

Abstract
Recent studies have shown a surprising phenomenon, whereby orthologous regulatory regions from different species drive similar expression levels despite being highly diverged in sequence. Here, we investigated this phenomenon by genomically integrating hundreds of ribosomal protein (RP) promoters from nine different yeast species into S. cerevisiae and accurately measuring their activity. We found that orthologous RP promoters have extreme expression conservation even across evolutionarily distinct yeast species. Notably, our measurements reveal two distinct mechanisms that underlie this conservation and which act in different regions of the promoter. In the core promoter region, we found compensatory changes, whereby effects of sequence variations in one part of the core promoter were reversed by variations in another part. In contrast, we observed robustness in Rap1 transcription factor binding sites, whereby significant sequence variations had little effect on promoter activity. Finally, cases in which orthologous promoter activities were not conserved could largely be explained by the sequence variation within the core promoter. Together, our results provide novel insights into the mechanisms by which expression is conserved throughout evolution across diverged promoter sequences.
Gene expression variation is greatly affected by the evolution and divergence of cis-regulatory sequences, both within the same species and across different species (Wittkopp et al. 2004, 2008; Dixon et al. 2007; Landry et al. 2007; Veyrieras et al. 2008; Tirosh et al. 2009). In particular, sequence variation within known regulatory elements, such as transcription factor (TF) binding sites, TATA boxes, and sequences that affect nucleosome positioning, has been suggested as an important determinant of expression variation (Landry et al. 2007; Tirosh et al. 2008; Choi and Kim 2009). In spite of the above, an unexpected phenomenon was recently observed, whereby some highly diverged orthologous regulatory sequences were shown to drive similar gene expression levels and localization patterns when transferred from one species to another (for review, see Weirauch and Hughes 2010). In one example, the RET gene is conserved between human and zebrafish, but the orthologous regulatory regions that control its expression are strikingly different. In spite of this, when the regulatory regions of the human RET gene were inserted into zebrafish cells, they were functional, and drove the expression of the fish RET gene similarly to the native fish regulatory elements (Fisher et al. 2006). In another example, transferring highly diverged orthologous even skipped stripe 2 enhancers from several different fly species into D. melanogaster resulted in patterns of expression similar to the native D. melanogaster enhancer (Ludwig et al. 1998). This phenomenon was also demonstrated on a chromosomal scale, with mouse hepatocyte cells that carry human chromosome 21 recapitulating gene expression levels observed in human hepatocytes (Wilson et al. 2008). However, the mechanisms by which function is conserved across highly diverged sequences are still not well understood. Previous studies mainly focused on enhancers; therefore, binding site conservation and turnover were predominantly suggested to be the mechanisms involved (Hare et al. 2008; Weirauch and Hughes 2010; Martinez et al. 2014).
Here, we set out to study the mechanisms that underlie this phenomenon in yeast by expressing in S. cerevisiae 668 ribosomal protein (RP) gene promoters taken from nine yeast species and by further examining several pairs of orthologous promoters through a library of 91 chimeric promoters. Our results shed new light on how yeast promoters can evolve in sequence while preserving their expression.
Results
Conservation of promoter activity despite high promoter sequence divergence
In order to test the extent to which a promoter sequence can change without affecting its activity, we constructed a library of 668 native RP promoters from nine different yeast species, including S. cerevisiae, and further ranging from S. paradoxus (ancestor common to it and to S. cerevisiae existed 0.4–3.4 million years ago) (Liti et al. 2006) to S. pombe (common ancestor 430–1000 million years ago) (Hedges 2002; Galagan et al. 2005). We included most of the RP promoters from the Saccharomyces sensu stricto species as well as from S. kluyveri and K. lactis, whereas from the more distant D. hansenii, Y. lipolytica, and S. pombe, we took 14–15 representatives each (see Methods for how the promoters were identified). Our choice to focus on the RP genes is due to their tight coregulation, constitutive high expression, and, within the Saccharomyces sensu stricto genus, their regulation by the same TFs (Tanay et al. 2005; Hogues et al. 2008; Wapinski et al. 2010). All promoters were then inserted into a fixed genomic location within S. cerevisiae, immediately upstream of a YFP reporter (Figs. 1, 2A; Supplemental Fig. 1), using a method described previously (Zeevi et al. 2011). The YFP expression levels driven by each promoter were then measured in vivo with high accuracy, allowing us to distinguish between activities of two promoters that differ by as little as 10% (see Supplemental Table 1).
Figure 1.
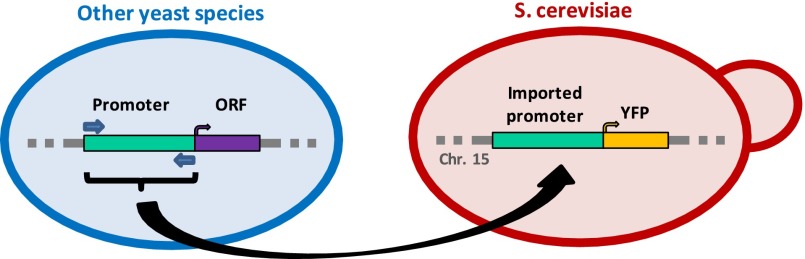
Our experimental system for measuring promoter activity conservation across species. Native RP promoters from nine different yeast species (including S. cerevisiae) were extracted and inserted into a fixed genomic location on chromosome 15 of S. cerevisiae, immediately upstream of a YFP reporter gene. YFP levels were measured in vivo, enabling promoter activity differences of as little as 10% to be significantly detected (see Zeevi et al. 2011).
Figure 2.

Promoter sequence and activity divergence for orthologous yeast RP promoters. (A) The mean orthologous sequence identity to S. cerevisiae along RP promoters of different yeast species, computed based on pairwise alignments (smoothed using a 21-bp sliding window). The color of each track corresponds to the color of the yeast species name appearing in the left phylogenetic tree. The black track is the result of aligning S. cerevisiae promoters to their own randomly shuffled sequence. The big dip in the black track toward its downstream edge is due to the extremely high A content of the S. cerevisiae RP promoters just upstream of the ORF (see Supplemental Fig. 2). (B) Measured native RP promoter activities for each species. Median values for each species appear in red. (C) Each dot plot compares the measured activities of RP promoters from one species versus their S. cerevisiae orthologs.
During evolution, the promoters of the 78 RP genes of these nine species have greatly diverged (Fig. 2A; Supplemental Fig. 1). D. hansenii, Y. lipolytica, and S. pombe, the three most distant species from S. cerevisiae, show no global or local alignment similarity to their orthologous S. cerevisiae promoters beyond the first ∼15 bps upstream of the translation start site (corresponding to the 5′UTR of these genes in S. cerevisiae). These promoters in these species are also controlled by different transcription factors (TFs) compared to S. cerevisiae (see Supplemental Figs. 3, 4; Tanay et al. 2005; Hogues et al. 2008). As expected from the above described complete divergence of both trans and cis regulation, for the two most distant species (Y. lipolytica and S. pombe), we measured no activity from their 15 RP promoters that we inserted into S. cerevisiae (Fig. 2B). Surprisingly, we observed some promoter activity for the D. hansenii species, since two of its 14 tested RP promoters showed non-negligible activity when inserted into S. cerevisiae (Fig. 2B), although D. hansenii diverged from S. cerevisiae somewhere between 150 and 850 million years ago (Heckman et al. 2001; Hedges 2002; Galagan et al. 2005).
The next two species, S. kluyveri and K. lactis, diverged much later, after the divergence of D. hansenii and prior to the whole genome duplication event that occurred ∼100 million years ago in the ancestor of the Saccharomyces sensu stricto genus (Kellis et al. 2004). As for the previous three species, the S. kluyveri and K. lactis promoters cannot be aligned to their S. cerevisiae orthologs (Fig. 2A). Remarkably, however, all 76 K. lactis and 79 S. kluyveri RP promoters that we tested were highly active when inserted into S. cerevisiae (Fig. 2B), with promoter activity levels significantly correlated to those of their S. cerevisiae orthologs (r = 0.41 in both species) (Fig. 2C). This discrepancy may be explained in part by the fact that we do identify TF binding sites of the S. cerevisiae RP regulators (e.g., Rap1, the main regulator of RP transcription) (Lieb et al. 2001) in some S. kluyveri and K. lactis promoters, although with somewhat different architectures than in S. cerevisiae. For example, the Rap1 binding sites in K. lactis are on average 100–200 bps upstream of their average locations in S. cerevisiae (Supplemental Fig. 4). This is in line with a previous work showing that RP promoters in these species are mostly regulated by the same TFs as in S. cerevisiae (Tanay et al. 2005) and also with our observation that there is yet a considerable conservation between the protein sequences of these TFs, especially in their DNA binding domains (Supplemental Fig. 3). For example, the Rap1 DNA binding domain (residues 358–601) (Henry et al. 1990; Matot et al. 2012) in S. kluyveri is 69% identical to that of S. cerevisiae.
The most striking case of promoter activity conservation despite sequence divergence was in the Saccharomyces sensu stricto species that we tested, namely S. cerevisiae, S. paradoxus, S. mikatae, and S. bayanus. In each of these species, we identified 137 RP genes that encode for the 78 ribosomal proteins, with the exact same composition of the 59 duplicate pairs and 19 single copy genes known from S. cerevisiae (Zeevi et al. 2011). This indicates that all changes in copy number occurred prior to the divergence of S. bayanus from this group, ∼10 million years ago (calculated using Liti et al. 2006; Scannell et al. 2011). Regarding their trans regulation, not only do they share the same regulators, but a comparison of the Rap1 DNA binding domain of S. cerevisiae to the other three species shows an almost perfect protein conservation of >98% identity (Supplemental Fig. 3). Similarly, for the RP regulator Fhl1 (Zeevi et al. 2011), the DNA binding domain (residues 451–555 in S. cerevisiae) (Hermann-Le Denmat et al. 1994) shows 99% identity to S. paradoxus and S. mikatae and 94% to S. bayanus. Therefore, we expect the promoters imported from these three species to be regulated by practically identical regulatory proteins when in S. cerevisiae as compared to their native environment.
In contrast to this high sequence identity of the regulatory proteins, the cis-regulatory sequence of these species underwent substantial divergence. When the RP promoters from S. paradoxus, S. mikatae, and S. bayanus are globally aligned to their orthologous promoters from S. cerevisiae, they show an identity of 77%, 67%, and 63%, respectively, gradually declining in accordance with their evolutionary distances (Supplemental Fig. 1). Strikingly, despite this gradual decline in conservation, the 120 orthologous promoters that we tested from each of these species showed no such decrease in their measured activities when inserted into S. cerevisiae, with median promoter activities of 1.48, 1.46, 1.46, and 1.46 for the promoters of S. cerevisiae, S. paradoxus, S. mikatae, and S. bayanus, respectively (Fig. 2B). More importantly, we found the promoter activities to be highly conserved at the single promoter level. S. paradoxus, S. mikatae, and S. bayanus all show the same high correlation of promoter activity to S. cerevisiae (r = 0.9) (Fig. 2C). Moreover, for most genes the absolute levels of promoter activity were highly similar between species (with a median coefficient of variation value of 0.08), compared to the large spectrum of promoter activity levels spanned for all RP genes within each of the four species (with coefficients of variation of 0.36, 0.33, 0.35, and 0.38).
Together, these results demonstrate extreme conservation of RP promoter activity levels within the sensu stricto species, despite a high degree of sequence divergence and up to ∼10 million years of separation from S. cerevisiae.
Sequence variation in transcription factor binding sites is not correlated with variation in promoter activity
How, then, can the highly diverged sensu stricto RP promoters drive highly similar promoter activity levels? One possible explanation is that most of the promoter activity is determined by short transcription factor binding sites; and thus, only variation in binding sites should be correlated with variation in promoter activity. To test this, we identified strong binding sites of the known RP regulators, Rap1, Fhl1, Sfp1, and the TATA box (Zeevi et al. 2011), using TF binding affinity models derived from in vitro experiments carried out on S. cerevisiae, where conservation information was not used (Badis et al. 2008; Zhu et al. 2009). First, as expected, we observed that these predicted TF binding sites are significantly more conserved between the four species than their immediate flanking regions (Fig. 3A). In line with a previous study of yeast TF binding sites (Moses et al. 2003), the mean conservation per position and the information content per position were correlated (Supplemental Fig. 6). These results suggest that many of the predicted TF binding sites that we identified are functional since they are under evolutionary constraint. Notably, however, for all four TFs, variation in binding site sequence was not significantly correlated to variation in promoter activity (r < 0.3; P > 0.07) (Fig. 3B). Although in the case of Fhl1 and the TATA box the observed lack of correlation might be due to the low number of involved genes, for Rap1 and Sfp1, there were enough genes involved to allow high statistical power (the power to detect a 0.4 correlation is 0.99 and 0.98, respectively).
Figure 3.

The magnitude and effect of sensu stricto TF binding sites variation. (A) Mean sensu stricto sequence conservation of positions within annotated TF binding sites (in blue) (see Supplemental Note) and of their flanking positions (in red, double the length of the binding site). For each TF, each position of its motif logo appears below the mean conservation of that position. For all TFs, conservation of TF binding site positions was significantly higher than that of flanking positions (rank-sum P-values written in blue). (B) Sensu stricto TF binding site variation (see Supplemental Note) versus promoter activity variation (the coefficient of variation of the four orthologous promoter activities).
One may speculate that the variation in orthologous binding sites was such that it did not significantly alter the strength of the binding sites, according to the known binding affinity models. However, a comparison of predicted Rap1 binding sites between orthologous RP promoters of the sensu stricto species revealed that this is not the case, such that for >95% of Rap1-regulated RPs, there are orthologous promoters where sequence variation led to variation in predicted strength of orthologous Rap1 binding sites. In particular, many Rap1 binding sites are strong only in a subset of the four orthologs, whereas in the other orthologous species they are significantly weaker or even deleted. The RP promoters of S. cerevisiae are known to predominately contain Rap1 binding sites in a tandem formation of two adjacent sites (Lascaris et al. 1999; Zeevi et al. 2011), and it was previously shown that a deletion of even one of these sites can significantly reduce expression (Mencía et al. 2002). Of the 120 orthologous RP promoter groups that we tested, 86 contained tandem Rap1 binding sites, and in 12 of them, one of the sites was lost in at least one of the four sensu stricto species (Supplemental Fig. 7A). Notably, in all 12 of these cases, we did not observe any change in promoter activity above our experimental noise in the species that lost the site.
Variation of core promoter sequence is highly correlated to variation in promoter activity
Since sequence variation of the entire promoter as well as in TF binding sites does not correlate with promoter activity variation, we examined other regions of the promoter to see whether sequence variation in these regions is correlated with activity variation.
Based on the pairwise alignment of our library’s 120 orthologous RP promoters from S. cerevisiae and each of the sensu stricto species, we identified that the [-200,-1] region (relative to the translation start site) is on average more conserved than the rest of the promoter (Fig. 2A). In yeast, unlike in metazoans, following the recruitment of the RNA polymerase II (pol II) preinitiation complex (PIC) and the unwinding of the DNA, the pol II additionally performs a downstream scan until it selects one of multiple alternative transcription start sites, resulting in typical core promoter lengths of 100–200 bps (Smale and Kadonaga 2003; Lubliner et al. 2013). Thus, the [-200,-1] region mainly consists of the core promoter, in particular in RP promoters (Rhee and Pugh 2012). Within this region, the highest conservation is at the [-15,-1] region (contained within the 5′UTRs of the S. cerevisiae RP genes) (Nagalakshmi et al. 2008) and another peak of conservation around the -115 position, corresponding to the expected location of PIC recruitment (Rhee and Pugh 2012). We then found that not only are these regions more conserved, but that in cases in which they are less conserved, orthologous promoters show higher variability in their measured promoter activity. We ordered the sensu stricto 120 orthologous gene groups (with four genes in each group) according to their within-group variation of measured activity and calculated the sequence conservation along the promoters of each group.
As shown in Figure 4A and Supplemental Figure 8, an orthologous promoter group with higher sequence variation within the [-200,-1] region tends to have higher variation in promoter activity. Quantitatively, using a 50-bp long sliding window (with a 25-bp step), we computed the correlation between the sequence variation and the expression variation for windows within the [-400,-1] region (Fig. 4B). Only windows falling within the [-200,-1] region showed a significant correlation (controlled for allowing a false discovery rate of 5%), with two peaks of correlation around positions -100 and -25 (r = 0.38 and r = 0.53, respectively), corresponding to the peaks of sequence conservation that we previously observed (Fig. 2A). Further zooming in on the [-50,-1] region (Fig. 4B), we found that the sequence variation of all 10-bp windows within this region is significantly correlated to the orthologous promoter activity variation, with correlation as high as 0.66 for the [-15,-6] window.
Figure 4.

The magnitude and effect of sensu stricto core promoter variation. (A) For each RP gene, its sensu stricto conservation track (see Supplemental Note) along the 200 bps upstream of the translation start appears in the left heatmap. Correspondingly, its sensu stricto promoter activity variation (the coefficient of variation of the four orthologous promoter activities) appears in the right bar plot. Genes were sorted by promoter activity variation. (B) For different promoter windows, we computed the Pearson correlation between the mean window sequence variation (1-conservation) and the promoter activity variation. For each window, the resulting correlation value (y-axis) is plotted above the window center position (x-axis). Significant correlations (controlled for having a false discovery rate of 5%) are marked by red circles. On the left are correlations for 50-bp windows (with a 25-bp step) within the 400 bps upstream of the translation start site. On the right we zoom in on the last 50 bps upstream of the translation start site and show correlations for 10-bp windows (with a 5-bp step).
To further study the effects of different parts of the promoter on activity at a much higher resolution, we fit linear models that predict promoter activity from promoter sequence features (see Methods). To this end, we computed a large set of sequence features falling within different windows along the full length of the sensu stricto promoters. These features included base content, k-mer presence and counts, features based on hits of position-specific scoring matrices (PSSMs) of known RP regulators (Rap1, Fhl1, Sfp1) (Pachkov et al. 2013) and the TATA box (Basehoar et al. 2004), and features of the predicted intrinsic nucleosome occupancy, computed using a published model (Kaplan et al. 2009). Notably, for all promoter windows that do not overlap with the [-200,-1] region, the models explain < 7% of the variance in promoter activity, whereas for windows that contain the [-200,-1] region, the models explain 65% of this variance (Supplemental Fig. 10). This strongly suggests that the promoter activity of the sensu stricto RP promoters is greatly determined by the ∼200 bps immediately upstream of the translation start site and is in line with our findings that this region is more conserved, and that sequence variation within it correlates with variation in activity of orthologous promoters. It is also in line with our recent study which highlights the important role of the core promoter sequence in determining maximal promoter activity (Lubliner et al. 2013). Finally, in Supplemental Figure 11, we show the results over the entire [-600,-1] promoter window, detailing the features that played an important role in predicting promoter activity.
Core promoter compensatory evolution and TF binding site robustness act to conserve promoter activity
In search for explanations to the high conservation of activity in the presence of promoter sequence divergence, we designed a library of synthetic promoters. Although not necessarily representative of all RP promoters, for a more in-depth exploration, we selected five pairs of orthologous RP promoters with conserved promoter activity yet with sequence divergence falling within known functional elements (Fig. 5), including lost or significantly weakened Rap1 binding sites, changes in lengths of poly(dA)/poly(dT) tracts, and core promoter polymorphisms (e.g., single nucleotide polymorphisms [SNPs]; see Supplemental Note for how elements were defined). We then designed the synthetic library such that it included chimeric promoters, where orthologous sequence elements or regions were reciprocally replaced between pairs of orthologous promoters. The sizes of the replaced sequences varied from single nucleotides and up to half of the promoter. We will henceforth refer to mutating a sequence by using the orthologous counterpart by the term orthologous mutation. All library sequences and measurements appear in Supplemental Table 2. Illustrations of the chimeric promoters and a comparison of their activities appear in Supplemental Figures 12–16. Importantly, the effects of the orthologous mutations on promoter activity were not due to the degree of introduced sequence variation (compared to the wild type), as shown in Supplemental Figure 17.
Figure 5.

Native pairs of orthologous RP promoters from which we constructed a library of orthologous mutations. Shown are five promoter pairs that we used to construct a library of mutated RP promoters, in which each mutation was generated by replacing an element or region in one native promoter with its orthologous counterpart in the other (see Supplemental Figs. 12–16).
First, we targeted the core promoter regions. We performed 22 orthologous mutations within core promoters (Supplemental Figs. 12i, 13r, 14k–l, 15c–f, 16e–h,k–t), and 55% (12/22) resulted in significant promoter activity changes (P < 10−4) (Fig. 6). This shows that sequence divergence in core promoters is not only correlated to promoter activity variation (shown above) but is in fact causal and actually affects promoter activity.
Figure 6.

The effect of various types of orthologous mutations on promoter activity. Most orthologous mutations of RP promoters fall into four categories: poly(dA)/poly(dT) tracts adjacent to Rap1 binding sites; chimeras of Rap1-regulated RP promoters, where entire promoter prefixes upstream of the core promoter region were replaced between orthologs; Rap1 binding sites (and possibly their adjacent context); and core promoter mutations. For each mutation, its absolute percent difference in promoter activity from the wild type is shown by a blue circle. Red circles show median values. A chimera was regarded to have a significant promoter activity difference only if its promoter activity was significantly different from those of both native RP promoters. Below each category name appears the total number of mutations (black font) and the number of those with promoter activity significantly different from the wild type (purple font). For each category, a significant mutation enrichment P-value (one-sided hyper-geometric test) is in gray.
Although specific core promoter orthologous mutations considerably changed promoter activity, the native promoter pairs had almost identical activities, suggesting that the effect of these specific sequence variations between the native orthologous promoters must be compensated for by sequence variations elsewhere within them. Indeed, we found support for this hypothesis. For example, for the pair of S. cerevisiae’s and S. bayanus’s orthologous RPL5 promoters, the first 20 bps upstream of the translation start site are identical, but in the next 10 bps, there are five SNPs between the two species. When we replaced these 5 nucleotides in the promoter of S. cerevisiae with the ones from S. bayanus, we measured an increase of 26% in promoter activity (Supplemental Fig. 15e). The reciprocal change resulted in a significant decrease of 9% in activity (Supplemental Fig. 15f). Replacing a longer sequence of 103 bps upstream of the translation start site, which includes additional 24 SNPs, gave similar results: An increase of 23% and a reciprocal decrease of 8% (Supplemental Fig. 15n,m, respectively). However, when we replaced an even longer sequence of 128 bps that also includes a poly(dA) sequence, which is different in length between the two species, the difference in activity reversed and the mutated promoters showed activity similar to the native ones (an increase of 1% and a decrease of 6%, both within the 95% confidence intervals of the native promoter activity values) (see Supplemental Fig. 15l,k, respectively).
We demonstrate this phenomenon further using orthologous promoters of RPL4A, a very short (∼200 bps) but very potent promoter. We created 23 constructs that exchange short sequence blocks between the RPL4A promoters of S. paradoxus and S. mikatae (Fig. 7; Supplemental Fig. 16; Supplemental Note). Several of these orthologous mutations showed a significant reciprocal effect on activity (e.g., +32% versus −37% when we replaced a poly(dA) which has different lengths in the two species and −22% versus +18% when we replaced a block of 30 bps directly downstream from the TATA box) (see Fig. 7). Since the promoter activities of the two native RPL4A promoters are essentially identical (1% measured difference), these results demonstrate compensation between sequence variations in different parts of the core promoter.
Figure 7.
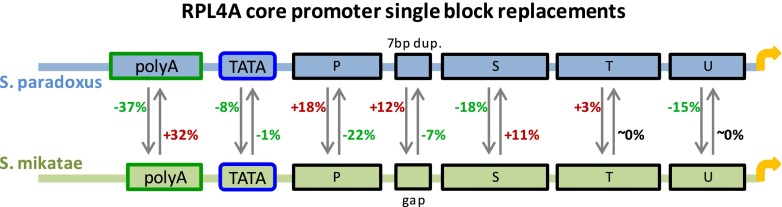
Results of single core promoter block replacements between RPL4A orthologs. Shown are the results of replacing 14 orthologous core promoter sequence blocks between the RPL4A promoters of S. paradoxus and S. mikatae. Percentage next to an arrow shows the decrease (green) or increase (red) in promoter activity relative to the wild type. For instance, inserting the S. paradoxus poly(dA) into the S. mikatae promoter, instead of the S. mikatae native poly(dA), results in a 37% drop compared to the S. mikatae wild type promoter activity.
Overall, these results demonstrate a previously undocumented mode of compensatory evolution, whereby mutations in one part of the core promoter reverse the effects of mutations in another part in order to preserve gene expression level.
Next, we targeted Rap1 binding sites that are upstream of the core promoter region, seeking to explore the effects of sequence variation within these sites. Since the Rap1 protein is the main regulator of the RP genes, its binding sites were previously thoroughly studied in vitro to produce well-defined DNA binding preferences (Badis et al. 2008; Zhu et al. 2009). We examined four pairs of orthologous promoters (RPL37B, RPS27B, RPL43A, and RPL5) (see Fig. 5) with similar activity but with notable differences in their Rap1 sites. We conducted orthologous mutations in which we replaced the Rap1 sites between these orthologous promoter pairs. In some cases, this resulted in a deletion of a site due to the orthologous promoter lacking a site at the same position. The reciprocal mutations resulted in promoters with an additional site not occurring naturally. Other orthologous mutations resulted in strengthening or weakening of sites. Overall, we created 27 promoters with different possible combinations from zero to three Rap1 sites (Supplemental Figs. 12a–f, 13a–e,g–n, 14a–f, 15a–b). Past studies showed that deletion of Rap1 binding sites significantly reduces, and may even completely abolish, RP gene expression (Rotenberg and Woolford 1986; Woudt et al. 1986; Klein and Struhl 1994). Significant reduction was observed even when only one of two tandem Rap1 sites was deleted (Mencía et al. 2002). Therefore, we expected to see a large variation in promoter activity between the different promoters that we constructed.
To our surprise, in 78% (21/27) of the cases orthologous mutations resulted in little to no change compared to the wild type promoter activity levels (P = 0.69) (Fig. 6). There are two possible explanations for this observation. The first is that the sites we manipulated had no effect on promoter activity to begin with. However, several lines of evidence strongly support otherwise. First, in some cases we deleted what seems to be the only known strong site of Rap1 in the promoter (Pachkov et al. 2013), and Rap1 was shown to preferably bind the strongest site on the promoter (Rhee and Pugh 2011). Second, to verify that strong sites according to the Rap1 PSSM are in fact functional, we used the Rap1 PSSM to annotate tandem pairs of Rap1 sites in the promoters of several other RP genes (RPL23B, RPL27B, RPS1B, RPS10A, RPS23A); and in each promoter we introduced a couple of SNPs into each site that were predicted to delete it according to the PSSM. Indeed, this resulted in substantial reduction in promoter activity (4.1-, 6.7-, 4-, 1.6-, and 28.2-fold reduction, respectively) (see Supplemental Table 3). Third, in the case of the RPL37B orthologous promoters, two cases of Rap1 mutations substantially reduced expression but in a context-dependent manner (on the S. mikatae promoter) (see Supplemental Fig. 12b,f), demonstrating that the mutated sites were functional. Fourth, most orthologous Rap1 site gains/losses in our mutation library were of Rap1 sites that existed in three of the four examined sensu stricto species (see Supplemental Fig. 7). An alternative explanation is that the promoter is much more robust to mutations in Rap1 binding sites than was previously known, in particular to the orthologous mutations that we examined. Such mutations seem to delete the Rap1 site but may in fact be tolerated, not affecting promoter activity levels.
Finally, in order to study the effect of other differences between orthologous promoters, we created 21 chimeras from orthologous promoter pairs by replacing entire segments of DNA that are upstream of the core promoter region (Supplemental Figs. 12k–p, 13s–x, 14m,o–r, 15g–j). None of these 21 chimeras showed significant promoter activity changes from wild type levels (P = 1) (Fig. 6). We observed a similar phenomenon when we tested orthologous replacements of poly(dA)/poly(dT) tracts that are adjacent to Rap1 binding sites (Supplemental Figs. 12g–h, 13o–p, 14i–j). In the six orthologous replacements that we conducted, none showed a significant change in promoter activity (P = 1) (Fig. 6). Similarly to the Rap1 sites, these sequences and their lengths are known to influence promoter activity from previous studies (Zeevi et al. 2011; Raveh-Sadka et al. 2012; Sharon et al. 2012), but the specific changes that occurred in these sequences in orthologous promoters were such that they did not affect promoter activities.
Discussion
Here we studied the phenomenon whereby highly diverged yeast orthologous RP promoters drive highly similar levels of promoter activity. For that purpose, we built a system for introducing native promoters from different yeast species into the yeast S. cerevisiae and measured the activity of 668 native RP promoters from nine yeast species with high accuracy. We further designed and measured promoter activity for 91 synthetically mutated promoters in order to test our hypotheses.
We found extreme conservation of promoter activity between orthologous promoters, to the extent that some orthologous promoters from species that diverged hundreds of millions of years ago maintained significant promoter activity when integrated into S. cerevisiae, and orthologous promoters from the closer species of the sensu stricto genus, which diverged from S. cerevisiae up to 10 million years ago, showed almost identical activities.
Although our observations were based on measurements in Synthetic Complete medium conditions (SCD), past evidence suggests that they are not condition specific. In S. cerevisiae, we previously showed that RP promoter activities are highly correlated between different conditions (Zeevi et al. 2011), in line with the fact that they scale linearly across conditions (Keren et al. 2013). Other studies show that RP genes are tightly coexpressed across species, and that in particular, within the Saccharomyces sensu stricto genus they are tightly coregulated and coexpressed across different conditions (Tanay et al. 2005; Tirosh et al. 2006; Wapinski et al. 2010).
A simple explanation of the observed orthologous promoter activity conservation is that most of the determinants of promoter activity are concentrated in short conserved sequences such as TF binding sites, and that the rest of the promoter has little effect on its activity. However, our measurements show the opposite.
Although we found known TF binding sites to be more conserved than their surrounding sequences, variation in these sites was not correlated to variation in promoter activity. Moreover, when we introduced orthologous mutations in binding sites of Rap1 (the main regulator of RP promoters), that according to published DNA binding affinity models (Badis et al. 2008; Zhu et al. 2009; Pachkov et al. 2013) were supposed to completely delete sites or introduce new ones, in most cases they resulted in no significant change to promoter activity. Another possible explanation for our observations is Rap1 binding site turnover, whereby the gain of a new site relaxed the fitness constraint on the later deletion of an old site. Using orthologously mutated promoters, we tested this hypothesis in cases in which such turnover seemed to have occurred. To each native promoter we added an orthologous site and then mutated its original one with the orthologous sequence lacking the site, and also conducted the opposite trajectory where we first mutated the site, and then added the new one. In the three orthologous pairs that we tested this way (RPL37B, RPS27B, and RPL43A) (Supplemental Figs. 12–14), we saw no difference in activity between the different trajectories, and the promoter activity was maintained, arguing against the binding site turnover hypothesis, contrary to what may have been expected by past studies (Hare et al. 2008; Weirauch and Hughes 2010; Martinez et al. 2014).
Our results support an alternative explanation, whereby RP promoters exhibit significant robustness to Rap1 mutations, such as those that naturally arose in evolution during which the orthologous sequences diverged. This is supported by a recent study claiming TF binding site mutational robustness to be the rule rather than an exception (Payne and Wagner 2014). Such robustness may also be conferred by the existence of additional weak Rap1 binding sites that may slip under the radar of the known binding affinity models, as was suggested by Tanay (2006). These results might also explain our previously published observation that the number of Rap1 sites and their overall strength are not correlated to promoter activity among the promoters of the RP genes in S. cerevisiae (Zeevi et al. 2011).
In addition, our results support an evolutionary compensation mechanism in a different part of the promoter—the core promoter region, within ∼200 bps upstream of the translation start sites. First, we observed that core promoter sequence variation between orthologous sensu stricto promoters is indeed correlated to variation in promoter activities. Accordingly, when we performed orthologous mutations in the core promoter region, we found that most changed promoter activity significantly. Moreover, orthologous mutations in different core promoter parts showed opposite effects on activity, implying that variations in different parts of the core promoter compensated for each other to preserve identical promoter activities to the wild type. We speculate that the evolutionary dynamics underlying our observations are as follows: Since the yeast core promoter is the region where the transcriptional machinery docks, scans, and initiates transcription (Smale and Kadonaga 2003), it is a large and continuous region where many bases can increase or decrease promoter activity if mutated. Such changes in promoter activity are probably at most weakly deleterious, unlike coding regions where mutations can easily cause frame shifts, premature stop codons, and truncated nonfunctional proteins. Therefore, when a mutation that decreases promoter activity occurs in the core promoter, we speculate that a following mutation within the core promoter that counters the effect of the first one will be selected for. Since this region is quite large, a reverse mutation in the exact same position as the original mutation is much less likely than another mutation elsewhere in the core promoter that has a similar reversing effect on the level of activity. Such evolutionary dynamics were suggested by a previous study that identified that local G/C content in intergenic regions is maintained through compensatory evolution, in which SNPs that alter G/C content tend to be followed by a compensatory SNP within a few bases that brings back G/C content to the previous level (Kenigsberg et al. 2010).
Overall, our results emphasize the critical role of the core promoter region in determining the levels of promoter activity. We also show for the first time, to the best of our knowledge, experimental evidence supporting a compensatory evolution mechanism that takes place in this region to maintain the wild type activity level of the promoter.
Methods
Orthologous promoter identification
We defined a promoter in S. cerevisiae as starting immediately upstream of a gene’s translation start site and ending at the upstream gene or 1200 bps, whichever is shorter. In order to identify the orthologous promoters in the sensu stricto species, we used the BLAST+ algorithm (Camacho et al. 2009) to compare a block of sequence from S. cerevisiae that contains the target gene, the intergenic region upstream of it (the promoter), and the upstream gene, to the genome of S. paradoxus, S. mikatae, and S. bayanus (Kellis et al. 2003) (http://fungalgenomes.org). The orthologous block that was identified was then pairwise aligned to the RP and upstream genes from S. cerevisiae using the Needleman–Wunsch algorithm (Needleman and Wunsch 1970), and the intergenic region between the identified orthologous genes was defined as the promoter in the target species. For the species S. kluyveri, K. lactis, D. hansenii and Y. lipolytica, we used the orthologous RP and upstream genes annotated by Génolevures (Sherman et al. 2009); and for S. pombe we used PomBase (Wood et al. 2012).
Library construction
All promoter libraries were constructed using a method developed previously in our laboratory and described elsewhere (Zeevi et al. 2011). Briefly, in order to introduce orthologous promoters into S. cerevisiae, we extracted genomic DNA from each species and amplified by PCR the desired promoters. Each promoter was linked (Linshiz et al. 2008) to a URA3 selection marker and integrated into the genome of a yeast master strain, upstream of a YFP reporter gene. This master strain also contained a control promoter driving mCherry, which functions as an identical promoter in all strains to estimate the system’s sensitivity and identify strains with general cellular machinery malfunctions. Integration into the genome was performed by standard homologous recombination protocols (Gietz and Schiestl 2007). See all sequences (promoters, primers, linkers, recombination sites, and master strain) in Supplemental Table 1 and in Zeevi et al. (2011).
In order to introduce desired mutations for the synthetic library, we first amplified by PCR the URA3-promoter sequences from the natural orthologous library. Amplification was performed in two overlapping segments, with an overlap of 35 bases covering the sequence designated for mutation. The primers had at their 3′ end a region of 20–25 bps perfectly matching the original promoter, and a 35-bp tail at their 5′ end, which included the desired mutation(s). The two amplified segments were then linked through their matching tails (Linshiz et al. 2008) and integrated into the genome of the master strain similarly to the orthologous wild type library.
Promoter activity measurements
Cells were inoculated from stocks of −80°C into SCD (180 μL, 96-well plate) and left to grow for 48 h at 30°C, reaching complete saturation. Next, 8 μL were passed into 180 μL of fresh medium; and optical density, YFP fluorescence, and mCherry fluorescence were measured every ∼20 min using a robotic system (Tecan Freedom EVO) with a plate reader (Tecan Infinite F500). All strains were grown and measured at least four times. For each promoter strain, we calculated the average promoter activity per cell per sec over the exponential phase by dividing the total amount of YFP produced during the exponential phase by the integral of the OD levels during the same time interval. For a detailed description of the pipeline for the measurements, growth phase detection, and calculation of promoter activities, see Zeevi et al. (2011).
Learning linear models that predict promoter activity from promoter sequence features
Linear models that predict promoter activities were learned from various features of the native sensu stricto promoters in a cross validated manner. Feature types are detailed in the main text (see above). A complete description of the cross validated linear model learning scheme appears in the Supplemental Note.
Supplementary Material
Acknowledgments
We thank Amos Tanay for valuable discussions. This work was supported by grants from the European Research Council (ERC) and the U.S. National Institutes of Health (NIH) to E.S.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.179259.114.
References
- Badis G, Chan ET, van Bakel H, Pena-Castillo L, Tillo D, Tsui K, Carlson CD, Gossett AJ, Hasinoff MJ, Warren CL, et al. 2008. A library of yeast transcription factor motifs reveals a widespread function for Rsc3 in targeting nucleosome exclusion at promoters. Mol Cell 32: 878–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basehoar AD, Zanton SJ, Pugh BF. 2004. Identification and distinct regulation of yeast TATA box-containing genes. Cell 116: 699–709. [DOI] [PubMed] [Google Scholar]
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. 2009. BLAST+: architecture and applications. BMC Bioinformatics 10: 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JK, Kim YJ. 2009. Intrinsic variability of gene expression encoded in nucleosome positioning sequences. Nat Genet 41: 498–503. [DOI] [PubMed] [Google Scholar]
- Dixon AL, Liang L, Moffatt MF, Chen W, Heath S, Wong KCC, Taylor J, Burnett E, Gut I, Farrall M, et al. 2007. A genome-wide association study of global gene expression. Nat Genet 39: 1202–1207. [DOI] [PubMed] [Google Scholar]
- Fisher S, Grice EA, Vinton RM, Bessling SL, McCallion AS. 2006. Conservation of RET regulatory function from human to zebrafish without sequence similarity. Science 312: 276–279. [DOI] [PubMed] [Google Scholar]
- Galagan JE, Henn MR, Ma LJ, Cuomo CA, Birren B. 2005. Genomics of the fungal kingdom: insights into eukaryotic biology. Genome Res 15: 1620–1631. [DOI] [PubMed] [Google Scholar]
- Gietz RD, Schiestl RH. 2007. Microtiter plate transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc 2: 5–8. [DOI] [PubMed] [Google Scholar]
- Hare EE, Peterson BK, Iyer VN, Meier R, Eisen MB. 2008. Sepsid even-skipped enhancers are functionally conserved in Drosophila despite lack of sequence conservation. PLoS Genet 4: e1000106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckman DS, Geiser DM, Eidell BR, Stauffer RL, Kardos NL, Hedges SB. 2001. Molecular evidence for the early colonization of land by fungi and plants. Science 293: 1129–1133. [DOI] [PubMed] [Google Scholar]
- Hedges SB. 2002. The origin and evolution of model organisms. Nat Rev Genet 3: 838–849. [DOI] [PubMed] [Google Scholar]
- Henry YA, Chambers A, Tsang JS, Kingsman AJ, Kingsman SM. 1990. Characterisation of the DNA binding domain of the yeast RAP1 protein. Nucleic Acids Res 18: 2617–2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann-Le Denmat S, Werner M, Sentenac A, Thuriaux P. 1994. Suppression of yeast RNA polymerase III mutations by FHL1, a gene coding for a fork head protein involved in rRNA processing. Mol Cell Biol 14: 2905–2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogues H, Lavoie H, Sellam A, Mangos M, Roemer T, Purisima E, Nantel A, Whiteway M. 2008. Transcription factor substitution during the evolution of fungal ribosome regulation. Mol Cell 29: 552–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan N, Moore IK, Fondufe-Mittendorf Y, Gossett AJ, Tillo D, Field Y, LeProust EM, Hughes TR, Lieb JD, Widom J, et al. 2009. The DNA-encoded nucleosome organization of a eukaryotic genome. Nature 458: 362–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellis M, Patterson N, Endrizzi M, Birren B, Lander ES. 2003. Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature 423: 241–254. [DOI] [PubMed] [Google Scholar]
- Kellis M, Birren BW, Lander ES. 2004. Proof and evolutionary analysis of ancient genome duplication in the yeast Saccharomyces cerevisiae. Nature 428: 617–624. [DOI] [PubMed] [Google Scholar]
- Kenigsberg E, Bar A, Segal E, Tanay A. 2010. Widespread compensatory evolution conserves DNA-encoded nucleosome organization in yeast. PLoS Comput Biol 6: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren L, Zackay O, Lotan-Pompan M, Barenholz U, Dekel E, Sasson V, Aidelberg G, Bren A, Zeevi D, Weinberger A, et al. 2013. Promoters maintain their relative activity levels under different growth conditions. Mol Syst Biol 9: 701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein C, Struhl K. 1994. Protein kinase A mediates growth-regulated expression of yeast ribosomal protein genes by modulating RAP1 transcriptional activity. Mol Cell Biol 14: 1920–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landry CR, Lemos B, Rifkin SA, Dickinson WJ, Hartl DL. 2007. Genetic properties influencing the evolvability of gene expression. Science 317: 118–121. [DOI] [PubMed] [Google Scholar]
- Lascaris RF, Mager WH, Planta RJ. 1999. DNA-binding requirements of the yeast protein Rap1p as selected in silico from ribosomal protein gene promoter sequences. Bioinformatics 15: 267–277. [DOI] [PubMed] [Google Scholar]
- Lieb JD, Liu X, Botstein D, Brown PO. 2001. Promoter-specific binding of Rap1 revealed by genome-wide maps of protein-DNA association. Nat Genet 28: 327–334. [DOI] [PubMed] [Google Scholar]
- Linshiz G, Ben Yehezkel T, Kaplan S, Gronau I, Ravid S, Adar R, Shapiro E. 2008. Recursive construction of perfect DNA molecules from imperfect oligonucleotides. Mol Syst Biol 4: 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liti G, Barton DBH, Louis EJ. 2006. Sequence diversity, reproductive isolation and species concepts in Saccharomyces. Genetics 174: 839–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubliner S, Keren L, Segal E. 2013. Sequence features of yeast and human core promoters that are predictive of maximal promoter activity. Nucleic Acids Res 41: 5569–5581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig MZ, Patel NH, Kreitman M. 1998. Functional analysis of eve stripe 2 enhancer evolution in Drosophila: rules governing conservation and change. Development 125: 949–958. [DOI] [PubMed] [Google Scholar]
- Martinez C, Rest JS, Kim AR, Ludwig M, Kreitman M, White K, Reinitz J. 2014. Ancestral resurrection of the Drosophila S2E enhancer reveals accessible evolutionary paths through compensatory change. Mol Biol Evol 31: 903–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matot B, Le Bihan YV, Lescasse R, Pérez J, Miron S, David G, Castaing B, Weber P, Raynal B, Zinn-Justin S, et al. 2012. The orientation of the C-terminal domain of the Saccharomyces cerevisiae Rap1 protein is determined by its binding to DNA. Nucleic Acids Res 40: 3197–3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mencía M, Moqtaderi Z, Geisberg JV, Kuras L, Struhl K. 2002. Activator-specific recruitment of TFIID and regulation of ribosomal protein genes in yeast. Mol Cell 9: 823–833. [DOI] [PubMed] [Google Scholar]
- Moses AM, Chiang DY, Kellis M, Lander ES, Eisen MB. 2003. Position specific variation in the rate of evolution in transcription factor binding sites. BMC Evol Biol 3: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, Snyder M. 2008. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320: 1344–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Needleman SB, Wunsch CD. 1970. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol 48: 443–453. [DOI] [PubMed] [Google Scholar]
- Pachkov M, Balwierz PJ, Arnold P, Ozonov E, van Nimwegen E. 2013. SwissRegulon, a database of genome-wide annotations of regulatory sites: recent updates. Nucleic Acids Res 41: D214–D220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne JL, Wagner A. 2014. The robustness and evolvability of transcription factor binding sites. Science 343: 875–877. [DOI] [PubMed] [Google Scholar]
- Raveh-Sadka T, Levo M, Shabi U, Shany B, Keren L, Lotan-Pompan M, Zeevi D, Sharon E, Weinberger A, Segal E. 2012. Manipulating nucleosome disfavoring sequences allows fine-tune regulation of gene expression in yeast. Nat Genet 44: 743–750. [DOI] [PubMed] [Google Scholar]
- Rhee HS, Pugh BF. 2011. Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell 147: 1408–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee HS, Pugh BF. 2012. Genome-wide structure and organization of eukaryotic pre-initiation complexes. Nature 483: 295–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotenberg MO, Woolford JL. 1986. Tripartite upstream promoter element essential for expression of Saccharomyces cerevisiae ribosomal protein genes. Mol Cell Biol 6: 674–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scannell DR, Zill OA, Rokas A, Payen C, Dunham MJ, Eisen MB, Rine J, Johnston M, Hittinger CT. 2011. The awesome power of yeast evolutionary genetics: new genome sequences and strain resources for the Saccharomyces sensu stricto genus. G3 (Bethesda) 1: 11–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon E, Kalma Y, Sharp A, Raveh-Sadka T, Levo M, Zeevi D, Keren L, Yakhini Z, Weinberger A, Segal E. 2012. Inferring gene regulatory logic from high-throughput measurements of thousands of systematically designed promoters. Nat Biotechnol 30: 521–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman DJ, Martin T, Nikolski M, Cayla C, Souciet J-L, Durrens P. 2009. Génolevures: protein families and synteny among complete hemiascomycetous yeast proteomes and genomes. Nucleic Acids Res 37: D550–D554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smale ST, Kadonaga JT. 2003. The RNA polymerase II core promoter. Annu Rev Biochem 72: 449–479. [DOI] [PubMed] [Google Scholar]
- Tanay A. 2006. Extensive low-affinity transcriptional interactions in the yeast genome. Genome Res 16: 962–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanay A, Regev A, Shamir R. 2005. Conservation and evolvability in regulatory networks: the evolution of ribosomal regulation in yeast. Proc Natl Acad Sci 102: 7203–7208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Weinberger A, Carmi M, Barkai N. 2006. A genetic signature of interspecies variations in gene expression. Nat Genet 38: 830–834. [DOI] [PubMed] [Google Scholar]
- Tirosh I, Weinberger A, Bezalel D, Kaganovich M, Barkai N. 2008. On the relation between promoter divergence and gene expression evolution. Mol Syst Biol 4: 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Reikhav S, Levy AA, Barkai N. 2009. A yeast hybrid provides insight into the evolution of gene expression regulation. Science 324: 659–662. [DOI] [PubMed] [Google Scholar]
- Veyrieras JB, Kudaravalli S, Kim SY, Dermitzakis ET, Gilad Y, Stephens M, Pritchard JK. 2008. High-resolution mapping of expression-QTLs yields insight into human gene regulation. PLoS Genet 4: e1000214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wapinski I, Pfiffner J, French C, Socha A, Thompson DA, Regev A. 2010. Gene duplication and the evolution of ribosomal protein gene regulation in yeast. Proc Natl Acad Sci 107: 5505–5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weirauch MT, Hughes TR. 2010. Conserved expression without conserved regulatory sequence: the more things change, the more they stay the same. Trends Genet 26: 66–74. [DOI] [PubMed] [Google Scholar]
- Wilson MD, Barbosa-Morais NL, Schmidt D, Conboy CM, Vanes L, Tybulewicz VLJ, Fisher EMC, Tavaré S, Odom DT. 2008. Species-specific transcription in mice carrying human chromosome 21. Science 322: 434–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittkopp PJ, Haerum BK, Clark AG. 2004. Evolutionary changes in cis and trans gene regulation. Nature 430: 85–88. [DOI] [PubMed] [Google Scholar]
- Wittkopp PJ, Haerum BK, Clark AG. 2008. Regulatory changes underlying expression differences within and between Drosophila species. Nat Genet 40: 346–350. [DOI] [PubMed] [Google Scholar]
- Wood V, Harris MA, McDowall MD, Rutherford K, Vaughan BW, Staines DM, Aslett M, Lock A, Bähler J, Kersey PJ, et al. 2012. PomBase: a comprehensive online resource for fission yeast. Nucleic Acids Res 40: D695–D699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woudt LP, Smit AB, Mager WH, Planta RJ. 1986. Conserved sequence elements upstream of the gene encoding yeast ribosomal protein L25 are involved in transcription activation. EMBO J 5: 1037–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeevi D, Sharon E, Lotan-Pompan M, Lubling Y, Shipony Z, Raveh-Sadka T, Keren L, Levo M, Weinberger A, Segal E. 2011. Compensation for differences in gene copy number among yeast ribosomal proteins is encoded within their promoters. Genome Res 21: 2114–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C, Byers KJRP, McCord RP, Shi Z, Berger MF, Newburger DE, Saulrieta K, Smith Z, Shah MV, Radhakrishnan M, et al. 2009. High-resolution DNA-binding specificity analysis of yeast transcription factors. Genome Res 19: 556–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.