–491. doi: 10.1016/j.semcancer.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 11.Adams LA, Lymp JF, St Sauver J, Sanderson SO, Lindor KD, Feldstein A, Angulo P. The natural history of nonalcoholic fatty liver disease: a population-based cohort study. Gastroenterology. 2005;129:113–121. doi: 10.1053/j.gastro.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 12.Jansen PL. Non-alcoholic steatohepatitis. Eur J Gastroenterol Hepatol. 2004;16:1079–1085. doi: 10.1097/00042737-200411000-00001. [DOI] [PubMed] [Google Scholar]
- 13.Nagaoki Y, Hyogo H, Aikata H, Tanaka M, Naeshiro N, Nakahara T, Honda Y, Miyaki D, Kawaoka T, Takaki S, et al. Recent trend of clinical features in patients with hepatocellular carcinoma. Hepatol Res. 2012;42:368–375. doi: 10.1111/j.1872-034X.2011.00929.x. [DOI] [PubMed] [Google Scholar]
- 14.Hashimoto E, Yatsuji S, Tobari M, Taniai M, Torii N, Tokushige K, Shiratori K. Hepatocellular carcinoma in patients with nonalcoholic steatohepatitis. J Gastroenterol. 2009;44 Suppl 19:89–95. doi: 10.1007/s00535-008-2262-x. [DOI] [PubMed] [Google Scholar]
- 15.Sanyal A, Poklepovic A, Moyneur E, Barghout V. Population-based risk factors and resource utilization for HCC: US perspective. Curr Med Res Opin. 2010;26:2183–2191. doi: 10.1185/03007995.2010.506375. [DOI] [PubMed] [Google Scholar]
- 16.Starley BQ, Calcagno CJ, Harrison SA. Nonalcoholic fatty liver disease and hepatocellular carcinoma: a weighty connection. Hepatology. 2010;51:1820–1832. doi: 10.1002/hep.23594. [DOI] [PubMed] [Google Scholar]
- 17.Kawada N, Imanaka K, Kawaguchi T, Tamai C, Ishihara R, Matsunaga T, Gotoh K, Yamada T, Tomita Y. Hepatocellular carcinoma arising from non-cirrhotic nonalcoholic steatohepatitis. J Gastroenterol. 2009;44:1190–1194. doi: 10.1007/s00535-009-0112-0. [DOI] [PubMed] [Google Scholar]
- 18.Karagozian R, Derdák Z, Baffy G. Obesity-associated mechanisms of hepatocarcinogenesis. Metabolism. 2014;63:607–617. doi: 10.1016/j.metabol.2014.01.011. [DOI] [PubMed] [Google Scholar]
- 19.Schett G, Elewaut D, McInnes IB, Dayer JM, Neurath MF. How cytokine networks fuel inflammation: Toward a cytokine-based disease taxonomy. Nat Med. 2013;19:822–824. doi: 10.1038/nm.3260. [DOI] [PubMed] [Google Scholar]
- 20.Maddur MS, Miossec P, Kaveri SV, Bayry J. Th17 cells: biology, pathogenesis of autoimmune and inflammatory diseases, and therapeutic strategies. Am J Pathol. 2012;181:8–18. doi: 10.1016/j.ajpath.2012.03.044. [DOI] [PubMed] [Google Scholar]
- 21.Grivennikov SI, Karin M. Inflammatory cytokines in cancer: tumour necrosis factor and interleukin 6 take the stage. Ann Rheum Dis. 2011;70 Suppl 1:i104–i108. doi: 10.1136/ard.2010.140145. [DOI] [PubMed] [Google Scholar]
- 22.Fukata M, Abreu MT. Role of Toll-like receptors in gastrointestinal malignancies. Oncogene. 2008;27:234–243. doi: 10.1038/sj.onc.1210908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berasain C, Castillo J, Perugorria MJ, Latasa MU, Prieto J, Avila MA. Inflammation and liver cancer: new molecular links. Ann N Y Acad Sci. 2009;1155:206–221. doi: 10.1111/j.1749-6632.2009.03704.x. [DOI] [PubMed] [Google Scholar]
- 24.He G, Karin M. NF-κB and STAT3 - key players in liver inflammation and cancer. Cell Res. 2011;21:159–168. doi: 10.1038/cr.2010.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakagawa H, Maeda S. Inflammation- and stress-related signaling pathways in hepatocarcinogenesis. World J Gastroenterol. 2012;18:4071–4081. doi: 10.3748/wjg.v18.i31.4071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121:977–990. doi: 10.1016/j.cell.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 27.Hirosumi J, Tuncman G, Chang L, Görgün CZ, Uysal KT, Maeda K, Karin M, Hotamisligil GS. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 28.Schattenberg JM, Singh R, Wang Y, Lefkowitch JH, Rigoli RM, Scherer PE, Czaja MJ. JNK1 but not JNK2 promotes the development of steatohepatitis in mice. Hepatology. 2006;43:163–172. doi: 10.1002/hep.20999. [DOI] [PubMed] [Google Scholar]
- 29.Luedde T, Beraza N, Kotsikoris V, van Loo G, Nenci A, De Vos R, Roskams T, Trautwein C, Pasparakis M. Deletion of NEMO/IKKgamma in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell. 2007;11:119–132. doi: 10.1016/j.ccr.2006.12.016. [DOI] [PubMed] [Google Scholar]
- 30.Siddique A, Kowdley KV. Insulin resistance and other metabolic risk factors in the pathogenesis of hepatocellular carcinoma. Clin Liver Dis. 2011;15:281–96, vii-x. doi: 10.1016/j.cld.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.He G, Yu GY, Temkin V, Ogata H, Kuntzen C, Sakurai T, Sieghart W, Peck-Radosavljevic M, Leffert HL, Karin M. Hepatocyte IKKbeta/NF-kappaB inhibits tumor promotion and progression by preventing oxidative stress-driven STAT3 activation. Cancer Cell. 2010;17:286–297. doi: 10.1016/j.ccr.2009.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wieckowska A, Papouchado BG, Li Z, Lopez R, Zein NN, Feldstein AE. Increased hepatic and circulating interleukin-6 levels in human nonalcoholic steatohepatitis. Am J Gastroenterol. 2008;103:1372–1379. doi: 10.1111/j.1572-0241.2007.01774.x. [DOI] [PubMed] [Google Scholar]
- 33.Cai D, Yuan M, Frantz DF, Melendez PA, Hansen L, Lee J, Shoelson SE. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med. 2005;11:183–190. doi: 10.1038/nm1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maione D, Di Carlo E, Li W, Musiani P, Modesti A, Peters M, Rose-John S, Della Rocca C, Tripodi M, Lazzaro D, et al. Coexpression of IL-6 and soluble IL-6R causes nodular regenerative hyperplasia and adenomas of the liver. EMBO J. 1998;17:5588–5597. doi: 10.1093/emboj/17.19.5588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, Elsharkawy AM, Karin M. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317:121–124. doi: 10.1126/science.1140485. [DOI] [PubMed] [Google Scholar]
- 36.Naugler WE, Karin M. The wolf in sheep’s clothing: the role of interleukin-6 in immunity, inflammation and cancer. Trends Mol Med. 2008;14:109–119. doi: 10.1016/j.molmed.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 37.Elinav E, Ali M, Bruck R, Brazowski E, Phillips A, Shapira Y, Katz M, Solomon G, Halpern Z, Gertler A. Competitive inhibition of leptin signaling results in amelioration of liver fibrosis through modulation of stellate cell function. Hepatology. 2009;49:278–286. doi: 10.1002/hep.22584. [DOI] [PubMed] [Google Scholar]
- 38.Cao Q, Mak KM, Ren C, Lieber CS. Leptin stimulates tissue inhibitor of metalloproteinase-1 in human hepatic stellate cells: respective roles of the JAK/STAT and JAK-mediated H2O2-dependant MAPK pathways. J Biol Chem. 2004;279:4292–4304. doi: 10.1074/jbc.M308351200. [DOI] [PubMed] [Google Scholar]
- 39.Cao Q, Mak KM, Lieber CS. Leptin enhances alpha1(I) collagen gene expression in LX-2 human hepatic stellate cells through JAK-mediated H2O2-dependent MAPK pathways. J Cell Biochem. 2006;97:188–197. doi: 10.1002/jcb.20622. [DOI] [PubMed] [Google Scholar]
- 40.Leclercq IA, Farrell GC, Schriemer R, Robertson GR. Leptin is essential for the hepatic fibrogenic response to chronic liver injury. J Hepatol. 2002;37:206–213. doi: 10.1016/s0168-8278(02)00102-2. [DOI] [PubMed] [Google Scholar]
- 41.Chitturi S, Farrell G, Frost L, Kriketos A, Lin R, Fung C, Liddle C, Samarasinghe D, George J. Serum leptin in NASH correlates with hepatic steatosis but not fibrosis: a manifestation of lipotoxicity? Hepatology. 2002;36:403–409. doi: 10.1053/jhep.2002.34738. [DOI] [PubMed] [Google Scholar]
- 42.Angulo P, Alba LM, Petrovic LM, Adams LA, Lindor KD, Jensen MD. Leptin, insulin resistance, and liver fibrosis in human nonalcoholic fatty liver disease. J Hepatol. 2004;41:943–949. doi: 10.1016/j.jhep.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 43.Sierra-Honigmann MR, Nath AK, Murakami C, García-Cardeña G, Papapetropoulos A, Sessa WC, Madge LA, Schechner JS, Schwabb MB, Polverini PJ, et al. Biological action of leptin as an angiogenic factor. Science. 1998;281:1683–1686. doi: 10.1126/science.281.5383.1683. [DOI] [PubMed] [Google Scholar]
- 44.Kitade M, Yoshiji H, Kojima H, Ikenaka Y, Noguchi R, Kaji K, Yoshii J, Yanase K, Namisaki T, Asada K, et al. Leptin-mediated neovascularization is a prerequisite for progression of nonalcoholic steatohepatitis in rats. Hepatology. 2006;44:983–991. doi: 10.1002/hep.21338. [DOI] [PubMed] [Google Scholar]
- 45.Ribatti D, Belloni AS, Nico B, Di Comite M, Crivellato E, Vacca A. Leptin-leptin receptor are involved in angiogenesis in human hepatocellular carcinoma. Peptides. 2008;29:1596–1602. doi: 10.1016/j.peptides.2008.05.011. [DOI] [PubMed] [Google Scholar]
- 46.Saxena NK, Sharma D, Ding X, Lin S, Marra F, Merlin D, Anania FA. Concomitant activation of the JAK/STAT, PI3K/AKT, and ERK signaling is involved in leptin-mediated promotion of invasion and migration of hepatocellular carcinoma cells. Cancer Res. 2007;67:2497–2507. doi: 10.1158/0008-5472.CAN-06-3075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hebbard L, Ranscht B. Multifaceted roles of adiponectin in cancer. Best Pract Res Clin Endocrinol Metab. 2014;28:59–69. doi: 10.1016/j.beem.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yokomizo T, Kita S, Sugiyama T, Miyagishi M, Hara K, Tsunoda M, et al. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature. 2003;423:762–769. doi: 10.1038/nature01705. [DOI] [PubMed] [Google Scholar]
- 49.Horton JD. Sterol regulatory element-binding proteins: transcriptional activators of lipid synthesis. Biochem Soc Trans. 2002;30:1091–1095. doi: 10.1042/bst0301091. [DOI] [PubMed] [Google Scholar]
- 50.Ouchi N, Kihara S, Arita Y, Okamoto Y, Maeda K, Kuriyama H, Hotta K, Nishida M, Takahashi M, Muraguchi M, et al. Adiponectin, an adipocyte-derived plasma protein, inhibits endothelial NF-kappaB signaling through a cAMP-dependent pathway. Circulation. 2000;102:1296–1301. doi: 10.1161/01.cir.102.11.1296. [DOI] [PubMed] [Google Scholar]
- 51.Kobashi C, Urakaze M, Kishida M, Kibayashi E, Kobayashi H, Kihara S, Funahashi T, Takata M, Temaru R, Sato A, et al. Adiponectin inhibits endothelial synthesis of interleukin-8. Circ Res. 2005;97:1245–1252. doi: 10.1161/01.RES.0000194328.57164.36. [DOI] [PubMed] [Google Scholar]
- 52.Yokota T, Oritani K, Takahashi I, Ishikawa J, Matsuyama A, Ouchi N, Kihara S, Funahashi T, Tenner AJ, Tomiyama Y, et al. Adiponectin, a new member of the family of soluble defense collagens, negatively regulates the growth of myelomonocytic progenitors and the functions of macrophages. Blood. 2000;96:1723–1732. [PubMed] [Google Scholar]
- 53.Hui JM, Hodge A, Farrell GC, Kench JG, Kriketos A, George J. Beyond insulin resistance in NASH: TNF-alpha or adiponectin? Hepatology. 2004;40:46–54. doi: 10.1002/hep.20280. [DOI] [PubMed] [Google Scholar]
- 54.Asano T, Watanabe K, Kubota N, Gunji T, Omata M, Kadowaki T, Ohnishi S. Adiponectin knockout mice on high fat diet develop fibrosing steatohepatitis. J Gastroenterol Hepatol. 2009;24:1669–1676. doi: 10.1111/j.1440-1746.2009.06039.x. [DOI] [PubMed] [Google Scholar]
- 55.Kamada Y, Tamura S, Kiso S, Matsumoto H, Saji Y, Yoshida Y, Fukui K, Maeda N, Nishizawa H, Nagaretani H, et al. Enhanced carbon tetrachloride-induced liver fibrosis in mice lacking adiponectin. Gastroenterology. 2003;125:1796–1807. doi: 10.1053/j.gastro.2003.08.029. [DOI] [PubMed] [Google Scholar]
- 56.Zhou M, Xu A, Tam PK, Lam KS, Chan L, Hoo RL, Liu J, Chow KH, Wang Y. Mitochondrial dysfunction contributes to the increased vulnerabilities of adiponectin knockout mice to liver injury. Hepatology. 2008;48:1087–1096. doi: 10.1002/hep.22444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Masaki T, Chiba S, Tatsukawa H, Yasuda T, Noguchi H, Seike M, Yoshimatsu H. Adiponectin protects LPS-induced liver injury through modulation of TNF-alpha in KK-Ay obese mice. Hepatology. 2004;40:177–184. doi: 10.1002/hep.20282. [DOI] [PubMed] [Google Scholar]
- 58.Xu A, Wang Y, Keshaw H, Xu LY, Lam KS, Cooper GJ. The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J Clin Invest. 2003;112:91–100. doi: 10.1172/JCI17797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu CJ, Chen PJ, Lai MY, Liu CH, Chen CL, Kao JH, Chen DS. High serum adiponectin correlates with advanced liver disease in patients with chronic hepatitis B virus infection. Hepatol Int. 2009;3:364–370. doi: 10.1007/s12072-008-9111-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tietge UJ, Böker KH, Manns MP, Bahr MJ. Elevated circulating adiponectin levels in liver cirrhosis are associated with reduced liver function and altered hepatic hemodynamics. Am J Physiol Endocrinol Metab. 2004;287:E82–E89. doi: 10.1152/ajpendo.00494.2003. [DOI] [PubMed] [Google Scholar]
- 61.Chen MJ, Yeh YT, Lee KT, Tsai CJ, Lee HH, Wang SN. The promoting effect of adiponectin in hepatocellular carcinoma. J Surg Oncol. 2012;106:181–187. doi: 10.1002/jso.23059. [DOI] [PubMed] [Google Scholar]
- 62.van der Poorten D, Samer CF, Ramezani-Moghadam M, Coulter S, Kacevska M, Schrijnders D, Wu LE, McLeod D, Bugianesi E, Komuta M, et al. Hepatic fat loss in advanced nonalcoholic steatohepatitis: are alterations in serum adiponectin the cause? Hepatology. 2013;57:2180–2188. doi: 10.1002/hep.26072. [DOI] [PubMed] [Google Scholar]
- 63.Saxena NK, Fu PP, Nagalingam A, Wang J, Handy J, Cohen C, Tighiouart M, Sharma D, Anania FA. Adiponectin modulates C-jun N-terminal kinase and mammalian target of rapamycin and inhibits hepatocellular carcinoma. Gastroenterology. 2010;139:1762–173, 1762-173. doi: 10.1053/j.gastro.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Man K, Ng KT, Xu A, Cheng Q, Lo CM, Xiao JW, Sun BS, Lim ZX, Cheung JS, Wu EX, et al. Suppression of liver tumor growth and metastasis by adiponectin in nude mice through inhibition of tumor angiogenesis and downregulation of Rho kinase/IFN-inducible protein 10/matrix metalloproteinase 9 signaling. Clin Cancer Res. 2010;16:967–977. doi: 10.1158/1078-0432.CCR-09-1487. [DOI] [PubMed] [Google Scholar]
- 65.Hebbard L, George J. Animal models of nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol. 2011;8:35–44. doi: 10.1038/nrgastro.2010.191. [DOI] [PubMed] [Google Scholar]
- 66.Wigg AJ, Roberts-Thomson IC, Dymock RB, McCarthy PJ, Grose RH, Cummins AG. The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxaemia, and tumour necrosis factor alpha in the pathogenesis of non-alcoholic steatohepatitis. Gut. 2001;48:206–211. doi: 10.1136/gut.48.2.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shanab AA, Scully P, Crosbie O, Buckley M, O’Mahony L, Shanahan F, Gazareen S, Murphy E, Quigley EM. Small intestinal bacterial overgrowth in nonalcoholic steatohepatitis: association with toll-like receptor 4 expression and plasma levels of interleukin 8. Dig Dis Sci. 2011;56:1524–1534. doi: 10.1007/s10620-010-1447-3. [DOI] [PubMed] [Google Scholar]
- 68.Mouzaki M, Comelli EM, Arendt BM, Bonengel J, Fung SK, Fischer SE, McGilvray ID, Allard JP. Intestinal microbiota in patients with nonalcoholic fatty liver disease. Hepatology. 2013;58:120–127. doi: 10.1002/hep.26319. [DOI] [PubMed] [Google Scholar]
- 69.Zhu L, Baker SS, Gill C, Liu W, Alkhouri R, Baker RD, Gill SR. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: a connection between endogenous alcohol and NASH. Hepatology. 2013;57:601–609. doi: 10.1002/hep.26093. [DOI] [PubMed] [Google Scholar]
- 70.Velayudham A, Dolganiuc A, Ellis M, Petrasek J, Kodys K, Mandrekar P, Szabo G. VSL#3 probiotic treatment attenuates fibrosis without changes in steatohepatitis in a diet-induced nonalcoholic steatohepatitis model in mice. Hepatology. 2009;49:989–997. doi: 10.1002/hep.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mencarelli A, Cipriani S, Renga B, Bruno A, D’Amore C, Distrutti E, Fiorucci S. VSL#3 resets insulin signaling and protects against NASH and atherosclerosis in a model of genetic dyslipidemia and intestinal inflammation. PLoS One. 2012;7:e45425. doi: 10.1371/journal.pone.0045425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Endo H, Niioka M, Kobayashi N, Tanaka M, Watanabe T. Butyrate-producing probiotics reduce nonalcoholic fatty liver disease progression in rats: new insight into the probiotics for the gut-liver axis. PLoS One. 2013;8:e63388. doi: 10.1371/journal.pone.0063388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wong VW, Won GL, Chim AM, Chu WC, Yeung DK, Li KC, Chan HL. Treatment of nonalcoholic steatohepatitis with probiotics. A proof-of-concept study. Ann Hepatol. 2013;12:256–262. [PubMed] [Google Scholar]
- 74.Malaguarnera M, Vacante M, Antic T, Giordano M, Chisari G, Acquaviva R, Mastrojeni S, Malaguarnera G, Mistretta A, Li Volti G, et al. Bifidobacterium longum with fructo-oligosaccharides in patients with non alcoholic steatohepatitis. Dig Dis Sci. 2012;57:545–553. doi: 10.1007/s10620-011-1887-4. [DOI] [PubMed] [Google Scholar]
- 75.Brun P, Castagliuolo I, Di Leo V, Buda A, Pinzani M, Palù G, Martines D. Increased intestinal permeability in obese mice: new evidence in the pathogenesis of nonalcoholic steatohepatitis. Am J Physiol Gastrointest Liver Physiol. 2007;292:G518–G525. doi: 10.1152/ajpgi.00024.2006. [DOI] [PubMed] [Google Scholar]
- 76.Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, Burcelin R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57:1470–1481. doi: 10.2337/db07-1403. [DOI] [PubMed] [Google Scholar]
- 77.Imajo K, Fujita K, Yoneda M, Nozaki Y, Ogawa Y, Shinohara Y, Kato S, Mawatari H, Shibata W, Kitani H, et al. Hyperresponsivity to low-dose endotoxin during progression to nonalcoholic steatohepatitis is regulated by leptin-mediated signaling. Cell Metab. 2012;16:44–54. doi: 10.1016/j.cmet.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 78.Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, Thaiss CA, Kau AL, Eisenbarth SC, Jurczak MJ, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482:179–185. doi: 10.1038/nature10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dapito DH, Mencin A, Gwak GY, Pradere JP, Jang MK, Mederacke I, Caviglia JM, Khiabanian H, Adeyemi A, Bataller R, et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell. 2012;21:504–516. doi: 10.1016/j.ccr.2012.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ridlon JM, Alves JM, Hylemon PB, Bajaj JS. Cirrhosis, bile acids and gut microbiota: unraveling a complex relationship. Gut Microbes. 2013;4:382–387. doi: 10.4161/gmic.25723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, Iwakura Y, Oshima K, Morita H, Hattori M, et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature. 2013;499:97–101. doi: 10.1038/nature12347. [DOI] [PubMed] [Google Scholar]
- 82.Jonker JW, Liddle C, Downes M. FXR and PXR: potential therapeutic targets in cholestasis. J Steroid Biochem Mol Biol. 2012;130:147–158. doi: 10.1016/j.jsbmb.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cariou B, van Harmelen K, Duran-Sandoval D, van Dijk TH, Grefhorst A, Abdelkarim M, Caron S, Torpier G, Fruchart JC, Gonzalez FJ, et al. The farnesoid X receptor modulates adiposity and peripheral insulin sensitivity in mice. J Biol Chem. 2006;281:11039–11049. doi: 10.1074/jbc.M510258200. [DOI] [PubMed] [Google Scholar]
- 84.Hollman DA, Milona A, van Erpecum KJ, van Mil SW. Anti-inflammatory and metabolic actions of FXR: insights into molecular mechanisms. Biochim Biophys Acta. 2012;1821:1443–1452. doi: 10.1016/j.bbalip.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 85.Zhang S, Wang J, Liu Q, Harnish DC. Farnesoid X receptor agonist WAY-362450 attenuates liver inflammation and fibrosis in murine model of non-alcoholic steatohepatitis. J Hepatol. 2009;51:380–388. doi: 10.1016/j.jhep.2009.03.025. [DOI] [PubMed] [Google Scholar]
- 86.Mudaliar S, Henry RR, Sanyal AJ, Morrow L, Marschall HU, Kipnes M, Adorini L, Sciacca CI, Clopton P, Castelloe E, et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology. 2013;145:574–82.e1. doi: 10.1053/j.gastro.2013.05.042. [DOI] [PubMed] [Google Scholar]
- 87.Yang F, Huang X, Yi T, Yen Y, Moore DD, Huang W. Spontaneous development of liver tumors in the absence of the bile acid receptor farnesoid X receptor. Cancer Res. 2007;67:863–867. doi: 10.1158/0008-5472.CAN-06-1078. [DOI] [PubMed] [Google Scholar]
- 88.Wolfe A, Thomas A, Edwards G, Jaseja R, Guo GL, Apte U. Increased activation of the Wnt/β-catenin pathway in spontaneous hepatocellular carcinoma observed in farnesoid X receptor knockout mice. J Pharmacol Exp Ther. 2011;338:12–21. doi: 10.1124/jpet.111.179390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Prawitt J, Caron S, Staels B. Bile acid metabolism and the pathogenesis of type 2 diabetes. Curr Diab Rep. 2011;11:160–166. doi: 10.1007/s11892-011-0187-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Thomas C, Gioiello A, Noriega L, Strehle A, Oury J, Rizzo G, Macchiarulo A, Yamamoto H, Mataki C, Pruzanski M, et al. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009;10:167–177. doi: 10.1016/j.cmet.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang YD, Chen WD, Yu D, Forman BM, Huang W. The G-protein-coupled bile acid receptor, Gpbar1 (TGR5), negatively regulates hepatic inflammatory response through antagonizing nuclear factor κ light-chain enhancer of activated B cells (NF-κB) in mice. Hepatology. 2011;54:1421–1432. doi: 10.1002/hep.24525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Keitel V, Donner M, Winandy S, Kubitz R, Häussinger D. Expression and function of the bile acid receptor TGR5 in Kupffer cells. Biochem Biophys Res Commun. 2008;372:78–84. doi: 10.1016/j.bbrc.2008.04.171. [DOI] [PubMed] [Google Scholar]
- 93.Péan N, Doignon I, Garcin I, Besnard A, Julien B, Liu B, Branchereau S, Spraul A, Guettier C, Humbert L, et al. The receptor TGR5 protects the liver from bile acid overload during liver regeneration in mice. Hepatology. 2013;58:1451–1460. doi: 10.1002/hep.26463. [DOI] [PubMed] [Google Scholar]