

Abstract
Gene expression levels are determined by the balance between rates of mRNA transcription and decay, and genetic variation in either of these processes can result in heritable differences in transcript abundance. Although the genetics of gene expression has been a subject of intense interest, the contribution of heritable variation in mRNA decay rates to gene expression variation has received far less attention. To this end, we developed a novel statistical framework and measured allele-specific differences in mRNA decay rates in a diploid yeast hybrid created by mating two genetically diverse parental strains. We estimate that 31% of genes exhibit allelic differences in mRNA decay rates, of which 350 can be identified at a false discovery rate of 10%. Genes with significant allele-specific differences in mRNA decay rates have higher levels of polymorphism compared to other genes, with all gene regions contributing to allelic differences in mRNA decay rates. Strikingly, we find widespread evidence for compensatory evolution, such that variants influencing transcriptional initiation and decay have opposite effects, suggesting that steady-state gene expression levels are subject to pervasive stabilizing selection. Our results demonstrate that heritable differences in mRNA decay rates are widespread and are an important target for natural selection to maintain or fine-tune steady-state gene expression levels.
Variation in gene expression levels constitutes a significant source of phenotypic diversity among individuals within populations and contributes to the evolutionary divergence between species (Skelly et al. 2009; Jones et al. 2012). In humans, regulatory variants affecting gene expression influence susceptibility to autoimmune, infectious, neoplastic, neurodegenerative, and psychiatric diseases (Skelly et al. 2009). In Darwin’s finches, regulatory variation affecting beak morphology likely played a role in their speciation (Abzhanov et al. 2004). Likewise, gene expression variation underlies the skeletal morphology differences in stickleback fish that distinguish saltwater from freshwater species (Jones et al. 2012).
Heritable regulatory variation can broadly be classified as either acting in cis or trans (Skelly et al. 2009). While trans-regulatory effects on gene expression are undoubtedly important, studies in several eukaryotic organisms, including yeast, fruit flies, mice, rats, and humans, suggest that cis-regulatory effects constitute a substantially higher proportion of the genetic variance in gene expression within species than do trans effects (Schadt et al. 2003; Hughes et al. 2006; Petretto et al. 2006; Emilsson et al. 2008; Pickrell et al. 2010; Skelly et al. 2011). In the budding yeast Saccharomyces cerevisiae, for example, nearly 80% of the genes that have transcribed polymorphisms between two diverse strains exhibit allele-specific expression differences (Skelly et al. 2011). In humans, it has been estimated that ∼90% of single nucleotide polymorphisms influencing gene expression levels are due to cis-regulatory mechanisms (Pickrell et al. 2010). Furthermore, cis-regulatory differences accumulate at a faster rate than trans-regulatory differences between closely related species (Wittkopp et al. 2008; Tirosh et al. 2009; Romero et al. 2012).
The balance between mRNA synthesis and decay determines steady-state levels of transcript abundance, and genetic variation affecting either of these processes can contribute to heritable differences in transcript abundance. However, to date, most research has concentrated on genetic variation affecting steady-state mRNA levels, failing to distinguish regulatory variation affecting transcription from that affecting decay (Skelly et al. 2009). Studies that have explored different classes of heritable variation underlying differences in steady-state gene expression focus primarily on transcription initiation, cataloging variation both within and between species in transcription factor binding sites, chromatin structure, and DNA methylation sites (Gerstein et al. 2010; The modENCODE Consortium et al. 2010; The ENCODE Project Consortium 2012; Connelly et al. 2014). In contrast, regulatory variants underlying differences in mRNA decay rate have received considerably less attention (Dori-Bachash et al. 2011; Pai et al. 2012).
To better delimit the contribution of cis-regulatory variation to heritable differences in mRNA decay rates, we developed a novel statistical framework and measured allele-specific differences in decay in a diploid hybrid created from two genetically diverse strains of the budding yeast, S. cerevisiae. We demonstrate that allelic differences in mRNA decay rates are widespread, affecting the expression levels of nearly 31% of measurable genes. Interestingly, we observe that a significant proportion of changes in decay rate are coupled to opposing changes in transcriptional initiation, suggesting pervasive compensatory evolution to stabilize or fine-tune steady-state gene expression levels. Our results also provide insights into the mechanisms through which cis-regulatory variation acts to influence mRNA decay rates, highlighting an important role for variants that affect mRNA secondary structure.
Results
Overview of experimental design
We measured rates of allele-specific mRNA decay (ASD) in a diploid yeast produced by
mating two genetically diverse haploid Saccharomyces cerevisiae
strains: the laboratory strain BY4716 (BY), which is isogenic to the reference
sequence strain S288C, and the wild Californian vineyard strain RM11-1a (RM) (Liti et al. 2009). A schematic of the
experimental design is shown in Figure 1.
Briefly, we introduced rpb1-1, a temperature-sensitive mutation in
an RNA polymerase II subunit encoded by the gene RPO21, to each of
the haploid yeast strains, mated the strains, and grew the resulting hybrid diploid
to mid-log phase at 24°C, before rapidly shifting the culture to 37°C to
inhibit transcription (Fig. 1A; Nonet et al. 1987). RNA-seq was performed on
culture samples taken at 0, 6, 12, 18, 24, and 42 min subsequent to the temperature
shift (Fig. 1A). To identify ASD, we used
transcribed polymorphisms to distinguish between parental transcripts and compared
the relative levels of transcript abundance over the time course (Fig. 1B). Note, this experimental design internally
controls for trans-acting regulatory variation as well as
environmental factors. Under the null hypothesis of no ASD, the proportion of reads
from the BY transcript 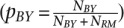 observed over the time course remains unchanged (Fig. 1B). However, genes with ASD will exhibit an
increasing or decreasing proportion of BY reads as a function of time (Fig. 1B). In total, we measured ASD from three
independent biological replicates.
observed over the time course remains unchanged (Fig. 1B). However, genes with ASD will exhibit an
increasing or decreasing proportion of BY reads as a function of time (Fig. 1B). In total, we measured ASD from three
independent biological replicates.
Figure 1.

Overview of experimental design. (A) We replaced the wild-type allele of the RPO21 (also known as RPB1) gene with the temperature-sensitive rpb1-1 allele in both the BY4716 (BY) and the RM11-1a (RM) strains of S. cerevisiae (Nonet et al. 1987). We mated these two haploid strains to produce a diploid hybrid and grew the diploid to mid-log phase at the permissive temperature of 24°C. We rapidly shifted the temperature of the culture to 37°C, halting transcription. Immediately following the temperature shift, and at 6, 12, 18, 24, and 42 min after the shift, we isolated mRNA and performed RNA-seq. (B) By quantifying the relative levels of the BY and RM alleles for each gene, we estimated pBY, the proportion of transcripts derived from BY, at each time point. Under the null hypothesis (H0; dashed line) of no allele-specific differences in mRNA decay rates, pBY remains constant. Under the alternative hypothesis (HA; solid line) of allelic differences in mRNA decay, we expect pBY to change as a function of time. For the gene represented by the solid line in the example pictured, pBY decreases significantly over time, indicating that the BY allele is decaying more quickly than the RM allele of this gene.
Statistical modeling of allele-specific mRNA decay
We developed a novel statistical framework to identify ASD. In brief, we use a linear
logistic model to measure the change in the proportion,
pBY, of reads derived from the BY
transcript as a function of time. To assess statistical significance, we use a
Bayesian hierarchical Markov chain Monte Carlo model (see Methods). In this model,
the prior probability of the alternative hypothesis (i.e., that a gene exhibits ASD)
is determined from the totality of data. We also estimate the mean and variance of
the decay rate differences under the alternative hypothesis from the data (see
Methods). The primary motivation for developing this more sophisticated framework is
that it accounts for genes that exhibit small departures from nonconstancy due to
high read counts in a more principled manner than alternative approaches (Dori-Bachash et al. 2011). We evaluated the power
and operating characteristics of our statistical framework through extensive
simulations and found that it generally has higher power and more accurately
estimates 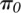 (the proportion of genes consistent with the null hypothesis of no allelic
differences in mRNA decay) compared to alternative approaches under a wide variety of
parameters (see Supplemental Methods and Results; Supplemental Fig. S1; Supplemental
Table S1).
(the proportion of genes consistent with the null hypothesis of no allelic
differences in mRNA decay) compared to alternative approaches under a wide variety of
parameters (see Supplemental Methods and Results; Supplemental Fig. S1; Supplemental
Table S1).
Pervasive influence of cis-regulatory variation on mRNA decay rates
Through a careful filtering pipeline, which included whole-genome sequencing of RM to mitigate read mapping bias (Degner et al. 2009; see Methods), we identified 27,569 transcribed single nucleotide variants (SNVs) in 4381 genes that could be used to assign whether individual RNA-seq reads derived from the BY or the RM allele of each gene. Of the ∼222 million RNA-seq reads we obtained across all replicates and all time points in our study, 13.57 million reads, averaging 2.26 ± 0.65 million reads per time point, were informative, such that they mapped to a variant site and could unambiguously be assigned as originating from BY or RM.
We applied the statistical inference framework described above to 3544 genes that
passed our filtering criteria (see Methods). From the Bayesian hierarchical MCMC
model, we estimated 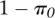 , the proportion of genes that exhibit ASD, to be 0.31. Thus,
∼31% of all measured genes are inferred to be inconsistent with the null
hypothesis and exhibit allelic differences in decay rates. Of these, we can identify
350 genes at a false discovery rate of 10% (Fig.
2A). Note, this corresponds to a false nondiscovery rate of 24%. The set of
genes called significant agrees well with a simpler approach of correcting for
multiple testing by the QVALUE software (Storey
2002; Storey and Tibshirani 2003;
Storey et al. 2004) and imposing a
threshold on the magnitude of effect sizes needed to be called as significant (see
Supplemental Methods and Results; Supplemental Fig. S2). We note that, in order to
inhibit transcription, we subjected the yeast to mild heat shock. The decay rates
observed in our data set are specific to the environmental condition that they were
measured in and, thus, may be different in other states, such as log phase growth
(Sun et al. 2012). Additionally, in theory,
some of the differences in decay rate that we measured could be due to
allele-specific transcriptional responses to heat shock, since there can be small
amounts of leaky transcription in the first 5–15 min following the temperature
shift (Nonet et al. 1987); however, such heat
shock-induced differences in allele-specific transcription are unlikely to comprise
most of the differences we observe.
, the proportion of genes that exhibit ASD, to be 0.31. Thus,
∼31% of all measured genes are inferred to be inconsistent with the null
hypothesis and exhibit allelic differences in decay rates. Of these, we can identify
350 genes at a false discovery rate of 10% (Fig.
2A). Note, this corresponds to a false nondiscovery rate of 24%. The set of
genes called significant agrees well with a simpler approach of correcting for
multiple testing by the QVALUE software (Storey
2002; Storey and Tibshirani 2003;
Storey et al. 2004) and imposing a
threshold on the magnitude of effect sizes needed to be called as significant (see
Supplemental Methods and Results; Supplemental Fig. S2). We note that, in order to
inhibit transcription, we subjected the yeast to mild heat shock. The decay rates
observed in our data set are specific to the environmental condition that they were
measured in and, thus, may be different in other states, such as log phase growth
(Sun et al. 2012). Additionally, in theory,
some of the differences in decay rate that we measured could be due to
allele-specific transcriptional responses to heat shock, since there can be small
amounts of leaky transcription in the first 5–15 min following the temperature
shift (Nonet et al. 1987); however, such heat
shock-induced differences in allele-specific transcription are unlikely to comprise
most of the differences we observe.
Figure 2.

Characteristics of genes that exhibit allele-specific mRNA decay. (A) Posterior probability that a gene exhibits allele-specific mRNA decay rates, as calculated from our Bayesian hierarchical Markov chain Monte Carlo model. The dashed line at posterior probability = 0.67 corresponds to the threshold we used to call genes as exhibiting significant allele-specific mRNA decay rates. (B) Histogram of the slope calculated from the linear logistic model for the 350 genes with significant (FDR = 10%) allele-specific mRNA decay rates. The exponential of the slope, which estimates λBY – λRM, is the change in the odds of observing a BY mRNA allele given a 1-min increase in time. (C) Decay rate time courses of all genes in which the RM allele decays significantly faster than the BY allele (left) and the BY allele decays significantly faster than the RM allele (right). The gray lines represent the decay rate time courses of each of the individual genes. The black lines represent the mean decay rate time courses for all of the genes included in each plot. (D) Correspondence of λBY – λRM to half-life differences between the BY and RM alleles of a gene. The dashed black lines represent the positive and negative of the median effect size, where effect size is defined as |λBY – λRM|, observed among the genes we identified with significant allele-specific differences in decay rates.
The exponential of the slope of the linear logistic model fit to each gene is a direct estimate of the difference in mRNA decay rate between the BY and RM alleles of that gene (see Methods). Among the genes with significant ASD, the effect size of the decay rate difference ranges from a 1.81 × 10−3 to a 5.62 × 10−2 change in the odds of observing an mRNA allele of the BY strain given a 1-min increase in time, with a median difference of 1.01 × 10−2 (Fig. 2B). This median difference corresponds to an ∼83% increase over 1 hr in the odds of observing an mRNA allele of the BY strain. The BY allele decays more quickly than the RM allele in 161 genes, while the RM allele decays more quickly than the BY allele in 189 genes. Genes with allelic differences in mRNA decay rates spanned a broad range of gene ontology terms, and we did not detect significant enrichment for particular functions or biological processes after correcting for multiple comparisons.
Allelic differences in mRNA decay reveal widespread compensatory evolution
To investigate the relationship between ASD and steady-state gene expression levels, we first inferred allele-specific expression (ASE) at the 0-min time point in our time course, which is a reasonable proxy for steady-state levels of transcript abundance. Using the method developed by Connelly et al. (2014), we find that 1137 genes exhibit ASE (posterior probability > 0.95) (Fig. 3). Five hundred and ninety-five of the 1137 genes (52.3%) that exhibit steady-state ASE have higher levels of the RM transcript, and 542 genes (47.7%) have higher levels of the BY transcript. The median log2-fold change for all genes with allele-specific steady-state expression differences is 0.43.
Figure 3.

mRNA decay rates in yeast are often coupled to opposite changes in transcription initiation. (Left) Venn diagram showing the overlap of genes with significant allelic differences in steady-state gene expression (ASE) and decay (ASD). (Right) Scatter plot showing estimates of differences in decay rates between BY and RM (x-axis) versus the proportion of transcripts from the BY allele (pBY) inferred from the 0-min time point for genes with both ASE and ASD. The shaded gray rectangles represent quadrants where magnitudes of ASD and ASE are discordant, suggesting compensatory evolution.
Of the 350 genes with significant ASD, 182 (52.0%) also exhibit ASE (Fig. 3). Strikingly, of the 182 genes with both ASD and ASE, 129 (70.9%) have increased decay rates in the allele with higher levels of steady-state expression, suggesting that there are variants influencing rates of transcriptional initiation with opposite effects to those influencing decay (Fig. 3). Similarly, the 168 genes that exhibit ASD but not ASE (Fig. 3) are also likely enriched for variants with opposing effects on transcriptional initiation and decay, since the difference in decay rate does not produce a corresponding difference in steady state levels. Thus, these data suggest that changes in mRNA decay rates in yeast are often coupled with opposite changes in transcription, consistent with pervasive compensatory evolution to stabilize or fine-tune steady-state gene expression levels.
Patterns of genetic diversity across transcripts with allelic differences in mRNA decay
Previous studies in yeast have found that genes with ASE exhibited higher levels of genetic diversity compared to those without such differences (Ronald et al. 2005). To explore patterns of diversity in our data set, we first compared overall levels of variation among four classes of genes: those with only ASD, those with only ASE in steady-state levels, those with both ASD and ASE, and those with no allele-specific differences. Limiting our analysis to the 2954 genes with reliable UTR annotations (Nagalakshmi et al. 2008), we found that genes with any type of allele-specific difference have 1.4-fold more variants than genes without ASD or ASE (4.62 and 3.32 variants/kb, respectively; Mann-Whitney U test, P-value < 2.20 × 10−16) (Fig. 4). Moreover, genes with ASD have 1.3-fold higher levels of variation compared to genes with only ASE (5.48 variants/kb compared to 4.31 variants/kb, respectively; Mann-Whitney U test, P-value = 2.26 × 10−10) (Fig. 4). One complication in interpreting these findings is that genes with larger numbers of variants tend to have more informative reads, and, therefore, there is greater power to detect allelic differences in expression and decay. Indeed, variant density is significantly correlated with the number of informative reads (r2 = 0.158, P-value < 2.20 × 10−16). To more formally explore whether differences in SNV density are simply related to power, we performed logistic regression where the predictor variables were coded as zero if a gene did not show ASD and one if it did. We found that a model that included both the number of variants/kb and the number of informative reads as covariates fits the data significantly better than a model using just the number of informative reads alone (ANOVA P-value < 2.20 × 10−16), suggesting that the increased levels of variation in genes with ASD and ASE are not simply a consequence of discovery bias.
Figure 4.

Levels of genetic diversity in genes with and without allele-specific differences in mRNA decay rates and steady-state expression levels. Bar plots show the mean number of single nucleotide variants (SNVs) between BY and RM per kilobase across the whole gene (left) or across each gene region separately (right). Error bars correspond to the 95% confidence interval of the mean. ASE and ASD denote allele-specific expression and allele-specific decay, respectively.
To identify regions that may be enriched for variants that influence decay rates, we compared levels of genetic variation in the 5′ UTR, coding region, and 3′ UTR among the four classes of genes described above. Levels of variation are significantly elevated across all genic regions for genes with ASD compared to genes with no allelic differences (Fig. 4). Genes with both ASD and ASE have 1.46-fold, 1.68-fold, and 1.74-fold higher levels of variation than genes with no allelic differences in the 5′ UTR, coding region, and 3′ UTR, respectively (Mann-Whitney U test, P-value = 1.43 × 10−2, < 2.20 × 10−16, and 1.47 × 10−4). Genes with ASD only have 1.88-fold, 1.64-fold, and 2.12-fold higher levels of variation than genes with no allelic differences in the 5′ UTR, coding region, and 3′ UTR, respectively (Mann-Whitney U test, P-value = 1.58 × 10−7, 1.12 × 10−15, and 6.77 × 10−4). Thus, allele-specific differences in mRNA decay rate are likely driven by variants positioned throughout the transcript. Consistent with this hypothesis, genes that only contain SNVs in either their coding region or UTR are less likely to exhibit ASD compared to genes with variants in both their coding region and UTR (Fisher’s exact test, P-value = 1.35 × 10−10 and 1.55 × 10−4, respectively).
Genes with ASD are enriched for variants that influence mRNA structure
To test the hypothesis that variation in mRNA secondary structure contributes to allelic differences in mRNA decay rates, we compared the minimum Gibb’s free energy (ΔG) associated with the predicted secondary structures of the BY and RM alleles for each mRNA transcript. Specifically, following standard practices (Tuller et al. 2010), we computed the ΔG of the predicted secondary structures for each of the 27,569 variants that we identified between BY and RM (Fig. 5A). We then calculated the absolute value of the difference in free energy between alleles, |ΔΔG| = |ΔGBY – ΔGRM|, and for each gene, we recorded the maximum |ΔΔG| of all its variants. Variants with larger values of |ΔΔG| are predicted to have more severe structural consequences (Fig. 5A). Genes that exhibit ASD are enriched for variants with larger predicted effects on mRNA secondary structure as compared to genes without any allelic differences in decay or steady-state expression (1.32-fold increase in the maximum |ΔΔG| observed in genes with ASD; Mann-Whitney U test, P-value = 5.48 × 10−15) (Fig. 5B). Although in silico predictions of differences in mRNA secondary structure are not perfect proxies for structures that occur in vivo, our observations are consistent with the hypothesis that allelic variation in mRNA secondary structure contributes to heritable variation in mRNA decay rates. Interestingly, the gene HSP78, which encodes a mitochondrial matrix chaperone, exhibits ASD and only contains a single variant (Supplemental Table S2), which has a large predicted effect on mRNA secondary structure (Fig. 5C), suggesting that allelic differences in decay of this gene are likely mediated by structural differences.
Figure 5.
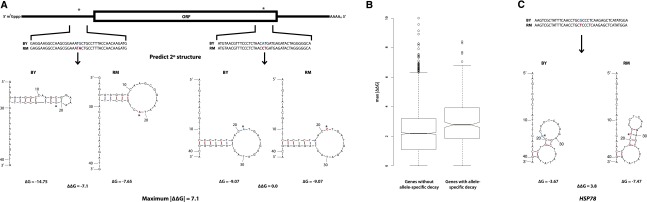
Genes with significant allele-specific decay are enriched for predicted differences in mRNA structure. (A) Calculation of ΔΔG for SNVs across a transcript. We determined the mRNA sequence for both the BY and RM alleles of each gene. For each SNV (denoted by *), we calculated the predicted minimum Gibb’s free energy (ΔG) associated with the mRNA secondary structures of each allele using 41-bp regions of the mRNA transcript centered on each of the SNVs. The difference in ΔG (ΔΔG) between the BY and RM alleles for each 41-bp window was measured by subtracting the ΔG calculated for the RM allele from the ΔG calculated for the BY allele. We then calculated the absolute value of the difference in free energy between alleles, |ΔΔG| = |ΔGBY – ΔGRM|, for each variant. For each gene, we recorded the maximum |ΔΔG| observed among all its variants. (B) The maximum |ΔΔG| of genes with and without allele-specific decay rate differences are significantly different (Mann-Whitney U test, P-value = 4.47 × 10−7), suggesting that genes with allele-specific decay rates are significantly enriched for variants with larger predicted effects on mRNA secondary structure compared to genes without allelic differences in decay. (C) Predicted mRNA secondary structures surrounding an SNV in the gene HSP78, which encodes a mitochondrial matrix chaperone. The location of the SNV in each transcript allele is denoted by *; the BY and RM alleles at the variant site are colored blue and red, respectively. Note, HSP78 only contains one SNV.
No widespread evidence for coupling between decay rates and translational efficiency
Previous reports measuring mRNA decay rate in one or a few genes have suggested that the translation efficiency of an mRNA might be directly coupled to mRNA decay rate (for review, see Garneau et al. 2007 and Schoenberg and Maquat 2012). To evaluate whether such coupling is common, we compared the types of codon changes occurring between BY and RM in genes with and without decay rate differences. If mRNA decay rate is coupled to translation efficiency, we would expect that genes with ASD would have higher proportions of codon changes that impact translation efficiency, as compared to genes without such differences. To this end, we compared the proportion of preferred to unpreferred synonymous codon changes in genes with and without ASD and found that genes with ASD have a slightly lower proportion of codon preference changes, although this is not statistically significant (68.1% versus 68.7%; Mann-Whitney U test, P-value = 8.60 × 10−2). Thus, we do not find widespread evidence that mRNA translation efficiency is directly coupled to mRNA decay rate. A caveat of this analysis is that more sensitive measures of translational efficiency may be needed to detect coupling. Moreover, these findings do not preclude the possibility that coupling exists for a subset of genes, which we may not have power to detect.
Discussion
We developed a novel statistical framework to measure allele-specific differences in mRNA decay rate in a diploid yeast hybrid created by mating two genetically diverse parental strains. A particular strength of our statistical approach is its ability to deal with genes that exhibit small departures from nonconstancy due to high read counts in a more principled manner than alternative approaches (Dori-Bachash et al. 2011). Using our statistical framework, we demonstrate the pervasive influence of cis-regulatory variation on mRNA decay rates, estimating that >30% of measurable genes exhibit ASD. Our results suggest that variation in mRNA decay rate is widespread across the genome, as well as among individuals within a species. Overall, our study provides further evidence of the importance of post-transcriptional processes in determining heritable differences in gene expression levels, which, in turn, impact phenotypic diversity among individuals within populations. Additionally, the novel statistical framework we developed has broad applications for future work in testing hypotheses of differential expression.
A striking feature of the data is that differences in mRNA decay rates are often coupled with opposite changes in transcription. It is difficult to precisely estimate the proportion of genes with significant ASD that is in the opposite direction of steady-state expression levels because of differences in the statistical power of detecting ASE and ASD. However, a naïve estimate suggests that up to 85% of genes with significant ASD are coupled with opposing effects on transcription (Fig. 3). These findings agree with previous studies, which observed that roughly 80% of differences between yeast species and 50% of differences among humans in mRNA decay rate are coupled to opposing differences in transcription (Dori-Bachash et al. 2011; Pai et al. 2012). Interestingly, in the remaining 15% of genes with significant ASD, there is no significant correlation (r2 = 0.255, P-value = 6.53 × 10−2) between the magnitude of the decay rate difference and the magnitude of the gene expression difference between the alleles. Collectively, these results suggest that steady-state gene expression levels are subject to strong stabilizing selection, and that heritable differences in mRNA decay rates are an important target for natural selection to maintain or fine-tune steady-state gene expression levels.
To explore which regions of the mRNA transcript are most important in determining mRNA decay rate differences, we compared the levels of genetic variation in the 5′ UTR, coding region, and 3′ UTR in genes with and without ASD. We hypothesized that the 3′ UTR would be the most important region governing ASD, and therefore, that genes exhibiting ASD would be especially enriched for polymorphisms between BY and RM in the 3′ UTR compared to genes without ASD or ASE. Instead, we observed that all three regions exhibited significantly more variation in genes with ASD compared to genes without ASD or ASE (Fig. 4). One explanation for these results is that the 3′ UTR contains the lowest overall amount of variation, suggesting that it is under significant functional constraint. If the 3′ UTR contains the highest density of cis-elements affecting mRNA decay rate, then changes to this region perhaps have a larger effect on mRNA decay rate, and therefore, are more likely to be removed by purifying selection. Conversely, changes in the 5′ UTR or coding region may cause differences in mRNA decay rates of a smaller effect size, and therefore be subject to less intense purifying selection. Thus, all three gene regions may be important determinants to within-species differences in mRNA decay rate. Our observation that genes with SNVs only in the coding region or, alternatively, only in the UTRs are less likely to exhibit ASD, also suggests that variation in all parts of the transcript can potentially impact decay rate.
Identifying allelic differences in mRNA decay rate is only the first step in the ultimate goal of identifying causal regulatory alleles and the mechanisms that they act through. To this end, it is interesting to note that 13 genes with ASD possess a single variant between the BY and RM alleles of the transcript (11 in coding regions and two in UTRs) (see Supplemental Table S2). These variants are strong candidate causal alleles, and as shown for HSP78 (Fig. 5C), enable mechanistic hypotheses to be formulated and ultimately tested. Furthermore, it will be important to consider additional processes that could influence allele-specific decay. For example, nonsense-mediated decay (NMD) can be triggered by AUG codons in the 5′ UTR, and if a SNV introduced or disrupted a 5′ UTR AUG, it could influence mRNA decay rates between the two alleles. In the 2954 genes that have reliable UTR annotations, 34 contain SNVs in the 5′ UTR that introduce or disrupt an AUG codon. Of these 34 genes, nine exhibit significant ASD, which is significantly more than we would expect by chance (Fisher’s exact test, P-value = 5.16 × 10−3); however, only three of the nine genes show decay rate differences in the direction expected if allelic differences in mRNA decay rate were mediated by nonsense-mediated decay. Thus, this process likely makes a minor contribution to patterns of ASD in our data. More generally, dissecting the mechanistic basis of allelic variation in mRNA decay rates will facilitate the robust prediction of causal regulatory alleles from sequence data.
Another critical area of research will be exploring the interactions of genetic variation with the environment. Our study was conducted in yeast undergoing exponential growth in a rich medium that underwent mild heat shock at the time of transcriptional shut-off; however, we would expect that patterns of ASD would differ markedly under differing growth conditions, such as nutrient-limited media or the presence of high concentrations of chemicals like ethanol or the various heavy metals. Additionally, the effects of cis-regulatory variation on mRNA decay are also likely to vary between different stages of the yeast life cycle, including during meiosis and during vegetative growth. Another important limitation of our study is that it only examines allele-specific differences in mRNA decay rate between two diverse yeast strains. Nonetheless, our results highlight the important contribution that heritable variation in mRNA decay rates make to buffer steady-state differences in gene expression and suggest that additional post-transcriptional processes should be studied in greater detail for a more comprehensive understanding of mechanisms contributing to transcriptional diversity within and between species.
Methods
Yeast strains
For the purposes of this study, we replaced the wild-type copy of the RPO21 (also known as RPB1) gene in the haploid S. cerevisiae strains BY4716 (BY) and RM11-1a (RM) (for detailed descriptions of these two strains, see Brem et al. 2002) with the rpb1-1 mutant allele (Nonet et al. 1987). We began by identifying the mutations that make rpb1-1 differ from wild-type RPO21 via standard Sanger sequencing of the RP021 locus in the strain Y262 (described in Herrick et al. 1990). We identified two mutations: a C to T substitution 206 bp after the translation start site, and a G to A substitution 4310 bp after the translation start site; both mutations are nonsynonymous. To substitute RPO21 with rpb1-1 in RM, we used a “pop-in, pop-out” strategy (Rothstein 1995; Duff and Huxley 1996). Specifically, in the “pop-in” step, we linearized a plasmid containing the URA3 selectable marker and the rpb1-1 mutant allele with a restriction enzyme that cut in the rpb1-1 sequence segment. We then transformed the linearized plasmid into RM cells and selected for cells in which the plasmid had recombined into the genome using uracil prototrophy. At the completion of the “pop-in” step, RM carried a duplication of the target genomic sequence segment, in which one duplicate contained the RPO21 wild-type allele and one duplicate contained the rpb1-1 mutant allele; the plasmid sequences and URA3 lay between the two duplicates. In the “pop-out” step, we added uracil back to the medium so that the URA3 gene was no longer required for viability, allowing spontaneous recombination events to occur between the duplicated target sequences. To select for recombination events, we used 5-fluororotic acid (5-FOA), which is metabolized by URA3 into a toxic compound. A recombination event will result in either retention of the mutant rpb1-1 allele or reversion to the wild-type RPO21 allele. Using this strategy, we first replaced the C located 206 bp after the translation start site in RPO21 with a T, and then, subsequently, we replaced the G located 4310 bp after the translation start site in RPO21 with an A. To confirm successful substitution of the wild-type allele with the mutant allele at both sites, we used standard Sanger sequencing.
To replace RPO21 with rpb1-1 in BY, we employed a backcrossing strategy. We could not use the “pop-in, pop-out” strategy because BY already contained the URA3 selectable marker. More specifically, we crossed BY to Y262, sporulated the hybrid diploid, and screened the resultant offspring for inability to grow at 37°C (Note: rpb1-1 mutants do not replicate at this temperature). We then performed four more rounds of backcrossing between the hybrid offspring and BY, such that the resulting yeast strain carried the rpb1-1 allele on an ∼97% BY genetic background. We confirmed that the final product of our backcross carried the two single nucleotide variants that make rpb1-1 mutants different from the wild-type RPO21 by standard Sanger sequencing. We mated the BY and RM rpb1-1 temperature-sensitive mutant strains and selected for the diploid hybrid by visually screening for BY and RM cells that had mated. We confirmed that our candidate diploid hybrids identified in our screen were, in fact, diploid using a standard Halo Mating Type Assay.
Measuring mRNA decay rates
mRNA decay rate time course sample collection
The BY × RM hybrid diploid we generated was grown at 24°C to mid-log phase (OD600 0.8–1.0) in 60 mL of yeast extract peptone dextrose (YEPD). We abruptly shifted the culture to 37°C via addition of 60 mL of 50°C YEPD. Immediately following, and at 6, 12, 18, 24, and 42 min after addition of the 50°C medium, we collected 20-mL aliquots of the culture using vacuum filtration. To maintain the increased temperature of the culture, we housed it in a 37°C shaking incubator between collection time points. The collected yeast cells from each time point were flash-frozen in liquid nitrogen and then stored at −80°C for no more than 2 d before we extracted total RNA from the cells using a standard phenol-chloroform preparation. In total, we collected three replicates of our time course.
RNA sequencing
We used a TruSeq RNA Sample Prep v2 Kit (Illumina) to create a sequencing library from the total RNA collected for each decay rate time course time point from each replicate. Per the protocol for the kit, we isolated mRNA from the total RNA using two rounds of poly(A) selection, then fragmented the isolated mRNA into ∼150 base pair (bp) fragments, and finally, used random hexamer primers to produce cDNA. Poly(A) selection, by definition, retains only mRNA with intact poly(A) tails in the resultant RNA pool. The most commonly used pathway of mRNA decay, referred to as deadenylation-dependent decapping, begins with shortening of the poly(A) tail by deadenylases, followed by removal of the 5′ cap structure by decapping enzymes, and, finally, 5′ to 3′ exonucleolytic degradation of the decapped intermediate (for review, see Wilusz et al. 2001 and Garneau et al. 2007). Thus, our experimental design is unable to detect allelic differences that affect the decapping rate or rate of exonucleolytic degradation. We chose to use poly(A) selection to isolate mRNA despite its inability to detect differences in the later stages of mRNA decay because previous studies of deadenylation-dependent decapping have demonstrated that mRNA decay proceeds very rapidly following deadenylation, and that deadenylation, as opposed to decapping or exonucleolytic degradation, is the rate-limiting step in the mRNA decay process (for review, see Wilusz et al. 2001 and Garneau et al. 2007). We created barcoded sequencing libraries from the cDNA from each sample and, in an effort to minimize technical variation between the data acquired from different decay rate time points, all samples from all replicates were sequenced in the same lane on an Illumina HiSeq 2000 (50-bp paired-end reads).
Whole-genome sequencing of RM
For whole genome sequencing of the S. cerevisiae strain RM, we inoculated the strain from −80°C freezer stock into 5 mL YEPD and grew the culture at 30°C to saturation. We pelleted the cells from the culture by centrifugation, decanted the supernatant, and froze the cells at −80°C. We extracted DNA using a Genomic-tip 100/G Kit (Qiagen) and then concentrated the sample using a standard ethanol precipitation. We prepared a DNA sequencing library using a TruSeq DNA Sample Prep v2 Kit (Illumina). Per the protocol for the kit, we used a Covaris sonicator to shear the DNA into ∼300- to 400-bp fragments, and, after ligating adaptors onto the DNA fragments, we additionally size-selected for 300- to 400-bp fragments by running the ligation products out on an agarose gel and gel-extracting the appropriate band. We performed whole-genome sequencing using an Illumina MiSeq (151-bp paired-end reads).
Read mapping
We obtained complete genome sequences for BY from the Saccharomyces Genome Database (version R64-1-1, released February 3, 2011; http://www.yeastgenome.org) (Engel et al. 2013) and for RM from the Broad Institute (http://www.broadinstitute.org). We used BWA version 0.5.9 (Li and Durbin 2009) to map both the DNA and RNA sequence reads to the BY genome and, separately, the RM genome. After mapping reads, we sorted BAM files and marked duplicate reads using Picard version 1.43 (http://picard.sourceforge.net).
Identification of variant sites for assigning the allele of individual RNA-seq reads
To obtain a set of variants for allele-specific read calling in the BY × RM diploid, we used LASTZ (http://www.bx.psu.edu/ miller_lab) to infer alignment scoring parameters appropriate for aligning the BY and RM genomes and to generate pairwise alignments between all chromosomes of the two strains. We then used threaded blockset aligner (TBA) (Blanchette et al. 2004) to compute a whole-genome alignment that is not biased in favor of a particular reference genome. We cataloged all SNVs, as well as all indels, identified in the alignment. As we were only interested in transcribed differences between the BY and RM genomes, we removed from our variant list all sites not within annotated BY open reading frames (obtained from the Saccharomyces Genome Database; http://www.yeastgenome.org) and their corresponding untranslated regions (UTRs) (UTR lengths were determined from Nagalakshmi et al. 2008). Manual review of the remaining variant sites using the program Integrated Genome Viewer (http://www.broadinstitute.org/igv/) revealed that many of the indels identified from the BY and RM alignment produced by TBA, as well as the SNVs closely flanking these indels, were miscalled. Therefore, we removed all indels and all SNVs within 10 bp of an indel from our variant list. Likewise, because we suspected that most or all of the SNVs we identified in genes with unusually high numbers of variants per kb were artifacts of alignment errors, we discarded all SNVs located in genes that exhibited greater than fivefold the average variant density of all genes across the genome. Due to the difficulty in distinguishing which gene an RNA-sequencing read that aligns to a location in which two yeast genes overlap derives from, we also threw out any variants that overlapped more than one annotated yeast gene. Finally, we removed any variants to which reads obtained from whole genome sequencing of RM were assigned more often to the BY allele of the variant than to the RM allele by the method developed by Skelly et al. (2011) (briefly described below) to assign whether individual RNA-seq reads derived from the BY or the RM allele of each gene.
Assignment of the allele of individual RNA sequencing reads
We performed assignment of individual RNA-seq reads as originating from either the BY or the RM allele of each gene as described in Skelly et al. (2011), with the following two changes. First, any read with an alignment to one genome that scores higher had to overlap one of the SNVs between BY and RM that were identified as described above. Second, we did not perform a correction for GC content.
Measuring allele-specific differences in mRNA decay rate
To determine whether a gene exhibited allele-specific mRNA decay rate differences, we developed a novel linear logistic model that we applied in conjunction with a quasi-likelihood ratio test to measure the change across our time course in the calculated proportion of reads deriving from the BY allele as compared to the total number of informative reads at each gene. Specifically, in our model, we let Nj(t) be the number of mRNA transcripts for strain j, j = 1, 2 (representing BY and RM) at time t. We then assumed that the rate of decay is dNj/dt = −λjt, with λj > 0, so that Nj(t) = N0j exp(−λjt), where N0j is the count at time 0 for strain j. For each time point, t, the number of RNA-seq reads that we can assign to a strain, nj(t), is a fraction, ft, of the total number of mRNA transcripts for that strain, such that nj(t) = ft Nj(t).
We then assumed the model
Under this model, the distribution of the counts for strain 1 (BY) given the total is binomial [we could make a binomial approximation since n(t) ≪ N(t)] with denominator n1(t) + n2(t) and probability (of strain 1):
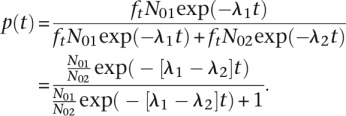 |
Taking the logit gives:
Although different fractions of the total mRNA transcript pool are sampled at each
time point, these fractions cancel in the above calculation, so that we compute the
relative proportion of strain 1 (BY) alleles in the pool from which we have sampled
at each time point. The exp(α) is the odds that we observe an
mRNA allele of strain 1 at time t = 0. In our linear logistic
model, we estimate α; however, from the above derivation we
know that these odds are
N01/N02,
but this proportion is unobserved. The parameter exp(β) is the
change in the odds of observing an mRNA allele of the strain 1 type given a 1-min
increase in time. Thus, exp(60 × β) is the change in the
odds of observing an mRNA allele of strain 1 given a 1-hr increase in time. For
example, if exp(60 × β) = 2, then the odds of
observing an mRNA allele of strain 1, when compared to the odds of observing an mRNA
allele of strain 2 (RM), doubles over 1 hr. If decay rates are the same in both
strains, then λ1 =
λ2, which is equivalent to
β = 0 in the logistic model. The parameter estimate
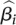 , along
with the associated standard error, are subsequently used within a hierarchical
model, as detailed shortly.
, along
with the associated standard error, are subsequently used within a hierarchical
model, as detailed shortly.
The null can be rejected with small departures from nonconstancy due to high counts,
if a frequentist test (such as a quasi-likelihood test) is used. This is a recognized
problem with frequentist testing in which power is not accounted for in the setting
of significance thresholds. Hence, to determine if β was
significantly different from 0, and therefore, whether a gene exhibited allele
specific differences in mRNA decay rate, we used a Bayesian hierarchical model. In
our model, we let Yi be the estimate of
the slope 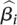 for the ith gene, and
for the ith gene, and 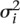 be the variance of this estimate. We
then assumed Yi |
be the variance of this estimate. We
then assumed Yi |
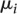 ∼ ind
N(
∼ ind
N(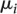 ,
, 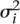 ), i = 1, …, m,
where m is the number of genes. We specified a mixture model for the
collection [
), i = 1, …, m,
where m is the number of genes. We specified a mixture model for the
collection [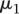 , …,
, …, 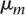 ], with
], with
The second mixture component contains the non-null genes. We integrated out over
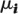 to
obtain a three-stage model, and we use mixture component indicators
Hi = 0/1 to denote the
zero/normal membership model for transcript i. The model is:
to
obtain a three-stage model, and we use mixture component indicators
Hi = 0/1 to denote the
zero/normal membership model for transcript i. The model is:
- Stage 1:
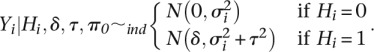
- Stage 2:
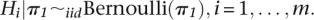
- Stage 3:
with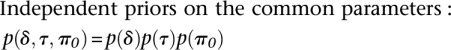
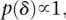
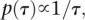
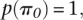
so that we had improper priors for δ and τ2. This model is appealing since we deal with overdispersion in the data using a reliable and distribution-free frequentist method and then take the information on the parameter of interest only (the differences), namely the estimate and its associated standard error, to model within the hierarchy. By only concentrating on the key parameters, we avoid having to make model assumptions concerning parameters of no interest.
We implemented this model via a Markov chain Monte Carlo algorithm in which we introduced indicator variable ωi to denote the mixture component of gene i. For our analysis, we only evaluated the 3544 genes that had at least 10 informative reads at each of the six time points, as well as less than a 50-fold difference in expression of the two alleles at the 0-min time point. General background of this testing framework can be found in Wakefield (2013).
To formally determine whether gene i exhibited allele-specific mRNA decay rate differences, we placed a threshold of 0.67 on the posterior probability ri = Pr(Hi = 1| data) of being non-null. At this threshold, the false discovery rate (FDR) is 0.099 and the false nondiscovery rate (FNDR) is 0.244. The FDR and FNDR are model-based estimates and are calculated as follows. For the list of R (say) genes i that pass the threshold, we calculate the sum of 1 – ri (i.e., the posterior probability of no difference in mRNA decay rate) and divide by the total number of “noteworthy” genes, R, to give the FDR. For all the remaining (3544 – R) non-noteworthy genes, we sum the ri (i.e., the posterior probability of a difference in mRNA decay rate) and then divide by (3544 – R) to give the FNDR.
Gene Ontology analysis
To assess whether there was any significant enrichment for genes involved in a particular molecular function, cellular component, or biological process in the set of genes we identified with allelic differences in mRNA decay, or in the two smaller subsets of genes in which one allele or the other decayed more quickly, we submitted each set of genes to AmiGO’s GO Term Enrichment Tool (http://amigo1.geneontology.org/cgi-bin/amigo/term_enrichment). We co-submitted all 3544 genes we analyzed for allele-specific mRNA decay rate differences as the input background set and selected SGD as the database filter. We chose 0.01 as our maximum P-value threshold and two as the minimum number of gene products.
Measuring allele-specific differences in mRNA steady-state levels
Using the numbers of mRNA transcripts from BY and RM for each gene at time point t = 0 min as a proxy for steady-state expression levels, we determined whether a gene exhibited allele-specific steady-state expression differences by performing the cis test exactly as described in Connelly et al. (2014), with the following modification: The test was performed with three, rather than two, replicates. Our primary motivation for choosing this method, as opposed to alternative approaches (Skelly et al. 2011), is that its statistical framework is most closely related to the framework we implemented for identifying allele-specific mRNA decay rate differences.
Classification of genes by type of allele-specific differences
For comparison between genes with allele-specific mRNA decay rate differences, allele-specific steady-state expression differences, and no allele-specific differences, we divided the genes into four classes: those with allele-specific differences in mRNA decay rate only, those with allele-specific differences in steady-state levels only, those with allele-specific differences in both mRNA decay rate and steady-state levels, and those with no allele-specific differences. Specifically, we categorized genes with a posterior probability greater than 0.67 in our Bayesian hierarchical Markov chain Monte Carlo model, but with a posterior probability less than 0.95 in our test for allele-specific steady-state expression differences as only having allele-specific differences in mRNA decay rate. We considered genes with a posterior probability greater than 0.67 in our Bayesian hierarchical Markov chain Monte Carlo model and a posterior probability greater than 0.95 in our test for allele-specific steady-state expression differences as exhibiting both allele-specific differences in mRNA decay rate and allele-specific differences in steady-state expression levels. Genes with a posterior probability greater than 0.95 in our test for allele-specific steady-state expression differences, but which did not have a posterior probability greater than 0.67 in our Bayesian hierarchical Markov chain Monte Carlo model, were classified as only having allele-specific differences in steady-state expression levels. For our final category of genes with no allelic differences, we grouped together genes with a posterior probability less than 0.30 in our Bayesian hierarchical Markov chain Monte Carlo model and a posterior probability less than 0.95 in our test for allele-specific steady-state expression differences. We choose 0.30 rather than 0.67 as the cut-off for the posterior probability in our Bayesian hierarchical Markov chain Monte Carlo model for this group of genes in an effort to minimize the number of false negatives (for allele-specific differences in mRNA decay rate) in this group.
Secondary structure analysis
To evaluate the differences in mRNA secondary structure between the BY and RM alleles of each gene, we began by determining the mRNA sequence for both the BY and RM alleles of each gene using the set of 27,569 variants we identified between BY and RM, the BY genome sequence (from the Saccharomyces Genome Database, version R64-1-1, released February 3, 2011; http://www.yeastgenome.org) (Engel et al. 2013), annotations of the BY open reading frames (from the Saccharomyces Genome Database; http://www.yeastgenome.org), and the predicted untranslated region lengths (UTRs) for the BY open reading frames (Nagalakshmi et al. 2008). We then used the UNAFold software package’s hybrid-ss-min tool to compute the predicted minimum Gibb’s free energy (ΔG) associated with the mRNA secondary structures of each allele of each transcript at 30°C (we chose to use 30°C because this is the standard temperature at which yeast are grown in the laboratory) (Markham and Zuker 2005, 2008). More specifically, following standard practices (Tuller et al. 2010), we calculated the ΔG of a 41-bp mRNA region surrounding each of the 27,569 SNVs in our data set (Markham and Zuker 2005, 2008). The variant of interest was placed at the center of each 41-bp window; however, if the variant was <20 bp from the end of the mRNA transcript, the 41-bp window was shifted such that the beginning or the end coincided with the beginning or the end of the mRNA transcript, as appropriate. The difference in ΔG (ΔΔG) between the BY and RM alleles for each 41-bp window was measured by simply subtracting the ΔG calculated for the RM allele from the ΔG calculated for the BY allele. We then calculated the absolute value of the difference in free energy between alleles, |ΔΔG| = |ΔGBY – ΔGRM|, for each variant. For each gene, we recorded the maximum |ΔΔG| we observed among all its variants.
Data access
Sequencing data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE60617.
Supplementary Material
Acknowledgments
We thank Jenny Madeoy and Marnie Johansson for expert technical assistance in strain construction, the Northwest Genomics Center for sequencing, and members of the Akey laboratory for helpful discussions and feedback on the manuscript. This work was supported by NIH grants 5R01GM094810 and 1R01GM098360 to J.M.A.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.175802.114.
References
- Abzhanov A, Protas M, Grant BR, Grant PR, Tabin CJ. 2004. Bmp4 and morphological variation of beaks in Darwin’s finches. Science 305: 1462–1465. [DOI] [PubMed] [Google Scholar]
- Blanchette M, Kent WJ, Riemer C, Elnitski L, Smit AF, Roskin KM, Baertsch R, Rosenbloom K, Clawson H, Green ED, et al. . 2004. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res 14: 708–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brem RB, Yvert G, Clinton R, Kruglyak L. 2002. Genetic dissection of transcriptional regulation in budding yeast. Science 436: 701–703. [DOI] [PubMed] [Google Scholar]
- Connelly CF, Wakefield J, Akey JM. 2014. Evolution and genetic architecture of chromatin accessibility and function in yeast. PLoS Genet 10: e1004427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degner JF, Marioni JC, Pai AA, Pickrell JK, Nkadori E, Gilad Y, Pritchard JK. 2009. Effect of read-mapping biases on detecting allele-specific expression from RNA-sequencing data. Bioinformatics 25: 3207–3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dori-Bachash M, Shema E, Tirosh I. 2011. Coupled evolution of transcription and mRNA degradation. PLoS Biol 9: e1001106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duff K, Huxley C. 1996. Targeting mutations to YACs by homologous recombination. Methods Mol Biol 54: 187–198. [DOI] [PubMed] [Google Scholar]
- Emilsson V, Thorleifsson G, Zhang B, Leonardson AS, Zink F, Zhu J, Carlson S, Helgason A, Walters GB, Gunnarsdottir S, et al. . 2008. Genetics of gene expression and its effect on disease. Nature 452: 423–428. [DOI] [PubMed] [Google Scholar]
- The ENCODE Project Consortium 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel SR, Dietrich FS, Fisk DG, Binkley G, Balakrishnan R, Costanzo MC, Dwight SS, Hitz BC, Karra K, Nash RS, et al. . 2013. The reference genome sequence of Saccharomyces cerevisiae: then and now. G3 4: 389–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garneau NL, Wilusz J, Wilusz CJ. 2007. The highways and byways of mRNA decay. Nat Rev Mol Cell Biol 8: 113–126. [DOI] [PubMed] [Google Scholar]
- Gerstein MB, Lu ZJ, Van Nostrand EL, Cheng C, Arshinoff BI, Liu T, Yip KY, Robilotto R, Rechtsteiner A, Ikegami K, et al. . 2010. Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science 330: 1775–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrick D, Parker R, Jacobson A. 1990. Identification and comparison of stable and unstable mRNAs in Saccharomyces cerevisiae. Mol Cell Biol 10: 2269–2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes KA, Ayroles JF, Reedy MM, Drnevich JM, Rowe KC, Ruedi EA, Cáceres CE, Paige KN. 2006. Segregating variation in the transcriptome: cis regulation and additivity of effects. Genetics 173: 1347–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones FC, Grabherr MG, Chan YF, Russell P, Mauceli E, Johnson J, Swofford R, Pirun M, Zody MC, White S, et al. . 2012. The genomic basis of adaptive evolution in threespine sticklebacks. Nature 484: 55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liti G, Carter DM, Moses AM, Warringer J, Parts L, James SA, Davey RP, Roberts IN, Burt A, Koufopanou V, et al. . 2009. Population genomics of domestic and wild yeasts. Nature 458: 337–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markham NR, Zuker M. 2005. DINAMelt web server for nucleic acid melting prediction. Nucleic Acids Res 33: W577–W581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markham NR, Zuker M. 2008. UNAFold: software for nucleic acid folding and hybridization. Methods Mol Biol 453: 3–31. [DOI] [PubMed] [Google Scholar]
- The modENCODE Consortium, Roy S, Ernst J, Kharchenko PV, Kheradpour P, Negre N, Eaton ML, Landolin JM, Bristow CA, Ma L, et al. . 2010. Identification of functional elements and regulatory circuits by Drosophila modENCODE. Science 330: 1787–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, Snyder M. 2008. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320: 1344–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonet M, Scafe C, Sexton J, Young R. 1987. Eucaryotic RNA polymerase conditional mutant that rapidly ceases mRNA synthesis. Mol Cell Biol 7: 1602–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pai AA, Cain CE, Mizrahi-Man O, De Leon S, Lewellen N, Veyrieras JB, Degner JF, Gaffney DJ, Pickrell JK, Stephens M, et al. . 2012. The contribution of RNA decay quantitative trait loci to inter-individual variation in steady-state gene expression levels. PLoS Genet 8: e1003000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petretto E, Mangion J, Dickens NJ, Cook SA, Kumaran MK, Lu H, Fischer J, Maatz H, Kren V, Pravenec M, et al. . 2006. Heritability and tissue specificity of expression quantitative trait loci. PLoS Genet 2: e172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell JK, Marioni JC, Pai AA, Degner JF, Engelhardt BE, Nkadori E, Veyrieras JB, Stephens M, Gilad Y, Pritchard JK. 2010. Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature 464: 768–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero IG, Ruvinsky I, Gilad Y. 2012. Comparative studies of gene expression and the evolution of gene regulation. Nat Rev Genet 13: 505–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronald J, Brem RB, Whittle J, Kruglyak L. 2005. Local regulatory variation in Saccharomyces cerevisiae. PLoS Genet 1: e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein R. 1995. Targeting, disruption, replacement, and allele rescue: integrative DNA transformation in yeast. In Guide to yeast genetics and molecular biology (ed. Guthrie C, Fink GR), pp. 281–301. Academic Press, San Diego. [Google Scholar]
- Schadt EE, Monks SA, Drake TA, Lusis AJ, Che N, Colinayo V, Ruff TG, Milligan SB, Lamb JR, Cavet G, et al. . 2003. Genetics of gene expression surveyed in maize, mouse and man. Nature 422: 297–302. [DOI] [PubMed] [Google Scholar]
- Schoenberg DR, Maquat LE. 2012. Regulation of cytoplasmic mRNA decay. Nat Rev Genet 13: 246–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skelly DA, Ronald J, Akey JM. 2009. Inherited variation in gene expression. Annu Rev Genomics Hum Genet 10: 313–332. [DOI] [PubMed] [Google Scholar]
- Skelly DA, Johansson M, Madeoy J, Wakefield J, Akey JM. 2011. A powerful and flexible statistical framework for testing hypotheses of allele-specific gene expression from RNA-seq data. Genome Res 21: 1728–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey JD. 2002. A direct approach to false discovery rates. J R Stat Soc Ser B Methodol 64: 479–498. [Google Scholar]
- Storey JD, Tibshirani R. 2003. Statistical significance for genome-wide experiments. Proc Natl Acad Sci 100: 9440–9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey JD, Taylor JE, Siegmund D. 2004. Strong control, conservative point estimation, and simultaneous conservative consistency of false discovery rates: a unified approach. J R Stat Soc Ser B Methodol 66: 187–205. [Google Scholar]
- Sun M, Schwalb B, Schulz D, Pirkl N, Etzold S, Larivière L, Maier KC, Seizl M, Tresch A, Cramer P. 2012. Comparative dynamic transcriptome analysis (cDTA) reveals mutual feedback between mRNA synthesis and degradation. Genome Res 22: 1350–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Reikhav S, Levy AA, Barkai N. 2009. A yeast hybrid provides insight into the evolution of gene expression regulation. Science 324: 659–662. [DOI] [PubMed] [Google Scholar]
- Tuller T, Waldman YY, Kupiec M, Ruppin E. 2010. Translation efficiency is determined by both codon bias and folding energy. Proc Natl Acad Sci 107: 3645–3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakefield J. 2013. Hypothesis testing and variable selection. In Bayesian and frequentist regression methods, pp. 157–200. Springer, New York. [Google Scholar]
- Wilusz CJ, Wormington M, Peltz SW. 2001. The cap-to-tail guide to mRNA turnover. Nat Rev Mol Cell Biol 2: 237–246. [DOI] [PubMed] [Google Scholar]
- Wittkopp PJ, Haerum BK, Clark AG. 2008. Regulatory changes underlying expression differences within and between Drosophila species. Nat Genet 40: 346–350. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.