

Abstract
The recent advent of ribosome profiling—sequencing of short ribosome-bound fragments of mRNA—has offered an unprecedented opportunity to interrogate the sequence features responsible for modulating translational rates. Nevertheless, numerous analyses of the first riboprofiling data set have produced equivocal and often incompatible results. Here we analyze three independent yeast riboprofiling data sets, including two with much higher coverage than previously available, and find that all three show substantial technical sequence biases that confound interpretations of ribosomal occupancy. After accounting for these biases, we find no effect of previously implicated factors on ribosomal pausing. Rather, we find that incorporation of proline, whose unique side-chain stalls peptide synthesis in vitro, also slows the ribosome in vivo. We also reanalyze a method that implicated positively charged amino acids as the major determinant of ribosomal stalling and demonstrate that it produces false signals of stalling in low-coverage data. Our results suggest that any analysis of riboprofiling data should account for sequencing biases and sparse coverage. To this end, we establish a robust methodology that enables analysis of ribosome profiling data without prior assumptions regarding which positions spanned by the ribosome cause stalling.
Translation of messenger RNAs into polypeptides by ribosomes is a fundamental process common to all life, and its dysregulation has been implicated in a wide range of diseases (Scheper et al. 2007). This has prompted a wealth of research into understanding the molecular underpinnings of translational dynamics. For instance, it has long been known that the frequency of codon usage in coding sequences (CDSs) is nonrandom, suggesting the action of natural selection on the efficiency and/or accuracy of translational elongation (Kanaya et al. 2001; Plotkin and Kudla 2011).
The origins of uneven codon usage have been studied extensively both experimentally and theoretically, implicating a number of different, nonmutually exclusive mechanisms—though all remain controversial (Gingold and Pilpel 2011; Plotkin and Kudla 2011). Much attention has been focused on the relationship between the cellular abundances of tRNAs and the frequencies of their cognate codons. Studies have found a strong correlation between gene expression levels and codon usage bias (CUB), revealing that highly expressed genes tend to use codons corresponding to the most abundant tRNAs in bacteria (Grantham et al. 1981), fungi (Bennetzen and Hall 1982), and metazoa (Shields et al. 1988; Stenico et al. 1994; Duret and Mouchiroud 1999); however, the abundances of charged tRNAs may be more important than total tRNA levels (Welch et al. 2009). As in vitro studies have shown that the rate of translation varies in a codon-specific manner, with the most rapid rates occurring at codons with highly abundant tRNAs (Varenne et al. 1984), it has long been presumed that CUB reflects selection for a high translational rate in highly expressed transcripts, minimizing sequestration of ribosomes at slowly translated codons (Andersson and Kurland 1990).
Other factors thought to slow translation rates include the presence of mRNA secondary structure, which must be “unwound” by ribosomes (Namy et al. 2006; Wen et al. 2008); wobble base-pairing, which can introduce nonoptimal geometries in codon–anticodon interactions (Thomas et al. 1988; Kato et al. 1990); codons encoding positively charged amino acids, which may participate in electrostatic interactions with the negatively charged ribosomal exit tunnel (Lu et al. 2007; Lu and Deutsch 2008; Tuller et al. 2011; Charneski and Hurst 2013); and proline, which is inefficiently incorporated into polypeptides due to the unique structure of its imino side-chain (Muto and Ito 2008; Wohlgemuth et al. 2008; Pavlov et al. 2009; Johansson et al. 2011; Doerfel et al. 2013; Gutierrez et al. 2013; Ude et al. 2013; Zinshteyn and Gilbert 2013). Interpretation of the relative contributions of these factors has been challenging, as their effects have typically been studied in conditions not normally encountered in living cells—such as within genes with low CUB but extremely high mRNA levels (Gingold and Pilpel 2011; Plotkin and Kudla 2011).
However, this situation has changed radically with the recent development of ribosome profiling, an in vivo technique for monitoring transcriptome-wide rates of translation (Ingolia et al. 2009). By isolating and sequencing short fragments of mRNA bound by actively translating ribosomes, “riboprofiling” provides nucleotide-resolution, quantitative information about the abundance and position of ribosomes on individual RNAs. When normalized for gene expression levels obtained by sequencing unprotected mRNA, increased ribosome-protected read coverage is expected from regions where ribosomes spend a greater fraction of their time, thereby identifying sequences that contribute to differences in rates of elongation (Ingolia et al. 2009, 2011).
Nevertheless, a number of recent studies that have analyzed the same yeast riboprofiling data (Ingolia et al. 2009) have come to contradictory conclusions regarding the major determinants of translation rate, including whether nonpreferred codons, RNA secondary structure, or particular amino acids stall translation (Kertesz et al. 2010; Siwiak and Zielenkiewicz 2010; Tuller et al. 2010a,b, 2011; Qian et al. 2012; Zur and Tuller 2012; Charneski and Hurst 2013; Wallace et al. 2013; Rouskin et al. 2014; Yang et al. 2014). Unfortunately, direct comparison of these analyses is challenging due to the differing assumptions made by each—such as the precise location of active sites in ribosome-protected fragments or the effects of sequences near ribosome-protected fragments.
An additional consideration is the possibility of sequence biases introduced during riboprofiling library construction. For example, such biases have been well documented in the case of RNA-seq library preparation, where local base composition of RNAs can produce undesirable secondary structure, bias reverse transcription priming, and interfere with enzymatic steps such as ligation (Zheng et al. 2011). Such effects manifest themselves as protocol-specific biases in read coverage along transcripts, leading to over- or underrepresentation of certain sequences (Bullard et al. 2010; Hansen et al. 2010; Li et al. 2010; Srivastava and Chen 2010; Zheng et al. 2011). In studies of ribosome-protected fragments, such biases could confound identification of the actual biological factors affecting translational rate. However, the riboprofiling protocol itself provides a means to mitigate technical biases introduced during library construction: As the sequencing libraries generated from both unprotected mRNA (the “mRNA” fraction) and ribosome-protected mRNA fragments (the “Ribo” fraction) differ only in the method used to isolate RNA, shared biases between the two are likely to represent technical artifacts (Qian et al. 2012).
In order to more thoroughly investigate factors that lead to increased ribosomal occupancy, we took advantage of two recently published yeast riboprofiling data sets that provide much higher coverage data than was previously available (Artieri and Fraser 2014; McManus et al. 2014) and compared them to the data of Ingolia et al. (2009). We observed consistent biases across data sets that could be attributed to library construction. Controlling for these artifacts identified codons uniquely enriched in the Ribo fractions of the high-coverage data sets, suggesting that they may contribute to ribosomal stalling in vivo.
Results
Riboprofiling data show consistent nucleotide biases
In order to explore how controlling for biases in library construction may affect our interpretation of sequences affecting translational rate, we analyzed two recently published Saccharomyces cerevisiae riboprofiling data sets (Artieri and Fraser 2014; McManus et al. 2014), hereafter, the “Artieri” and “McManus” data (Supplemental Table S1). These data sets have ∼28× and ∼7× greater sequencing depth than was previously available (Ingolia et al. 2009), respectively. As most previous studies of ribosomal occupancy (Kertesz et al. 2010; Siwiak and Zielenkiewicz 2010; Tuller et al. 2010a,b, 2011; Qian et al. 2012; Zur and Tuller 2012; Charneski and Hurst 2013; Wallace et al. 2013; Rouskin et al. 2014) reanalyzed the S. cerevisiae data generated by Ingolia et al. (2009) (the “Ingolia” data), we also included these data. The Ingolia data include two different growth conditions: rich and amino-acid-starved media (analysis of the starved data are in the Supplemental Material).
Reads from all samples were mapped to the S. cerevisiae genome (see Methods). Expression level estimates agreed well among replicates within each data set (Spearman’s ρ = 0.96–0.99 and 0.92–0.99 for the Ribo and mRNA fractions, respectively), as well as between data sets (ρ = 0.94–0.95 and 0.84–0.92 for the Ribo and mRNA fractions, respectively) (Supplemental Figs. S1, S2). The Ribo fractions of all three data sets showed an enrichment of reads mapping at 28–29 nucleotides (nt), as expected based on the size of the ribosome-protected fragment (Ingolia et al. 2009); however, the degree of enrichment varied among data sets (Supplemental Fig. S3; see Supplemental Material).
A larger proportion of the 5′ ends of Ribo fraction reads map to the first reading frame of codons compared with the second or third (Ingolia et al. 2009), suggesting that there may be differences among reads mapping to different reading frames. Therefore we analyzed the nucleotide content of the mRNA and Ribo fraction reads separately for those mapping to the first, second, or third frame of codons (Fig. 1).
Figure 1.
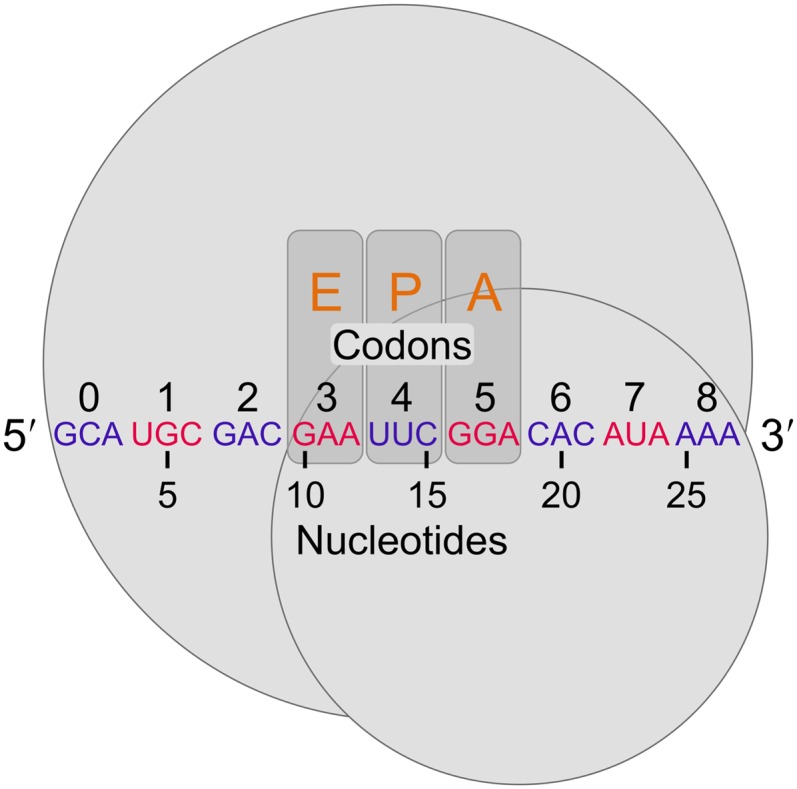
Defining positions relative to the 5′ end of riboprofiling reads. Following the mapping approach of Ingolia (2010), ribosomes (large and small subunits represented by gray circles) protect at least 27 nt of mRNA, corresponding to at least nine codons. Nucleotides and in-frame codons were counted from 5′ to 3′ as shown (arbitrary codons are indicated in alternating blue and red for clarity). In the figure, the ribosome-protected fragment begins in the first reading frame within a codon. However, for reads mapping to the second or third reading frames, while nucleotide counting begins at the first nucleotide, codon counting remains in-frame with the first codon, 0, corresponding to the one containing the first nucleotide. For reference, the orange letters indicate the codons that previous studies have indicated as the exit-tRNA (E-site), the peptidyl-tRNA (P-site), and aminoacyl-tRNA (A-site) sites, respectively (Ingolia et al. 2009; Stadler and Fire 2011; Li et al. 2012; Qian et al. 2012; Zinshteyn and Gilbert 2013).
We observed three general patterns of sequence bias. First, all data sets shared substantial biases in the 5′ ends of non-rRNA reads in both fractions, The most consistent of these is a preference for adenine in the 5′-most position, especially among first-frame mappers (Fig. 2; Supplemental Figs. S4, S5). In the case of the Ribo fraction of the Ingolia data, 66% of reads begin with adenine—twofold greater than the adenine content within CDSs (32.6%). In comparison, 34% of the Artieri and 33% of the McManus Ribo fraction reads begin with adenine (Supplemental Table S2; Supplemental Material). This 5′ bias is likely an artifact of library construction. Second, all data sets showed some depletion of cytosines at position 4, which was generally more pronounced in the Ribo fractions (see Supplemental Material; Supplemental Figs. S4, S5). Third, the 3′ termini of reads in the Artieri and Ingolia data sets showed a general preference for adenine, particularly in the mRNA fractions (Fig. 2). This is likely a consequence of the use of polyadenylation as a template to prime reverse transcription; the McManus data were generated with an alternative approach, which appears to mitigate this bias (see Supplemental Material).
Figure 2.

Patterns of nucleotide and codon representation across the three data sets. Reads were separated into those whose 5′ ends map to the first, second, or third reading frame within codons (frame 1, 2, or 3). The fold enrichment of each nucleotide was determined by dividing its number of counts at each position by the mean number of counts at positions within the same reading frame across the 27 nucleotides analyzed, thereby accounting for differences in expected nucleotide proportions among reading frames within codons. Enrichment is plotted in log2 scale: red, adenine; blue, cytosine; green, guanine; and yellow, thymine. Each codon position overlapped by each read was also determined by identifying the nine consecutive codons beginning from the 5′ end, as indicated in Figure 1. The gray bars indicate the coefficient of variation (CV) as a measure of the degree to which each position deviates from the expected background frequency of the 61 sense codons; codon position 4 is indicated for reference.
We assessed to what extent these sequence biases affected codon usage by identifying the nine in-frame codons spanned by each read (labeled positions 0–8 beginning from the 5′ end of mapping reads in Fig. 1) and determining the relative abundance of each of the 61 sense codons compared with its expected frequency across all reads (see Methods). We then calculated the coefficient of variation (CV) among the relative abundances at each of the nine positions, where higher CVs indicate a greater deviation from the expected codon frequencies (Fig. 2; Supplemental Figs. S4, S5). Both fractions of all three data sets showed strong biases in position 0, as was expected from the observed sequence biases.
Interestingly, the Ribo fractions of the Artieri and McManus data showed strong biases at internal codon positions relative to the mRNA fraction—particularly in the case of reads mapping to the first reading frame—coinciding with the expected location of active ribosomal sites (Fig. 1), suggesting that these may reflect a biological signal of ribosome stalling. A similar pattern was observed in the Ingolia data, though this was overshadowed by the stronger biases at 5′ codons (Fig. 2). We also noted that 28-nt reads, corresponding to the expected length of the ribosome-protected footprint, showed stronger internal codon biases in all three data sets compared with other mapping lengths (Supplemental Figs. S6, S7; Supplemental Material). In contrast, the less common second-frame mappers showed less pronounced internal codon biases. Interestingly, reads mapping to the third reading frame of codons in all three data sets were offset by +1 codon, indicating that the ribosome was likely positioned one codon downstream compared with first- and second-frame mappers.
Ribosome occupancy is associated with proline codons
Sequences that contribute to ribosome stalling should be enriched only in the Ribo fraction, whereas the identical methodology applied to library construction in both fractions will lead to shared technical artifacts (Ingolia et al. 2011; Stadler and Fire 2011; Qian et al. 2012). Therefore, we normalized Ribo fraction coverage by that of the mRNA fraction (hereafter, “corrected Ribo coverage”) as outlined in Figure 3 and Supplemental Figure S8. Unlike previous studies (Ingolia et al. 2009; Stadler and Fire 2011; Li et al. 2012; Qian et al. 2012; Zinshteyn and Gilbert 2013), we did not attempt to define specific positions within the ribosome-protected fragments corresponding to the ribosomal active sites to avoid any assumptions as to which position(s) were responsible for stalling. Instead, we analyzed a window from eight codons upstream of the 5′ end of Ribo fraction reads to eight codons downstream (labeled positions −8 to +8, with position 0 corresponding to the in-frame codon to which the 5′ end of the read mapped) (Supplemental Fig. S8); including codons upstream of the reads may reveal effects of already-incorporated amino acids on translation, such as interactions between positively charged residues and the exit tunnel (Charneski and Hurst 2013). The log2-transformed enrichment of each codon at each of the 17 positions was scaled by the mean value of all codons at the same position, such that codons with positive values were enriched and those with negative values were depleted (Fig. 3; see Methods). Due to differences in 5′ biases and coverage, we focused our analysis on first-frame mappers in the two higher-coverage data sets (for analysis of second- and third-frame mappers and the Ingolia data, see Supplemental Material).
Figure 3.

Steps in our calculation of corrected Ribo coverage. We analyzed Ribo fraction reads in a position-specific manner that controlled for shared biases between the two fractions while making no a priori assumptions about which codon position(s) may be most important in explaining patterns of coverage. (i) The 5′ ends of reads were mapped and codon-level coverage determined from each fraction separately. Only sites with data from both fractions were considered (excluded codons are indicated in gray). (ii) To account for coverage differences among genes, codon-level coverage values were scaled by the mean codon-level coverage of analyzed codons within each gene. (iii) These scaled values were used to calculate a log2(Ribo/mRNA) coverage ratio for each codon, thereby accounting for shared biases between the two fractions. (iv) Because increased coverage at the 5′ position of ribosome-protected fragments could be driven by sequence factors upstream or downstream, the log2(Ribo/mRNA) coverage at position 0 (green arrow) was recorded for all codons from −8 to +8 relative to the 5′ end for each analyzed site. The expected position of the ribosome is indicated for reference. (v) We repeated this across all analyzed codons in the transcriptome, generating a distribution for each of the 61 nonstop codons at each of the 17 positions, representing its position-specific relative contribution to ribosomal occupancy. (vi) Finally, the relative enrichment of each codon at each position was determined by scaling its mean log2(Ribo/mRNA) coverage value by the mean value of all 61 sense codons at that position, such that codons with positive log2 values were enriched relative to expectations and those with negative values were depleted (as plotted as in Fig. 4A).
The scaled enrichment values of all 61 sense codons in the Artieri data are shown in Figure 4A, revealing that the strongest enrichments occurred at position 4, the position with the most strongly biased codon representation specific to Ribo fraction reads (Fig. 2; for the values plotted in all heatmaps, see Supplemental Table S3). Nearly identical results were also observed in the McManus data (Supplemental Fig. S9). We note that position 0 also showed considerable enrichment, especially in the Ingolia data (Supplemental Figs. S10, S11). However, this is most likely due to the remaining effects of library construction (see Supplemental Material). In addition, there is no evidence of enrichment among upstream codons (−8 to −1), as would be expected if positive amino acids slow translation as they pass through the negatively charged ribosome exit tunnel (see Supplemental Material; Lu et al. 2007; Charneski and Hurst 2013). This pattern disappeared completely in both data sets when the order of codons was randomly shuffled within each gene, preserving the relative positions of mapped reads, indicating that it was not an artifact of the relationship between codon order and patterns of read mapping positions within transcripts (Supplemental Fig. S12). Furthermore, observed patterns were robust to differences in 5′-to-3′ coverage biases between fractions introduced by oligo-dT selection on the mRNA fraction (Supplemental Fig. S13; Zheng et al. 2011).
Figure 4.

The corrected Ribo coverage reveals a strong enrichment of proline codons. (A) Heatmap of the mean-scaled log2 enrichment of codon positions −8 to 8 in the Artieri data (the McManus data are similar) (Supplemental Fig. S9). All 61 sense codons are shown in alphabetical order indicated by their sequences on the left. Enriched codons are indicated by an increasing intensity of yellow color, while depleted codons are blue. Colored boxes to the right of each row indicate the biochemical category to which the codon belongs (color key is at the top of panel B). Codons associated with the E, P, and A active sites of the ribosome (positions 3, 4, and 5, respectively) are indicated. (B) Bar plots indicating the log2 enrichment values at position 4 of both the Artieri and McManus data sets. Codons are organized by amino acid using single-letter designations below the panel and grouped by biochemical type as indicated at the top of the panel. Individual codons for each amino acid are in alphabetical order. The 95% confidence intervals around the scaled enrichment values are indicated at the top of each bar. The asterisks indicate that proline (P) codons are more enriched than any other amino acid (Kruskal-Wallis rank sum test, P < 10−15).
As position 4 showed the strongest degree of preference for particular codons among internal positions (Figs. 2, 4A), we focused on this position, which has been defined by previous studies as the P-site (Ingolia et al. 2009; Stadler and Fire 2011; Li et al. 2012; Qian et al. 2012; Zinshteyn and Gilbert 2013). We first explored whether any biochemical properties of amino acids (i.e., positive, negative, polar, or hydrophobic) were significantly enriched (Fig. 4B). No category showed consistent enrichment, although both data sets did show a slight paucity of coverage among codons for hydrophobic amino acids.
Among individual amino acids, both data sets showed greater enrichment among the proline codons (CCN) than for any other amino acid (Kruskal-Wallis rank sum test, P < 10−15) (Fig. 4B). The four proline-encoding codons were among the five most enriched codons in both data sets (the fifth, CGG, encodes arginine; see below). We observed this enrichment at all gene expression levels (Supplemental Fig. S14), as well as in two additional riboprofiling data sets from the closely related species Saccharomyces paradoxus (Supplemental Figs. S15–S17; Artieri and Fraser 2014; McManus et al. 2014). These results are consistent with proline’s previously implicated role in translational pausing in vitro (Wohlgemuth et al. 2008; Pavlov et al. 2009; Johansson et al. 2011).
We next tested whether other factors were associated with ribosomal occupancy. Previous analyses of riboprofiling data have suggested that mRNA secondary structure can slow translation (Tuller et al. 2010b, 2011; Charneski and Hurst 2013; Yang et al. 2014). Therefore, we searched for evidence of increased corrected Ribo coverage upstream of regions of mRNA secondary structure (Ouyang et al. 2013). However, we observed that secondary structure had stronger correlations with terminal adenine biases than with any signal of ribosome stalling (Supplemental Fig. S18; Supplemental Material). In addition, although G:U wobble base-pairing has been associated with pausing in nematodes and humans (Stadler and Fire 2011), we observed no such pattern in yeast (Supplemental Fig. S19).
Finally, supporting previous riboprofiling-based observations made in yeast (Qian et al. 2012; Zinshteyn and Gilbert 2013), Escherichia coli (Li et al. 2012), and mouse (Ingolia et al. 2011), we found no correlation between corrected Ribo coverage and nonoptimality of the codons at either position 4 (P-site) or position 5 (A-site) using three separate measures of codon optimality (Supplemental Figs. S20, S21; Supplemental Material). Interestingly, the rarest codon in S. cerevisiae, CGG (encoding arginine), showed a substantial level of enrichment in both data sets (Fig. 4B). However, this may not be related to its rarity, as similarly rare codons (CGC and CGA, also encoding arginine), showed no such enrichment.
Patterns of bias and enrichment in riboprofiling data from other species
To test whether the sequence biases that we observed were a general feature of riboprofiling data, we applied our analysis method to data sets from two additional species: one generated in the nematode Caenorhabditis elegans (Stadler and Fire 2013) and the other in the zebrafish Danio rerio (see Supplemental Material; Supplemental Table S4; Bazzini et al. 2014).
Patterns of sequence bias in the C. elegans data were similar to those observed in yeast, which was not surprising as it was generated using a nearly identical protocol (Supplemental Fig. S22). Perhaps because the per-base coverage was ∼20-fold lower than the Artieri data, codon enrichments were weak (Supplemental Fig. S23). However, we did observe increased ribosomal occupancy among G:U wobble-pairing codons at position 4 (Supplemental Fig. S24), similar to Stadler and Fire (2011), suggesting that the different patterns observed in yeast are not simply a result of analytical differences.
In contrast to the other species, the zebrafish data showed completely different patterns of sequence bias that were also largely fraction specific (Supplemental Fig. S25), likely reflecting the investigators’ use of a different method for both mRNA isolation and monosome purification. Nevertheless, despite these differences, biases were pronounced in both the 5′ and 3′ ends of reads. As was the case in the yeast data, biases at internal codon positions were unique to the Ribo fractions, though the position of strongest bias was shifted by +1 codon (position 5), which may reflect differences between species in the size of the ribosome-protected fragment as well as the specific positioning of the ribosomal active sites (Stadler and Fire 2011).
In order to increase our power to detect enriched codons, we separately pooled all mRNA and all Ribo fraction reads for analysis (resulting in about fourfold lower per-base coverage than the Artieri data) (see Supplemental Methods). Consistent with our observations in yeast, all four proline (P) codons were enriched at position 4 (P-site) (Supplemental Fig S26). Furthermore, three of the four proline codons were also enriched at position 5, which is the position showing the strongest deviation from expected codon frequencies (Supplemental Fig. S25). Therefore the stalling effect of proline incorporation appears to be conserved between yeast and vertebrates.
Revisiting the effects of positively charged amino acids
A recent reanalysis of the Ingolia data concluded that positively charged amino acids were the primary determinant of ribosomal velocity (Charneski and Hurst 2013). Their approach assumed that upon encountering a sequence feature causing ribosomal stalling (such as a positive amino acid), the ribosome slows, leading to an accumulation of Ribo fraction reads immediately downstream from the feature. By comparing the magnitude of this accumulation to read coverage upstream of the stalling sequence—where the rate of translation was presumed to be unhindered—they generated a normalized metric of stalling as shown in Figure 5. Specifically, to test the effect of a codon at position 0, the occupancy of all codon positions (rpos) from 30 codons upstream to 30 codons downstream was divided by the mean occupancy of upstream codons −30 to −1 (rprec30), producing a normalized pausing value (rpos/rprec30), where a value of one represents the average rate of translation. The area under the curve (AUC) of the mean-normalized occupancy values from position 0 until the position where mean occupancy returned to the average was used as a measure of the stalling effect, if positive (Fig. 5).
Figure 5.

The rpos/rprec30 method of Charneski and Hurst. (A) As a measure of the stalling effect of a codon (or group of codons beginning) at position 0, the occupancy of all codon positions (rpos) from 30 codons upstream (position −30) to 30 codons downstream (position 30) of the putative stalling codon was divided by the mean occupancy of upstream codons −30 to −1 (rprec30, indicated by the bracket). (B) This produced a normalized pausing value (rpos/rprec30), where a value of one represents the average rate of translation. (C) After averaging the rpos/rprec30 values among all similar groups of codons, the AUC (indicated by the shaded purple area) of the mean-normalized occupancy values from position 0 until the position where mean occupancy returned to the average was used as a measure of the stalling effect (if positive).
We sought to test if the stalling effect of positive amino acids was also detected in the higher-coverage Artieri and McManus data sets. We first replicated the pattern of increased stalling with increasingly large clusters of positive amino acids (Fig. 5 in Charneski and Hurst 2013) using the Ingolia data, confirming that the same methods were being used (Supplemental Fig. S27). However, applying this approach to both higher-coverage data sets showed no such trend (Fig. 6A,B). Similarly, using our analysis framework we also found no enrichment of positive amino acids among upstream codons (position −8 to −1) in any of the S. cerevisiae riboprofiling data sets (Supplemental Fig. S28; Supplemental Material).
Figure 6.

No evidence of stalling at positive amino acids. We recalculated Charneski and Hurst’s (2013) Figure 5 using either the Artieri (A) or the McManus (B) data. Following the published approach, clusters of increasing numbers of positive amino acid encoding codons were identified within the range bounded by pairs of inverted triangles. The horizontal gray line indicates the average rate of translation. Error bars, ±SEM. No additive effect is observed in either high-coverage data set, in contrast to the Ingolia data (Supplemental Fig. S27); the AUCs for one, two, three, four or five, and six or more positive charge clusters were 7.89, 12.83, −0.71, −1.36, and −2.75 for the Artieri data, and 6.46, 0.08, −0.59, 0.04, and 0.09 for the McManus data, respectively. (C) The data from Charneski and Hurst’s (2013) Figure 5 (black) compared to the mean rpos/rprec30 generated from 100 random samplings of 61-codon windows devoid of any positive amino acid encoding codons (red). The average stalling pattern of windows lacking any positive charges is stronger than that observed in any of the clusters (Kruskal-Wallis rank sum test of distributions’ AUC values, P < 10−15 for all clusters except for six or more positive charges, where P = 0.02 after Bonferroni correction for multiple tests). Therefore the observed stalling effect of positive amino acids is not greater than what would be expected by chance within the Ingolia data.
To further investigate this discrepancy, we performed an important control not reported in the original analysis (Charneski and Hurst 2013): levels of apparent stalling in the absence of any positive amino acids, using the same data set (see Methods) (Ingolia et al. 2009). We found that the median apparent stalling effect was actually stronger in the absence of any positively charged residues than in any sized clusters of positive charges (Fig. 6C). We observed a similar pattern of stalling when averaging over all possible 61-codon windows in all genes (Supplemental Fig. S29), suggesting that the apparent pattern of stalling is unlikely to be related to the presence of positively charged amino acids.
We then explored whether read coverage could affect these patterns even in the absence of any stalling by generating simulated data at a range of coverage levels. Indeed, we observed stalling in low-coverage but not high-coverage windows (Supplemental Fig. S29; Supplemental Material). Since the simulated data contained no actual stalling, we concluded that the rpos/rprec30 method detects stalling in any series of windows with sparse read coverage. As a further test, we down-sampled the higher-coverage data to the level used in the original analysis and found that overall patterns of stalling indeed increased (Supplemental Fig. S30).
Discussion
Library construction biases
The relative importance of various factors in influencing the rate of translation has remained controversial, despite recent advances in our ability to measure translation rates at the level of individual codons (Gingold and Pilpel 2011; Plotkin and Kudla 2011). Most of these factors were originally identified using in vitro approaches, which may not accurately represent intracellular conditions. As an in vivo method, riboprofiling has offered an unprecedented opportunity to study translational dynamics in living cells; yet a number of different studies reanalyzing the same riboprofiling data (Ingolia et al. 2009) have produced incompatible findings, based on differing assumptions and methods of analysis (Kertesz et al. 2010; Siwiak and Zielenkiewicz 2010; Tuller et al. 2010a,b, 2011; Qian et al. 2012; Zur and Tuller 2012; Charneski and Hurst 2013; Wallace et al. 2013; Rouskin et al. 2014; Yang et al. 2014).
Our approach presents a number of improvements over previous analyses. First, we have explicitly taken into account shared technical biases between the Ribo and mRNA fractions. Second, we made no a priori assumptions regarding which codon positions near the ribosome-protected fragments were responsible for rate variation, but rather focused on codon position 4 in yeast because it was a clear outlier in terms of enrichment in corrected Ribo coverage. Third, we analyzed two independently generated, high-coverage yeast data sets (Artieri and Fraser 2014; McManus et al. 2014) and found strong agreement between them. And fourth, we found sequence biases in riboprofiling data from other species, as well as conservation of the stalling effect of proline in zebrafish.
Our analysis revealed that like other next-generation sequencing methods (Bullard et al. 2010; Hansen et al. 2010; Li et al. 2010; Srivastava and Chen 2010; Zheng et al. 2011), riboprofiling is subject to library construction biases that may confound any analysis of ribosomal occupancy. In particular, non-rRNA mapping reads from both fractions of all yeast data sets, as well as the C. elegans data (Stadler and Fire 2013), showed a substantial preference for adenine bases at the 5′ ends of reads (and in some instances, the 3′ ends as well), as well as a paucity of cytosines four bases from the 5′ end (Fig. 2; Supplemental Material). Furthermore, zebrafish libraries generated with another method show evidence of their own distinct biases (Supplemental Fig. S25). As the majority of reads from the Ribo fraction mapped to the first reading frame of codons, this produces a skewed representation of reads mapping to codons that begin with these bases. In the Ingolia data in particular, the biases at the 5′ ends of reads overwhelmed those of all other positions, suggesting that patterns of coverage are strongly influenced by this library construction bias (Fig. 2; Supplemental Fig. S28; Supplemental Material).
It is also important to note that an additional caveat applicable to all riboprofiling data sets discussed in this article is the use of cycloheximide to arrest translation immediately prior to RNA extraction (Ingolia et al. 2009; Stadler and Fire 2013; Zinshteyn and Gilbert 2013; Artieri and Fraser 2014; Bazzini et al. 2014; McManus et al. 2014). Cycloheximide binds to the occupied E-site of the ribosome and prevents translocation by inhibiting the release of the uncharged tRNA (Obrig et al. 1971; Schneider-Poetsch et al. 2010). This has the effect of stabilizing ribosomes during a specific phase of the elongation cycle, which may obscure the effects of sequences that exert their effect during other steps of elongation (Lareau et al. 2014). Furthermore, it is unknown whether cycloheximide shows preferences for particular tRNAs or local sequence context, but if it does, this could produce artifactual signals of ribosome accumulation that mask true biological signals of ribosome stalling.
Proline codons are enriched in corrected Ribo coverage
Of the features previously implicated in modulating the rate of translation in yeast, we observed consistent enrichment of Ribo coverage only at proline codons (Fig. 4B): In both S. cerevisiae and S. paradoxus, all four proline codons (CCN) were among the most significantly enriched at codon position 4 in both the Artieri and McManus data. Furthermore, we also observed enrichment of proline codons at positions 4 and 5 in zebrafish (Supplemental Fig. S26). Interestingly, position 4 corresponds to what previous studies have defined as the P-site (Ingolia et al. 2009; Stadler and Fire 2011; Li et al. 2012; Zinshteyn and Gilbert 2013), where the imino side-chain of proline is known to act as a particularly poor substrate in the peptidyl transfer reaction. This is likely due to its restricted conformational flexibility, which may limit the rate of translational elongation (Wohlgemuth et al. 2008; Pavlov et al. 2009). Proline’s ribosomal pausing effect is known to play an important role in programmed stalling (Garza-Sanchez et al. 2006; Tanner et al. 2009), and previous riboprofiling studies have found an enrichment of proline codons in the context of multi-amino-acid motifs (PPE [Ingolia et al. 2011] and PG [Zinshteyn and Gilbert 2013]), even in cells not treated with cycloheximide (Ingolia et al. 2011).
No evidence that positive amino acids stall ribosomes
Though several recent studies have suggested that positively charged amino acids may impede the progress of the peptide chain through the negatively charged ribosomal exit tunnel (Lu et al. 2007; Lu and Deutsch 2008), we observed no consistent enrichment for codons encoding positive amino acids in corrected Ribo coverage either within or upstream of the footprints in any data sets (Fig. 4B; Supplemental Figs. S9, S10, S16, S23, S26, S28). Two previous studies found an association between riboprofiling read coverage and the presence of positive amino acids in yeast—both based on reanalysis of the data of Ingolia et al. (2009). The first (Tuller et al. 2011) noted an association between ribosomal occupancy at the 5′ ends of CDSs and codons encoding positive amino acids; however, this can be explained entirely by the requirements of hydrophilic N-termini of transmembrane proteins (Charneski and Hurst 2014). The results of the second study (Charneski and Hurst 2013) were not supported by either high-coverage data set (Fig. 6). Furthermore, upon reanalysis of the method previously employed, we found that it led to false signals of stalling in low-coverage windows, indicating apparent pausing even in simulated data where no pausing was present, and produced the strongest signals of ribosome pausing in regions containing no positive codons at all (Fig. 6C). Therefore we conclude that there is no evidence for a stalling effect of positive amino acids in vivo.
Other factors associated with ribosomal stalling
Multiple studies have shown that mRNA secondary structure plays an important role in regulating translational initiation (Schauder and McCarthy 1989; Kudla at al. 2009; Goodman et al. 2013; Shah et al. 2013). However, its importance in affecting the rate of ribosomal elongation remains controversial. For instance, a recent study concluded that yeast mRNA secondary structure is far less extensive in vivo than in vitro and is poorly predicted by computational methods (Rouskin et al. 2014). Furthermore, analyses of the effects of structure using yeast riboprofiling data have been inconclusive (Tuller et al. 2011; Zur and Tuller 2012; Charneski and Hurst 2013; Yang et al. 2014). Because mRNA structure is influenced by base content (since G:C bonds are stronger than A:U bonds), biases including the enrichment of adenines at both termini of reads overwhelms any potential signal of increased Ribo occupancy near regions of secondary structure. Therefore, riboprofiling data may not be ideal for studies of the effect of mRNA structure in the absence of methodological developments that control for biases introduced during library construction.
Analysis of base-level riboprofiling data
Riboprofiling represents a significant advance over previous methods of translational analysis by enabling measurements of ribosomal occupancy across the transcriptome. While this approach has dramatically increased our knowledge of translational regulation and evolution (Ingolia et al. 2009, 2011; Brar et al. 2012; Li et al. 2012; Stadler and Fire 2013; Artieri and Fraser 2014; Bazzini et al. 2014; Lareau et al. 2014; McManus et al. 2014), inconsistent interpretation of nucleotide-level data has produced contradictory results and made direct comparisons between studies challenging. We conclude that mitigating technical biases in riboprofiling—either experimentally or computationally—will likely reveal additional features of mRNAs that are most relevant to translational biology.
Methods
Riboprofiling data
The Saccharomyces riboprofiling data used in this study were obtained from Artieri and Fraser (2014), McManus et al. (2014), and Ingolia et al. (2009) (Gene Expression Omnibus [GEO] entries GSE50049, GSE52119, and GSE13750, respectively). In the case of the Artieri data, some of these samples were sequenced by mixing riboprofiling libraries generated from both S. cerevisiae and the closely related species S. paradoxus (Supplemental Table S5). Therefore we independently sequenced the S. cerevisiae Ribo fraction replicate 1 sample (deposited in the NCBI Sequence Read Archive [SRA] entry SRS514738) and mapped the reads (see below) in parallel to the sample generated by sequencing the mixed species libraries (GEO sample GSM1278062). The strong congruence of estimated RPKMs between the individual and the multiplexed sequencing samples (rho = 0.995, P < 10−15) (Supplemental Fig. S31), as well as patterns of nucleotide and codon enrichment between the mixed and nonmixed biological replicates (Supplemental Figs. S5, S32), indicated that the stringent mapping method successfully identified S. cerevisiae reads from the mixed sample. Supplemental Table S5 indicates the sources of the individual replicates.
Riboprofiling library mapping
Reads from both fractions of all yeast data sets were mapped in a strand-specific manner using the iterative method described in Ingolia (2010). We first excluded reads that mapped to the complete rDNA sequence of S. cerevisiae when trimmed to a length of 23 nt from the 5′ end using Bowtie version 0.12 (Langmead et al. 2009), allowing three mismatches and a maximum of 40 mapping locations. Remaining reads were mapped to the S. cerevisiae strain S288c genome (R61-1-1, June 2008), allowing no multimappers and no mismatches. Unmapped reads were then subjected to a second round of mapping to a reference consisting of the CDSs of nonmitochondrial, nondubious genes present in annotation R61-1-1 in order to account for splice-junction spanning reads. We observed that allowing mismatches when mapping to the genome distorted the pattern of the first reading frame preference of Ribo fraction reads, consistent with the known property of reverse transcriptase to add untemplated bases the 5′ end during cDNA synthesis (Supplemental Fig. S33; Supplemental Material; Zajac et al. 2013). Because fewer reads mapped with mismatches than without, and because these reads showed inconsistent patterns of reading frame distortion among samples, we chose to retain only nonmismatch mapping reads. However, we note that retaining mismatch mapping reads does not change our conclusions regarding the biological factors causing ribosome stalling (Supplemental Fig. S34). Nonmismatch mapping reads were filtered such that no more than 30 bp and no less than 27 bp mapped. Uniquely mapping reads were then assigned to the CDSs if their 5′-most base mapped at or between the 16th codon and 16 codons before the end, in order to avoid effects of ribosomes paused near the start and stop codons (Ingolia et al. 2009, 2011). Only protein-coding genes with 40 or more codons were analyzed.
The read mapping length distribution (Supplemental Fig. S3) was determined using the iterative trimming method as above on all non-rRNA mapping reads but instead began with reads trimmed to 35 bp (the shortest read length generated among all three data sets) and trimming 1 nt at a time until reaching 23 nt, retaining the longest mapping read length. Barplots were then generated by determining the percentage of reads mapping at each length among all mapping reads.
Identifying technical biases in riboprofiling data
Non-rRNA mapped reads were separated into categories based on whether their 5′ ends mapped to the first, second, or third reading frame of codons. The relative proportion of each base among reads was calculated for the first 27 nt of each read (corresponding to the minimum mapping read length) (the number of nucleotides analyzed was extended accordingly for Supplemental Fig. S7). Nucleotide bias was then determined by scaling the proportions of each base within each reading frame by its mean proportion across all of the same positions within codons (i.e., all first, second, or third positions) in the 27 nt, thereby accounting for codon-position–specific differences in expected base compositions. The ratios were log2-transformed for the purpose of plotting. In order to determine the degree of overrepresentation of adenines at the 5′ ends of reads, the proportion of adenines in the first nucleotide position of mapping reads was compared to the proportion of adenines within the CDSs of analyzed genes (see also Supplemental Table 2).
The corresponding codon bias was determined by a fraction-specific method analogous to that presented in Figure 3 and Supplemental Figure S8: The 5′ ends of reads from each fraction were mapped separately, and the codon-level coverage was determined, retaining only codons with 5′ mapped reads in both fractions for analysis. Within each gene, codon-level coverage values for each fraction were separately scaled by the mean codon-level coverage of analyzed codons in order to account for coverage differences among genes. These scaled values were then log2-transformed (e.g., log2[scaled mRNA coverage] or log2[scaled Ribo coverage]) and then applied to the 5′ mapping codon and to the eight consecutive codons downstream (labeled 0–8; representing the minimum number of codons overlapped by a short read), producing a coverage value for each codon at each position. In this manner, the mean log2 coverage value for each of the 61 sense codons at each position was determined. We then asked whether the codons at each position were over- or underrepresented relative to all nine positions by scaling the log2 coverage value of each codon at each position by the mean log2 coverage value across all nine positions, producing a new value that represents the degree to which each of the 61 sense codons deviates from its mean representation across the length of the read. To represent the degree to which each position deviated from expected codon frequencies in a graphical manner, we calculated the coefficient of variation (CV)—the standard deviation expressed as a percentage of the mean—across 61 sense codons at each position, where higher CVs indicate positions with a greater deviation from expected codon proportions.
Determination of position-specific corrected Ribo coverage
In order to account for mapping biases shared between the mRNA and Ribo fractions in a position-specific manner, Ribo fraction occupancy was scaled by that of the mRNA fraction in the manner outlined in Figure 3 and Supplemental Figure S8: The 5′ ends of reads from both fractions were mapped as detailed above, and the codon-level coverage was determined for each fraction separately, retaining only codons with 5′ mapped reads from both fractions for analysis. Within each gene, codon-level coverage values were scaled by the mean codon-level coverage of analyzed codons. These scaled values were used to calculate the log2(Ribo/mRNA coverage) for each codon, accounting for shared biases between the two fractions. This log2(Ribo/mRNA coverage) at position 0 was then recorded in a codon and position-specific manner (from −8 to +8 codons relative to the codon overlapped by the 5′ end, representing 17 codons in total). Performing this analysis over all positions with data within the coding transcriptome produced a distribution of log2(Ribo/mRNA coverage) values for each of the 61 nonstop codons at each of the 17 positions representing that codon’s contribution to ribosomal pausing, given its position relative to the ribosome-protected fragment (represented in tabular format by the mean log2[Ribo/mRNA coverage] of each codon at each position). The relative enrichment of each codon at each position was determined by scaling its mean log2(Ribo/mRNA coverage) value by the mean value of all codons at that position such that codons with positive log2 values were enriched relative to expectations and those with negative values were depleted. In cases where a codon was not represented at a particular position, which only occurred when data were down-sampled or divided into low-coverage subgroups, the codon was given a log2(Ribo/mRNA coverage) value of zero at that position. Supplemental File 1 contains scripts and code required to map riboprofiling data and perform the analyses noted above.
As a negative control, the analysis was rerun 100 times on data sets in which the genomic coordinates of the 5′ ends of mapping reads were preserved but the order of the codons within each gene was shuffled at random. The start and stop codons were always excluded from the shuffling by virtue of the exclusion of codons at the beginning and ends of transcripts (see above).
Analysis of factors implicated in affecting rates of translation
Codons were grouped into standard biochemical categories (i.e., positively charged, negatively charged, polar noncharged, and hydrophobic) plus an additional “special” category containing cytosine, glycine, and proline. Wobble base positions in S. cerevisiae were obtained from Percudani and Ottonello (1999). Positions within mRNAs in either single-stranded or double-stranded conformation were obtained from Ouyang et al. (2013). The three different optimality measures used were relative synonymous codon usage (RSCU) (Sharp and Li 1987), absolute adaptiveness (Wi) (dos Reis et al. 2004), and the normalized translational efficiency scale (nTE) (Pechmann and Frydman 2013).
Application of the Charneski and Hurst method
The rpos/rprec30 values for 61-codon windows centered on the first positive amino acid encoding codon of a cluster of positive charges were determined as indicated in Charneski and Hurst (2013). The number of positive amino acids in each cluster (one, two, three, four or five, and six or more), as well as the maximum number of codons spanned by a cluster, were also defined as in Charneski and Hurst (2013). Codon level coverage was calculated as the mean nucleotide coverage within a codon. To reproduce the results of the original analysis, we combined the replicate data as per their method: taking the average of the coverage in each replicate. Note however, that unlike the original analysis, we did not map reads to the mitochondrial transcriptome as it is unclear whether translational dynamics are affected by differences between the cytoplasmic and mitochondrial ribosomes and tRNA pools.
To determine whether a stalling effect was observed within regions without positive charges, we identified all 61-codon windows that do not contain any positive amino acids and treated the center codon as the focal position for calculating the rpos/rprec30 values. As many such windows are immediately adjacent to one another (e.g., a run of 70 nonpositive amino acids will contain 10 possible 61-codon windows), we subsampled a number of positions equivalent to the number of “1 positive charge” clusters used to draw panel 1 of Supplemental Fig. S27 at random 100 times from all possible windows lacking any positive amino acids, and averaged the rpos/rprec30 values over the replicate subsamples. To test whether the stalling effect of subsampled data was significantly different from the observed data, we performed Kruskal-Wallis rank sum tests (see below) on the distribution of AUC values from all of the positions analyzed in the actual data in comparison to the mean AUCs of the 100 randomly sampled replicates.
In order to explore how lower read coverage influenced the appearance of ribosomal slowing in 61-codon windows, we simulated either 10, 100, or 1000 reads per CDS with random mapping location and an equal probability of any length from 27 to 30 nt. The start and stop positions of the CDSs were based on the definition of CDS mapping reads used in Charneski and Hurst (2013): The 5′ end of reads mapped between 16 nt before the start and 14 nt before the end of the annotated CDSs.
Statistics
All statistics were performed using R version 2.14.0 (R Core Team 2013) in addition to custom Perl scripts. The 95% confidence intervals were empirically determined from the distribution of log2(Ribo/mRNA coverage) values from the data using the “boot” package (Davison and Hinkley 1997). Kruskal-Wallis tests were performed using 10,000 permutations of the data as implemented in the “coin” package (Hothorn et al. 2008).
Data access
The raw sequencing reads generated in this study have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra) under accession number SRS514738. The locations of all other data are indicated in the Methods and Supplemental Table S3.
Supplementary Material
Acknowledgments
We thank members of the Fraser laboratory, the Stanford Whole Genome Sequencing group, and three anonymous reviewers for useful comments. We also thank Catherine Charneski for helpful discussions and sharing computational code for analysis. This work was supported by a Natural Sciences and Engineering Research Council of Canada Postdoctoral Fellowship to C.G.A. and NIH grant 1R01GM097171-01A1. H.B.F. is a Sloan Fellow and Pew Scholar. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author contributions: C.G.A. and H.B.F. designed the study. C.G.A. generated the experimental data and performed the analysis. C.G.A. and H.B.F. wrote the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.175893.114.
References
- Andersson SG, Kurland CG. 1990. Codon preferences in free-living microorganisms. Microbiol Rev 54: 198–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artieri CG, Fraser HB. 2014. Evolution at two levels of gene expression in yeast. Genome Res 24: 411–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazzini AA, Johnstone TG, Christiano R, Mackowiak SD, Obermayer B, Fleming ES, Vejnar CE, Lee MT, Rajewsky N, Walther TC, et al. 2014. Identification of small ORFs in vertebrates using ribosome footprinting and evolutionary conservation. EMBO J 33: 981–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennetzen JL, Hall BD. 1982. Codon selection in yeast. J Biol Chem 257: 3026–3031. [PubMed] [Google Scholar]
- Brar GA, Yassour M, Friedman N, Regev A, Ingolia NT, Weissman JS.. 2012. High-resolution view of the yeast meiotic program revealed by ribosome profiling. Science 335: 552–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullard JH, Purdom E, Hansen KD, Dudoit S. 2010. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinformatics 11: 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charneski CA, Hurst LD. 2013. Positively charged residues are the major determinants of ribosomal velocity. PLoS Biol 11: e1001508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charneski CA, Hurst LD. 2014. Positive charge loading at protein termini is due to membrane protein topology, not a translational ramp. Mol Biol Evol 31: 70–84. [DOI] [PubMed] [Google Scholar]
- Davison AC, Hinkley DV. 1997. Bootstrap methods and their application. Cambridge University Press, Cambridge, UK. [Google Scholar]
- Doerfel LK, Wohlgemuth I, Kothe C, Peske F, Urlaub H, Rodnina MV. 2013. EF-P is essential for rapid synthesis of proteins containing consecutive proline residues. Science 339: 85–88. [DOI] [PubMed] [Google Scholar]
- dos Reis M, Savva R, Wernisch L. 2004. Solving the riddle of codon usage preferences: a test for translational selection. Nucleic Acids Res 32: 5036–5044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duret L, Mouchiroud D. 1999. Expression pattern and, surprisingly, gene length shape codon usage in Caenorhabditis, Drosophila, and Arabidopsis. Proc Natl Acad Sci 96: 4482–4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garza-Sánchez F, Janssen BD, Hayes CS. 2006. Prolyl-tRNAPro in the A-site of SecM-arrested ribosomes inhibits the recruitment of transfer-messenger RNA. J Biol Chem 281: 34258–34268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingold H, Pilpel Y. 2011. Determinants of translation efficiency and accuracy. Mol Syst Biol 7: 481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman DB, Church GM, Kosuri S. 2013. Causes and effects of N-terminal codon bias in bacterial genes. Science 342: 475–479. [DOI] [PubMed] [Google Scholar]
- Grantham R, Gautier C, Gouy M, Jacobzone M, Mercier R. 1981. Codon catalog usage is a genome strategy modulated for gene expressivity. Nucleic Acids Res 9: r43–r74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez E, Shin BS, Woolstenhulme CJ, Kim JR, Saini P, Buskirk AR, Dever TE. 2013. eIF5A promotes translation of polyproline motifs. Mol Cell 51: 35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KD, Brenner SE, Dudoit S. 2010. Biases in Illumina transcriptome sequencing caused by random hexamer priming. Nucleic Acids Res 38: e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hothorn T, Hornik K, van de Wiel MA, Zeileis A. 2008. Implementing a class of permutation tests: the coin package. J Stat Softw 28: 1–23. [Google Scholar]
- Ingolia NT. 2010. Genome-wide translational profiling by ribosome footprinting. Methods Enzymol 470: 119–142. [DOI] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS. 2009. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324: 218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Lareau LF, Weissman JS. 2011. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 147: 789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson M, Ieong KW, Trobro S, Strazewski P, Åqvist J, Pavlov MY, Ehrenberg M. 2011. pH-sensitivity of the ribosomal peptidyl transfer reaction dependent on the identity of the A-site aminoacyl-tRNA. Proc Natl Acad Sci 108: 79–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaya S, Yamada Y, Kinouchi M, Kudo Y, Ikemura T. 2001. Codon usage and tRNA genes in eukaryotes: correlation of codon usage diversity with translation efficiency and with CG-dinucleotide usage as assessed by multivariate analysis. J Mol Evol 53: 290–298. [DOI] [PubMed] [Google Scholar]
- Kato M, Nishikawa K, Uritani M, Miyazaki M, Takemura S. 1990. The difference in the type of codon-anticodon base pairing at the ribosomal P-site is one of the determinants of the translational rate. J Biochem 107: 242–247. [DOI] [PubMed] [Google Scholar]
- Kertesz M, Wan Y, Mazor E, Rinn JL, Nutter RC, Chang HY, Segal E. 2010. Genome-wide measurement of RNA secondary structure in yeast. Nature 467: 103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudla G, Murray AW, Tollervey D, Plotkin JB. 2009. Coding-sequence determinants of gene expression in Escherichia coli. Science 324: 255–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lareau LF, Hite DH, Hogan GJ, Brown PO. 2014. Distinct stages of the translation elongation cycle revealed by sequencing ribosome-protected mRNA fragments. eLife 3: e01257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Jiang H, Wong WH. 2010. Modeling non-uniformity in short-read rates in RNA-Seq data. Genome Biol 11: R50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li GW, Oh E, Weissman JS. 2012. The anti-Shine-Dalgarno sequence drives translational pausing and codon choice in bacteria. Nature 484: 538–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Deutsch C. 2008. Electrostatics in the ribosomal tunnel modulate chain elongation rates. J Mol Biol 384: 73–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Kobertz WR, Deutsch C. 2007. Mapping the electrostatic potential within the ribosomal exit tunnel. J Mol Biol 371: 1378–1391. [DOI] [PubMed] [Google Scholar]
- McManus J, May G, Spealman P, Shteyman A. 2014. Ribosome profiling reveals post-transcriptional buffering of divergent gene expression in yeast. Genome Res 24: 422–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muto H, Ito K. 2008. Peptidyl-prolyl-tRNA at the ribosomal P-site reacts poorly with puromycin. Biochem Biophys Res Commun 366: 1043–1047. [DOI] [PubMed] [Google Scholar]
- Namy O, Moran SJ, Stuart DI, Gilbert RJ, Brierley I. 2006. A mechanical explanation of RNA pseudoknot function in programmed ribosomal frameshifting. Nature 441: 244–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obrig TG, Culp WJ, McKeehan WL, Hardesty B. 1971. The mechanism by which cycloheximide and related glutarimide antibiotics inhibit peptide synthesis on reticulocyte ribosomes. J Biol Chem 246: 174–181. [PubMed] [Google Scholar]
- Ouyang Z, Snyder MP, Chang HY. 2013. SeqFold: genome-scale reconstruction of RNA secondary structure integrating high-throughput sequencing data. Genome Res 23: 377–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlov MY, Watts RE, Tan Z, Cornish VW, Ehrenberg M, Forster AC. 2009. Slow peptide bond formation by proline and other N-alkylamino acids in translation. Proc Natl Acad Sci 106: 50–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pechmann S, Frydman J. 2013. Evolutionary conservation of codon optimality reveals hidden signatures of cotranslational folding. Nat Struct Mol Biol 20: 237–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percudani R, Ottonello S. 1999. Selection at the wobble position of codons read by the same tRNA in Saccharomyces cerevisiae. Mol Biol Evol 16: 1752–1762. [DOI] [PubMed] [Google Scholar]
- Plotkin JB, Kudla G. 2011. Synonymous but not the same: the causes and consequences of codon bias. Nat Rev Genet 12: 32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian W, Yang JR, Pearson NM, Maclean C, Zhang J. 2012. Balanced codon usage optimizes eukaryotic translational efficiency. PLoS Genet 8: e1002603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team 2013. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- Rouskin S, Zubradt M, Washietl S, Kellis M, Weissman JS. 2014. Genome-wide probing of RNA structure reveals active unfolding of mRNA structures in vivo. Nature 505: 701–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauder B, McCarthy JE. 1989. The role of bases upstream of the Shine-Dalgarno region and in the coding sequence in the control of gene expression in Escherichia coli: translation and stability of mRNAs in vivo. Gene 78: 59–72. [DOI] [PubMed] [Google Scholar]
- Scheper GC, van der Knaap MS, Proud CG. 2007. Translation matters: protein synthesis defects in inherited disease. Nat Rev Genet 8: 711–723. [DOI] [PubMed] [Google Scholar]
- Schneider-Poetsch T, Ju J, Eyler DE, Dang Y, Bhat S, Merrick WC, Green R, Shen B, Liu JO. 2010. Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nat Chem Biol 6: 209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah P, Ding Y, Niemczyk M, Kudla G, Plotkin JB. 2013. Rate-limiting steps in yeast protein translation. Cell 153: 1589–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp PM, Li WH. 1987. The codon Adaptation Index: a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res 15: 1281–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields DC, Sharp PM, Higgins DG, Wright F. 1988. “Silent” sites in Drosophila genes are not neutral: evidence of selection among synonymous codons. Mol Biol Evol 5: 704–716. [DOI] [PubMed] [Google Scholar]
- Siwiak M, Zielenkiewicz P. 2010. A comprehensive, quantitative, and genome-wide model of translation. PLoS Comput Biol 6: e1000865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, Chen L. 2010. A two-parameter generalized Poisson model to improve the analysis of RNA-seq data. Nucleic Acids Res 38: e170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler M, Fire A. 2011. Wobble base-pairing slows in vivo translation elongation in metazoans. RNA 17: 2063–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler M, Fire A. 2013. Conserved translatome remodeling in nematode species executing a shared developmental transition. PLoS Genet 9: e1003739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenico M, Lloyd AT, Sharp PM. 1994. Codon usage in Caenorhabditis elegans: delineation of translational selection and mutational biases. Nucleic Acids Res 22: 2437–2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner DR, Cariello DA, Woolstenhulme CJ, Broadbent MA, Buskirk AR. 2009. Genetic identification of nascent peptides that induce ribosome stalling. J Biol Chem 284: 34809–34818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas LK, Dix DB, Thompson RC. 1988. Codon choice and gene expression: synonymous codons differ in their ability to direct aminoacylated-transfer RNA binding to ribosomes in vitro. Proc Natl Acad Sci 85: 4242–4246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuller T, Carmi A, Vestsigian K, Navon S, Dorfan Y, Zaborske J, Pan T, Dahan O, Furman I, Pilpel Y. 2010a. An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell 141: 344–354. [DOI] [PubMed] [Google Scholar]
- Tuller T, Waldman YY, Kupiec M, Ruppin E. 2010b. Translation efficiency is determined by both codon bias and folding energy. Proc Natl Acad Sci 107: 3645–3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuller T, Veksler-Lublinsky I, Gazit N, Kupiec M, Ruppin E, Ziv-Ukelson M. 2011. Composite effects of gene determinants on the translation speed and density of ribosomes. Genome Biol 12: R110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ude S, Lassak J, Starosta AL, Kraxenberger T, Wilson DN, Jung K. 2013. Translation elongation factor EF-P alleviates ribosome stalling at polyproline stretches. Science 339: 82–85. [DOI] [PubMed] [Google Scholar]
- Varenne S, Buc J, Lloubes R, Lazdunski C. 1984. Translation is a non-uniform process. Effect of tRNA availability on the rate of elongation of nascent polypeptide chains. J Mol Biol 180: 549–576. [DOI] [PubMed] [Google Scholar]
- Wallace EW, Airoldi EM, Drummond DA. 2013. Estimating selection on synonymous codon usage from noisy experimental data. Mol Biol Evol 30: 1438–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch M, Govindarajan S, Ness JE, Villalobos A, Gurney A, Minshull J, Gustafsson C. 2009. Design parameters to control synthetic gene expression in Escherichia coli. PLoS ONE 4: e7002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen JD, Lancaster L, Hodges C, Zeri AC, Yoshimura SH, Noller HF, Bustamante C, Tinoco I. 2008. Following translation by single ribosomes one codon at a time. Nature 452: 598–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlgemuth I, Brenner S, Beringer M, Rodnina MV. 2008. Modulation of the rate of peptidyl transfer on the ribosome by the nature of substrates. J Biol Chem 283: 32229–32235. [DOI] [PubMed] [Google Scholar]
- Yang JR, Chen X, Zhang J. 2014. Codon-by-codon modulation of translational speed and accuracy via mRNA folding. PLoS Biol 12: e1001910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zajac P, Islam S, Hochgerner H, Lönnerberg P, Linnarsson S. 2013. PLoS ONE Base preferences in non-templated nucleotide incorporation by MMLV-derived reverse transcriptases. 8: e85270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W, Chung LM, Zhao H. 2011. Bias detection and correction in RNA-Sequencing data. BMC Bioinformatics 12: 290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinshteyn B, Gilbert WV. 2013. Loss of a conserved tRNA anticodon modification perturbs cellular signaling. PLoS Genet 9: e1003675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zur H, Tuller T. 2012. Strong association between mRNA folding strength and protein abundance in S. cerevisiae. EMBO Rep 13: 272–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.