

Increased PI3K signaling in pancreatic ductal adenocarcinoma (PDAC) correlates with poor prognosis. Baer et al. show that inactivation of the p110α PI3K isoform blocked the irreversible transition of exocrine acinar cells into pancreatic preneoplastic ductal lesions by oncogenic Kras and/or pancreatic injury. p110α signaling through small GTPase Rho and actin cytoskeleton controlled the reprogramming of acinar cells. p110α was necessary for pancreatic ductal cancers to arise from Kras-induced preneoplastic lesions by increasing epithelial cell proliferation in the context of mutated p53.
Keywords: genetic mouse models, PI3K isoforms, pancreas, oncogenes, signaling, transdifferentiation
Abstract
Increased PI 3-kinase (PI3K) signaling in pancreatic ductal adenocarcinoma (PDAC) correlates with poor prognosis, but the role of class I PI3K isoforms during its induction remains unclear. Using genetically engineered mice and pharmacological isoform-selective inhibitors, we found that the p110α PI3K isoform is a major signaling enzyme for PDAC development induced by a combination of genetic and nongenetic factors. Inactivation of this single isoform blocked the irreversible transition of exocrine acinar cells into pancreatic preneoplastic ductal lesions by oncogenic Kras and/or pancreatic injury. Hitting the other ubiquitous isoform, p110β, did not prevent preneoplastic lesion initiation. p110α signaling through small GTPase Rho and actin cytoskeleton controls the reprogramming of acinar cells and regulates cell morphology in vivo and in vitro. Finally, p110α was necessary for pancreatic ductal cancers to arise from Kras-induced preneoplastic lesions by increasing epithelial cell proliferation in the context of mutated p53. Here we identify an in vivo context in which p110α cellular output differs depending on the epithelial transformation stage and demonstrate that the PI3K p110α is required for PDAC induced by oncogenic Kras, the key driver mutation of PDAC. These data are critical for a better understanding of the development of this lethal disease that is currently without efficient treatment.
Mammals have four isoforms of class I PI 3-kinase (PI3K; α, β, γ, and δ) that generate the phosphatidylinositol triphosphate (PIP3) lipid upon cell stimulation by cell surface receptors. These heterodimeric enzymes consist of a p110 catalytic subunit complexed to a regulatory subunit. Despite their high sequence and activity similarities, class I PI3K isoforms have nonredundant roles under physiological, nondisease conditions (Vanhaesebroeck et al. 2010). In cancer, this nonredundancy of PI3K isoforms appears more complex. Indeed, cancer cell signaling is complicated as a consequence of genetic alterations, autocrine/paracrine signaling, and cancer cell-extrinsic signals. These parameters are thought to be different in each tissue, possibly explaining the tissue-specific context of PI3K isoform function in cancer initiation and progression (Jia et al. 2008; Berenjeno et al. 2012; Rodon et al. 2013). Based on this paradigm, current clinical strategies target all isoforms through pan-PI3K or pan-PI3K/mTOR inhibitors. However, these drugs often exhibit severe toxicity when combined with other therapies, raising the possibility that isoform-specific PI3K inhibitors could be a better approach for the treatment of solid cancers. It is thus crucial to delineate and understand which PI3K isoform to therapeutically target for each cancer type.
Pancreatic ductal adenocarcinoma (PDAC) is one of the most lethal cancers, with a 5-yr death rate of 95% of diagnosed patients. No efficient chemotherapy is currently available (Hidalgo 2010). Several genes (e.g., KRAS, INK4A/ARF, p53, and SMAD4) and signaling pathways (e.g., epithelial growth factor [EGF], transforming growth factor α [TGFα], Notch, and Hedgehog) have been implicated in pancreatic cancer progression, but mutation of KRAS is the driving force of this pathology and is detected in early premalignant lesions, termed pancreatic intraepithelial neoplasia (PanIN). In this cancer, phosphorylation of the PI3K signaling pathway component Akt is increased and correlates with a poor prognosis (Schlieman et al. 2003; Yamamoto et al. 2004). Activating mutations in PI3K genes such as p110α (PIK3CA) are very rarely found or are not detected in PDAC (Jones et al. 2008; Biankin et al. 2012). However, the expression of activated p110α in the pancreas mimics mutated Kras cancerogenesis (Eser et al. 2013), indicative that this PI3K isoform could be a signaling hub in PDAC formation downstream from mutated Kras.
In addition to genetic factors such as p53 mutation, nongenetic factors such as pancreatitis contribute to mutated Kras-induced pancreatic cancerogenesis. While it is clear that inflammation amplifies oncogenic Kras-induced neoplastic transformation (Guerra et al. 2007; Carriere et al. 2009), the molecular basis of such nongenetic risk factors in the induction of pancreatic cancer is still poorly characterized (Yadav and Lowenfels 2013). In human acute or chronic pancreatitis, pancreatic epithelial cells change their differentiation state from mature acinar cells secreting digestive enzymes to “duct-like” structures presenting a decreased expression of acinar markers (such as digestive enzymes trypsin or amylase), inappropriate expression of ductal markers (such as cytokeratin CK19), and a transition to a duct-like morphology (Strobel et al. 2007; Fendrich et al. 2008); this process is termed acinar-to-ductal metaplasia (ADM). The cholecystokinin analog caerulein induces secretion of pancreatic enzymes, which provokes pancreatic injury, pancreatitis, and ADM formation. Both pancreatic injury and oncogenes induce pancreatic inflammation and ADM. ADM is a precursor of PanIN lesions that progress rapidly to PDAC only on a Kras mutant background.
In this study, we sought to investigate the role of p110α signaling in the induction of preneoplastic lesions and PDAC downstream from mutated Kras by pharmacological and genetic inactivation of p110α.
Results
p110α is expressed and PI3K signaling is activated in human and murine PDAC
Of the four class I PI3K isoforms, only p110α and p110β were found to be expressed in the normal murine pancreas, in particular in the murine pancreatic exocrine acinar compartment (Fig. 1A). In normal human pancreas adjacent to tumor samples, p110α, p110β, and p110γ were expressed, whereas in human PDAC, all class I PI3K isoforms were overexpressed (Fig. 1B). Murine pancreatic PanIN cells or cancer cell lines from murine or human PDAC also expressed varying levels of all class I PI3K isoforms without a clear correlation with the phosphorylation status of the downstream effector AKT (Supplemental Fig. 1A–D). These data revealed that p110α was present in all pancreatic tissue samples or cell lines tested. Most importantly, PI3Ks were able to activate their downstream effectors such as Akt in human PDAC (Fig. 1B,C).
Figure 1.
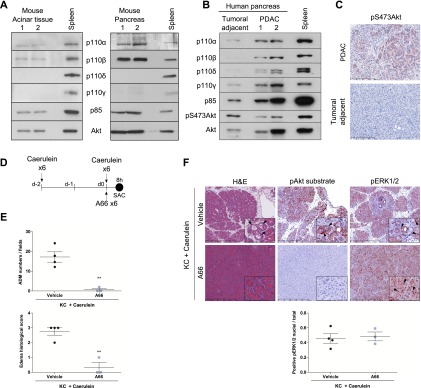
PI3K/Akt pathway activation in pancreatic cancer and pharmacological inhibition of p110α catalytic activity prevent the development of pancreatic ADM lesions induced by injury in the context of mutated Kras. (A) Analysis of p110 isoforms and its regulatory subunit expression by Western blot in microdissected murine acinar cells (left; N = 4) or whole-pancreas lysates (right; N = 4). Total spleen was used as a control for p110δ and p110γ expression (isoforms overexpressed in immune cells). p85 and total Akt correspond to loading controls. (B) p110s and p85 expression in human samples. N = 3. (C) Levels of pAkt in human PDAC. Scale, 200 μm. (D) Scheme. (E,F) Quantification of ADM numbers/fields and edema histological score (E) and representative hematoxylin and eosin (H&E) and immunohistochemistry (IHC) stainings (F; insets are in high magnification) of young KC mice injected with caerulein in the presence of the specific p110α inhibitor A66 (N = 3) or vehicle (N = 4) and sacrificed 8 h post-injections. ADM numbers were analyzed on six random 20×-magnification fields per representative slide for each mouse; edema scoring was achieved on a representative slide for each mouse. Mean ± SEM; (**) P < 0.001, Student’s t-test. IHC using indicated antibodies. Quantification of nuclear pERK staining was performed on five 20× magnification fields in each mouse normalized with the total number of nuclei. Mean ± SEM; (NS) Student’s t-test. Scale, 500 or 90 μm.
Because of its expression across pancreatic cancerogenesis, we studied the role of p110α in the initiation of preneoplastic lesions by mutated Kras and the initiation of pancreatic cancer by mutated Kras and p53.
p110α is critical for the initiation of preneoplastic ADM pancreatic lesions by tissue injury in the context of Kras mutation
The KrasLSL-G12D/+;Pdx1Cre/+ mouse model (referred to hereafter as KC) reproducibly shows limited formation of ADM and PanIN precursor lesions at ∼8 wk of age (Hingorani et al. 2003). Such early stages progress with long latency (>1 yr) to invasive metastatic PDAC. Induction of pancreatic injury by caerulein dramatically accelerates the induction of pancreatic preneoplastic ADM lesions by mutated Kras and increases their outcome as PanINs and cancer (Guerra et al. 2007; Carriere et al. 2009; Kopp et al. 2012). However, in the absence of oncogenic Kras, these lesions do not progress to PanIN. The activation status of the PI3K/Akt pathway during pancreatic injury in the context of mutated Kras is unknown.
In young KC mice, caerulein treatment induced edema and massive ADM formation (Fig. 1D–F). The PI3K and Raf/Mek/ERK pathways were activated in acinar cells and ADM as assessed by Akt substrate and ERK phosphorylation, respectively (Fig. 1F). Systemic pharmacological inhibition of p110α using the p110α-selective inhibitor A66 inhibited Akt substrate phosphorylation and completely blocked the appearance of massive ADM induced by tissue injury in the presence of mutated Kras (Fig. 1E,F). Interestingly, the activity of ERK, as assessed by nuclear translocation or phosphorylation of ERK, was not altered by p110α inhibition (Fig. 1F).
Altogether, these data suggest that PI3K signaling is critically involved in pancreatic preneoplastic lesion initiation associated with pancreatic injury. Activation of the ERK pathway without signaling via p110α is not sufficient to induce ADM. Preneoplastic ADM initiation induced by tissue injury in the context of mutated Kras is sensitive to specific pharmacological inhibition of p110α.
Pancreas-specific inactivation of p110α prevents initiation of pancreatic preneoplastic lesions and cancer by mutated Kras
To identify the role of p110α in pancreatic pathophysiology, we next genetically inactivated the kinase domain of p110α in the pancreas using a conditional targeting strategy (p110αlox/lox) (Graupera et al. 2008). These mice were crossed with the Pdx1Cre/+ pancreas-specific Cre-expressing mouse model (abbreviated as C). Recombination of the p110α gene exons encoding its catalytic activity results in a kinase-dead p110α enzyme, mimicking cell-autonomous, pharmacological blockade of p110α (Supplemental Fig. 2A–C). This approach is superior to a full p110α gene knockout strategy: Besides removing a potential scaffolding role of p110α, PI3K knockout strategies are also known to also induce compensatory catalytic and regulatory subunit expression leading to off-target effects (Vanhaesebroeck et al. 2005). Indeed, genetic inactivation of the kinase domain of pancreatic p110α in mice (referred to as C;p110αlox/lox) exhibited a decrease of basal phosphorylation of Akt but conserved expression levels of p110α and p110β and their regulatory subunit, p85 (Supplemental Fig. 2D). No lethality or changes in pancreatic morphology and endocrine function were observed (Supplemental Fig. 3A–C).
To test whether pancreatic epithelial p110α activity is required for pancreatic preneoplastic lesion and cancer formation, we examined the effects of genetic p110α inhibition in the KC model. Concomitant induction of the activating mutation of Kras and genetic inactivation of one or both p110α alleles were achieved by expression of the Cre recombinase in the pancreas in mice called KC;p110α+/lox and KC;p110αlox/lox, respectively (Fig. 2A).
Figure 2.

Genetic inactivation of p110α catalytic activity in the pancreas prevents the development of mutated Kras-induced pancreatic preneoplastic and neoplastic lesions. (A) Experimental setup to express kinase-dead p110α and oncogenic KrasG12D in the pancreas. (B) Survival curve. Expected Mendelian ratios for Cre+;p110αlox/lox were found; total number of pups was 155. N > 10 per genotype. (C) Percentage of 6-mo-old mice harboring no lesions, ADM, or PanINs depending on genotypes. (D) Representative H&E stains (insets show representative areas in high magnification; scale, 500 or 100 μm) and indicated IHC analysis on paraffin-embedded KC, KC;p110α+/lox, and KC;p110αlox/lox pancreata. (Arrowheads) Pancreatic lobules with stromal Ki67-positive cells present in areas of ADM ; (em) early metaplastic transitions; (*) ADM lesions; (#) low-grade PanIns; (red arrowheads) CK19-positive normal ducts. CK19 is a well-defined ductal cell marker overexpressed first at the basolateral membrane of acinar cells (em) (Zhu et al. 2007) that undergo ADM. N = 4. (E) Pancreatic sections of KC, KC;p110α+/lox, and KC;p110αlox/lox animals were analyzed for the presence of pS473Akt, pAkt substrate, and pThr202/Tyr204ERK1/2 (cytoplasmic and nuclear staining) by IHC on serial sections (4 μm). Slides were counterstained with hematoxylin. Representative pictures are shown (N = 4). Scale, 100 μm. (F) Experimental setup to express kinase-dead p110β and oncogenic KrasG12D in the pancreas. (G) Percentage of 6-mo-old mice harboring no lesions, ADM, or PanINs depending on genotypes. (H) H&E.
p110α inactivation dose-dependently prevented mouse lethality and the occurrence of all types of lesions induced by mutated Kras (Fig. 2B,C). Careful analysis of serial sections spaced throughout the pancreata showed that while all KC and most KC;p110α+/lox animals (littermate controls) presented ADM and PanIN lesions and the replacement of normal pancreatic tissue architecture by tumorous tissue, the appearance of all types of pancreatic lesions was blocked by homozygous genetic inactivation of p110α, with most of these mice having no histological alterations and no appearance of Ki67-positive cells and ectopic CK19 staining (Fig. 2D; Supplemental Fig. 3D). Indeed, only four KC;p110αlox/lox animals out of 14 exhibited a very limited number and area of ADM and PanIN lesions (Fig. 2C; Supplemental Fig. 3D,E), one of them being an escapee without complete recombination of pik3ca (Supplemental Fig. 3F). Interestingly, the few PanIN lesions that developed under full pik3ca recombination were not surrounded by stromal reaction and presented no pAkt staining and decreased levels of pERK (Supplemental Fig. 3E), indicating a role of p110α in tumor stroma interactions and in the maintenance of pERK signals in all preneoplastic PanIn lesions.
In KC and KC;p110α+/lox littermate pancreata presenting lesions, the PI3K pathway was activated (Fig. 2E). In contrast, the absence of pancreatic lesions in KC;p110αlox/lox pancreata was associated with the loss of activation of both the PI3K and ERK pathways (Fig. 2E). These data show at the genetic level that PI3K activity of pancreatic p110α is necessary for the cancerogenesis initiated by oncogenic Kras. In opposition, genetic ablation of the other PI3K isoform expressed in acinar cells, p110β, did not prevent dose-dependently the apparition of preneoplastic lesions induced by mutated Kras (Fig. 2F–H). We thus searched for the specific role of p110α in the initiating events of pancreatic cancer.
Acinar cell-autonomous p110α activity is required for acinar-to-ductal plasticity in the presence of tissue injury and mutated Kras
We next explored the role of acinar p110α during pancreatic injury and preneoplastic lesion formation in more detail. We aimed, in particular, to dissect the role of p110α in pancreatic tissue injury alone or in the context of injury in the presence of mutated Kras (Fig. 3A). In all genotypes tested, caerulein induced a depletion of amylase granules in acinar cells, confirming the activity of this agent in all mice tested (Supplemental Fig. 4A). p110α inactivation in non-Kras-mutated pancreatic epithelial cells (C;p110αlox/lox) reduced the pancreatic lesions and blocked ADM formation induced by caerulein after 1 d of treatment (Fig. 3B,D). Strikingly, the worsening of the caerulein-induced lesions observed in the mutated Kras genetic context was completely blocked by full p110α inactivation in the epithelial cells (Fig. 3C,E, KC;p110αlox/lox vs. KC;p110α+/lox and KC).
Figure 3.

Acinar p110α activity is crucial for induction and maintenance of pancreatic ADM lesions by tissue injury, also in the genetic context of mutated Kras. (A) Experimental setup. (B,C) Representative H&E staining (insets in high magnification) of caerulein- or vehicle-injected 8-wk-old wild-type (WT) or C;p110αlox/lox (B) and KC, KC;p110α+/lox or KC;p110αlox/lox (C) mice 1 or 5 d after the last injection of caerulein. Dashed lines in insets surround a mixed acinar–ductal metaplasic structure. (#) Normal acinar cells containing characteristic zymogen granules. Scale, 4–50–500 μm. (D,E) Quantification of ADM or PanINs per field (magnification, 20×; six random fields per mouse; normalization per field). Each point represents a single mouse, and horizontal bars represent the mean percentage for each group. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001, Student’s t-test, KC;p110α+/lox or KC;p110αlox/lox versus KC. (F) Phase contrast images of Ptf1aCre; KrasG12D acinar cells 5 d after isolation and treatment with 50 ng/mL TGF-α in the presence of increasing doses of p110α-specific inhibitor A66 (1–10 μM) or vehicle. Quantification of the percentage of ductal structures after 5 d in culture. N = 3 ex vivo acinar cultures from independent mice; (*) P < 0.05; (**) P < 0.01, Student’s t-test). Scale, 50 μm. (G) Rat AR4-2J B13 acinar cells were subjected to DEX+EGF (1 μM + 20 ng/mL) with the indicated treatment for 5 d to induce ADM. Immunofluorescence staining was performed as indicated, and representative confocal images are presented (N ≥ 2). Scale, 10 μm.
p110α activity is required for the induction of preneoplastic lesions upon pancreatic injury in a Kras mutant background
Pancreatic tissue is highly plastic and physiologically regenerates; indeed, 5 d after caerulein injection, both wild-type and p110α activity-deficient pancreata presented a normal histology (Fig. 3B,D). Strikingly, p110α blockade allowed also the complete prevention of pancreatic lesion induction of the pancreas after 1 or 5 d of caerulein treatment in a Kras mutated genetic background (Fig. 3C,E), while these lesions were maintained in KC and KC;p110α+/lox pancreata. Besides a normal cytoarchitecture, acinar cell proliferation in KC;p110αlox/lox pancreata, which was decreased at day 1 compared with KC and KC;p110α+/lox, was increased compared with PBS-treated animals 5 d after caerulein treatment, indicative of a regeneration process and demonstrating that p110α activity-deficient cells retain the capacity to proliferate (Supplemental Fig. 4B). Recruitment of immune cells to KC;p110αlox/lox pancreata was also reduced upon full pancreatic p110α inactivation (Supplemental Fig. 4C).
Our data demonstrate that p110α prevents the induction of preneoplastic lesions induced by caerulein in a mutated Kras background (scheme in Supplemental Fig. 4E) and allowed the maintenance of immune cell infiltration surrounding the acinar cells.
p110α activity in epithelial cells is required for acinar-to-ductal plasticity ex vivo
Our data show that p110α activity is necessary for cancer initiation through regulation of the plasticity of acinar cells in vivo. To assess whether p110α mediates ductal reprogramming of acinar cells in a cell-autonomous manner, we next used a physiological cell culture model of acinar-to-ductal transdifferentiation. Previous studies had demonstrated that TGFα, through signaling via the EGF receptor (EGFR), induces the formation of tubular structures from acinar clusters embedded in collagen isolated from adult pancreata expressing mutated Kras (Means et al. 2005; Morris et al. 2010).
To test whether TGFα–EGFR/Kras-induced ADM formation depends on p110α activity, we isolated acini from Ptf1aCre/+; KrasG12D/+ pancreata and performed an in vitro ADM assay. Consistent with the in vivo caerulein-induced short-term acinar-to-ductal transdifferentiation model, we observed that pharmacological inactivation of p110α dose-dependently blocked ADM in the presence of TGFα and mutated Kras (Fig. 3F). Similar results were obtained with genetic inactivation of p110α (Supplemental Fig. 5). These observations were confirmed in an in vitro two-dimensional (2D) model (Fig. 3G), demonstrating that this role of p110α is independent of cell polarity and Kras mutation. Indeed, in the amylase-expressing rat acinar cell line AR4-2J/B13, where Kras is not mutated, the 5-d reprogramming treatment with dexamethasone + EGF (DEX+EGF) (Al-Adsani et al. 2010) induced the loss of expression of acinar-specific digestive enzymes such as trypsin together with the increase of expression of the ductal marker cytokeratin CK19 (Fig. 3G). Interestingly, pharmacological inhibition of p110α with A66 delayed the DEX+EGF-induced decrease in trypsin expression and largely prevented the induction of ductal marker expression of CK19 by DEX+EGF (Fig. 3G).
Taken together, our data show that reprogramming of acinar cells into ductal cells induced by a range of intrinsic or extrinsic stimuli is dependent on p110α activity.
p110α activity in acinar cells is not required for the induction of the embryonic progenitor transcription factor Pdx1 during pancreatic injury
Our data show that p110α inactivation blocks acinar-to-ductal plasticity in an acinar cell-autonomous way. Transcription factors such as Pdx1 that are associated with pancreatic progenitors and the maintenance of an undifferentiated status are re-expressed during this process (Jensen et al. 2005). However, their relevance for injury- and Kras-driven ADM and PanIN formation remains unclear. We therefore monitored the expression of pancreatic progenitor markers in the cellular transitions from acinar cells to ducts and PanINs.
We detected expression of Pdx1 in established ADM lesions induced by injury in the wild type and the KC model as well as in PanIN lesions (Supplemental Fig. 4C). Complete inactivation of p110α, while repressing the induction of preneoplastic ADM lesions in the presence of injury, did not inhibit Pdx1 reinduction (Supplemental Fig. 4D).
Our data support the conclusion that p110α does not control the activation of the reprogramming transcription factor Pdx1 in pancreatic epithelial cells upon injury and/or mutation of Kras. This argues that Pdx1 up-regulation is dispensable for ADM and PanIN formation.
Cytoskeleton remodeling during injury-driven acinar-to-ductal transition is blocked by p110α inactivation
In order to understand how blockade of p110α could interfere with a change of acinar cytoarchitecture as early as 8 h after induction of injury (Fig. 1E,F), we next analyzed the actin cytoskeleton during this process as a possible rapid acinar cell-intrinsic mechanism that could mediate the cell shape modification from acinar to ductal cells. We thus analyzed the kinetics of the actin fiber architecture during this process. We show for the first time that the cortical actin network is relocalized during caerulein-induced acinar-to-ductal transition (Fig. 4A; Supplemental Fig. 6A,B). Upon caerulein treatment, we observed an early depolymerization of actin in the apical and lateral membranes followed by an intensification of the subcortical network (apical, lateral, and basal) in late ADM structures and PanINs (Fig. 4A, shown schematically in B). Only a complete genetic p110α blockade, but not of p110β, was found to prevent this depolymerization of the actin apical–lateral network in acinar cells upon caerulein treatment (Fig. 4A; Supplemental Fig. 6A). Similarly, pharmacological inactivation of p110α but not of p110β in acinar cell line cultures prevented the modification of F-actin localization and the formation and quantity of cytosolic F-actin stress fibers concomitantly with an absence of cell spreading during acinar-to-ductal transdifferentiation (Fig. 4C; Supplemental Fig. 6B).
Figure 4.

p110α activity stimulates in vivo actin cytoskeleton remodeling and activation of Rho small GTPases during pancreatic cancer initiation. (A) Confocal microscopic images of actin staining on paraffin-embedded pancreata in the indicated treatments and genotypes 1 d after the last caerulein injection (N ≥ 3 per genotype). White arrowheads show cortical actin staining, and white asterisks indicate the lumen, the diameter of which is enlarged during acinar-to-ductal transition. Scale, 10 μm. (B) Representative scheme of actin staining during ADM. (C) F-actin staining of A66 (5 μM), TGX-221 (0.5 μM), or vehicle-treated AR4-2J B13 acinar cells that were transdifferentiated in “ductal-like” cells by DEX+EGF for 5 d. Scale, 10 μm. Intensity of signal quantified by epifluorescence on five fields per condition. N = 3; (***) P < 0.001, Student’s t-test. (E, top panel) Western blot analysis of pancreatic lysates from KC compared with KC;p110αlox/lox or KC;p110βlox/lox mice (N ≥ 3) sacrificed 1 d after the last caerulein (Cae.) or PBS injection. Relative quantification is shown in the bottom panel. N ≥ 3; (**) P < 0.01; (*) P < 0.05, Student’s t-test. (F) Rho and Rac1 in vivo activities from pancreatic lysates of the indicated genotypes 1 d after caerulein treatment. N ≥ 2; (*) P < 0.05, Student’s t-test. Positive and negative controls correspond to GTP and GDP loading, respectively (not shown). (G) Overexpression of constitutively active RhoA or wild-type (WT) RhoA in AR4-2J B13 acinar cells treated as indicated. Scale, 10 μm. Transduction of acinar cells with GFP-expressing vectors leads to a >95% rate of transduction and was similarly affected by treatments compared with wild-type AR4-2J cells (not shown). RhoA overexpression was verified by Western blot; p-Cofilin was used as a control of constitutive activity of RhoA (not shown).
In order to gain further mechanistic insight, we first confirmed that PI3K signaling was inhibited both at the basal level and after caerulein treatment, as assessed by a decrease of the phosphorylation of downstream effectors pS473–Akt or pS240/244–S6 upon inactivation of both p110α and p110β at day 1 after caerulein treatment (Fig. 4E; Supplemental Fig. 6C). The activity of the small GTPases RhoA, Rac1, and cdc42 is regulated by class I PI3K activity and controls F-actin dynamics (Heasman and Ridley 2008). Rac1 is known to inhibit Rho activity. In vivo complete p110α inactivation prevented caerulein-induced activation of Rho GTPases but not the activation of Rac1 (Fig. 4F), while Cdc42 is known not to be expressed in the mouse exocrine pancreas (Kowluru et al. 1996). Overexpression of a constitutively active RhoA in acinar cells but not of a wild-type RhoA rescued the phenotype induced by A66 treatment on acinar cytoskeletal reorganization and cell spreading during their transdifferentiation (Fig. 4G).
Taken together, these data show that p110α is necessary for F-actin network remodeling during injury-driven ADM and for activation of the Rho small GTPases.
p110α activity is required for in vivo superactivation of mutated Kras and other signaling pathways and for the induction of stroma surrounding acinar-to-ductal lesions
We next aimed to gain insight into the long-term fate of the acinar cells during pancreatic injury in the presence of the Kras mutation. The current model proposes that mutated Kras activity is further increased during ADM through activation of NF-κB (Daniluk et al. 2012). Signals that contribute to the initiation or progression of mutated Kras-induced ADM lesions to PanINs include EGFR overexpression combined with downstream ERK activation (Ardito et al. 2012; Navas et al. 2012) or IL6 secretion combined with downstream STAT3 activation (Corcoran et al. 2011; Fukuda et al. 2011; Lesina et al. 2011). Concomitant kinetic activation and termination of all of these signals in pancreatic lesion progression has not been described in vivo. We analyzed the molecular events activated during early pancreatic cancerogenesis in acinar cells associated with stromal environment modifications (scheme in Supplemental Fig. 4E).
Genetic p110α but not p110β inactivation completely prevented caerulein-induced oncogenic Kras activation in vivo (Fig. 5A; Supplemental Fig. 7A). Genetic p110α inactivation also prevented expression and nuclear localization of NF-κB/p65 in ADM (Fig. 5B). We confirmed blockade of p65 activation by pharmacological inhibition of p110α in vivo (Supplemental Fig. 7B). STAT3 activation measured by phosphorylation at Y705 occurred in the same number of cells in KC;p110α+/lox and KC;p110αlox/lox mice (Fig. 5B; Supplemental Fig. 7C), albeit the global level of pSTAT3 was significantly decreased as compared with control animals at day 1 as assessed by Western blotting (Fig. 5C; Supplemental Fig. 7D). On day 5, however, pSTAT3 was completely abrogated by p110α inactivation, while the signal was still partially maintained in KC;p110α+/lox animals compared with KC animals (Fig. 5B; Supplemental Fig. 7C). This defect in STAT3 phosphorylation and nuclear localization correlated with the absence of IL6 and αSMA-positive cells surrounding the acini, as detected in caerulein-treated control animals by immunohistochemistry (IHC) as early as day 1 after pancreatic injury (Fig. 5B).
Figure 5.

p110α activity triggers the maintenance of inflammatory and proliferative pathways in the pancreas. (A) Ras in vivo activity from pancreatic lysates of KC compared with KC;p110αlox/lox and KC;p110βlox/lox mice (N ≥ 3) sacrificed 1 d after the last caerulein (Cae.) injection. The relative quantification of all mice analyzed is shown below. (*) P < 0.05, Student’s t-test. Positive and negative controls correspond to GTP and GDP loading of Raf1-RBD, respectively (data not shown). (B) Immunohistochemical analysis of p65, pY705STAT3, pERK1/2, IL6, and αSMA on KC and KC;p110α+/lox compared with KC;p110αlox/lox (N = 3) pancreata 1 or 5 d after the last caerulein or vehicle injection. Black arrowheads show acinar (pERK, pSTAT3, or p65) or stromal (IL6 or αSMA) positively stained cells. Scale, 100 μm. (C) Western blot analysis for STAT3 phosphorylation and EGFR expression of pancreatic lysates from KC compared with KC;p110αlox/lox mice sacrificed 1 d after the last caerulein or PBS injection. (D) EGFR IHC as indicated. Arrowheads show membrane staining. Scale, 50 μm. N = 3. (E) IHC analysis of pS473Akt, pAkt substrate, and pERK1/2 in caerulein-injected KC mice sacrificed 4 mo after the last injection. Black arrowheads show positive ADM or PanIN structures as indicated. N = 3. Scale, 100 μm.
ERK phosphorylation and nuclear localization were induced in acinar cells on day 1 upon injury, independently of the p110α activity status, but this signal was not maintained at a later time point when p110α activity was absent from pancreata together with the absence of any lesions (Fig. 5B). Genetic inactivation of p110α did not prevent EGFR overexpression upon caerulein treatment but dose-dependently prevented EGFR membrane expression in ADM lesions (Fig. 5C,D; Supplemental Fig. 7D).
The levels of activation of these signals and their activating stimuli in KC;p110α+/lox and KC;p110αlox/lox mice were correlated with the time course of immune cell infiltration within pancreatic tissue. Although present, infiltrating immune cells had a tendency to exhibit decreased recruitment in pancreata lacking p110α activity (Supplemental Fig. 4C). Immune cell recruitment/maintenance was significantly decreased on day 5 only if p110α activity was absent, as assessed by the absence of immune cells residing at this time point.
KC pancreata are heterogeneous, presenting different stages of lesions. When comparing the PI3K pathway to ERK activation (Fig. 5E), we observed that the strongest staining intensity of key surrogates of PI3K signaling (pS473–Akt or p-Akt–substrate amino acid motif) was observed in acini before they underwent the cellular phenotypic transformation into ducts. The STAT3 signaling pathway was also increased as early as ADM compared with PI3K signaling, albeit not in all cells, but STAT3 stimulation was maintained in early PanINs, while the increase of PI3K activity was surprisingly not completely sustained in late ADM and low-grade PanIN lesions. Activation of ERK signaling was progressively observed in ADM structures and PanINs (Fig. 5E).
Taken together, our data show that, in vivo, pancreatic p110α activity is required for (1) the superactivation of oncogenic Kras, (2) the rapid induction of nuclear NF-κB/p65, (3) the secretion of IL6, (4) the induction of full STAT3 and ERK activation in ADM, (5) the maintenance of these signals in PanINs, and (6) the activation of the stroma surrounding the ADM structures (the presence of αSMA-positive cells or immune cells). Partial inactivation of p110α decreases the intensity of these markers at a late time point, in line with the fact that KC;p110α+/lox pancreata present a significantly decreased incidence of preneoplastic lesions (Fig. 2C). In other words, p110α appears to be the signaling hub controlling the signals necessary for induction of the ADM and PanIN stages and maintaining these signals in preneoplastic lesions.
p110α is required for PDAC progression induced by combined mutation of Kras and p53
Our data shown above demonstrate that signals emanating from p110α initiate the induction of ADM and PanINs by mutated Kras. Such Ras-induced preneoplastic lesions, which progress to PDAC with long latency (>1 yr), can be greatly accelerated by concomitant inactivation of the p53 tumor suppressor. This can be achieved by crossing KC mice with a LSL-p53R172H allele, allowing expression of mutated p53 when the Cre recombinase is expressed, giving rise to a more aggressive model of pancreatic cancerogenesis (referred to as the KPC model) (Fig. 6A; Hingorani et al. 2005).
Figure 6.

p110α activity is required for the transition from preneoplastic lesions to adenocarcinoma induced by oncogenic Kras and mutated p53. (A) Experimental setup. (B) Survival curves. N ≥ 6 for each genotype. (C) Percentage of 4.5-mo-old mice harboring no lesions, ADM/PanIN1, PanIN2, or high-grade PanIN3/PDAC pancreatic lesions depending on genotypes. (D) Quantification of total pancreatic lesions in Pdx1-Cre; LSL-Tp53R172H; LSL-KrasG12D (KPC; n = 9), KPC;p110α+/lox (n = 8), and KPC;p110αlox/lox (n = 8) mice. (*) P < 0.05, Mann-Whitney test. (E) Representative H&E stainings and IHC using the indicated antibodies (insets show representative areas in high magnification) of KPC, KPC;p110α+/lox, and KPC;p110αlox/lox mice. Scale, 2–100–200 μm. (F) MEF cells were stably transfected with murine shp53 with or without KrasG12V. Cell proliferation during 4 d in the presence of A66 (10 μM) or vehicle was analyzed. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001, two-way ANOVA.
KPC lethality was completely prevented by pancreas-specific genetic inactivation of p110α (Fig. 6B). KPC; p110αlox/lox animals presented some low-grade PanIN lesions, the surface of pancreatic tissue replaced by disrupted tissue being dose-dependently significantly reduced compared with KPC pancreata (Fig. 6C–E). Importantly, KPC;p110αlox/lox mice did not harbor any high-grade or cancer lesions (PanIN3 or adenocarcinoma), showing that inactivation of p110α prevents the progression of low-grade PanIN toward high-grade PanIN and adenocarcinoma (Fig. 6C). Genetic inactivation of p110α dose-dependently led to lesions with a decreased index of proliferation of both epithelial cells and stromal cells (Fig. 6E). Interestingly, the few PanIN lesions that developed under full pik3ca recombination presented decreased levels of pAkt and pERK (Supplemental Fig. 8A,B), further pointing to an active role of p110α in tumor stroma interactions and the maintenance of pERK signals in preneoplastic PanIn lesions. These data were also consistent with the fact that murine embryonic fibroblasts (MEFs), immortalized with p53 shRNA with or without overexpression of oncogenic mutated Kras, were sensitive to p110α inhibitor (Fig. 6F; Supplemental Fig. 9). In other words, p110α plays a critical role in the increase of proliferation of transformed cells.
Taken together, our data show that p110α activity is dose-dependently crucial for the appearance of high-grade proliferative lesions in an epithelial cell-autonomous manner.
Discussion
A better understanding of the molecular events involved in pancreatic cancer will help to design new therapeutic strategies for this lethal pathology. Here, we show that inactivation of a single isoform of PI3K, p110α, is sufficient to prevent the initiation of pancreatic cancer even in the context of inflammation or p53 inactivation and that this kinase activity is critical for the initiation of proliferative advanced lesions and PDAC (Fig. 7). This is the first genetic demonstration of the physiopathological implication of one PI3K isoform in PDAC in vivo. Next-generation sequencing studies clearly showed that mutationally activated Kras is the signature event in PDAC that drives PDAC initiation and progression. We show that nonmutated p110α is downstream from oncogenic Kras in PDAC and is critical for Kras-driven carcinogenesis.
Figure 7.

p110α activity is required for pancreatic cancerogenesis. Schematic representation. (Lof) Loss of function.
Involvement of PI3K activity in pancreatic cancerogenesis had previously been suggested by the protumorigenic effect of pancreatic-restricted loss of the PTEN enzyme, which reverses PI3K activity (Stanger et al. 2005; Hill et al. 2010; Ying et al. 2011; Mann et al. 2012). In tissue other than the pancreas, Pten deletion-induced tumorigenesis was shown to be PI3K isoform-specific. The p110α isoform is critical for thyroid cancer induction (Berenjeno et al. 2012), and p110β is critical for prostate cancer development (Jia et al. 2008; Berenjeno et al. 2012). Deletion of Pten alone in the pancreas induces an increase of ADM and the number of acinoductal cells (Stanger et al. 2005). Combined with our data on the role of p110α in ADM, we propose that Pten deletion-induced pancreatic cancer initiation is likely to be mediated by its effect on p110α signaling rather than p110β. In line with our data, p110α but not p110β is directly activated by mutated Kras (Gupta et al. 2007; Fritsch et al. 2013), and p110β dependency of Pten loss-induced ovarian cancer initiation is overridden in favor of p110α in the genetic context of mutated Kras (Schmit et al. 2014).
The dynamics of the cell cytoskeleton network during acinar cell transdifferentiation are poorly understood. We described for the first time the kinetics of actin rearrangement during ADM and found that p110α is critical for actin depolymerization/repolymerization, explaining the lack of pancreatic plasticity upon p110α inactivation. This is in line with the positive role of cortical actin rearrangements during Ras-induced cellular transformation in vitro (Rodriguez-Viciana et al. 1997). Similarly, loss of PTEN or oncogenic p110α/PIK3CA disrupts breast epithelial cell architecture in three-dimensional (3D) cultures (Berglund et al. 2013). Among the small GTPases that regulate the actin cytoskeleton (Heasman and Ridley 2008), Rho GTPase overexpression is known to increase the quantity of intracellular actin stress fibers. Pancreatic p110α inactivation prevents in vivo Rho activation and actin stress fiber polymerization induced by inflammation and oncogenic Kras, thereby blocking ADM and pancreatic cancer initiation. p110α is involved in different steps of pancreatic cancerogenesis, corresponding to different cellular contexts. While p110α activity-deficient acinar cells can proliferate during pancreatic regeneration after injury (Supplemental Figure 4B), this enzyme is important for proliferation in later stages of pancreatic carcinogenesis (Fig. 6E). It contributes to oncogenic Kras hyperactivation in preneoplastic stages and to the maintenance of essential signaling nodes, including STAT3, NF-κB, and ERK, known to be critical for progression toward cancer lesions (Corcoran et al. 2011; Fukuda et al. 2011; Lesina et al. 2011); similarly, in cancer cells, PI3K was found to regulate nonmutated Kras activity and further signaling through MAPK/ERK (Will et al. 2014). In pancreatic cancer, such hyperactivation of mutated Kras is thought to be regulated by NF-κB activation (Daniluk et al. 2012), which is indeed controlled by p110α. The cellular effects of p110α-driven and PI3K-driven signals depend on the cell type involved in association with the genetic and nongenetic context driving the kinetic of cell transformation.
In PDAC, PI3K activation is increased and correlated with a poor prognosis (Schlieman et al. 2003; Yamamoto et al. 2004). p110α controls the initiation of proliferative cancer lesions, while preclinical data show that inhibitors hitting all PI3Ks decrease cancer cell proliferation in vivo (Navas et al. 2012; Eser et al. 2013). In human PDAC, treatment with mTOR inhibitors (downstream target of PI3K) does not give any significant improvements of patient survival or well-being (Wolpin et al. 2009), while panPI3K/mTOR phase I/II trials on patients with solid cancer, including pancreatic cancer, are ongoing. Interestingly, preclinical anti-tumoral activity of the mTORC1 inhibitor is found to be dependent on the genetic background inducing the pancreatic cancer (Morran et al. 2014), with PDAC arising from pten-deficient background being more sensitive to the drug. In this study, we showed that hitting p110α activity using a kinase-dead model and not hitting its expression is sufficient to completely prevent the initiation of pancreatic ductal cancers in a dose-dependent manner. Our data also support that p110α is also a target in p53 mutant PDAC (which represents the majority of PDAC cases). In contrast to EGFR deletion (Ardito et al. 2012; Navas et al. 2012), p110α inactivation completely protects from oncogenic Kras and mutated p53-induced PDAC and lethality.
An advantage of isoform-specific PI3K inhibitors is that they may be tolerated at doses resulting in more complete target inhibition with fewer adverse effects and resistance mechanisms. Combinative therapeutic strategies hitting two crucial pathways at the same time represent the future for most resistant cancers, such as metastatic nonresectable pancreatic cancers, but are very toxic (Javle et al. 2010). Increasing the selectivity of the drugs is crucial to decrease the toxicity. Understanding the basic biology of isoform specificity of PI3Ks will help to better design therapeutic strategies hitting these enzymes and choose the patient subset likely to respond to these targeted therapies.
Materials and methods
Reagents
Reagents were purchased as follows: A66 (32 nM for p110α, >12,500 nM for p110β, >12,500 nM for p110δ, 3480 nM for p110γ, and >5000 nM for mTOR in vitro IC50) (Jamieson et al. 2011) and TGX-221 (5000 nM for p110α, 5 nM for p110β, 100 nM for p110δ, and >10,000 nM for p110γ in vitro IC50) were from Axon Medchem, caerulein and dexamethasone were from Sigma, and EGF was from R&D Systems. Human pancreatic cell lines came from American Type Culture Collection (ATCC), and human PDAC samples were from Biochain (CliniSciences).
Mice
The LSL-KrasG12D and LSL-p53R172H knock-in (from D .Tuveson, Mouse Models of Human Cancers Consortium repository, National Cancer Institute-Frederick), Pdx1-cre (from D.A. Melton, Harvard University, Cambridge, MA), and p110αlox (from B. Vanhaesebroeck, University College London, London) strains were interbred on a mixed background (CD1/SV129/C57Bl6) to obtain compound mutant LSL-KrasG12D;Pdx1-Cre (named KC), LSL-KrasG12D;LSL-p53R172H;Pdx1-Cre (named KPC), LSL-KrasG12D;Pdx1-Cre;p110α+/lox (named KC;p110α+/lox) or p110αlox/lox (named KC;p110αlox/lox), and LSL-KrasG12D; LSL-p53R172H;Pdx1-Cre;p110α+/lox (named KPC;p110α+/lox) or p110αlox/lox (named KPC;p110αlox/lox). Pancreatic cell lines were isolated as described in Hingorani et al. (2005) and Eser et al. (2013), and recombination was verified as described in Hingorani et al. (2005). Littermates not expressing Cre as well as Pdx1-Cre and p110α+/lox mice of the same age were used as controls. The absence of a histopathological phenotype was checked in Pdx1-cre;p110α+/lox and Pdx1-cre; p110αlox/lox mice. All procedures and animal housing conformed to the regulatory standards and were approved by the ethical committee according to European legislation translated to French law as Décret 2013-118 on February 1, 2013. Genotyping was performed as referenced in Supplemental Figure 1. Pancreatic injury was induced in young mice (8–12 wk) by a series of six hourly intraperitoneal injections of caerulein (75 μg per kilogram of body weight) that was repeated after 48 h in the presence or absence of 50 mg/kg A66. Animals were euthanized 8 h or 1, 5, or 120 d later, and recombination was verified before analysis as described in Graupera et al. (2008) and Guillermet-Guibert et al. (2008). To set up the conditions of pancreatic injury, plasmatic amylase dosage was measured (Phadebas Magle).
Histology and immunostaining
All pancreata were analyzed in blinded fashion. Pancreases were fixed in 10% neutral-buffered formalin and embedded in paraffin. For histopathological analysis, pancreata were serially sectioned (4 μm), and every five sections were stained with hematoxylin and eosin (H&E). Histopathological scoring of pancreatic lesions was performed using serial H&E-stained sections (100 μm apart, three sections per pancreas). One representative slide per mouse was imaged using a Hamamatsu Nanozoomer 2 slide scanner (Hamamatsu Photonics), ADM/PanIN lesions were counted, and the damaged pancreatic tissue, ADM, or PanIN area was measured on the entire section with the Nanozoomer Digital Pathology view software (Hamamatsu).
Immunostainings were conducted using standard methods on formalin-fixed, paraffin-embedded tissues. Antigen retrieval and antibody dilution were carried out as described in the Supplemental Material. All phospho-specific antibodies were revealed using a Cell Signaling Signal Boost system followed by AEC or DAB incubation. Corresponding blocking phosphopeptide (Cell Signaling Technologies) and λ-phosphatase (New England Biolabs) treatments were used for validation of antibody specificity. For immunofluorescence, a Zeiss LSM780 confocal microscope was used.
Western blots and Kras, Rac1, and Rho activity assays
Arcturus laser capture microdissection (4-μm cryosections) was used. Western blotting was conducted using standard methods with antibodies as described in the Supplemental Material. Kras, Rac1, and Rho pull-down activity assays were performed on frozen samples of pancreas (Millipore, 17-218, 17-283, and 17-294).
Ex vivo and in vitro culture of acinar cells
Acinar cells were isolated as described and cultured for 5 d in collagen (Eser et al. 2013). AR42J-B13 cells (a rat pancreatic acinar cell line) from ATCC were cultured in Dulbecco’s modified Eagle’s medium (Sigma) containing 10% FBS (Sigma). For immunostaining, cells were grown on sterile glass coverslips in six-well culture (62.5 × 103 cells per well) for 24 h. Acinar-to-ductal transdifferentiation was induced with 1 μM dexamethasone and 20 ng/mL EGF (for 5 d with or without 5 μM A66). Transduction with wild-type RhoA or constitutive active RhoA adenoviruses was performed 1 d before the beginning of the transdifferentiation treatment. The culture medium with the inhibitor and drugs was changed every 48 h. Data were obtained using a Zeiss LSM780 confocal or epifluorescence microscope.
Proliferation
MEFs were isolated as described in Guillermet-Guibert et al. (2008). Proliferation of immortalized and/or KrasG12V transformed MEFs was assessed by cell counting (0.1 × 106 cells per six-well plate).
Supplementary Material
Acknowledgments
We thank R. Anesia for mouse genotyping; undergraduate students and members of CRCT for technical help; P. Clerc for help on mouse experiment setup; V. Gigoux, M. Fanjul, and S. Lowe for reagent supply (respectively, anti-ratCK19 antibody, anti-CA2, and anti-trypsin vectors for MEFs); M.B. Delisle and her staff for the histology platform (Centre de Microscopie Électronique Appliquée à la Biologie [CMEAB], Federative Structure of Bio-Medical Research for Toulouse [IFR-BMT], University of Toulouse III); UMS006-Anexplo for mouse husbandry and histological preparation of samples; Astrid Canivet; Sophie Allart (Le Centre de Physiopathologie de Toulouse-Purpan [CPTP], Inserm); Romina D’Angelo (I2MC) for the imaging TRI platform; and Lucie Fontaine (I2MC, Inserm) for histology preparation. R.B. is funded by LNCC (JG/VSP-8949). C.C. is funded by LNCC (GB/MA/VSP-10443). L.P. is funded by EU-ERG FP7 (270696-PaCa/PI3K) and GSOEmergence R14040BB/RPL14007BBA. The research of J.G.-G. is funded by EU-ERG FP7 (270696-PaCa/PI3K), LNCC (7FI10193LQJY), Arc (SFI20101201616), Labelisation LNCC Team6-CRCT, GSOEmergence R14040BB/RPL14007BBA, and Université Toulouse III Emergence. D.S. is funded by Deutsche Forschungsgemeinschaft SA 1374/4-1, SFB824 TPC9, and Helmholtz Alliance Preclinical Comprehensive Cancer Center. B.V. is a consultant for Retroscreen (London, UK) and Karus Therapeutics (Oxford, UK).
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.249409.114.
References
- Al-Adsani A, Burke ZD, Eberhard D, Lawrence KL, Shen CN, Rustgi AK, Sakaue H, Farrant JM, Tosh D. 2010. Dexamethasone treatment induces the reprogramming of pancreatic acinar cells to hepatocytes and ductal cells. PLoS ONE 5: e13650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardito CM, Gruner BM, Takeuchi KK, Lubeseder-Martellato C, Teichmann N, Mazur PK, Delgiorno KE, Carpenter ES, Halbrook CJ, Hall JC, et al. 2012. EGF receptor is required for KRAS-induced pancreatic tumorigenesis. Cancer Cell 22: 304–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berenjeno IM, Guillermet-Guibert J, Pearce W, Gray A, Fleming S, Vanhaesebroeck B. 2012. Both p110α and p110β isoforms of PI3K can modulate the impact of loss-of-function of the PTEN tumour suppressor. Biochem J 442: 151–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berglund FM, Weerasinghe NR, Davidson L, Lim JC, Eickholt BJ, Leslie NR. 2013. Disruption of epithelial architecture caused by loss of PTEN or by oncogenic mutant p110α/PIK3CA but not by HER2 or mutant AKT1. Oncogene 32: 4417–4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL, Miller DK, Wilson PJ, Patch AM, Wu J, et al. 2012. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 491: 399–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carriere C, Young AL, Gunn JR, Longnecker DS, Korc M. 2009. Acute pancreatitis markedly accelerates pancreatic cancer progression in mice expressing oncogenic Kras. Biochem Biophys Res Commun 382: 561–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran RB, Contino G, Deshpande V, Tzatsos A, Conrad C, Benes CH, Levy DE, Settleman J, Engelman JA, Bardeesy N. 2011. STAT3 plays a critical role in KRAS-induced pancreatic tumorigenesis. Cancer Res 71: 5020–5029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniluk J, Liu Y, Deng D, Chu J, Huang H, Gaiser S, Cruz-Monserrate Z, Wang H, Ji B, Logsdon CD. 2012. An NF-κB pathway-mediated positive feedback loop amplifies Ras activity to pathological levels in mice. J Clin Invest 122: 1519–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eser S, Reiff N, Messer M, Seidler B, Gottschalk K, Dobler M, Hieber M, Arbeiter A, Klein S, Kong B, et al. 2013. Selective requirement of PI3K/PDK1 signaling for Kras oncogene-driven pancreatic cell plasticity and cancer. Cancer Cell 23: 406–420. [DOI] [PubMed] [Google Scholar]
- Fendrich V, Esni F, Garay MV, Feldmann G, Habbe N, Jensen JN, Dor Y, Stoffers D, Jensen J, Leach SD, et al. 2008. Hedgehog signaling is required for effective regeneration of exocrine pancreas. Gastroenterology 135: 621–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritsch R, de Krijger I, Fritsch K, George R, Reason B, Kumar MS, Diefenbacher M, Stamp G, Downward J. 2013. RAS and RHO families of GTPases directly regulate distinct phosphoinositide 3-kinase isoforms. Cell 153: 1050–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda A, Wang SC, Morris JP 4th, Folias AE, Liou A, Kim GE, Akira S, Boucher KM, Firpo MA, Mulvihill SJ et al. 2011. Stat3 and MMP7 contribute to pancreatic ductal adenocarcinoma initiation and progression. Cancer Cell 19: 441–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graupera M, Guillermet-Guibert J, Foukas LC, Phng L-K, Cain RJ, Salpekar A, Pearce W, Meek S, Millan J, Cutillas PR, et al. 2008. Angiogenesis selectively requires the p110α isoform of PI3K to control endothelial cell migration. Nature 453: 662–669. [DOI] [PubMed] [Google Scholar]
- Guerra C, Schuhmacher AJ, Canamero M, Grippo PJ, Verdaguer L, Perez-Gallego L, Dubus P, Sandgren EP, Barbacid M. 2007. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 11: 291–302. [DOI] [PubMed] [Google Scholar]
- Guillermet-Guibert J, Bjorklof K, Salpekar A, Gonella C, Ramadani F, Bilancio A, Meek S, Smith AJH, Okkenhaug K, Vanhaesebroeck B. 2008. The p110β isoform of phosphoinositide 3-kinase signals downstream of G protein-coupled receptors and is functionally redundant with p110γ. Proc Natl Acad Sci 105: 8292–8297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Ramjaun AR, Haiko P, Wang Y, Warne PH, Nicke B, Nye E, Stamp G, Alitalo K, Downward J. 2007. Binding of ras to phosphoinositide 3-kinase p110α is required for ras-driven tumorigenesis in mice. Cell 129: 957–968. [DOI] [PubMed] [Google Scholar]
- Heasman SJ, Ridley AJ. 2008. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol 9: 690–701. [DOI] [PubMed] [Google Scholar]
- Hidalgo M. 2010. Pancreatic cancer. N Engl J Med 362: 1605–1617. [DOI] [PubMed] [Google Scholar]
- Hill R, Calvopina JH, Kim C, Wang Y, Dawson DW, Donahue TR, Dry S, Wu H. 2010. PTEN loss accelerates KrasG12D-induced pancreatic cancer development. Cancer Res 70: 7114–7124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, et al. 2003. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 4: 437–450. [DOI] [PubMed] [Google Scholar]
- Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, Tuveson DA. 2005. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 7: 469–483. [DOI] [PubMed] [Google Scholar]
- Jamieson S, Flanagan JU, Kolekar S, Buchanan C, Kendall JD, Lee WJ, Rewcastle GW, Denny WA, Singh R, Dickson J, et al. 2011. A drug targeting only p110α can block phosphoinositide 3-kinase signalling and tumour growth in certain cell types. Biochem J 438: 53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javle MM, Shroff RT, Xiong H, Varadhachary GA, Fogelman D, Reddy SA, Davis D, Zhang Y, Wolff RA, Abbruzzese JL. 2010. Inhibition of the mammalian target of rapamycin (mTOR) in advanced pancreatic cancer: results of two phase II studies. BMC Cancer 10: 368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen JN, Cameron E, Garay MV, Starkey TW, Gianani R, Jensen J. 2005. Recapitulation of elements of embryonic development in adult mouse pancreatic regeneration. Gastroenterology 128: 728–741. [DOI] [PubMed] [Google Scholar]
- Jia S, Liu Z, Zhang S, Liu P, Zhang L, Lee SH, Zhang J, Signoretti S, Loda M, Roberts TM, et al. 2008. Essential roles of PI(3)K-p110β in cell growth, metabolism and tumorigenesis. Nature 454: 776–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, et al. 2008. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 321: 1801–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp JL, von Figura G, Mayes E, Liu FF, Dubois CL, Morris JP 4th, Pan FC, Akiyama H, Wright CV, Jensen K, et al. 2012. Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell 22: 737–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowluru A, Seavey SE, Li G, Sorenson RL, Weinhaus AJ, Nesher R, Rabaglia ME, Vadakekalam J, Metz SA. 1996. Glucose- and GTP-dependent stimulation of the carboxyl methylation of CDC42 in rodent and human pancreatic islets and pure β cells. Evidence for an essential role of GTP-binding proteins in nutrient-induced insulin secretion. J Clin Invest 98: 540–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesina M, Kurkowski MU, Ludes K, Rose-John S, Treiber M, Kloppel G, Yoshimura A, Reindl W, Sipos B, Akira S, et al. 2011. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell 19: 456–469. [DOI] [PubMed] [Google Scholar]
- Mann KM, Ward JM, Yew CC, Kovochich A, Dawson DW, Black MA, Brett BT, Sheetz TE, Dupuy AJ, Australian Pancreatic Cancer Genome Initiative, et al. 2012. Sleeping Beauty mutagenesis reveals cooperating mutations and pathways in pancreatic adenocarcinoma. Proc Natl Acad Sci 109: 5934–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Means AL, Meszoely IM, Suzuki K, Miyamoto Y, Rustgi AK, Coffey RJ Jr, Wright CV, Stoffers DA, Leach SD. 2005. Pancreatic epithelial plasticity mediated by acinar cell transdifferentiation and generation of nestin-positive intermediates. Development 132: 3767–3776. [DOI] [PubMed] [Google Scholar]
- Morran DC, Wu J, Jamieson NB, Mrowinska A, Kalna G, Karim SA, Au AY, Scarlett CJ, Chang DK, Pajak MZ, et al. 2014. Targeting mTOR dependency in pancreatic cancer. Gut 63: 1481–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JP 4th, Cano DA, Sekine S, Wang SC, Hebrok M. 2010. β-Catenin blocks Kras-dependent reprogramming of acini into pancreatic cancer precursor lesions in mice. J Clin Invest 120: 508–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navas C, Hernandez-Porras I, Schuhmacher AJ, Sibilia M, Guerra C, Barbacid M. 2012. EGF receptor signaling is essential for k-ras oncogene-driven pancreatic ductal adenocarcinoma. Cancer Cell 22: 318–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodon J, Dienstmann R, Serra V, Tabernero J. 2013. Development of PI3K inhibitors: lessons learned from early clinical trials. Nat Rev Clin Oncol 10: 143–153. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Viciana P, Warne PH, Khwaja A, Marte BM, Pappin D, Das P, Waterfield MD, Ridley A, Downward J. 1997. Role of phosphoinositide 3-OH kinase in cell transformation and control of the actin cytoskeleton by Ras. Cell 89: 457–467. [DOI] [PubMed] [Google Scholar]
- Schlieman MG, Fahy BN, Ramsamooj R, Beckett L, Bold RJ. 2003. Incidence, mechanism and prognostic value of activated AKT in pancreas cancer. Br J Cancer 89: 2110–2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmit F, Utermark T, Zhang S, Wang Q, Von T, Roberts TM, Zhao JJ. 2014. PI3K isoform dependence of PTEN-deficient tumors can be altered by the genetic context. Proc Natl Acad Sci 111: 6395–6400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanger BZ, Stiles B, Lauwers GY, Bardeesy N, Mendoza M, Wang Y, Greenwood A, Cheng KH, McLaughlin M, Brown D, et al. 2005. Pten constrains centroacinar cell expansion and malignant transformation in the pancreas. Cancer Cell 8: 185–195. [DOI] [PubMed] [Google Scholar]
- Strobel O, Dor Y, Alsina J, Stirman A, Lauwers G, Trainor A, Castillo CF, Warshaw AL, Thayer SP. 2007. In vivo lineage tracing defines the role of acinar-to-ductal transdifferentiation in inflammatory ductal metaplasia. Gastroenterology 133: 1999–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhaesebroeck B, Ali K, Bilancio A, Geering B, Foukas LC. 2005. Signalling by PI3K isoforms: insights from gene-targeted mice. Trends Biochem Sci 30: 194–204. [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. 2010. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol 11: 329–341. [DOI] [PubMed] [Google Scholar]
- Will M, Qin AC, Toy W, Yao Z, Rodrik-Outmezguine V, Schneider C, Huang X, Monian P, Jiang X, de Stanchina E et al. 2014. Rapid induction of apoptosis by PI3K inhibitors is dependent upon their transient inhibition of RAS–ERK signaling. Cancer Discov 4: 334–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolpin BM, Hezel AF, Abrams T, Blaszkowsky LS, Meyerhardt JA, Chan JA, Enzinger PC, Allen B, Clark JW, Ryan DP, et al. 2009. Oral mTOR inhibitor everolimus in patients with gemcitabine-refractory metastatic pancreatic cancer. J Clin Oncol 27: 193–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav D, Lowenfels AB. 2013. The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology 144: 1252–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto S, Tomita Y, Hoshida Y, Morooka T, Nagano H, Dono K, Umeshita K, Sakon M, Ishikawa O, Ohigashi H, et al. 2004. Prognostic significance of activated Akt expression in pancreatic ductal adenocarcinoma. Clin Cancer Res 10: 2846–2850. [DOI] [PubMed] [Google Scholar]
- Ying H, Elpek KG, Vinjamoori A, Zimmerman SM, Chu GC, Yan H, Fletcher-Sananikone E, Zhang H, Liu Y, Wang W et al. 2011. PTEN is a major tumor suppressor in pancreatic ductal adenocarcinoma and regulates an NF-κB–cytokine network. Cancer Discov 1: 158–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, Shi G, Schmidt CM, Hruban RH, Konieczny SF. 2007. Acinar cells contribute to the molecular heterogeneity of pancreatic intraepithelial neoplasia. Am J Pathol 171: 263–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.