

Here, Li et al. show that inflammatory signaling regulates embryonic hematopoietic stem and progenitor cell (HSPC) formation. HSCs from aorta/gonad/mesonephros (AGM) regions of midgestation mouse embryos expressed a robust innate immune/inflammatory signature. Mouse embryos lacking interferon γ (IFN-γ )or IFN-α signaling and zebrafish lacking IFN-γ and IFN-ϕ activity had fewer AGM HSPCs. IRF2-occupied genes identified in human fetal liver CD34+ HSPCs were actively transcribed in human and mouse HSPCs.
Keywords: interferon, hematopoiesis, lymphoid progenitor, hematopoietic stem cell, mouse, zebrafish
Abstract
Identifying signaling pathways that regulate hematopoietic stem and progenitor cell (HSPC) formation in the embryo will guide efforts to produce and expand HSPCs ex vivo. Here we show that sterile tonic inflammatory signaling regulates embryonic HSPC formation. Expression profiling of progenitors with lymphoid potential and hematopoietic stem cells (HSCs) from aorta/gonad/mesonephros (AGM) regions of midgestation mouse embryos revealed a robust innate immune/inflammatory signature. Mouse embryos lacking interferon γ (IFN-γ) or IFN-α signaling and zebrafish morphants lacking IFN-γ and IFN-ϕ activity had significantly fewer AGM HSPCs. Conversely, knockdown of IFN regulatory factor 2 (IRF2), a negative regulator of IFN signaling, increased expression of IFN target genes and HSPC production in zebrafish. Chromatin immunoprecipitation (ChIP) combined with sequencing (ChIP-seq) and expression analyses demonstrated that IRF2-occupied genes identified in human fetal liver CD34+ HSPCs are actively transcribed in human and mouse HSPCs. Furthermore, we demonstrate that the primitive myeloid population contributes to the local inflammatory response to impact the scale of HSPC production in the AGM region. Thus, sterile inflammatory signaling is an evolutionarily conserved pathway regulating the production of HSPCs during embryonic development.
Hematopoiesis in the embryo begins with the emergence of committed progenitors followed by hematopoietic stem cells (HSCs). Primitive erythrocytes differentiate in the yolk sac (YS) of mice from erythrocyte and erythrocyte/megakaryocyte progenitors that emerge at embryonic day 7.0 (E7.0) (Palis et al. 1999; Tober et al. 2007). Macrophage progenitors appear in the YS at E8.0, and macrophages subsequently migrate throughout the embryo (Palis et al. 1999; Bertrand et al. 2005). Definitive erythrocytes and myeloid lineage cells differentiate in the YS starting at E8.25 from a committed erythro–myeloid progenitor (EMP) (Bertrand et al. 2007; Frame et al. 2013). Cells with lymphoid potential (B and T) can be found in the YS and para-aortic splanchopleura at E8.5–E9.5 (Liu and Auerbach 1991; Godin et al. 1995; Yokota et al. 2006; Yoshimoto et al. 2012). HSCs first appear at E10.5–E11.5 in the aorta/gonad/mesonephros (AGM) region and umbilical and vitelline arteries (Müller et al. 1994; de Bruijn et al. 2000). Progenitors and HSCs differentiate directly from hemogenic endothelium and briefly accumulate in clusters in the aortic lumen before release into the circulation to colonize the fetal liver (FL) (Swiers et al. 2013). How the embryo designates production of committed progenitors with limited potential versus HSCs from hemogenic endothelium is not known.
Although lymphoid progenitors are detectable by E8.5–E9.5 in mouse embryos and by 4 d post-fertilization (dpf) in the thymus of zebrafish embryos, a functional adaptive immune system is not present in mice until after birth and is not present in zebrafish until 3 wk of age (Lam et al. 2004; Page et al. 2013). However, as early as 1 dpf, zebrafish embryos have a functional innate immune system that can respond to bacterial and viral pathogens in their environment (Davis et al. 2002). The cellular effectors of innate immunity in the zebrafish embryo are primitive macrophages and granulocytes, and molecular components include pattern recognition receptor Toll-like receptors (TLRs) and nucleotide-binding oligomerization domain (NOD) proteins, cytokines such as interleukin 1β (IL-1β) and interferon γ (IFN-γ), and transcription factors such as nuclear factor κB (NF-κB) and IFN regulatory factors (IRFs) (Oehlers et al. 2011). The mammalian fetus is likewise protected from pathogens by a robust innate immune system at the maternal/fetal interface, the maternal decidual components of which are stromal and immune cells (Abrahams 2008). Macrophages, which are of maternal origin initially and are replaced at E9.5 by macrophages that are YS-derived, also play an important role in fetal innate immune responses (Bertrand et al. 2005). Activation of the innate immune system results in the synthesis of eicosanoids, such as leukotrienes and prostanoids, and inflammatory cytokines, including tumor necrosis factor (TNF), IL-1β, IL-6, IL-12, and type I and II IFNs.
Tonic innate immune signaling influences normal developmental processes. The paradigmatic example is establishment of dorsoventral patterning of the precellular Drosophila embryo by Toll and its downstream effector, the NF-κB homolog Dorsal (Anderson et al. 1985). Toll signaling also regulates the number of blood cells (hemocytes) in Drosophila and the differentiation of a particular hemocyte lineage, lamellocytes (Qiu et al. 1998). In mouse embryos, IL-1 signaling impacts hematopoietic stem and progenitor cells (HSPCs) in the AGM region (Orelio et al. 2008). However, across vertebrate species, a general role for innate immune or inflammatory signaling in HSPC production in the absence of a microbial challenge has not been proposed.
Here we show that progenitors with lymphoid potential (LPs) isolated from the major arteries (dorsal aorta, umbilical, and vitelline) of mouse embryos have a robust innate immune/inflammatory molecular signature. The number of LPs in mouse and zebrafish embryos is positively regulated by the inflammatory cytokines IFN-γ and IFN-α (IFN-ϕ in zebrafish), with IFN-γ signaling also impacting the number of functional HSCs. Furthermore, we demonstrate that inflammatory signaling is active in human fetal HSPCs based on the expression of known IFN target genes. Finally, we show that the primitive myeloid population contributes to the local inflammatory response to regulate the number of HSPCs. Together, our data indicate that sterile tonic inflammatory signaling regulates HSPC formation in the vertebrate embryo.
Results
Ly6a-GFP expression enriches for HSCs and cells with lymphoid potential
A role for inflammatory signaling in definitive hematopoiesis was uncovered while characterizing the expression of a transgenic HSC marker, Ly6a-GFP. Ly6a encodes the cell surface molecule Sca-1, which is found on all HSCs in the FL and bone marrow (BM) but on only a subset of newly emerging HSCs in the E11.5 AGM region (de Bruijn et al. 2002). In contrast, GFP expression from a multicopy Ly6a-GFP transgene marks all functional AGM HSCs, as determined by transplantation into adult recipient mice; thus, unlike cell surface Sca-1, the Ly6a-GFP transgene is a reliable marker for these cells (de Bruijn et al. 2002). To determine whether Ly6a-GFP expression could distinguish HSCs from earlier and more abundant YS-derived committed EMPs, we isolated CD45+ Ly6a-GFP+ and CD45+ Ly6a-GFP− cells and quantified EMPs in each population in methylcellulose colony-forming assays conducted in the presence of EPO, SCF, IL-6, and IL-3 (Fig. 1A). We found that most CD45+ cells and EMPs in the YS were Ly6a-GFP− (Fig. 1B,C). In addition, most progenitors in the E11.5 FL, which at that time are primarily YS-derived EMPs (Frame et al. 2013), were also Ly6a-GFP− (Fig. 1C).
Figure 1.
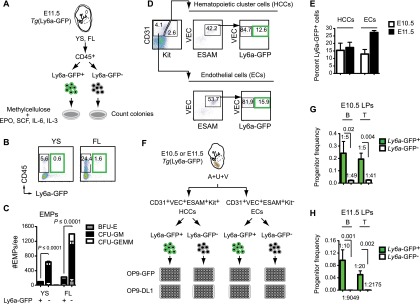
Ly6a-GFP expression marks LPs but not EMPs. (A) Scheme for isolating Ly6a-GFP+/− CD45+ cells from E11.5 embryos and methylcellulose colony-forming assays to quantify EMPs. (B) Scatter plots of representative sort samples for colony assays. (C) Number of EMPs per embryo equivalent (ee) of the indicated tissue. (BFU-E) Burst-forming units erythroid; (CFU-GM) colony-forming units granulocyte/monocyte; (CFU-GEMM) CFUs granulocyte/erythrocyte/monocyte/megakaryocyte. Error bars represent mean ± 95% CI. Data are from three replicates, using pooled embryos. P-values were calculated by t-test as described in the Materials and Methods. (D) Representative scatter plots of cell isolation for lymphoid progenitor assays. (E) Percentage of Ly6a-GFP+ cells in the hematopoietic cluster (HCC: CD31+VEC+ESAM+Kit+) and endothelial (EC: CD31+VEC+ESAM+Kit−) populations. Data are averaged from five to six litters of pooled embryos (mean ± SD). (F) Scheme for limiting dilution assay to enumerate progenitors with B and T potential (LPs) on OP9-GFP and OP9-DL1 stromal cells, respectively. B cells were identified as CD19+ B220+, and T cells were identified as CD25+ Thy1.1+. (G) Frequency of LPs in the Ly6a-GFP+ and Ly6a-GFP− fractions of HCCs from E10.5 embryos (mean ± SD). Progenitor frequency is indicated above columns. Data are from three experiments using pooled cells from Tg(Ly6a-GFP) embryos. Additional data, including cell numbers, are summarized in Supplemental Table S1. (H) Frequency of LPs at E11.5, as in G (n = 4 experiments) (see also Supplemental Table S1).
To determine whether Ly6a-GFP expression marks LPs, which would include committed lymphoid progenitors and multipotent HSPCs, we sorted cells from dissected AGM regions and umbilical and vitelline arteries (A+U+V) using three endothelial markers—CD31, vascular endothelial cadherin (VEC), and endothelial cell (EC) adhesion molecule (ESAM)—and further separated the cells into intra-arterial hematopoietic cluster cells (HCCs) and ECs using an antibody to Kit that specifically marks the HCCs. Both HCCs (CD31+VEC+ESAM+Kit+) and ECs (CD31+VEC+ESAM+Kit−) were then segregated into Ly6a-GFP+ and Ly6a-GFP− fractions (Fig. 1D). LPs with B lineage potential were enumerated by limiting dilution on OP9 stromal cells, and LPs with T potential were enumerated by limiting dilution on OP9 expressing the Notch ligand Delta-like 1 (DL1) in the presence of Flt3 ligand (FLT3L) and IL-7 (Fig. 1F). At E10.5, only 15% of HCCs and ECs were Ly6a-GFP+ (Fig. 1E). Notably, LPs were present at a much higher frequency in the Ly6a-GFP+ population of HCCs; approximately one in five Ly6a-GFP+ HCCs had in vitro B or T lineage potential, as compared with one in 49 Ly6a-GFP− HCCs (Fig. 1G; Supplemental Table S1). The segregation of LPs into the Ly6a-GFP+ fraction was even more pronounced at E11.5, with a 100-fold higher frequency in Ly6a-GFP+ versus Ly6a-GFP− HCCs (Fig. 1H; Supplemental Table S1). No LPs were found in Ly6a-GFP+/− ECs at either E10.5 or E11.5 (data not shown). In summary, Ly6a-GFP specifically marks HSCs (de Bruijn et al. 2002) and LPs within intra-arterial HCCs and does not mark EMPs in the YS or FL. As Ly6a-GFP+ cells represent only 15%–20% of the HCCs, HSCs and LPs can be greatly enriched by separating HCCs based on this marker.
Ly6a-GFP+ HCCs and ECs express genes involved in innate immunity and inflammation
We hypothesized that signaling pathways specifically active in Ly6a-GFP+ HCCs and ECs may regulate the formation of HSCs and LPs from hemogenic endothelium. To identify these pathways, we performed gene expression analyses comparing Ly6a-GFP+ and Ly6a-GFP− HCCs and ECs. We sorted CD31+VEC+ESAM+Kit+/− cells from A+U+V tissue dissected from E10.5 embryos, separated these populations into Ly6a-GFP+ and Ly6a-GFP− fractions, and examined gene expression of pooled replicate samples. We found 8222 differentially expressed genes across the four cell subtypes (P < 0.01, one-way ANOVA). Consensus clustering of these genes, varying the number of clusters (k) from two to 20, identified an optimal setting of k = 8 from cumulative distribution function (CDF) plots (Fig. 2A; Supplemental Fig. S1). Cluster 1 (C1), which consisted of 1305 genes with higher expression in Ly6a-GFP+ HCCs versus all other cells, generated the largest number of highly enriched gene ontology (GO) terms (Fig. 2B; Supplemental Fig. S2; Supplemental Tables S2, S3). By far the most highly enriched GO term associated with C1 was “innate immune response” (P = 10−533) (Fig. 2C). Other highly enriched GO terms in C1 were “immune response,” “inflammatory response,” “positive regulation of T-cell proliferation,” “response to bacterium,” “defense response to virus,” “TLR signaling pathway,” “defense response to protozoan,” and “positive regulation of TNF production” (Supplemental Table S2). C1 was also enriched for GO terms associated with lymphoid responses and differentiation (Supplemental Table S2), consistent with the enrichment of LPs in Ly6a-GFP+ HCCs. Examples of genes up-regulated in C1 include TLRs (Tlr1, Tlr2, Tlr7, and Tlr13), histocompatibility antigens (Hmh1), lymphocyte-associated genes (Cd69 and Fc receptor-like S [Fcrls]), and IFN-regulated genes (Irf1, Isg20, Psmb8, RnaseI, Tap2, and Tabpb) (Fig. 2D,E; Supplemental Table S3). Macrophages were excluded as the major source of the innate immune/inflammatory profile, as, by flow cytometry, <1% of Ly6a-GFP+ HCCs were F4/80+ (data not shown). Therefore, we conclude that the innate immune/inflammatory signature is intrinsic to Ly6a-GFP+ HCCs, which suggests inflammatory signaling has an active role in the specification or expansion of embryonic HSCs and LPs.
Figure 2.

Ly6a-GFP+ HCCs have an innate immune/inflammatory signature. (A) Consensus clustering of microarray data from Ly6a-GFP+ and Ly6a-GFP− HCCs and ECs. Sorts were performed as in Figure 1D. Duplicate RNA samples from each population (four total; ∼15,000 cells per sample) were analyzed by microarray. Each row represents one gene. Values in the key represent the frequency at which two genes were observed to cluster together. Cluster numbers, including gene number (n) within each set, are indicated at the right. C1 (boxed) is expanded in B. (B) Dendrogram of C1. Gene expression levels are normalized by Z-score transformation across microarray experiments. (H) HCCs; (E) ECs. (C) Top 10 enriched GO biological process terms among C1 genes. (D) Examples of innate immune/inflammatory genes with up-regulated expression in Ly6a-GFP+ HCCs. (E) qPCR analysis of several genes in D represented as fold difference relative to Hprt (n = 3, mean ± SD). Significance is by one-way ANOVA and Dunnett’s multiple comparison test with Ly6a-GFP+ HCCs as a comparator (#). (ns) Not significant.
Cluster 2 (C2) consisted of genes up-regulated in both Ly6a-GFP+ and Ly6a-GFP− HCCs relative to ECs and was enriched for the GO terms “transcription,” “DNA-dependent,” and “chromatin modification” (Supplemental Fig. S2; Supplemental Tables S2, S3). Hematopoietic-specific genes were not in C2, as they were more highly expressed in Ly6a-GFP+ HCCs and segregated to C1 (Supplemental Table S3). Cluster 3 (C3), which included 1417 genes more highly expressed in Ly6a-GFP+ ECs versus all other populations, was significantly enriched for GO terms related to angiogenesis and vasculogenesis. Cluster 4 (C4) contained 965 genes more highly expressed in Ly6a-GFP+ HCCs and ECs as compared with their Ly6a-GFP− counterparts and, like C1, generated GO terms associated with innate immune/inflammatory signaling (Supplemental Fig. S2; Supplemental Table S2). However, unlike C1, C4 lacked GO terms associated with lymphocyte differentiation and activation correlating with the absence of LPs in the EC population. Ly6a-GFP+ ECs and HCCs therefore share an innate immune/inflammatory signature, suggesting that inflammatory signaling pathways are activated in a subset of ECs that give rise to or stimulate production of HSCs and LPs.
Type I and II IFNs regulate lymphoid progenitors and HSCs in the fetus
Sca-1 is a well-known mediator of innate immune/inflammatory signaling in adult mice, and its expression is activated by proinflammatory cytokines (Luna et al. 2004; Flanagan et al. 2008; Essers et al. 2009; Purwanti et al. 2011; Shi et al. 2013). We therefore hypothesized that innate immune/inflammatory signaling may activate Ly6a-GFP in the embryo. To determine which inflammatory cytokines could further elevate its expression, we cultured explanted E9.5 Tg(Ly6a-GFP) embryos for 2 d in the presence of factors known to induce Sca-1 expression in the adult (Fig. 3). IL-1β, IL-4, and IL-6 had very little impact on Ly6a-GFP expression in embryo explants (Fig. 3B). TNFα caused a small but reproducible increase in the number of Ly6a-GFP+ HCCs, while type I and II IFN (IFN-α4 and IFN-γ) dramatically increased both Ly6a-GFP+ HCCs and ECs (Fig. 3B). The increase in Ly6a-GFP+ cells was caused by both higher expression of Ly6a-GFP, indicated by elevations in mean fluorescence intensity (MFI) in HCCs and ECs (Fig. 3C), and enhanced proliferation, seen as a higher percentage of Ki67+ cells in both populations (Fig. 3D). We conclude that HCCs and ECs in the embryo are wired to robustly respond directly or indirectly to IFN stimulation.
Figure 3.
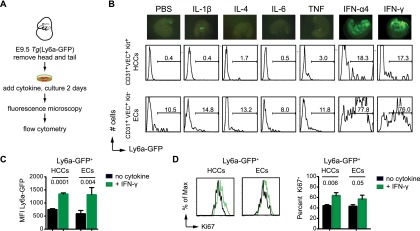
Type I and II IFNs induce Ly6a-GFP expression in embryo explants. (A) Experimental scheme to identify inflammatory cytokines capable of inducing Ly6a-GFP expression in 2-d embryo explant cultures. (B, top) Fluorescent images of embryo explants after 2 d of culture with the indicated cytokines (20 ng/mL). Representative histograms of GFP levels in HCCs (middle) and ECs (bottom). Data are representative of three experiments. (C) Increase in Ly6a-GFP MFI in HCCs and ECs following 2 d of explant culture. (n = 4; mean ± SD; t-test). (D) Increase in the percentage of Ki67+ cells in the Ly6a-GFP+ HCC and EC populations after 2 d of explant culture (n = 3; mean ± SD; t-test; representative histograms are at the left).
To determine whether the response to IFN-γ and IFN-α4 in HCCs and ECs was direct, we assessed expression of their receptors and Stat1 phosphorylation in response to ligand. The receptor for IFN-γ (IFNGR1) was present on >95% of CD31+Kit+ YS EMPs and >95% of HCCs and ECs in the A+U+V of E11.5 embryos, although the MFI was eightfold lower on ECs (Fig. 4A,B). We were unable to detect the IFN-α receptor on either HCCs or ECs for technical reasons but found Ifnar1 mRNA in both populations (Fig. 4C). Addition of IFN-γ or IFN-α4 to disaggregated A+U+V cells increased p-Stat1 in the majority of HCCs within 15 min but not in most ECs (Fig. 4D). Thus, HCCs, but not the bulk EC population, respond rapidly and robustly to IFNs.
Figure 4.

IFNγ signaling regulates embryonic hematopoiesis. (A) Flow cytometry for IFNGR1 on CD31+Kit+ YS EMPs (E11.5). (FMO) Fluorescence minus one control. (B) Flow cytometry for IFNGR1 on Ly6a-GFP+/− HCCs and ECs from the A+U+V (E11.5). (C) qPCR for Ifnar1 mRNA in Ly6a-GFP+/− HCCs (H) and ECs (E) (E10.5) shown as fold difference/Hprt. (D) Increase in p-Stat1 in HCCs (VEC+Kit+) but not ECs (VEC+Kit−) 15 min after addition of 50 ng/mL IFN-γ or IFN-α4 to disaggregated A+U+V cells (E10.5). (E) Number of EMPs per YS in E10.5 wild-type (WT) embryos and those deficient for IFN signaling (n = 7–14 embryos; mean ± SD; one-way ANOVA). (F) Number of LPs per A+U+V in E10.5 wild-type embryos and those deficient in IFN-γ or IFN-α signaling determined by limiting dilution on OP9 and OP9-DL1 stromal cells. P-values for total number of LPs (B+T) calculated by one-way ANOVA and Dunnett’s multiple comparison tests (n = 3–9 from two experiments; mean ± SD). (G) Determination of HSC numbers by limited dilution transplantation. Irradiated adult recipients (Ly5.1) were transplanted with the indicated fractions (1.0 or 0.3 embryo equivalents) of A+U+V cells (Ly5.2). The contribution of donor cells (positive engraftment scored as >1%) to CD34−Flt3−LSK BM was analyzed at 16 wk (n = 7–10 from eight experiments). Number of LT-HSCs per A+U+V and significance from wild type were determined by extreme limiting dilution analysis (Hu and Smyth 2009).
More relevant is whether hematopoiesis is affected by loss of IFN signaling. There was no decrease in YS EMP number in E10.5 Ifng−/−, Ifngr1−/−, or Ifnar1−/− embryos (Fig. 4E), correlating with the absence of Ly6a-GFP expression in EMPs. As >95% of EMPs are IFNGR1+ (Fig. 4A), this means that EMPs either are not exposed to IFN-γ in the YS or are unable to mount an IFN response, or their production is unaffected by IFN-γ signaling. In contrast, there was a twofold reduction in the frequency of LPs in the A+U+V of Ifng−/−, Ifngr1−/−, and Ifnar1−/− embryos at E10.5 (Fig. 4F), indicating that IFN signaling regulates the formation or numbers of LPs. To determine whether IFN signaling impacts the number of functional HSCs, we transplanted 1.0 and 0.3 embryo equivalents of A+U+V cells from E11.5 Ifng−/− and Ifngr1−/− embryos into adult mice and examined BM engraftment. There was a trend toward reduced HSC numbers per A+U+V in Ifng−/− embryos and a significant, fourfold decrease in Ifngr1−/− embryos (Fig. 4G), indicating that IFN-γ signaling regulates the number of functional HSCs in the A+U+V. Given that phenotypic long-term repopulating HSCs are reportedly present at normal frequencies in the adult BM of IFN-γ-deficient mice (Baldridge et al. 2010), the effects of IFN-γ signaling deficiency are either transient or compensated for by other factors, modifying the numbers of HSCs that emerge at E11.5 in the A+U+V but not HSC maintenance.
A role for IFN signaling is conserved in zebrafish
Zebrafish possess two genes homologous to mammalian Ifng (ifng1-1 and ifng1-2) and three genes that encode the Ifng receptor (crfb6, crfb13, and crfb17). Morpholinos (MOs) targeting both ifng1-1 and ifng1-2 (combination referred to as Ifng MO) decreased runx1 expression in the AGM region at 33 h post-fertilization (hpf), as did combined knockdown of crfb6, crfb13, and crfb17 (referred to as Ifngr MO) (Fig. 5A,B). Elevation of IFN-γ levels by injection of ifng1-1 mRNA into embryos increased runx1 expression in the AGM (Fig. 5A,B), indicating that IFN-γ signaling induces HSPC formation in vivo. The impact of MO-mediated Ifng/Ifngr loss was due to a decrease in total cell number rather than runx1 expression per cell, as the number of flk1:dsRed+cmyb:gfp+ AGM HSPCs (flk1:dsRed marks ECs and HSCs, and cmyb:gfp marks HSCs and myeloid progenitors) (Bertrand et al. 2010) was also reduced (Supplemental Fig. S3A,B). MOs targeting Ifng and Ifngr similarly decreased the number of cd41:gfp+ HSCs in the caudal hematopoietic territory (CHT), a site of colonization and expansion of AGM-derived cells, between 36 and 72 hpf (Fig. 5C,D). Expression of rag1 in the thymus and the number of rag2:dsRed+ thymocytes were subsequently diminished (Fig. 5E–G). In contrast, the percentage of lmo2:GFP+gata1:dsRed+ EMPs was not impacted (Supplemental Fig. S3C). The efficiency of the Ifng knockdown was confirmed by altered expression of several known IFN-γ target genes (Supplemental Fig. S3D). Vascular development, maturation, and circulation in zebrafish morphants (Supplemental Fig. S3E,F) as well as vascular morphology in IFN-γ signaling-deficient murine embryos (Supplemental Fig. S3G) appeared normal, indicating that reductions in HSPC number were not secondary to gross vascular defects.
Figure 5.

IFN signaling regulates hematopoiesis in zebrafish embryos. (A) Whole-mount in situ hybridization (WISH) for runx1 in the AGM region of representative embryos injected with MOs targeting Ifng (ifng1.1 plus ifng1.2) or the IFN-γ receptors (crfb6, crfb13, and crfb17) (Ifngr MO) or with mRNA for ifng1-1 compared with sibling controls at 33 hpf (n ≥ 100 embryos per condition, from four experiments). (B) Bar graph showing qualitative phenotypic distribution as percent of embryos from A scored with low, medium, or high runx1 expression in the AGM. (C) Representative effects of Ifng and Ifngr knockdown on cd41:gfp+ HSPCs in the CHT at 48 hpf. Boxed areas in the top panels are magnified in the panels below. (D) Absolute counts of cd41:gfp+ cells in the CHT were determined at fixed time intervals (n ≥ 17 embryos per condition). P-value, determined for each time point relative to control (#), was ≤0.0001 at all time windows. (E) WISH for rag1 expression in the thymi of representative embryos at 5 dpf following Ifng and Ifngr knockdown (n ≥ 7 embryos per condition). (F) Qualitative phenotypic distribution as percentage of embryos scored with low, medium, or high rag1 expression. (G) Percentage of rag2:dsRed+ cells in total 5-dpf fish embryos quantified by flow cytometry (ANOVA, n ≥ 4 replicates of five pooled embryos). (H) WISH for runx1 in the AGM of representative embryos (n ≥ 23 embryos per condition) injected with paired MOs targeting IFN-ϕs. (I) Phenotype distribution of embryos from H, scored as in B. (J) WISH for rag1 in the thymi of representative embryos (n ≥ 23 embryos per condition) injected with paired MOs targeting IFN-ϕs. (K) Phenotype distribution of embryos from J, scored as in F. (L) cd41:gfp+ cells in the CHT following MO knockdown of IFN-ϕs and crfb5 (n ≥ 19 embryos per condition). Error bars indicate SD. (ns) Not significant. (M) cd41:gfp+ cells in the CHT at 38 hpf following MO knockdown of Ifng and crfb5 alone or in combination (n = 8–20 embryos per condition).
IFN-α has no zebrafish equivalent, but there are four type I IFN genes in zebrafish not found in mammals, referred to as ifnphi 1–4 (Levraud et al. 2007). The dimeric receptor for each of the IFN-ϕ isoforms uses one specific (crfb1 or crfb2) and one common (crfb5) chain. MO knockdown of Ifnϕ1+4, Ifnϕ2+3, or the common receptor chain crfb5 decreased runx1 and rag1 expression in the AGM and thymus, respectively (Fig. 5H–K). Knockdown of IFN-ϕs and crfb5 decreased the number of cd41:gfp+ cells in the CHT (Fig. 5L) alone and in combination with Ifng-MO (Fig. 5M). Thus, as in mice, multiple IFNs regulate embryonic HSPC production in zebrafish.
IRF2 occupies enhancers in human CD34+ FL cells
To provide further evidence that sterile IFN signaling was active in embryonic HSPCs, we examined the expression of IFN target genes in mouse and human embryos. IFN target genes were identified by chromatin occupancy of the transcription factor IRF2 in human CD34+ FL HSPCs; IRF2 was selected out of the nine IRF genes in mice, as it is the only IRF known to function in adult HSCs, where it promotes quiescence and engraftment by dampening IFN signaling (Sato et al. 2009). We identified 2833 IRF2-bound peaks in hCD34+ FL cells (Fig. 6A). To systematically identify IRF2 targets, we used the genomics regions enrichment of annotation tool (GREAT). Although associating transcription factor chromatin immunoprecipitation (ChIP) combined with sequencing (ChIP-seq) peaks with their target genes is computationally challenging and all bioinformatic tools have limitations, GREAT is superior to the more commonly used nearest-gene strategy (McLean et al. 2010). GREAT revealed multiple significantly enriched (false discovery rate [FDR] Q-value < 0.05) GO terms and pathways, including IFN signaling (Fig. 6B; Supplemental Table S4). Genes within several of the enriched functional categories were more highly expressed in hCD34+ FL HSPCs compared with all genes as a whole (Fig. 6C,D) and relative to the core embryonic stem cell genes OCT4 and NANOG (Supplemental Fig. S4A), providing further evidence for their correct association as IRF2 target genes. Finally, to demonstrate relevance across systems, we showed that representative IRF2-occupied genes are expressed in mouse FL HSCs (Supplemental Fig. S4B).
Figure 6.

IRF2 target genes in human CD34+ FL HSPCs overlap with IFN pathway genes and C1 genes. (A) Venn diagram of IFN signaling genes expressed in hCD34+ FL HSPCs, C1 genes (expressed in Ly6a-GFP+ HCCs), and IRF2-bound genes in hCD34+ FL HSPCs. P-values for overlap between all three sets were computed using a hypergeometric test. P-values for overlap of IRF2-bound and expressed genes were computed using GREAT. (B) Enriched GO biological process terms for IRF2-bound genes determined by GREAT. (C) Expression levels of IRF2-bound targets relative to all genes in hCD34+ FL HSPCs enriched for GO terms. P-values (in parentheses) as computed by t-test. Gene expression was analyzed from previously published Affymetrix microarray data (Xu et al. 2012). Only genes annotated with GO terms that had significantly different expression compared with the genome background are shown. (D) Expression levels of all genes and IRF2-bound genes in hCD34+ FL HSPCs enriched for representative pathway terms, as in C. (E) ChIP-seq signals for representative IFN pathway genes bound by IRF2. (F) Phenotype distribution of runx1 expression by WISH (AGM region) for irf2a morphants generated with two independent MOs compared with sibling controls (n ≥ 40 embryos per condition). (G) Absolute number of cd41:gfp+ HSPCs in the CHT of irf2a morphants at 38 hpf (n = 17; mean ± SD; t-test). (H) Percent of rag2:dsRed+ cells in whole embryos at 5 dpf by flow cytometry in irf2a morphants (n = 7; mean ± SD; t-test). (I) qPCR for IRF2 target genes in irf2a morphants (n = 2, from four replicates; mean ± SD; t-test).
We compared the expression of IRF2-occupied genes in hCD34+ FL HSPCs with that of genes up-regulated in Ly6a-GFP+ HCCs (C1) and found significant overlap in the expression of orthologous IFN signaling-related genes (Fig. 6A; Supplemental Table S4). Expression of several of these genes in Ly6a-GFP+ HCCs was confirmed by quantitative PCR (qPCR) (Supplemental Fig. S4C). ChIP-seq signal plots illustrate IRF2 occupancy of representative genes that were expressed in both human FL CD34+ cells and mouse Ly6a-GFP+ HCCs (Fig. 6E). We conclude that IFN signaling, as evidenced by expression of IRF2-bound genes, is occurring in human HSPCs, mouse FL HSCs, and Ly6a-GFP+ HCCs.
Since IRF2 attenuates IFN signaling in adult HSCs, we asked whether IRF2 negatively regulates HSPC formation in the embryo. MO knockdown of irf2a in zebrafish embryos increased runx1 expression in the AGM (Fig. 6F; Supplemental Fig. S4E) and cd41:gfp+ cells in the CHT (Fig. 6G). Thymic rag1 expression (data not shown) and the number of rag2:dsRed+ cells (Fig. 6H) also increased in irf2a morphants, as did IRF2 target gene expression (Fig. 6I), including components of the IFN signaling cascade. Thus, IRF2 negatively regulates HSPC formation or numbers in zebrafish embryos, likely by dampening IFN signaling. These data confirm that IFN signaling is normally active during the window of HSPC production in the AGM.
Multiple cells in the embryo express inflammatory cytokines
As the number of HSPCs in both mouse and zebrafish embryos was reduced but not eliminated by loss of IFN signaling, we determined whether additional proinflammatory cytokines contribute to HSPC production alone or in combination with IFN. IL-1β was previously shown to modulate HSC formation in the murine AGM region (Orelio et al. 2008), and TNF caused a small increase in Ly6a-GFP expression in E9.5 embryo explants (Fig. 3B). In zebrafish embryos, both tnfa and il1b morphants had lower runx1 expression in the AGM (Fig. 7A) and fewer cd41-gfp+ cells in the CHT (Fig. 7B,D). Combinatorial MO knockdown of ifng and tnfa caused a greater decrease in runx1 expression than knockdown of either gene alone at 33 hpf (Fig. 7C) and sustained the effect of Ifng-MO at 72 hpf (Fig. 7D). Thus, multiple inflammatory cytokines can cooperate in the formation and expansion of embryonic HSPCs in the absence of pathological stimulation.
Figure 7.

Multiple inflammatory cytokines and primitive myeloid cells promote HSPC formation. (A) Phenotype distribution of runx1 expression by WISH (AGM region) for tnfa or il1b morphants compared with sibling controls (n ≥ 77 embryos per condition). (B) Absolute number of cd41:gfp+ HSPCs in the CHT of il1b morphants (n ≥ 17; mean ± SD; t-test). (C) Phenotype distribution of runx1 expression by WISH (AGM) for lnfg knockdown alone and in combination with tnfa MOs (n ≥ 54 embryos per condition). (D) Absolute counts of cd41:gfp+ cells in the CHT determined at fixed times intervals in embryos (n ≥ 18 embryos per condition) with lnfg MO ± tnfa MO. P-value, as in Figure 5D, was ≤0.0001 at each time point (one-way ANOVA; [ns] not significant). (E) Number of LPs per A+U+V in E10.5 Ifng+/− mouse embryos from wild-type (+/+), Ifng+/−, and Ifng−/− dams (n = 3; mean ± SD; t-test). (F) qPCR for proinflammatory cytokine mRNAs in E10.5 mouse ECs (CD31+VEC+Kit−), macrophages (F4/80+), granulocytes (Gr1+), and Ly6a-GFP+/− HCCs, normalized to Hprt (n = 3; mean ± SD). mRNA levels in professional inflammatory cells (F4/80+ Mac1+ Ter119− Gr1− macrophages) are shown for comparison. (G) qPCR for proinflammatory cytokine mRNAs from sorted zebrafish embryo fractions: EC (flk1+cmyb−), macrophage (flk1−mpeg+), HCC (flk1+cmyb+), and nonhematovascular (flk1−mpeg−) (normalized to 18S; n = 2 replicates in two experiments; mean ± SD; t-test). (H) qPCR for IFN pathway regulatory targets and transcription factors from sorted zebrafish embryo fractions, as in G. (I) qPCR for IFN-γ and IFN-ϕ receptor mRNAs from sorted zebrafish embryo fractions, as above. (J) Phenotype distribution of csf1ra expression by WISH in irf8 morphants (n ≥ 31 embryos per condition). (K) Phenotype distribution of runx1 expression by WISH (AGM) in irf8 morphants (n ≥ 28 embryos per condition). (L) Absolute counts of cd41:gfp+ cells in the CHT, as in D, following irf8 knockdown or combinatorial reductions of spi1a and spi1b (n ≥ 29 embryos per condition; one-way ANOVA). (M) Phenotype distribution of irf8 expression by WISH following MO knockdown of spi1a and spi1b alone and in combination (n ≥ 28 embryos per condition). (N) Phenotype distribution of mpo expression by WISH following MO knockdown, as in M (n ≥ 29 embryos per condition). (O) Phenotype distribution of runx1 expression by WISH (AGM) following MO knockdown (n ≥ 40 embryos per condition).
We next sought to identify cellular sources of inflammatory cytokines. To determine whether the source of IFN-γ in mice was embryonic or maternal, we measured the number of LPs in Ifng+/− fetuses from wild-type, Ifng+/−, or Ifng−/− dams. The number of LPs was not influenced by the genotype of the dam (Fig. 7E), indicating that the primary source of IFN-γ is embryonic. However, Ifng mRNA was undetectable in cells from whole A+U+V tissue (data not shown) or isolated A+U+V subsets (ECs, macrophages [F4/80+], granulocytes [Gr1+], and Ly6a-GFP+/− HCCs) but was present in macrophages from adult mouse spleens (F4/80+Mac1+Ter119−Gr1−) (Fig. 7F). Similarly, FACS-sorted ECs (flk1:dsRed+cmyb:GFP−) and macrophages (flk1:dsRed−mpeg:GFP+) from zebrafish embryos contained virtually undetectable levels of ifng1-1 or ifng1-2 mRNAs. In contrast, HSPCs (flk1:dsRed+cmyb:GFP+) expressed ifng1-2 (Fig. 7G) and possessed detectable levels of ifng1-1. ifng1-1 mRNA, which is deposited maternally and persists throughout zebrafish development (Sieger et al. 2009), was also present in disaggregated embryos from which ECs (including cmyb+ HSPCs) and macrophages were depleted (flk1−mpeg−) (Fig. 7G). In mouse embryos, placental embryonic trophoblasts secrete IFN-γ beginning at E10 (Platt and Hunt 1998). Thus, the relevant cellular sources of IFN-γ in zebrafish and mouse embryos may not be located in the immediate vicinity of the emerging HSPCs but rather originate from more distant sites. On the other hand, Ifna and ifnphi1 mRNAs were found in ECs in mouse and zebrafish embryos, respectively (Fig. 7F,G), and may function as a local source of IFN signaling during HSPC production from hemogenic endothelium; indeed, expression analysis of IRF2 morphant embryos indicated that ifnphi1+3 were significantly increased in the presence of elevated IFN-γ signaling (Supplemental Fig. S4F).
Although macrophages in zebrafish and mouse embryos express little or no IFN-γ, they contained high levels of TNF and IL1-β mRNAs (Fig. 7F,G), consistent with their well-characterized responses to IFN-mediated inflammatory stimulation. Zebrafish macrophages also expressed high levels of mRNA encoding receptors for IFNs (crfb13, crfb17, and crfb5) and transcriptional mediators of IFN signaling (stat1a, stat2, irf1b, irf2a, and irf8) (Fig. 7H,I), suggesting that they can be activated by IFN signaling and relay that signal to HSPCs. To determine whether primitive macrophages influenced HSPC formation, we depleted macrophages using MOs targeting irf8 or all primitive myeloid cells with combined MOs for spi1b and its paralog, spi1a. MOs against irf8 reduced the number of csf1ra+ macrophages (Fig. 7J), as expected, as well as expression of runx1 in the AGM (Fig. 7K) and the number of cd41:gfp+ cells in the CHT (Fig. 7L). Combinatorial knockdown of spi1b + spi1a, which decreased the production of both irf8+ macrophages and mpo+ granulocytes, dramatically reduced runx1 expression in the AGM (Fig. 7M–O; Supplemental Fig. S5A). Furthermore, spi1b/spi1a loss caused sustained reductions in cd41:gfp+ HSPCs in the CHT at 72 hpf (cf. Figs. 7L and 5D,L), implying that primitive myeloid cells serve to amplify proinflammatory inductive signals regulating HSPC numbers. In the AGM region of E10.5 mouse embryos, macrophages are located in the vicinity of intra-arterial HCCs (Supplemental Fig. S5B), including some observed with projections inserted between Ly6a-GFP+ ECs (Supplemental Fig. S5C,D). Therefore, primitive myeloid lineage cells may act as a local source of inflammatory signals to aid the production of vertebrate HSCs and LPs.
Discussion
Multiple signaling pathways work sequentially and in concert to promote hematopoietic cell formation in the embryo. Here we report that innate immune/inflammatory signaling is one of those pathways. We show that mouse embryos are capable of robustly responding to the inflammatory molecules IFN-α4 and IFN-γ. The numbers of LPs in mouse embryos deficient in IFN-γ or the IFN-γ and IFN-α receptors are significantly reduced, as are transplantable HSCs in embryos lacking the IFN-γ receptor. A role for IFN signaling in HSPC and LP formation and/or number is evolutionarily conserved, as MO knockdown of IFN-γ or its receptors and IFN-ϕ or its receptors decreased HSPCs and thymocytes in zebrafish embryos, while, conversely, knockdown of irf2a, a negative regulator of IFN-α signaling, had the opposite effect. In contrast, EMP number in mouse or zebrafish embryos was not affected by loss of IFN signaling. The selective response of LPs and HSCs to loss of IFN signaling correlated with a robust innate immune/inflammatory expression signature in these cells. IRF2 occupancy of enhancers in human CD34+ FL HSPCs and the expression of IRF2 target genes in those cells as well as in HCCs in mice provided further evidence for active IFN signaling in HSPCs of the conceptus. We conclude that tonic inflammatory signaling has a conserved developmental function to enhance formation of HSPCs in the embryo.
There appear to be multiple cellular sources of inflammatory cytokines in the developing embryo. For example, monocytes/macrophages, which are widely distributed in the embryo, including in the vicinity of blood-producing arteries, and in some cases intimately associated with Ly6a-GFP+ endothelium, express TNF and IL-1β in both mouse and fish embryos. Embryonic macrophages are competent innate immune cells: They express TLRs and can be stimulated to produce multiple cytokines and chemokines (Balounova et al. 2014). In fact, inflammatory responses in the absence of overt infections are actively restrained in fetal macrophages. Macrophages are the precursors for microglia cells in the brain, and mutations in a noncanonical NOD-like receptor, nlrc3-like, in the zebrafish embryo resulted in an unchecked inflammatory response in primitive macrophages that prevented them from differentiating into microglia (Shiau et al. 2013). Here, reduction in the number of macrophages and other primitive myeloid lineage cells through MO knockdown of their essential regulators (irf8 and spi1b/spi1a) lowered HSPC production and, in the case of combined spi1b/spi1a MOs, reduced HSPCs even more dramatically than the loss of individual cytokines. Thus, there is likely to be redundancy in and cooperativity between different classes of cytokines, which may explain why the removal of individual factors, in the absence of infection, had a relatively modest effect and did not completely inhibit HSPC formation. Furthermore, our data suggest that robust production of the primitive myeloid population may have developed or expanded in part to aid HSPC production rather than simply prepare the early embryo to fight potential infections.
Although it is plausible that exposure of nonmammalian embryos to environmental pathogens could play a role in stimulating innate immune/inflammatory signaling to drive a developmental process, we do not envision that the mammalian fetus would rely on maternal transmission of pathogens to convey such a signal. Rather, we hypothesize that normal developmental events would activate innate immune/inflammatory signaling. These events could include apoptosis or hypoxia, both of which are extensive in the embryo. The expression of endogenous retroviruses in the fetal extraembryonic tissues of placental mammals could also trigger innate immune/inflammatory signaling. An interesting question is why the embryo evolved to use innate immune/inflammatory signaling pathways to promote HSPC formation. One possibility is that tonic innate immune/inflammatory signaling in the fetus may function to prime genes in the emergent cells of the adaptive immune system, enabling the cells to mount an effective response to pathogenic challenge should or when the fetus or adult encounter one. A more fundamental, developmental role for inflammatory signaling might be to promote the vascular remodeling associated with HCC formation, the migration of HSPCs to the FL or CHT, extravasation into these sites of secondary colonization, and/or HSPC expansion, all of which are processes associated with classical inflammatory responses.
On a more practical note, attempts to generate HSCs from mouse or human embryonic stem cells have thus far failed, and perhaps careful modulation of inflammatory signaling could improve current in vitro differentiation protocols. We note that prostaglandin E2, which is a small inflammatory signaling molecule, increased HSPC formation in zebrafish embryos and engraftment of adult mouse and human HSCs (North et al. 2007; Hoggatt et al. 2009), consistent with this notion. Likewise, flow-mediated production of nitric oxide, also known to be secreted from inflammatory cells, has been shown to robustly induce HSPC production in vivo and in vitro (Adamo et al. 2009; North et al. 2009). Additional expression profiling of the primitive myeloid populations active in the AGM throughout the course of HSC and LP specification, production, and budding will further clarify the timing and complexities of inflammatory modulation in these processes; however, it is clear that inflammatory signaling has a previously unappreciated yet fundamental role in embryonic HSPC production.
Materials and methods
Mouse lines and embryo production
Tg(Ly6a-GFP) mice [B6.Cg-Tg(Ly6a-GFP)G5Dzk/J] were described previously (Ma et al. 2002). Ifnar1Tm1Agt mice were provided by Dr. Carolina Lopez. C57BL/6J, C57BL/6 × 129S1/SVImJ F1, Ifng−/− (B6.129S7-Ifngtm1Ts/J), and Ifngr1−/− (B6.129S7-Ifngr1tm1Agt/J) mice were from Jackson Laboratory, and B6.SJL-Ptprca Pepcb/BoyJ (CD45.1) mice from the National Cancer Institute. Tg(Ly6a-GFP) conceptuses were generated by crossing C57BL/6 × 129S1/SVImJ F1 females with Tg(Ly6a-GFP) males.
Zebrafish husbandry
Zebrafish were maintained according to Institutional Animal Care and Use Committee (IACUC)-approved protocols. Tg[-6.0itga2b(CD41):eGFP] (Lin et al. 2005), Tg[kdrl(flk1):dsRed2] (Kikuchi et al. 2011), Tg(cmyb:eGFP) (North et al. 2007), Tg(rag2:dsRed) (Ma et al. 2012), Tg(mpeg1:GFP) (Ellett et al. 2011), and Tg(kdrl:GFP) (Jin et al. 2005) lines were described previously.
MO and RNA injection
Optimized doses of MOs (50 µM to 0.25 mM; GeneTools) (Supplemental Table S5) were injected into one-cell stage embryos as described previously (North et al. 2009). ifng1-1 mRNA was synthesized using the manufacturer’s directions for Ambion mMessage mMachine, and 400 pg was injected into one-cell stage embryos.
Whole-mount in situ hybridization (WISH)
WISH was performed on 4% PFA-fixed embryos at the time points indicated using previously published probes (Li et al. 2011; Carroll et al. 2014) and established methods (http://zfin.org/ZFIN/Methods/ThisseProtocol.html). Qualitative phenotypes (two or more replicate clutches; n = 15–100 embryos per condition) were scored using a dissecting microscope (Zeiss) as high/medium/low expression compared with sibling controls.
Progenitor assays
Embryonic cells were prepared for flow cytometry and methylcellulose assays as described previously (North et al. 2002; Chen et al. 2009). OP9 and OP9-DL1 stromal (in αMEM containing 20% FBS and antibiotics) were plated into 96-well plates at a concentration of 5500 cells per well 1 d before dilutions of embryonic cells were added. Recombinant Flt3L and IL-7 were added (5 ng/mL and 10 ng/mL for OP9, respectively, or 5 ng/mL and 1 ng/mL for OP9-DL1) (Peprotech). Cells were analyzed by flow cytometry 12 d later. B cells were identified as CD19+ B220+, and T cells were identified as CD25+ Thy1.1+. The progenitor frequency was calculated using L-Calc software (StemCell Technologies).
Cell sorting and flow cytometry
For mice, the following antibodies were used: Alexa Fluor 647 anti-CD144 (eBioBV13), PE anti-CD45 (30-F11), PE-Cy7 anti-CD31 (390), APC- eFluor 780 anti-c-kit (2B8), PE-anti-ESAM (1G8/ESAM), APC anti-CD19 (1D3), PE-Cy7 anti-B220 (RA3-6B2), PE-Cy anti-Thy1.2 (53-2.1), APC-C7 anti-CD25 (PC61), PerCP-Cy5.5 anti-Gr-1 (RB6-8C5), PE anti-CD3ɛ (145-2C11), APC-Cy7 anti-Mac-1 (M1/70), FITC anti-CD45.2 (104), PE-Cy7 anti-CD45.1 (A20), PerCP-Cy5.5 anti-Sca1 (D7), eFluor 450 lineage-specific cocktail [(anti-TER119 (TER119), anti-Gr-1 (RB6-8C5), anti-B220 (RA3-6B2), anti- CD3 (17A2), and anti-Mac-1 (M1/70)], APC anti-CD34 (MEC14.7), PE anti-Flt3 (A2F10), PE anti-IFNgr1 (2E2, Abcam), APC anti-CD48 (HM48-1), and PE-Cy7 anti-CD150 (TC15-12F12.2). DAPI was used to exclude dead cells. Cells were sorted on a BD FACSvantage at low speed and low pressure (North et al. 2002) and analyzed on a BD LSR II flow cytometer; data were analyzed with FlowJo (Tree Star).
For zebrafish, transgenic embryos (five per sample with four or more replicates) were incubated in 0.5 mg/mL Liberase (Roche) solution with gentle agitation for 90 min at 37°C and then dissociated, filtered, and washed with PBS. Dead cells were excluded by 5 nM SYTOX red (Life Technologies) prior to analysis using a BD FACS Canto II or Beckman Coulter Gallios. Cell sorting was performed using a Beckman Coulter MoFlo XCP.
Microarray
Thirty-thousand Ly6a-GFP+ HCCs (CD31+ VEC+ ESAM+ Kit+), 130,000 Ly6a-GFP− HCCs, 70,000 Ly6a-GFP+ ECs (CD31+ VEC+ ESAM+ Kit−), and 300,000 Ly6a-GFP− ECs were sorted from 100 E10.5 Tg(Ly6a-GFP) embryos into TRIzol-LS (Invitrogen). RNA was isolated using Qiagen RNeasy minikit (Qiagen), split into two fractions, amplified by the Ovation RNA-seq system version 2 (NuGEN), and hybridized to an Agilent 4x44k Whole-Mouse Genome version 2 oligonucleotide microarray. The Gene Expression Omnibus accession number is GSE55493.
Embryo explant culture
Heads and tails were removed from E9.5 Tg(Ly6a-GFP) embryos, and the body was cultured in DMEM + 1% FBS in a 24-well plate at 37°C (Rojas et al. 2005). Recombinant mouse cytokines (20 ng/mL) were added to the culture. Embryos with beating hearts at 48 h were harvested for analysis by fluorescent microscopy and flow cytometry.
Transplant analyses
B6.SJL-PtprcaPepcb/BoyJ (CD45.1) mice were subjected to a split dose of 900 cGy 3 h apart. Each recipient received 1.0 or 0.3 embryo equivalents of E11.5 A+U+V with 2 × 105 carrier spleen cells by tail vein injection. Donor engraftment was assessed (CD45.2) in peripheral blood at weeks 3, 7, 12, and 16 and in BM at week 16 post-transplantation. HSC frequencies were determined by extreme limiting dilution analysis (ELDA) (Hu and Smyth 2009).
Whole-mount fluorescence microscopy
For mice, whole-mount confocal microscopy was performed as described (Yokomizo et al. 2012). Embryos were stained with rat anti-CD31 (MEC13.3, BD Biosciences), rabbit anti-Runx (EPR3099, Abcam), chicken anti-GFP (Invitrogen Molecular Probes), and rat anti-F4/80 (CI: A3-1, Abcam) primary antibodies and goat anti-rat Alexa Fluor 555 (Abcam), goat anti-rabbit Alexa Fluor 488, goat anti-chicken Alexa Fluor 647 (Jackson Immunoresearch), and goat anti-rat Alexa Fluor 647 (Invitrogen Molecular Probes) secondary antibodies. Specimens were analyzed using a Zeiss LSM 710 confocal microscope with 25× objectives using multitrack sequential mode. The pinhole was set at 1 airy unit; for three-dimensional (3D) reconstructions, steps were 5 µm per z-section.
For zebrafish, fluorescent reporter embryos were treated as indicated in the text and live-imaged under tricaine anesthesia as previously described (North et al. 2007) using a Zeiss SteREO Discovery V8 microscope.
Quantitative real-time PCR analysis
For mice, RNA was extracted using Qiagen RNeasy minikit (Qiagen) and reverse-transcribed into cDNA with High-Capacity cDNA reverse transcription kit (Applied Biosystems). qPCR was performed using Power SYBR Green PCR kit (Applied Biosystems) and run on a 7900HT Fast Real-Time PCR system (Applied Biosystems). Human and mouse qPCR primers are listed in Supplemental Tables S6 and S7.
For zebrafish, RNA was extracted using RNAqueous microkit (Life Technologies). cDNA was synthetized and amplified using Ovation Pico WTA system version 2 (Nugen) prior to qPCR. qPCR was performed on cDNA isolated from pooled embryos using the Bio-Rad CFX384 detection system. Zebrafish primer sequences are listed in Supplemental Table S8.
ChIP and ChIP-seq
ChIP-seq analysis of IRF2 was performed using chromatin prepared from primary human FL CD34+ HSPCs, as previously described (Xu et al. 2012). Chromatin was immunoprecipitated using anti-IRF2 antibodies (ABE115, Millipore; ab55331, Abcam) or normal rabbit IgG (sc-2027, Santa Cruz Biotechnology). ChIP DNA was isolated and processed for library generation followed by sequencing using the Illumina HiSeq2500 platform. Raw ChIP-seq data were processed using the Illumina software pipeline and aligned to reference human genome (USCS, hg19).
Intracellular and phospho-flow cytometry
Cells from dissociated E10.5 AGM regions were incubated for 1.5 h at 37°C at a concentration of 2 × 106 per milliliter in IMDM prior to cytokine stimulation. Recombinant mouse IFN-γ or IFN-α4 (50 ng/mL) was added, and cells were incubated for 15 min at 37°C. Cells were fixed with 1.5% paraformaldehyde for 10 min at room temperature and permeabilized with 90% methanol for 20 min at 4°C. Cells were then stained with PE anti-phospho stat1 (Y701) (4a, BD Biosciences), APC- eFluor 780 anti-c-kit, and APC anti-CD144 (VEC). To stain Ki67, cells were collected by FACS after E9.5 embryo explant culture with or without 20 ng/mL recombinant mouse IFN-γ for 2 d, fixed and permeabilized with Cytofix/Cytoperm kit (BD Biosciences), and stained with PE anti-Ki67 (SolA15, eBioscience).
Microarray data analysis
Raw Agilent microarray hybridization signals were normalized using quantile normalization in the R package limma (Smyth 2004). Differentially expressed genes were detected using one-way ANOVA. At the P-value cutoff of 0.01, we identified 8222 differentially expressed genes across the four cell types. We performed consensus clustering using the R package ConsensusClusterPlus (Wilkerson and Hayes 2010) that implements the Consensus Clustering algorithm by Monti et al. (2003). Level 3 GO annotation terms were used. Statistical significance of term enrichment was computed using hypergeometric distribution. P-values were corrected for multiple testing using the method of Benjamini and Hochberg (1995).
ChIP-seq data analysis
Model-based analysis for ChIP-seq (MACS) (Zhang et al. 2008), with its default parameter settings, was used to identify IRF2-binding peaks.
GREAT analysis
GREAT (McLean et al. 2010) was used to identify functional categories enriched among genes bound by IRF2. GREAT determines enrichment by associating the ChIP-seq peak regions with nearby genes and uses the gene annotations to compute enrichment. Association with gene annotation is a two-step process. First, every gene is assigned a regulatory domain; the default parameters (basal plus extension) were used to assign the regulatory domain for each gene. Next, each IRF2 peak was associated with all genes whose regulatory domains it overlapped. Enrichment P-values were computed using hypergeometric test. A FDR of 0.05 was used as the enrichment cutoff.
Statistics analysis
The significance of multivariable comparisons throughout the study was determined by one-way ANOVA and Dunnett’s multiple comparison test. Pairwise variation from control was determined by unpaired two-tailed Students t-test.
Supplementary Material
Acknowledgments
We thank B. Black for explant culture protocols, V. Mulero (University of Murcia) for ifng expression construct, and B. van Handel and H. Mikkola (University of California at Los Angeles) for hCD34+ FL cells. This work was supported by National Institutes of Health (NIH) R01HL091724 (N.A.S.), U01HL100405 (N.A.S.), R01HG006130 (K.T.), and P01-HL032259 (S.H.O.). Y.L. and J.T. are supported by the Leukemia and Lymphoma Society, A.D.Y. is supported by NIH 1F31HL120615, M.W.M. is supported by Rubicon grant 825.12.003, and J.X. is supported by K01DK093543. T.E.N. is supported by NIH 1R01DK098241 and the American Society of Hematology. Core services were provided by the Abramson Cancer Center (mice), the Harvard Stem Cell Institute, and Beth Israel Deaconess Medical Center (zebrafish).
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.253302.114.
References
- Abrahams VM. 2008. Pattern recognition at the maternal-fetal interface. Immunol Invest 37: 427–447. [DOI] [PubMed] [Google Scholar]
- Adamo L, Naveiras O, Wenzel PL, McKinney-Freeman S, Mack PJ, Gracia-Sancho J, Suchy-Dicey A, Yoshimoto M, Lensch MW, Yoder MC, et al. 2009. Biomechanical forces promote embryonic haematopoiesis. Nature 459: 1131–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson KV, Bokla L, Nusslein-Volhard C. 1985. Establishment of dorsal-ventral polarity in the Drosophila embryo: the induction of polarity by the Toll gene product. Cell 42: 791–798. [DOI] [PubMed] [Google Scholar]
- Baldridge MT, King KY, Boles NC, Weksberg DC, Goodell MA. 2010. Quiescent haematopoietic stem cells are activated by IFN-γ in response to chronic infection. Nature 465: 793–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balounova J, Vavrochova T, Benesova M, Ballek O, Kolar M, Filipp D. 2014. Toll-like receptors expressed on embryonic macrophages couple inflammatory signals to iron metabolism during early ontogenesis. Eur J Immunol 44: 1491–1502. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Royal Stat Soc Ser B 57: 289–300. [Google Scholar]
- Bertrand JY, Jalil A, Klaine M, Jung S, Cumano A, Godin I. 2005. Three pathways to mature macrophages in the early mouse yolk sac. Blood 106: 3004–3011. [DOI] [PubMed] [Google Scholar]
- Bertrand JY, Kim AD, Violette EP, Stachura DL, Cisson JL, Traver D. 2007. Definitive hematopoiesis initiates through a committed erythromyeloid progenitor in the zebrafish embryo. Development 134: 4147–4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand JY, Chi NC, Santoso B, Teng S, Stainier DY, Traver D. 2010. Haematopoietic stem cells derive directly from aortic endothelium during development. Nature 464: 108–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll KJ, Esain V, Garnaas MK, Cortes M, Dovey MC, Nissim S, Frechette GM, Liu SY, Kwan W, Cutting CC, et al. 2014. Estrogen defines the dorsal-ventral limit of VEGF regulation to specify the location of the hemogenic endothelial niche. Dev Cell 29: 437–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MJ, Yokomizo T, Zeigler BM, Dzierzak E, Speck NA. 2009. Runx1 is required for the endothelial to haematopoietic cell transition but not thereafter. Nature 457: 889–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JM, Clay H, Lewis JL, Ghori N, Herbomel P, Ramakrishnan L. 2002. Real-time visualization of mycobacterium–macrophage interactions leading to initiation of granuloma formation in zebrafish embryos. Immunity 17: 693–702. [DOI] [PubMed] [Google Scholar]
- de Bruijn MF, Speck NA, Peeters MC, Dzierzak E. 2000. Definitive hematopoietic stem cells first develop within the major arterial regions of the mouse embryo. EMBO J 19: 2465–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bruijn MF, Ma X, Robin C, Ottersbach K, Sanchez MJ, Dzierzak E. 2002. Hematopoietic stem cells localize to the endothelial cell layer in the midgestation mouse aorta. Immunity 16: 673–683. [DOI] [PubMed] [Google Scholar]
- Ellett F, Pase L, Hayman JW, Andrianopoulos A, Lieschke GJ. 2011. mpeg1 promoter transgenes direct macrophage-lineage expression in zebrafish. Blood 117: e49–e56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essers MA, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA, Trumpp A. 2009. IFNα activates dormant haematopoietic stem cells in vivo. Nature 458: 904–908. [DOI] [PubMed] [Google Scholar]
- Flanagan K, Modrusan Z, Cornelius J, Chavali A, Kasman I, Komuves L, Mo L, Diehl L. 2008. Intestinal epithelial cell up-regulation of LY6 molecules during colitis results in enhanced chemokine secretion. J Immunol 180: 3874–3881. [DOI] [PubMed] [Google Scholar]
- Frame JM, McGrath KE, Palis J. 2013. Erythro–myeloid progenitors: ‘definitive’ hematopoiesis in the conceptus prior to the emergence of hematopoietic stem cells. Blood Cells Mol Dis 51: 220–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godin I, Dieterlen-Lièvre F, Cumano A. 1995. Emergence of multipotent hematopoietic cells in the yolk sac and paraaortic splanchnopleura of 8.5 dpc mouse embryos. Proc Natl Acad Sci 92: 773–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoggatt J, Singh P, Sampath J, Pelus LM. 2009. Prostaglandin E2 enhances hematopoietic stem cell homing, survival, and proliferation. Blood 113: 5444–5455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Smyth GK. 2009. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods 347: 70–78. [DOI] [PubMed] [Google Scholar]
- Jin SW, Beis D, Mitchell T, Chen JN, Stainier DY. 2005. Cellular and molecular analyses of vascular tube and lumen formation in zebrafish. Development 132: 5199–5209. [DOI] [PubMed] [Google Scholar]
- Kikuchi K, Holdway JE, Major RJ, Blum N, Dahn RD, Begemann G, Poss KD. 2011. Retinoic acid production by endocardium and epicardium is an injury response essential for zebrafish heart regeneration. Dev Cell 20: 397–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam SH, Chua HL, Gong Z, Lam TJ, Sin YM. 2004. Development and maturation of the immune system in zebrafish, Danio rerio: a gene expression profiling, in situ hybridization and immunological study. Dev Comp Immunol 28: 9–28. [DOI] [PubMed] [Google Scholar]
- Levraud JP, Boudinot P, Colin I, Benmansour A, Peyrieras N, Herbomel P, Lutfalla G. 2007. Identification of the zebrafish IFN receptor: implications for the origin of the vertebrate IFN system. J Immunol 178: 4385–4394. [DOI] [PubMed] [Google Scholar]
- Li L, Jin H, Xu J, Shi Y, Wen Z. 2011. Irf8 regulates macrophage versus neutrophil fate during zebrafish primitive myelopoiesis. Blood 117: 1359–1369. [DOI] [PubMed] [Google Scholar]
- Lin HF, Traver D, Zhu H, Dooley K, Paw BH, Zon LI, Handin RI. 2005. Analysis of thrombocyte development in CD41-GFP transgenic zebrafish. Blood 106: 3803–3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CP, Auerbach R. 1991. In vitro development of murine T cells from prethymic and preliver embryonic yolk sac hematopoietic stem cells. Development 113: 1315–1323. [DOI] [PubMed] [Google Scholar]
- Luna G, Paez J, Cardier JE. 2004. Expression of the hematopoietic stem cell antigen Sca-1 (LY-6A/E) in liver sinusoidal endothelial cells: possible function of Sca-1 in endothelial cells. Stem Cells Dev 13: 528–535. [DOI] [PubMed] [Google Scholar]
- Ma X, Robin C, Ottersbach K, Dzierzak E. 2002. The Ly-6A (Sca-1) GFP transgene is expressed in all adult mouse hematopoietic stem cells. Stem Cells 20: 514–521. [DOI] [PubMed] [Google Scholar]
- Ma D, Wang L, Wang S, Gao Y, Wei Y, Liu F. 2012. Foxn1 maintains thymic epithelial cells to support T-cell development via mcm2 in zebrafish. Proc Natl Acad Sci 109: 21040–21045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. 2010. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol 28: 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monti S, Tamayo P, Mesirov J, Golub T. 2003. Consensus clustering: a resampling-based method for class discovery and visualization of gene expression microarray data. Mach Learn 52: 91–118. [Google Scholar]
- Müller AM, Medvinsky A, Strouboulis J, Grosveld F, Dzierzak E. 1994. Development of hematopoietic stem cell activity in the mouse embryo. Immunity 1: 291–301. [DOI] [PubMed] [Google Scholar]
- North TE, de Bruijn MF, Stacy T, Talebian L, Lind E, Robin C, Binder M, Dzierzak E, Speck NA. 2002. Runx1 expression marks long-term repopulating hematopoietic stem cells in the midgestation mouse embryo. Immunity 16: 661–672. [DOI] [PubMed] [Google Scholar]
- North TE, Goessling W, Walkley CR, Lengerke C, Kopani KR, Lord AM, Weber GJ, Bowman TV, Jang IH, Grosser T, et al. 2007. Prostaglandin E2 regulates vertebrate haematopoietic stem cell homeostasis. Nature 447: 1007–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North TE, Goessling W, Peeters M, Li P, Ceol C, Lord AM, Weber GJ, Harris J, Cutting CC, Huang P, et al. 2009. Hematopoietic stem cell development is dependent on blood flow. Cell 137: 736–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oehlers SH, Flores MV, Hall CJ, Swift S, Crosier KE, Crosier PS. 2011. The inflammatory bowel disease (IBD) susceptibility genes NOD1 and NOD2 have conserved anti-bacterial roles in zebrafish. Dis Model Mech 4: 832–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orelio C, Haak E, Peeters M, Dzierzak E. 2008. Interleukin-1-mediated hematopoietic cell regulation in the aorta–gonad–mesonephros region of the mouse embryo. Blood 112: 4895–4904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page DM, Wittamer V, Bertrand JY, Lewis KL, Pratt DN, Delgado N, Schale SE, McGue C, Jacobsen BH, Doty A, et al. 2013. An evolutionarily conserved program of B-cell development and activation in zebrafish. Blood 122: e1–e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palis J, Robertson S, Kennedy M, Wall C, Keller G. 1999. Development of erythroid and myeloid progenitors in the yolk sac and embryo proper of the mouse. Development 126: 5073–5084. [DOI] [PubMed] [Google Scholar]
- Platt JS, Hunt JS. 1998. Interferon-γ gene expression in cycling and pregnant mouse uterus: temporal aspects and cellular localization. J Leukoc Biol 64: 393–400. [DOI] [PubMed] [Google Scholar]
- Purwanti N, Karabasil MR, Matsuo S, Chen G, Javkhlan P, Azlina A, Hasegawa T, Yao C, Akamatsu T, Hosoi K. 2011. Induction of Sca-1 via activation of STAT3 system in the duct cells of the mouse submandibular gland by ligation of the main excretory duct. Am J Physiol Gastrointest Liver Physiol 301: G814–G824. [DOI] [PubMed] [Google Scholar]
- Qiu P, Pan PC, Govind S. 1998. A role for the Drosophila Toll/Cactus pathway in larval hematopoiesis. Development 125: 1909–1920. [DOI] [PubMed] [Google Scholar]
- Rojas A, De Val S, Heidt AB, Xu SM, Bristow J, Black BL. 2005. Gata4 expression in lateral mesoderm is downstream of BMP4 and is activated directly by Forkhead and GATA transcription factors through a distal enhancer element. Development 132: 3405–3417. [DOI] [PubMed] [Google Scholar]
- Sato T, Onai N, Yoshihara H, Arai F, Suda T, Ohteki T. 2009. Interferon regulatory factor-2 protects quiescent hematopoietic stem cells from type I interferon-dependent exhaustion. Nat Med 15: 696–700. [DOI] [PubMed] [Google Scholar]
- Shi X, Siggins RW, Stanford WL, Melvan JN, Basson MD, Zhang P. 2013. Toll-like receptor 4/stem cell antigen 1 signaling promotes hematopoietic precursor cell commitment to granulocyte development during the granulopoietic response to Escherichia coli bacteremia. Infect Immun 81: 2197–2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiau CE, Monk KR, Joo W, Talbot WS. 2013. An anti-inflammatory NOD-like receptor is required for microglia development. Cell Reports 5: 1342–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieger D, Stein C, Neifer D, van der Sar AM, Leptin M. 2009. The role of γ interferon in innate immunity in the zebrafish embryo. Dis Model Mech 2: 571–581. [DOI] [PubMed] [Google Scholar]
- Smyth GK. 2004. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3: 1544–6115. [DOI] [PubMed] [Google Scholar]
- Swiers G, Rode C, Azzoni E, de Bruijn MF. 2013. A short history of hemogenic endothelium. Blood Cells Mol Dis 51: 206–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tober J, Koniski A, McGrath KE, Vemishetti R, Emerson R, de Mesy-Bentley KK, Waugh R, Palis J. 2007. The megakaryocyte lineage originates from hemangioblast precursors and is an integral component both of primitive and of definitive hematopoiesis. Blood 109: 1433–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkerson MD, Hayes DN. 2010. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics 26: 1572–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Shao Z, Glass K, Bauer DE, Pinello L, Van Handel B, Hou S, Stamatoyannopoulos JA, Mikkola HK, Yuan GC, et al. 2012. Combinatorial assembly of developmental stage-specific enhancers controls gene expression programs during human erythropoiesis. Dev Cell 23: 796–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokomizo T, Yamada-Inagawa T, Yzaguirre AD, Chen MJ, Speck NA, Dzierzak E. 2012. Whole-mount three-dimensional imaging of internally localized immunostained cells within mouse embryos. Nat Protoc 7: 421–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokota T, Huang J, Tavian M, Nagai Y, Hirose J, Zuniga-Pflucker JC, Peault B, Kincade PW. 2006. Tracing the first waves of lymphopoiesis in mice. Development 133: 2041–2051. [DOI] [PubMed] [Google Scholar]
- Yoshimoto M, Porayette P, Glosson NL, Conway SJ, Carlesso N, Cardoso AA, Kaplan MH, Yoder MC. 2012. Autonomous murine T-cell progenitor production in the extra-embryonic yolk sac before HSC emergence. Blood 119: 5706–5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. 2008. Model-based analysis of ChIP-Seq (MACS). Genome Biol 9: R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.