

Rodenfels et al. show that the Drosophila intestine responds to nutrient availability by regulating production of a circulating lipoprotein-associated form of Hedgehog (Hh). Levels of circulating Hh tune the rates of growth and developmental timing in a coordinated fashion. Circulating Hh is especially important during starvation, when it is also required for mobilization of fat body triacylglycerol stores.
Keywords: Hedgehog, lipid metabolism, Drosophila, steroid hormones, growth
Abstract
In Drosophila larvae, growth and developmental timing are regulated by nutrition in a tightly coordinated fashion. The networks that couple these processes are far from understood. Here, we show that the intestine responds to nutrient availability by regulating production of a circulating lipoprotein-associated form of the signaling protein Hedgehog (Hh). Levels of circulating Hh tune the rates of growth and developmental timing in a coordinated fashion. Circulating Hh signals to the fat body to control larval growth. It regulates developmental timing by controlling ecdysteroid production in the prothoracic gland. Circulating Hh is especially important during starvation, when it is also required for mobilization of fat body triacylglycerol (TAG) stores. Thus, we demonstrate that Hh, previously known only for its local morphogenetic functions, also acts as a lipoprotein-associated endocrine hormone, coordinating the response of multiple tissues to nutrient availability.
Organismal growth and development are regulated by the availability of nutrients. The reaction of a developing animal to nutrient deprivation involves a complex interorgan response orchestrated by the nervous, endocrine, and digestive systems, with the goal to allow basic cellular metabolism but delay energy-demanding processes. A major physiological response to starvation is the utilization of stored energy and the delay of growth and development.
The Drosophila fat body, a tissue analogous to mammalian white adipose tissue and the liver, has an important role in coupling nutrient availability to growth. In response to changes in the availability of sugars, amino acids, and lipids, it releases secreted factors that regulate growth by modulating systemic insulin signaling (Arquier et al. 2008; Geminard et al. 2009; for review, see Hietakangas and Cohen 2009; Rajan and Perrimon 2012). It also acts as a depot for these nutrients and can release them in response to starvation (for review, see Telfer and Kunkel 1991; Arrese and Soulages 2010). The fat body directly senses circulating amino acid levels using the amino acid transporter Slimfast and the target of rapamycin (TOR) pathway (Colombani et al. 2003). However, how it senses the availability of other types of nutrients is unknown.
In addition to regulating growth, nutrient availability also controls developmental progression by influencing production of ecdysteroids in the prothoracic gland. During larval development, ecdysteroids are produced in pulses that regulate molting and initiate pupariation (for review, see Thummel 1996). These pulses are tightly coupled to organismal growth. A key developmental decision occurs once larvae achieve a “critical weight” (for review, see Mirth and Riddiford 2007). After this point, starvation no longer delays pupariation, although it affects the final size of resulting adults. Critical weight is associated with a small increase in ecdysteroid levels, which later rise dramatically to initiate pupariation (Warren et al. 2006). The commitment to and timing of pupariation are subject to multiple checkpoints and can be affected by not only nutrition but also damage to imaginal tissues, circadian signals, and temperature (for review, see Shingleton and Mirth 2012). Consistent with this, multiple signaling pathways in the prothoracic gland influence the decision to up-regulate ecdysteroid production (Colombani et al. 2005; Mirth et al. 2005; McBrayer et al. 2007; Rewitz et al. 2009; Cáceres et al. 2011; Gibbens et al. 2011; Ou et al. 2011). How these networks interact to couple ecdysteroids production to the size of the animal is far from understood.
In other organisms, the digestive system is an important node in the signaling network that controls metabolism. Signals originating from the stomach and the intestine activate neural and humoral pathways to regulate food intake and metabolic homeostasis (for review, see Cummings and Overduin 2007). Less is known about signals produced by the Drosophila gastrointestinal tract, although it harbors neuroendocrine cells that can synthesize peptide hormones and is connected to the nervous system (Cognigni et al. 2011; Miguel-Aliaga 2012; Lemaitre and Miguel-Aliaga 2013). The digestive system is well placed to directly sense nutrient availability as opposed to circulating nutrients. However, whether the Drosophila gastrointestinal tract conveys this type of nutritional information is not known.
Hedgehog (Hh) signaling is known to regulate metabolic processes such as fat storage and glucose uptake in mice (Suh et al. 2006; Pospisilik et al. 2010; Teperino et al. 2012). However, the source of the relevant Hh ligand has never been identified. Interestingly, we showed that circulating lipoproteins in flies and mammals carry not only lipid cargo but also the lipid-modified signaling molecule Hedgehog (Palm et al. 2013). The tissue source and function of circulating Hh is unknown. In this study, we investigate the source and function of lipoprotein-associated systemically circulating Hh in Drosophila.
Results
Midgut-derived Hh circulates systemically
To investigate the source of circulating Hh, we characterized the tissue expression patterns of a variety of different Gal4 drivers (Supplemental Fig. 1A) and used them to knock down Hh production. We then measured Hh levels in the larval hemolymph. Ubiquitous induction of hhRNAi using tubulin-Gal4 efficiently reduces the level of circulating Hh (Supplemental Fig. 1B). We next used en-Gal4, en105-Gal4, or hh-Gal4 to target hhRNAi to posterior compartments of larval ectoderm and imaginal discs, which express hh. These drivers are also active in the salivary glands. Knockdown using these drivers had no effect on the level of circulating Hh despite strongly reducing hh expression in imaginal discs. (Fig. 1A; Supplemental Fig. 1C). Thus, circulating Hh does not derive from the imaginal discs, larval ectoderm, or salivary gland. Therefore, we used immunofluorescence to identify other tissues that might express hh. We noted that Hh protein is present in the larval midgut and its attached gastric caeca (Fig. 1B). In the midgut, Hh localizes to the enterocytes (ECs), which function in nutrient absorption (Fig. 1C). To ask whether this region of the gut also transcribes hh, we performed in situ hybridization and RT–PCR on dissected midgut samples. These experiments showed that hh mRNA is present in the midgut and that it localizes to ECs but not gastric caeca (Fig. 1D,E). Thus, Drosophila midgut ECs transcribe and translate Hh. The Hh protein found in gastric ceaca must be derived from other sources.
Figure 1.

Midgut-derived Hh systemically circulates. (A) Immunoblot of hemolymph from larvae in which Hh is knocked down with en-Gal4, en(105)-Gal4, hh-Gal4, lpp-Gal4, elav-Gal4, and myo1A-Gal4. Knockdown with myo1A-Gal4 strongly reduces Hh levels in the hemolymph. (B,C) Immunofluorescence of second instar larval midguts of wild-type (B,C) and tub>hhRNAi (C) animals, stained for Hh, F-actin (phalloidin), and DNA (DAPI). (GC) Gastic ceace; (MG) midgut. Bars: B, 100 µm; C, 5 µm. Ubiquitous knockdown of hh with tub-Gal4 depletes EC-localized Hh. (D) RT–PCR for hh from second instar midgut samples. Hh message is present in dissected midgut samples. (E) In situ hybridization for hh mRNA on second instar midgut samples. Higher magnification of the marked areas are displayed at the right (in panels I,II) Bars: 100 µm and 5 µm in the marked areas. hh mRNA localizes to migut ECs.
To ask whether circulating Hh originates from the larval midgut, we used the myo1A-Gal4 driver. This driver is active in the midgut ECs and the salivary gland and is also expressed in a few cells in the fat body, CNS, and disc peripodial membrane (Fig. 1; Supplemental Fig. 1A). Driving hhRNAi with myo1A-Gal4 reduces, but does not completely eliminate, the amount of Hh in the larval hemolymph (Fig. 1A). To ask whether the activity of myo1A-Gal4 in the fat body or CNS underlies its ability to reduce Hh levels in the hemolymph, we induced hhRNAi in these tissues more completely using lpp-Gal4 or elav-Gal4 (which drive strong expression in the fat body and CNS, respectively). Neither reduced systemic Hh levels when used to drive hhRNAi (Fig. 1A). These results strongly suggest that midgut ECs are a major source of systemic Hh, although minor amounts may come from other tissues.
Circulating Hh inhibits systemic growth and slows developmental timing
Although larvae lacking circulating Hh can give rise to normally patterned adults, these animals are somewhat smaller than controls. We therefore wondered whether Hh might influence larval growth or developmental timing. To investigate this, we first examined the larval growth rate of animals homozygous for a temperature-sensitive mutation of hh (hhts2). At the restrictive temperature, hhts2 animals grow faster and pupariate earlier than two different wild-type controls (Supplemental Fig. 2A). They pupariate just as efficiently as wild-type animals (Supplemental Fig. 2D), and faster growth and earlier pupariation combine to produce hhts2 pupae of normal size (although hhts2 animals do not give rise to viable adults) (Supplemental Fig. 2A). Thus, in addition to its other patterning functions in the larva, Hh acts to slow the rate of larval growth and delay pupariation. To ask whether loss of circulating Hh was responsible for these effects, we asked whether the speedier growth and development of hhts2 larvae could be reversed by expression of hh in midgut ECs. Indeed, driving hh expression using npc-Gal4 in the hhts2 mutant background completely reverses the accelerated pupariation and growth of hhts2 larvae (Fig. 2A). In fact, these animals pupariate even later than wild-type controls. Pupal volume is indistinguishable from that of wild type in all cases, suggesting that delayed pupariation is accompanied by a reduction in the larval growth rate (Fig. 2B). Thus, hhts2 mutant larvae develop more rapidly than wild type because they lack hh expression in the midgut.
Figure 2.

Circulating Hh inhibits systemic growth and slows developmental timing. (A,C,E) Percentage of pupariated animals at different times (in hours) after egg laying (AEL). The time AEL at which 50% of larvae have pupariated (P50) was determined by nonlinear regression for each biological replicate. P-values are based on Student’s t-test comparing genotype-specific P50 values. Error bars represent standard deviation (SD). n = 3. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001. (B,D,F) Measurement of pupal volume of animals with increased or decreased circulating Hh levels. Error bars represent SD. n = 3. (*) P < 0.05.
To ask whether reducing Hh production in the midgut influenced developmental timing, we used myo1A-Gal4 to drive hhRNAi in midgut ECs and enhanced the efficiency of RNAi by coexpressing Dicer2 (Dcr2). This treatment reduces but does not eliminate circulating Hh. These animals pupariate significantly earlier than animals expressing Dcr2 alone (Fig. 2C; Supplemental Fig. 2B). However, myo1A>Dcr2 larvae develop more slowly than other control larvae. Thus, it was difficult to be sure that reduced Hh production in midgut ECs had increased the rate of larval development. To investigate this further, we asked whether removing one wild-type hh allele could further accelerate the development of myo1A>Dcr2, hhRNAi animals. Indeed, RNAi-mediated hh knockdown in a hhts2/+ heterozygous background caused significantly earlier pupariation than knockdown in a wild-type background. (Fig. 2C). Larvae pupariate at the same volume (Fig. 2D), suggesting that the growth rate is also increased by performing Hh knockdown in a hhts2/+ heterozygous background. Taken together, these results suggest that lowering production of hh in the midgut speeds the rate of larval growth and pupariation.
To ask whether elevating midgut Hh production could slow larval development of otherwise wild-type animals, we expressed hh under the control of two different Gal4 drivers: myo1A-Gal4 and npc1b-Gal4. Npc1b-Gal4-driven expression is strictly midgut-specific and strongly increases the level of circulating Hh. Myo1A-Gal4 is expressed more highly than npc1b-Gal4 in the midgut but is also active a few other tissues. Myo1A-Gal4-driven hh expression increases circulating Hh levels more dramatically than npc1b-Gal4 (Palm et al. 2013). Pupariation of npc1b>hh larvae is delayed by 14 h compared with control npc1b/+ larvae (Fig. 2E). Myo1A-driven hh has a stronger effect, delaying pupariation by 4 d compared with controls (Fig. 2E). Delayed pupariation in hh-overexpressing animals is accompanied by a proportional reduction in larval growth such that these animals pupariate at close to a normal size (Fig. 2F; Supplemental Fig. 2C).
Taken together, these data show that the amount of Hh circulating in the hemolymph controls both the larval growth rate and the speed of pupariation in a proportional fashion.
Circulating Hh promotes survival under starvation
Growth and developmental timing are modulated in response to nutrient availability. Circulating Hh is produced in the tissue in which nutrient absorption occurs. Therefore, we investigated whether its production responded to nutrient uptake. Quantitative RT–PCR (qRT–PCR) and Western blot analysis show that midgut hh expression is significantly elevated upon starvation in both pre- and post-critical weight larvae (Fig. 3A,B). To distinguish the effects of protein, sugar, and lipids on hh expression, we asked which nutrients could reverse the up-regulation of hh expression in starving larvae. Adding 5% sugar (fructose or glucose) to starvation medium (PBS/agar) did not reverse hh up-regulation (Fig. 3C). Adding 10% chloroform-extracted yeast autolysate as a source of amino acids and other water-soluble molecules partially reversed hh up-regulation (Fig. 3C). However, adding both sugar and protein consistently and completely reversed hh up-regulation in the midgut (Fig. 3C). These data suggest that midgut cells deprived of either sugar or amino acids increase hh expression. Interestingly, dietary lipids do not appear to be required to keep hh expression low in the midgut.
Figure 3.
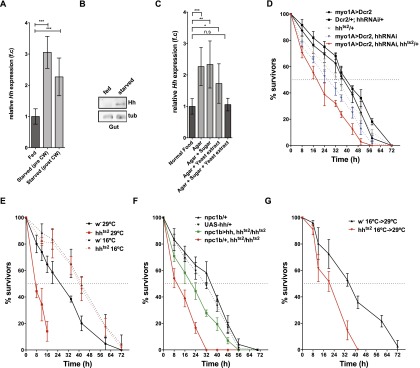
Midgut Hh promotes survival under starvation conditions. (A) Measurements of relative midgut hh mRNA expression by qRT–PCR of animals that were either fed or starved (pre- and post-critical weight). hh expression was normalized to rp49. Fold changes are relative to fed controls. n > 15. (***) P < 0.001. (B) Immunoblot for Hh of fed or starved midguts. Hh becomes enriched in starved midguts. (C) Measurements of relative midgut hh mRNA expression by qRT–PCR of animals that were either fed, starved, or fed sugar, chloroform-extracted yeast autolysate, or sugar plus chloroform-extracted yeast autolysate. hh expression was normalized to rp49. Fold changes are relative to fed controls. n = 6. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001. (D) Survival rate upon starvation of myo1A>Dcr2,Dcr2/+,hhRNAi/+,hhts2/+ myo1A>Dcr2,hhRNAi and myo1A>Dcr2,hhRNAi,hhts2/+ late second instar larvae cultured at 29°C. (E) Survival rate upon starvation of wild-type and hhts2 late second instar larvae cultured at permissive (16°C) or at restrictive (29°C) temperature after embryonic development until time of starvation. (F) Survival rate upon starvation of control (npc1b/+, UAS-hh/+) and npc1b/+, hhts2/hhts2 and npc1b>hh, hhts2/hhts2 cultured at restrictive temperature (29°C). (G) Survival rate upon starvation of wild type and hhts2 cultured at permissive temperature (16°C) until the late second instar followed by starvation at the restrictive temperature (29°C).
Control larvae can survive for 40 h when they are starved before reaching critical weight (Fig. 3D). To ask whether Hh production by the midgut was important for survival under starvation conditions, we reduced Hh function in larvae starved before critical weight by driving hhRNAi in the midgut with myo1A-Gal4. Although myo1A-driven hhRNAi does not reduce survival of normally fed animals (Supplemental Fig. 2E), it reduces survival on starvation from 40 to 24 h (Fig. 3D). To confirm that starvation sensitivity was caused by reduced Hh production in the midgut, we asked whether the phenotype could be enhanced in a hhts2/+ heterozygous background. Indeed, removing one functional copy of hh decreased survival even further (Fig. 3D). Thus, even the incomplete reduction of circulating Hh caused by myo1A-Gal4-induced hhRNAi strongly decreases survival upon starvation. This suggests that circulating Hh promotes survival under starvation conditions.
To reduce systemic Hh levels more completely, we blocked Hh function using the hhts2. hhts2 larvae cultured at the restrictive temperature (29°C) from hatching onward complete larval development with no increased lethality when fed normally but die during early pupal stages (Supplemental Fig. 2D). Thus, Hh function is not required for larval survival or pupariation of normally fed animals. However, when starved before critical weight, these animals die 38 h earlier than wild-type controls (Fig. 3E). This reduction in viability is due to the temperature-sensitive mutation because hhts2 larvae do not differ from wild-type controls when starved at the permissive temperature (16°C) (Fig. 3E).
To ask whether the starvation sensitivity of hhts2 homozygous larvae was due to lack of Hh production in the midgut, we asked whether midgut-specific Hh expression could rescue survival of starved hhts2 homozygous animals. Indeed, midgut-specific hh expression significantly improves survival on starvation. (Fig. 3F). Thus, midgut Hh expression increases survival in starving larvae.
We wondered whether circulating Hh acted at the time of starvation to increase larval survival. Alternatively, loss of circulating Hh during larval growth might affect nutrient storage, making them more sensitive to later starvation. To distinguish these possibilities, we shifted hhts2 larvae to 29°C coincident with starvation and monitored their survival. These animals die 24 h earlier than wild type (Fig. 3G). Taken together, these results show that increased production of circulating Hh by the midgut upon starvation is essential for normal survival under starvation conditions.
Hh produced in the midgut is not required for midgut homeostasis or midgut tracheal branching
We wondered whether Hh produced in the midgut was required locally to influence intestinal function. To ask whether altered Hh levels in the midgut might affect its structure or homeostasis, we assayed morphology, proliferation, and apoptosis in the midgut. We also counted the number of undifferentiated adult midgut precursor (AMP) cells within a cluster and total AMP clusters in a given region of interest. Neither hh overexpression nor RNAi-mediated knockdown of hh in the midgut induced apoptosis or changed EC cell size or morphology (Supplemental Fig. 3A). Furthermore, the total number of AMP clusters and the number within a given cluster remained constant (Supplemental Fig. 3A,B), consistent with previous results (Jiang and Edgar 2009). Thus, Hh produced by midgut ECs does not locally influence their morphology or homeostasis.
To more directly assess whether Hh signaling might affect the function of ECs or AMPs, we autonomously increased or decreased pathway activity in these cell types by expressing RNAi or dominant-negative/active constructs for Hh pathway components under the control of myo1A-Gal4 and esg-Gal4, respectively. The Hedgehog receptor Patched (Ptc) represses signaling when Hh is not present. Binding of Hh to Ptc blocks Ptc-mediated repression of the seven-pass transmembrane protein Smoothened (Smo), which can mediate both transcriptional and nontranscriptional pathway outputs (Alcedo et al. 1996; van den Heuvel and Ingham 1996; DeCamp et al. 2000; Chen et al. 2001; Zhang et al. 2001; Ogden et al. 2008). We either activated Hh signaling using ptcRNAi or inhibited it by smoRNAi in ECs or AMPs. None of these manipulations altered cell morphology, apoptosis, or proliferation in the midgut (Supplemental Fig. 3C,D). To further assess whether Hh signaling in ECs and AMPs could regulate systemic growth and developmental timing, we measured the timing of pupariation and the resulting pupal volume after increasing or decreasing Hh signaling activity in midgut ECs and AMPs. None of these manipulations affected pupal volume or the timing of pupariation (Supplemental Fig. 3E,F).
Taken together, these results strongly suggest that Hh produced in the midgut is not required locally in either ECs or AMPs to regulate systemic growth and developmental timing. We wondered whether Hh signaling in the midgut might influence the ability of larvae to cope with starvation. To test this, we either activated or inhibited Hh signaling in ECs and monitored survival upon starvation. Neither inhibition nor activation of Hh signaling in ECs affects starvation sensitivity (Supplemental Fig. 3G).
Recently, the regulated branching of midgut trachea has been implicated in the regulation of energy stores and their mobilization in response to nutrients (Linneweber et al. 2014). To test whether midgut Hh expression might regulate starvation sensitivity by affecting the structure of midgut trachea, we investigated their morphology in larvae with altered Hh expression in the midgut. Examining trachea in three different regions of the midgut revealed no effect of hh overexpression or knockdown on tracheal size or morphology (Supplemental Fig. 4A). Furthermore, activation or inhibition of hh signaling in trachea terminal cells did not affect survival upon starvation (Supplemental Fig. 4B). Thus, Hh produced by midgut ECs does not appear to influence the overlying trachea.
Circulating Hh does not influence local wing imaginal disc Hh signaling
Is circulating Hh required for normal Hh signaling in the imaginal discs? The growth of imaginal discs exerts strong nonautonomous effects on larval developmental timing (Colombani et al. 2012; Garelli et al. 2012). Changes in imaginal disc Hh signaling due to circulating Hh might thereby indirectly affect larval developmental timing. To ask whether endogenously circulating Hh was required for normal patterning in the wing disc, we reduced circulating Hh levels in the gut using myo1A-Gal4 and monitored Hh levels and pathway activity in the wing disc. Hh is locally expressed in the posterior compartment of the wing disc and spreads into the anterior compartment, where its receptor, ptc, is expressed. Hh signaling stabilizes the full-length form of the Cubitus interruptus (Ci) transcription factor (Ci155) and up-regulates transcription of target genes, including ptc (Tabata and Kornberg 1994; Aza-Blanc et al. 1997). Reducing Hh levels in the hemolymph did not affect the distribution of Hh in the wing disc (Fig. 4A,B) or change the range of Ci155 stabilization or ptc expression (Fig. 4A,B), and flies emerged with normally patterned wings (data not shown). Thus, endogenous levels of circulating Hh are not required for local patterning in wing imaginal discs. While we observed previously that strong elevation of circulating Hh levels perturbs local patterning in the wing imaginal disc (Palm et al. 2013), more moderate increases produced by expressing hh under the control of npc1b-Gal4 have no effect (Fig. 4A,B). Nevertheless, npc1b-driven hh expression reduces larval growth and delays pupariation (Fig. 2C). Thus, circulating Hh does not act indirectly through imaginal discs.
Figure 4.

Circulating Hh does not influence local wing imaginal disc Hh signaling. (A) Immunofluorescence of third instar wing disc stained for Hh, Ptc, and Ci155 from myo1A>Dcr2, myo1A>Dcr2,hhRNAi, and npc1b>hh animals. Bar, 30 µm. The anterior (A)/posterior (P) compartment boundary is marked with a yellow line. (B) Quantification of Hh (left), Ptc (middle), and Ci155 (right) levels of control (myo1A>Dcr2; black line) animals and animals with decreased (myo1A>Dcr2, hh-RNAi; blue line) and increased (npc1b>hh; red line) midgut Hh levels. n = 13. Dashed lines represent SD.
Circulating Hh does not influence the frequency of mouth hook contractions
The mammalian intestine produces hormones that act on the brain to regulate feeding behavior (Strader and Woods 2005; Sandoval et al. 2008).To ask whether circulating Hh produced by the midgut might influence larval feeding, we counted larval mouth hook contractions. Neither overexpression nor RNAi-mediated knockdown of hh in the midgut changed the frequency of larval mouth hook contractions (Supplemental Fig. 4C). Thus, changes in growth and developmental timing are not a simple consequence of less enthusiastic feeding.
Circulating Hedgehog promotes neutral lipid mobilization in the fat body upon starvation
Since Hh signaling can reduce triacylglycerol (TAG) levels in both flies and mammals, we wondered whether the influence of circulating Hh on starvation resistance, growth rate, or pupariation timing was mediated through effects on fat metabolism. We first examined how loss or gain of circulating Hedgehog influenced TAG levels in normally fed animals using shotgun mass spectrometry (MS). We observed no change in the amount of stored TAG under either condition in either the fat body or whole animals (Fig. 5A,B). Thus, altering the levels of circulating Hh is insufficient to change TAG metabolism in well-fed animals. These observations suggest that Hh influences growth and developmental timing by mechanisms that do not depend on TAG metabolism.
Figure 5.

Circulating Hedgehog promotes neutral lipid mobilization in the fat body upon starvation. (A,B) Whole-animal (A) and fat body (B) TAG levels of normal fed late second instar (68 h AEL) and third instar (96 h AEL) larvae with reduced or increased Hh levels in the midgut, quantified by MS (n = 3). TAG levels were normalized to the total amount of membrane lipids AEL. (C,D) Changes in whole-animal (C) and fat body (D) TAG levels upon starvation of late second instar larvae with reduced Hh levels in the midgut, quantified by MS (n = 3). (**) P < 0.01. Error bars represent SD.
We next investigated whether circulating Hh might influence TAG metabolism under starvation conditions. In control larvae, starvation initially increases levels of whole-larval TAG within 12 h by almost twofold (Fig. 5C). Interestingly, TAGs do not increase in the fat body at this time; this increase must occur in other tissues (Fig. 5D). Over the next 12 h, both whole body and fat body TAGs drop dramatically (Fig. 5C,D). Wild-type larvae normally die ∼24 h after TAG stores have dropped (48 h after starvation) (Fig. 3D).
Reducing the levels of circulating Hh during starvation completely blocks mobilization of fat body TAGs (Fig. 5D). However, it does not prevent the disappearance of TAGs from other tissues; whole body TAGs drop between 12 and 24 h to levels almost as low as those in control animals (Fig. 5C). Residual whole body TAG at this time likely reflects failed mobilization of fat body stores. These larvae die 24 h earlier than control larvae, right after the time that wild-type animals would have mobilized fat body TAG stores. Thus, circulating Hh is required to mobilize fat body TAG stores under starvation conditions. However, elevating circulating Hh in well-fed animals is not sufficient by itself to reduce TAGs in the fat body—other signals active during starvation must work together with Hh to accomplish this.
Fat body Hh signaling regulates starvation resistance and larval growth
We wondered whether some of the effects of circulating Hh might be mediated by direct signaling to the fat body; indeed, the fat body expresses Hh pathway components but not hh itself (Supplemental Fig. 5A; Suh et al. 2006; Pospisilik et al. 2010; Palm et al. 2013). To investigate this, we first asked whether Hh protein was detectable in the fat body despite the fact that it is not expressed there. Immunoblotting shows that the fat bodies of feeding third instar larvae indeed contain small amounts of Hh protein (Fig. 6A). Fat body Hh protein levels, but not hh transcription, increase upon starvation (Fig. 6A; Supplemental Fig. 5B). These are precisely the conditions under which Hh production is up-regulated in the midgut (Fig. 3A,B). To determine whether Hh in the fat body was derived from the midgut, we reduced Hh production there and monitored Hh protein in the fat body of starving larvae by Western blotting. Indeed, hh knockdown in the midgut reduces Hh protein levels in the fat body (Fig. 6B). Thus, the midgut is the source of Hh ligand found endogenously in the fat body. Furthermore, driving hh overexpression in the gut using npc1b-Gal4 elevates the amount of Hh in the fat body (Fig. 6A). Thus, the amount of Hh accumulating in the fat body directly reflects the amount of Hh produced by the midgut.
Figure 6.

Fat body Hh signaling regulates starvation resistance and larval growth. (A) Immunoblot for early third instar fat body Hh from animals that have been normally fed or starved or with increased hh expression in the midgut. (B) Immunoblot for fat body Hh upon starvation from animals with reduced midgut Hh expression. (C,D) Survival rate upon starvation of lpp>ptc∆loop2 (C) and lpp>ptcRNAi (D) and their respective control late second instar larvae. (E) Changes in larval volume and determination of the timing of pupariation of animals with increased (lpp>ptcRNAi) or decreased (lpp>smoRNAi) Hh signaling specifically in the fat body. The asterisk marks the white pupal stage and volume. (F) Measurement of the pupal volume of animals with increased or decreased Hh pathway activity in the fat body. (G) Measurements of the timing of pupariation of animals with increased or decreased Hh pathway activity in the fat body.
To ask whether Hh signaling in the fat body might mediate effects on growth, pupariation, and starvation resistance, we autonomously increased or decreased pathway activity in this tissue by expressing RNAi or dominant-negative/active constructs for Hh pathway components under the control of lpp-Gal4. We first examined whether Hh signaling in the fat body controlled TAG metabolism or starvation resistance. Although manipulation of the pathway at the level of the systemic Hh ligand does not alter TAG stores, autonomous pathway activation and inactivation in the fat body cause opposing changes in whole-animal TAG, consistent with previous reports (Supplemental Fig. 5C; Suh et al. 2006; Pospisilik et al. 2010). Interestingly, both pathway activation and pathway repression reduced survival upon starvation (Fig. 6C,D). These animals may die for opposite reasons. When Hh signaling is permanently activated in the fat body, larvae have less stored TAG to mobilize. When it is inactivated, they may be unable to mobilize it.
Is the fat body responsible for the alterations in growth and/or developmental timing caused by perturbing hh expression in the midgut? To test this, we monitored the timing of pupariation and resulting pupal volume after increasing or decreasing Hh signaling in the fat body. Activating the pathway by RNAi-mediated ptc knockdown decreased the larval growth rate (Fig. 6E) but did not affect pupariation timing (Fig. 6E,G). As a consequence, pupal volume was decreased (Fig. 6E,F). Pathway inhibition caused by smoRNAi increased larval growth rate (Fig. 6E), again without affecting the timing of pupariation (Fig. 6E,G), resulting in larger pupae (Fig. 6F). Similar results were obtained when Hh signaling was blocked by expression of the constitutively active ptc∆loop2 (Fig. 6F,G). Interestingly, these effects of Ptc and Smo in the fat body appear to be exerted by a noncanonical pathway that does not involve processing of the transcription factor Ci (Supplemental Material; Supplemental Fig. 6). Instead, up-regulation and down-regulation of Hh cause opposing changes in specific fat body diacylglycerol species (Supplemental Fig. 6H). Taken together, these data show that the fat body mediates the effects of circulating Hh on larval growth and does so by a noncanonical pathway. However, its effects on developmental timing must be exerted through a different tissue.
Hh signaling in the prothoracic gland regulates developmental timing
The timing of ecdysteroid production by the prothoracic gland regulates developmental transitions, including pupariation. We therefore asked whether Hh might signal to the prothoracic gland to control developmental timing. We first investigated whether Hh pathway components were present in this tissue using immunofluorescence. We detected Ptc, Smo, and Ci155 specifically in the prothoracic gland but not in the corpus allatum or corpus cardiacum, which produce other hormones (Fig. 7A,B). Furthermore, Lpp, which carries circulating Hh, also accumulates specifically on cells of the prothoracic gland (Fig. 7A,B). However, Hh itself could not be detected by immunofluorescence (data not shown). To ask whether Hh produced in the midgut was able to travel to the ring gland, we expressed either hh-gfp or membrane-tethered cd8-hh-gfp under the control of npc1b-Gal4 and stained ring glands with an antibody to GFP. Hh-GFP is clearly detected on cells of the prothoracic gland but not when the midgut expresses comparable levels of the membrane-tethered version (Fig. 7; Supplemental Fig. 7A). Thus, Hh made in the midgut can travel to the prothoracic gland, which expresses Hh pathway components.
Figure 7.

Prothoracic gland Hh signaling regulates ecdysteroid production. (A) Cartoon of a Drosophila ring gland. (PG) Prothoracic gland; (CC) corpus cardiac; (CA) corpus allatum. (B) Immunofluorescence of phm>CD8-GFP second instar ring gland stained for GFP, phalloidin, Ci155, Smo, Ptc, and Lpp. Bar, 20 µm. (C) Immunofluorescence of npc1b>cd8-hh-gfp and npc1B>hh-gfp second instar ring gland stained for GFP, Discs large (Dlg), and Lpp. (D) Percentage of larvae pupariating with inhibited (phm>gfp,smoRNAi) or activated (phm>gfp,ptcRNAi, phm>gfp, cd8-hh-gfp, phm>gfp,CiPKA) Hh signaling pathway in the prothoracic gland. Error bars represent SD. (E) Percentage of larvae pupariating with activated (phmGal4,Gal80ts>gfp,ptcSSD) Hh signaling pathway in the prothoracic gland. Error bars represent SD. (F) Immunofluorescence of phm>gfp, phm>gfp,smoRNAi, and phm>ptcRNAi second instar ring gland stained for GFP, DNA (DAPI), and cleaved caspase 3. Bar, 25 μm. (G) Quantitative ecdysteroid MS time course for 20E (E) and makisterone A from whole larva with inhibited (phm>gfp,smoRNAi) or activated (phm>gfp,ptcRNAi) Hh signaling pathway in the prothoracic gland or from control animals (phm>gfp). The asterisk marks the white pupal stage. Error bars indicate SD of biological triplicates. (H) qRT–PCR for phm, cyp6t3, spok, InsR, and toros at 96 h AEL from dissected brain–ring gland complexes (BRCs) with increased or decreased Hh pathway activity in the prothoracic gland relative to control (phm>gfp) brain–ring gland complexes. Error bars represent SD of biological triplicates. (*) P < 0.05; (***) P < 0.001.
To investigate the autonomous effects of Hh signaling in the prothoracic gland, we used phm-Gal4 to drive the expression of constructs that activate or inactivate Hh signaling. We then monitored the rate of larval growth, timing of pupariation, and starvation sensitivity. We used four methods to activate Hh signaling: RNAi-mediated knockdown of ptc, expression of the dominant-negative ptcSSD, expression of membrane-tethered Hh, and expression of dominant-active Ci (CiPKA). Each of these treatments inhibits pupariation, and loss of Ptc function blocks it almost completely (Fig. 7D,E). In contrast, inhibiting Hh signaling by smoRNAi expression only slightly delays puparation (8–12 h after wild-type controls), and larvae pupariate at the same size (Supplemental Fig. 7B). These data suggest that loss of Hh signaling alone is not sufficient to induce early pupariation but that increased Hh signaling in the prothoracic gland inhibits pupariation. In contrast to its function in the fat body, Hh signaling in the prothoracic gland does not strongly influence larval growth or starvation sensitivity (Supplemental Fig. 7C,D).
Taken together, these results suggest that the physiological effects of elevating circulating Hh can be accounted for by its combined action in the fat body and prothoracic gland; Hh signaling in the fat body reduces larval growth, and Hh signaling in the prothoracic gland delays pupariation.
How does Hh signaling in the prothoracic gland block pupariation? Expressing ptcRNAi or smoRNAi in the prothoracic gland does not disturb its morphology or cause caspase activation; thus, cell death in the prothoracic gland cannot account for the effects of ptcRNAi on pupariation (Fig. 7F). ptcRNAi reduces nuclear area by 20%, suggesting that these cells may be slightly less polyploid than those in wild-type prothoracic glands (Supplemental Fig. 7E).
Pupariation is normally initiated by a rise in the level of ecdysteroid hormones. At early wandering stages, the prothoracic gland increases transcription of ecdysteroid biosynthetic enzymes and elevates production of ecdysone, which is hydroxylated by other tissues to 20-OH ecdysone. To ask whether Hh signaling in the prothoracic gland blocked pupariation by preventing production of ecdysteroids, we quantified ecdysteroids over the course of the third instar and in early pupae using liquid chromatography and tandem MS (LC-MS/MS). In wild-type third instar larvae, we detected both 20-OH ecdysone and the related hormone makisterone A but not ecdysone or methyl-ecdysone (the precursor of makisterone A) (Fig. 7G). This suggests that 20-hydroxylation must be rapid and efficient at this stage. 20-OH ecdysone and makisterone A levels begin to rise 56 h after the molt from L2 to L3 and peak in white prepupae (Fig. 7G). In contrast, larvae with constitutively active Hh signaling in the prothoracic gland fail to produce high levels of either 20-OH ecdysone or makisterone A (Fig. 7G). Only a slight rise in hormone levels is apparent even 96 h after the molt from L2 to L3, long after wild-type larvae have pupariated. Thus, constitutive Hh signaling in the prothoracic gland blocks 20-OH ecdysone and makisterone A production. Reducing Hh signaling by smo knockdown in the prothoracic gland only slightly delays pupariation (Supplemental Fig. 7B). Consistent with this, these animals increase production of 20-OH ecdysone and makisterone A with slightly delayed kinetics compared with wild type (Fig. 7G).
qRT–PCR shows that expression of ptcRNAi and ptcSSD in the prothoracic gland reduces mRNA levels for the ecdysteroid biosynthetic enzymes phantom (phm) and spookier (spok), (Fig. 7H; Supplemental Fig. 7F; Warren et al. 2004). However, expression of cyp6t3, a potential rate-limiting enzyme, in ecdysteroid biosynthesis remains unchanged (Fig. 7H; Supplemental Fig. 7F). This suggests that reduced transcription of biosynthetic enzymes contributes to lower ecdysone/methyl-ecdysone biosynthesis in the prothoracic gland.
Both prothoracicotropic hormone (PTTH) and insulin signaling can regulate the onset of ecdysone production (Mirth et al. 2005; Rewitz et al. 2009). To ask whether Hh signaling might disturb either of these processes, we quantified expression of insulin receptor (InsR) or the PTTH receptor torso by qRT–PCR in brain–ring gland complexes. Neither ptcRNAi nor expression of ptcSSD significantly changed the expression of insR or torso, suggesting that Hh signaling does not act by reducing susceptibility to PTTH or Drosophila insulin-like peptides (Dilps) (Fig. 7H; Supplemental Fig. 7F). Thus, high Hh signaling reduces transcription of ecdysteroid biosynthetic enzymes and inhibits production of ecdysone and makisterone A, delaying pupariation.
Taken together, these data suggest that the increase in ecdysteroid biosynthesis that precedes and initiates pupariation does not occur if Hh signaling in the prothoracic gland is too high. However, reducing Hh signaling is not sufficient to initiate ecdysteroid biosynthesis. Lowered Hh signaling is likely only one of many inputs that regulate this important decision.
Discussion
The ability to couple nutrition to growth and developmental progression allows animals to thrive in unpredictable environments. Work in Drosophila has begun to outline how different organs communicate to coordinate these processes. Key interactions have been demonstrated between the fat body, prothoracic gland, and CNS (Colombani et al. 2005; Mirth et al. 2005; Geminard et al. 2009; Delanoue et al. 2010). Our results highlight the importance of the intestine as another node in the signaling network. The intestine is well placed to specifically sense intake of nutrients as opposed to their circulating levels, which may reflect other physiological conditions and not food availability alone.
We showed that Hh pathway activation in the prothoracic gland inhibits ecdysone production and delays pupariation, consistent with the delay in pupariation caused by increasing circulating Hh. In contrast, although reducing circulating Hh causes larvae to pupariate sooner, inhibiting Hh pathway activation in the prothoracic gland is not sufficient to do so. Clearly, the information directly provided to the prothoracic gland by lower-circulating Hh levels must be integrated with that of other signals. For example, insulin signaling, TGFβ signaling, and PTTH signaling are all required to increase ecdysteroid production (Colombani et al. 2005; Mirth et al. 2005; McBrayer et al. 2007; Rewitz et al. 2009; Gibbens et al. 2011). It seems likely that the drop in circulating Hh is sensed by other tissues that influence the production of such confirmatory signals.
Circulating Hh also signals to the fat body to slow larval growth and mobilize TAGs during nutrient deprivation. Its action in the fat body and prothoracic gland are independent of each other. Growth and pupariation timing can be altered separately by autonomously activating Hedgehog signaling in either of these tissues by itself. Thus, circulating Hh couples systemic growth and developmental timing by its independent actions in the fat body and the prothoracic gland. While Hh acts directly on both of these tissues, other studies have clearly shown that information can also flow back and forth between the ring gland, the fat body, and Dilp-producing neurons in the brain. Humoral signals such as Activin and the fat body-derived Leptin homolog Unpaired are transmitted to the brain and promote the release of Dilps. These increase insulin signaling in the ring gland, which elevates ecdysteroid production (Mirth et al. 2005; Geminard et al. 2009; Gibbens et al. 2011; Rajan and Perrimon 2012; Ghosh and O’Connor 2014). Conversely, moderately increased levels of ecdysteroids act on the fat body to inhibit myc activity, which slows larval growth, possibly by reducing Dilp release (Colombani et al. 2005; Delanoue et al. 2010). Taken together, the signals flowing between the gut, fat body, insulin-producing cells, and prothoracic gland represent a robust information processing network that produces sensible decisions about growth and development.
We highlighted the importance of intestinal-derived circulating Hedgehog to regulate lipid mobilization and couple systemic growth to developmental progression during nutritional stress. Interestingly, work by others shows that hh transcription is increased by bacterial infection in the gut and also by loss of DHR96 (Bujold et al. 2010; Chakrabarti et al. 2012). This nuclear hormone receptor mediates the response to xenobiotics and regulates TAG metabolism (King-Jones et al. 2006; Sieber and Thummel 2009). Thus, it would be interesting to test whether circulating Hh might regulate growth and development in response to other physiological stresses.
A variety of different Hh secretion forms have been reported (Zeng et al. 2001; Chen et al. 2004; Panáková et al. 2005; Tanaka et al. 2005; Creanga et al. 2012; Ohlig et al. 2012; Palm et al. 2013). We previously studied two different secretion forms: one that is sterol-modified and lipoprotein-associated and another that is monomeric and not sterol-modified (Palm et al. 2013). These different forms exert different and synergistic effects on the signaling pathway in Drosophila imaginal discs (Palm et al. 2013). In contrast, signaling in the fat body is mediated entirely by the Lpp-associated form. In discs, the Lpp form of Hh blocks the repressive activity of lipoprotein lipids on Smoothened stability, but even high levels do not influence target gene activation by Ci; this requires the non-sterol-modified form (Palm et al. 2013). Consistently, the action of lipoprotein-associated Hedgehog in the fat body requires Ptc and Smo but does not appear to act through Ci-dependent transcription. These findings emphasize the physiological importance of the lipoprotein-associated Hh form and suggest that the fat body will be a good system to further study its unique effects on the Hh pathway. It will be interesting to know whether Lpp-associated Hh might directly influence lipid storage or regulate the availability of signaling lipids in the fat body, as suggested by work in imaginal discs. (Khaliullina et al. 2009; Palm et al. 2013).
Recently, the noncanonical Hedgehog signaling pathway has been implicated in regulating lipid and sugar metabolism in adult animals, including mice and flies (Pospisilik et al. 2010; Teperino et al. 2012). However, whether this occurs in response to a Hedgehog ligand and what the source of such a ligand might be were completely unknown. Here we show that circulating Drosophila Hh, produced by the intestine, regulates lipid and steroid hormone metabolism and adapts the animal to its nutritional environment. Since the presence of Hedgehog in circulation is conserved (Palm et al. 2013), it may be that circulating mammalian Sonic hedgehog (Shh) exerts a similar function. It will be particularly interesting to also examine the impact of circulating Shh on adrenal steroidogenesis, given its function in Drosophila and the dramatic effects of glucocorticoids on lipid and sugar metabolism (Walker 2007). Interestingly, the production of Hh proteins in the gut is widely conserved, even in cnidarians (van den Brink 2007; Matus et al. 2008). Thus, it seems possible that a metabolic function for circulating and lipoprotein-associated Hedgehog proteins might have arisen early in evolution.
Material and methods
Fly stocks
The following stocks were used: lpp-Gal4 (Brankatschk and Eaton 2010), en(105)-Gal4, en-Gal4 (Eugster et al. 2007), hh-Gal4, tub-Gal4, myo1A-Gal4 (Jiang et al. 2009), npc1b-Gal4 (Voght et al. 2007), phm-Gal4 (Ono et al. 2006), hhts2, UAS-hh (Strigini and Cohen 1997), UAS-ptc∆loop2 (Briscoe et al. 2001),UAS-ptcSSD (gift from I. Guerrero), UAS-CiPKA (Méthot and Basler 1999), drsf-Gal4 (Gervais and Casanova 2011), esg-Gal4, and UAS-cd8-gfp (this study). Wild-type Oregon R (OreR) and w− were obtained from Bloomington Stock Center. UAS-Dicer2, smoRNAi, and ptcRNAi were obtained from Vienna Drosopholia RNAi Center (VDRC), and hhRNAi was obtained from Drosophila Genetic Resource Center (DGRC).
Survival upon starvation
To measure survival, larvae from 4-h egg collections were kept on normal food until 60 h after egg laying (AEL). For experiments with hhts2 animals, eggs were collected for 4 h at 22°C and kept for 48 h at permissive temperature (16°C) to allow embryonic development. Hatched hhts2 and control larvae were shifted to restrictive temperature either at 48-h AEL or just prior to starvation. Second instar larvae were washed in PBS and transferred to 24-well plates (a single larva per well) filled with 0.8% (w/v) agar/PBS. Dead larvae were counted every 8 h, and the percentage of surviving animals from individual 24-well plates was plotted over time. For each genotype, three 24-well plates were quantified.
Growth rates and developmental timing analysis
Growth rate and developmental timing analysis was performed as described (Colombani et al. 2005; Delanoue et al. 2010). In brief, larvae of different genotypes from a 4-h egg collection were synchronized at hatching (23 h AEL) and cultured in 6-cm petri dishes filled with normal food at a density of 50 larvae per dish. Digital images were captured every 24 h, and the larval or pupal volume was estimated by quantifying the area of the length and width of the animals in Fiji and then estimating the corresponding volume using the formula
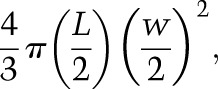 |
where L is larval length, and w is larval width. The average volume of a least 15 animals was plotted over time in biological triplicates. Timing of pupariation was defined as the time at which 50% of the larvae had pupariated.
Lipids extraction and shotgun MS
Lipids were extracted from the fat body and whole larvae and quantified by shotgun MS as described in Carvalho et al. (2012) with modifications. Briefly, samples were homogenized in 150 mM ammonium bicarbonate buffer. From each homogenate, a volume containing ∼2 nmol of lipid material was removed and extracted using a modified Folch protocol at 4°C (whole larva, n = 3; fat body, n = 4; fed to 12 h of starvation, n = 6 for 24 h of starvation experiment).
MS analyses were performed on a Q Exactive instrument (Thermo Fisher Scientific) equipped with a robotic nanoflow ion source TriVersa NanoMate (Advion BioSciences) using chips with 4.1-mm diameter spraying nozzles. The ion source was controlled by Chipsoft 8.1 software. Ionization voltage was set at +0.95 kV and −0.95 kV, and back pressure was set at 1.25 and 0.6 psi in positive and negative modes, respectively. The temperature of ion transfer capillary was 200°C; tube voltages were 90 V (MS+) and −150 V (MS−). Acquisitions were performed at the mass resolution Rm/z400 = 140,000. AGC control was set at 106 ions, and maximum injection time was 250 msec.
Dried lipid extracts were resuspended in 100 μL of 1:2 (v/v) chloroform–methanol mixture. For the analysis, 10 μL of samples were loaded onto a 96-well plate (Eppendorf) and diluted with either 13 μL of 13.3 mM ammonium acetate in 2-propanol for positive ion mode analysis or 10 μL of 0.01% methylamine in methanol for negative ion mode analysis. The plate was sealed with aluminum foil. Each sample was analyzed for ∼1 min in positive and negative ion modes: PE, PC, PC-O, TAG, CerPE, and DAG were quantified in positive mode, and PI, PS, PG, PE, PE-O, Cer, and HexCer were quantified in negative mode.
Lipids were identified by LipidXplorer (Herzog et al. 2011) software by matching the m/z of their monoisotopic peaks to the corresponding elemental composition constraints. Molecular Fragmentation Query Language (MFQL) queries compiled for all of the aforementioned lipid classes are available at the LipidXplorer wiki site (https://wiki.mpicbg.de/wiki/lipidx/index.php/Main_Page). Mass tolerance was 5 ppm, and intensity threshold was set according to the noise level reported by Xcalibur software (Thermo Scientific).
The content of glycerolipids was normalized per polar lipids and expressed in picomole of lipid x/picomole of polar lipids. Lipid standards were purchased from Avanti Polar Lipids (Alabaster). Solvents were purchased from Sigma-Aldrich.
Quantification of ecdysteroids by LC-MS/MS.
Larvae of different genotypes from a 4-h egg collection were synchronized at hatching (23 h AEL) and cultured in petri dishes filled with normal food at a density of 50 larvae each plate. Larvae were staged again at the second to third instar transition (AEL3). Every 4 h, groups of four animals were collected in 100 μL of H2O, smashed with a plastic pestle, and extracted overnight with 1 mL of cold (6°C) methanol. Samples were centrifuged, and supernatant was collected. The pellet was re-extracted twice with 1 mL of methanol for 1 h, and the extracts were combined, cleaned by solid-phase extraction, dried down, and analyzed by LC-MS/MS as described in Hikiba et al. (2013) with minor modifications. The detection limit of the method was ∼5 pg per individual ecdysteroid. Lipid standards were purchased from Avanti Polar Lipids. Solvents were purchased from Sigma-Aldrich.
For additional Materials and Methods, see the Supplemental Material
Supplementary Material
Acknowledgments
We are grateful to Bruce Edgar, Michael O’Connor, Isabell Guerrero, and the Bloomington Stock Center for fly stocks. We gratefully acknowledge Robert Holmgren and the Developmental Studies Hybridoma Bank for providing antibodies. We thank Sven Ssykor and Marko Brankatschk for help with transgenesis. We thank Andreas Sagner, Marko Brankatschk, and Jochen Rink for critical comments on the manuscript.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.249763.114.
References
- Alcedo J, Ayzenzon M, Ohlen Von T, Noll M, Hooper JE. 1996. The Drosophila smoothened gene encodes a seven-pass membrane protein, a putative receptor for the hedgehog signal. Cell 86: 221–232. [DOI] [PubMed] [Google Scholar]
- Arquier N, Geminard C, Bourouis M, Jarretou G, Honegger B, Paix A, Léopold P. 2008. Drosophila ALS regulates growth and metabolism through functional interaction with insulin-like peptides. Cell Metab 7: 333–338. [DOI] [PubMed] [Google Scholar]
- Arrese EL, Soulages JL. 2010. Insect fat body: energy, metabolism, and regulation. Annu Rev Entomol 55: 207–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aza-Blanc P, Ramírez-Weber FA, Laget MP, Schwartz C, Kornberg TB. 1997. Proteolysis that is inhibited by hedgehog targets Cubitus interruptus protein to the nucleus and converts it to a repressor. Cell 89: 1043–1053. [DOI] [PubMed] [Google Scholar]
- Brankatschk M, Eaton S. 2010. Lipoprotein particles cross the blood-brain barrier in Drosophila. J Neurosci 30: 10441–10447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briscoe J, Chen Y, Jessell TM, Struhl G. 2001. A hedgehog-insensitive form of patched provides evidence for direct long-range morphogen activity of sonic hedgehog in the neural tube. Mol Cell 7: 1279–1291. [DOI] [PubMed] [Google Scholar]
- Bujold M, Gopalakrishnan A, Nally E, King-Jones K. 2010. Nuclear receptor DHR96 acts as a sentinel for low cholesterol concentrations in Drosophila melanogaster. Mol Cell Biol 30: 793–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cáceres L, Necakov AS, Schwartz C, Kimber S, Roberts IJH, Krause HM. 2011. Nitric oxide coordinates metabolism, growth, and development via the nuclear receptor E75. Genes Dev 25: 1476–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho M, Sampaio JL, Palm W, Brankatschk M, Eaton S, Shevchenko A. 2012. Effects of diet and development on the Drosophila lipidome. Mol Syst Biol 8: 600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti S, Liehl P, Buchon N, Lemaitre B. 2012. Infection-induced host translational blockage inhibits immune responses and epithelial renewal in the Drosophila gut. Cell Host Microbe 12: 60–70. [DOI] [PubMed] [Google Scholar]
- Chen W, Burgess S, Hopkins N. 2001. Analysis of the zebrafish smoothened mutant reveals conserved and divergent functions of hedgehog activity. Development 128: 2385–2396. [DOI] [PubMed] [Google Scholar]
- Chen M-H, Li Y-J, Kawakami T, Xu S-M, Chuang P-T. 2004. Palmitoylation is required for the production of a soluble multimeric Hedgehog protein complex and long-range signaling in vertebrates. Genes Dev 18: 641–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cognigni P, Bailey AP, Miguel-Aliaga I. 2011. Enteric neurons and systemic signals couple nutritional and reproductive status with intestinal homeostasis. Cell Metab 13: 92–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombani J, Raisin S, Pantalacci S, Radimerski T, Montagne J, Léopold P. 2003. A nutrient sensor mechanism controls Drosophila growth. Cell 114: 739–749. [DOI] [PubMed] [Google Scholar]
- Colombani J, Bianchini L, Layalle S, Pondeville E, Dauphin-Villemant C, Antoniewski C, Carré C, Noselli S, Léopold P. 2005. Antagonistic actions of ecdysone and insulins determine final size in Drosophila. Science 310: 667–670. [DOI] [PubMed] [Google Scholar]
- Colombani J, Andersen DS, Léopold P. 2012. Secreted peptide Dilp8 coordinates Drosophila tissue growth with developmental timing. Science 336: 582–585. [DOI] [PubMed] [Google Scholar]
- Creanga A, Glenn TD, Mann RK, Saunders AM, Talbot WS, Beachy PA. 2012. Scube/You activity mediates release of dually lipid-modified Hedgehog signal in soluble form. Genes Dev 26: 1312–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings DE, Overduin J. 2007. Gastrointestinal regulation of food intake. J Clin Invest 117: 13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCamp DL, Thompson TM, de Sauvage FJ, Lerner MR. 2000. Smoothened activates Gαi-mediated signaling in frog melanophores. J Biol Chem 275: 26322–26327. [DOI] [PubMed] [Google Scholar]
- Delanoue R, Slaidina M, Léopold P. 2010. The steroid hormone ecdysone controls systemic growth by repressing dMyc function in Drosophila fat cells. Dev Cell 18: 1012–1021. [DOI] [PubMed] [Google Scholar]
- Eugster C, Panáková D, Mahmoud A, Eaton S. 2007. Lipoprotein–heparan sulfate interactions in the Hh pathway. Dev Cell 13: 57–71. [DOI] [PubMed] [Google Scholar]
- Garelli A, Gontijo AM, Miguela V, Caparros E, Dominguez M. 2012. Imaginal discs secrete insulin-like peptide 8 to mediate plasticity of growth and maturation. Science 336: 579–582. [DOI] [PubMed] [Google Scholar]
- Geminard C, Rulifson EJ, Léopold P. 2009. Remote control of insulin secretion by fat cells in Drosophila. Cell Metab 10: 199–207. [DOI] [PubMed] [Google Scholar]
- Gervais L, Casanova J. 2011. The Drosophila homologue of SRF acts as a boosting mechanism to sustain FGF-induced terminal branching in the tracheal system. Development 138: 1269–1274. [DOI] [PubMed] [Google Scholar]
- Ghosh AC, O’Connor MB. 2014. Systemic Activin signaling independently regulates sugar homeostasis, cellular metabolism, and pH balance in Drosophila melanogaster. Proc Natl Acad Sci 111: 5729–5734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbens YY, Warren JT, Gilbert LI, O’Connor MB. 2011. Neuroendocrine regulation of Drosophila metamorphosis requires TGFβ/Activin signaling. Development 138: 2693–2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog R, Schwudke D, Schuhmann K, Sampaio JL, Bornstein SR, Schroeder M, Shevchenko A. 2011. A novel informatics concept for high-throughput shotgun lipidomics based on the molecular fragmentation query language. Genome Biol 12: R8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hietakangas V, Cohen SM. 2009. Regulation of tissue growth through nutrient sensing. Annu Rev Genet 43: 389–410. [DOI] [PubMed] [Google Scholar]
- Hikiba J, Ogihara MH, Iga M, Saito K, Fujimoto Y, Suzuki M, Kataoka H. 2013. Simultaneous quantification of individual intermediate steroids in silkworm ecdysone biosynthesis by liquid chromatography-tandem mass spectrometry with multiple reaction monitoring. J Chromatogr B Analyt Technol Biomed Life Sci 915–916: 52–56. [DOI] [PubMed] [Google Scholar]
- Jiang H, Edgar BA. 2009. EGFR signaling regulates the proliferation of Drosophila adult midgut progenitors. Development 136: 483–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Patel PH, Kohlmaier A, Grenley MO. 2009. Cytokine/Jak/Stat signaling mediates regeneration and homeostasis in the Drosophila midgut. Cell 137: 1343–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaliullina H, Panáková D, Eugster C, Riedel F, Carvalho M, Eaton S. 2009. Patched regulates Smoothened trafficking using lipoprotein-derived lipids. Development 136: 4111–4121. [DOI] [PubMed] [Google Scholar]
- King-Jones K, Horner MA, Lam G, Thummel CS. 2006. The DHR96 nuclear receptor regulates xenobiotic responses in Drosophila. Cell Metab 4: 37–48. [DOI] [PubMed] [Google Scholar]
- Lemaitre B, Miguel-Aliaga I. 2013. The digestive tract of Drosophila melanogaster. Annu Rev Genet 47: 377–404. [DOI] [PubMed] [Google Scholar]
- Linneweber GA, Jacobson J, Busch KE, Hudry B, Christov CP, Dormann D, Yuan M, Otani T, Knust E, de Bono M, et al. 2014. Neuronal control of metabolism through nutrient-dependent modulation of tracheal branching. Cell 156: 69–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matus DQ, Magie CR, Pang K, Martindale MQ, Thomsen GH. 2008. The Hedgehog gene family of the cnidarian, Nematostella vectensis, and implications for understanding metazoan Hedgehog pathway evolution. Dev Biol 313: 501–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBrayer Z, Ono H, Shimell M, Parvy J-P, Beckstead RB, Warren JT, Thummel CS, Dauphin-Villemant C, Gilbert LI, O’Connor MB. 2007. Prothoracicotropic hormone regulates developmental timing and body size in Drosophila. Dev Cell 13: 857–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Méthot N, Basler K. 1999. Hedgehog controls limb development by regulating the activities of distinct transcriptional activator and repressor forms of Cubitus interruptus. Cell 96: 819–831. [DOI] [PubMed] [Google Scholar]
- Miguel-Aliaga I. 2012. Nerveless and gutsy: intestinal nutrient sensing from invertebrates to humans. Semin Cell Dev Biol 23: 614–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirth CK, Riddiford LM. 2007. Size assessment and growth control: how adult size is determined in insects. BioEssays 29: 344–355. [DOI] [PubMed] [Google Scholar]
- Mirth C, Truman JW, Riddiford LM. 2005. The role of the prothoracic gland in determining critical weight for metamorphosis in Drosophila melanogaster. Curr Biol 15: 1796–1807. [DOI] [PubMed] [Google Scholar]
- Ogden SK, Fei DL, Schilling NS, Ahmed YF, Hwa J, Robbins DJ. 2008. G protein Gαi functions immediately downstream of Smoothened in Hedgehog signalling. Nature 456: 967–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlig S, Pickhinke U, Sirko S, Bandari S, Hoffmann D, Dreier R, Farshi P, Gotz M, Grobe K. 2012. An emerging role of Sonic Hedgehog shedding as a modulator of heparan sulfate interactions. J Biol Chem 287: 43708–43719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono H, Rewitz KF, Shinoda T, Itoyama K, Petryk A, Rybczynski R, Jarcho M, Warren JT, Marqués G, Shimell MJ, et al. 2006. Spook and Spookier code for stage-specific components of the ecdysone biosynthetic pathway in Diptera. Dev Biol 298: 555–570. [DOI] [PubMed] [Google Scholar]
- Ou Q, Magico A, King-Jones K. 2011. Nuclear receptor DHR4 controls the timing of steroid hormone pulses during Drosophila development. PLoS Biol 9: e1001160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm W, Swierczynska MM, Kumari V, Ehrhart-Bornstein M, Bornstein SR, Eaton S. 2013. Secretion and signaling activities of lipoprotein-associated hedgehog and non-sterol-modified hedgehog in flies and mammals. PLoS Biol 11: e1001505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panáková D, Sprong H, Marois E, Thiele C, Eaton S. 2005. Lipoprotein particles are required for Hedgehog and Wingless signalling. Nature 435: 58–65. [DOI] [PubMed] [Google Scholar]
- Pospisilik JA, Schramek D, Schnidar H, Cronin SJF, Nehme NT, Zhang X, Knauf C, Cani PD, Aumayr K, Todoric J, et al. 2010. Drosophila genome-wide obesity screen reveals hedgehog as a determinant of brown versus white adipose cell fate. Cell 140: 148–160. [DOI] [PubMed] [Google Scholar]
- Rajan A, Perrimon N. 2012. Drosophila cytokine unpaired 2 regulates physiological homeostasis by remotely controlling insulin secretion. Cell 151: 123–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rewitz KF, Yamanaka N, Gilbert LI, O’Connor MB. 2009. The insect neuropeptide PTTH activates receptor tyrosine kinase torso to initiate metamorphosis. Science 326: 1403–1405. [DOI] [PubMed] [Google Scholar]
- Sandoval D, Cota D, Seeley RJ. 2008. The integrative role of CNS fuel-sensing mechanisms in energy balance and glucose regulation. Annu Rev Physiol 70: 513–535. [DOI] [PubMed] [Google Scholar]
- Shingleton AW, Mirth CK. 2012. Integrating body and organ size in Drosophila: recent advances and outstanding problems. Front Endocrinol (Lausanne) 3: 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieber MH, Thummel CS. 2009. The DHR96 nuclear receptor controls triacylglycerol homeostasis in Drosophila. Cell Metab 10: 481–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strader AD, Woods SC. 2005. Gastrointestinal hormones and food intake. Gastroenterology 128: 175–191. [DOI] [PubMed] [Google Scholar]
- Strigini M, Cohen SM. 1997. A Hedgehog activity gradient contributes to AP axial patterning of the Drosophila wing. Development 124: 4697–4705. [DOI] [PubMed] [Google Scholar]
- Suh JM, Gao X, McKay J, McKay R, Salo Z, Graff JM. 2006. Hedgehog signaling plays a conserved role in inhibiting fat formation. Cell Metab 3: 25–34. [DOI] [PubMed] [Google Scholar]
- Tabata T, Kornberg TB. 1994. Hedgehog is a signaling protein with a key role in patterning Drosophila imaginal discs. Cell 76: 89–102. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Okada Y, Hirokawa N. 2005. FGF-induced vesicular release of Sonic hedgehog and retinoic acid in leftward nodal flow is critical for left-right determination. Nature 435: 172–177. [DOI] [PubMed] [Google Scholar]
- Telfer WH, Kunkel JG. 1991. The function and evolution of insect storage hexamers. Annu Rev Entomol 36: 205–228. [DOI] [PubMed] [Google Scholar]
- Teperino R, Amann S, Bayer M, McGee SL, Loipetzberger A, Connor T, Jaeger C, Kammerer B, Winter L, Wiche G, et al. 2012. Hedgehog partial agonism drives Warburg-like metabolism in muscle and brown fat. Cell 151: 414–426. [DOI] [PubMed] [Google Scholar]
- Thummel CS. 1996. Flies on steroids—Drosophila metamorphosis and the mechanisms of steroid hormone action. Trends Genet 12: 306–310. [DOI] [PubMed] [Google Scholar]
- van den Brink GR. 2007. Hedgehog signaling in development and homeostasis of the gastrointestinal tract. Physiol Rev 87: 1343–1375. [DOI] [PubMed] [Google Scholar]
- van den Heuvel M, Ingham PW. 1996. smoothened encodes a receptor-like serpentine protein required for hedgehog signalling. Nature 382: 547–551. [DOI] [PubMed] [Google Scholar]
- Voght SP, Fluegel ML, Andrews LA, Pallanck LJ. 2007. Drosophila NPC1b promotes an early step in sterol absorption from the midgut epithelium. Cell Metab 5: 195–205. [DOI] [PubMed] [Google Scholar]
- Walker BR. 2007. Glucocorticoids and cardiovascular disease. Eur J Endocrinol 157: 545–559. [DOI] [PubMed] [Google Scholar]
- Warren JT, Petryk A, Marqués G, Parvy J-P. 2004. Phantom encodes the 25-hydroxylase of Drosophila melanogaster and Bombyx mori: a P450 enzyme critical in ecdysone biosynthesis. Insect Biochem Mol Biol 34: 991–1010. [DOI] [PubMed] [Google Scholar]
- Warren JT, Yerushalmi Y, Shimell MJ, O’Connor MB, Restifo LL, Gilbert LI. 2006. Discrete pulses of molting hormone, 20-hydroxyecdysone, during late larval development of Drosophila melanogaster: correlations with changes in gene activity. Dev Dyn 235: 315–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng X, Goetz JA, Suber LM, Scott WJ, Schreiner CM, Robbins DJ. 2001. A freely diffusible form of Sonic hedgehog mediates long-range signalling. Nature 411: 716–720. [DOI] [PubMed] [Google Scholar]
- Zhang XM, Ramalho-Santos M, McMahon AP. 2001. Smoothened mutants reveal redundant roles for Shh and Ihh signaling including regulation of L/R asymmetry by the mouse node. Cell 105: 781–792. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.