

Abstract
Background & objectives:
As there are no standard laboratory techniques for the rapid detection of Pneumocystis jirovecii in India, this study was undertaken to evaluate and establish an optimal and rapid technique for the detection of P. jirovecii by comparing three different techniques - staining technique, application of a real time polymerase chain reaction (RT-PCR) targeting kex 1 gene and application of nested PCR targeting mitochondrial large subunit (mtLSU) gene for rapid detection of P. jirovecii in HIV positive patients.
Methods:
One hundred and fifty sputum specimens from HIV positive (n = 75) and HIV negative (n = 75) patients were subjected to three different techniques -KOH/Calcoflour and Grocott methanamine silver staining (GMS), RT-PCR targeting kex1 gene, PCR targeting mtLSU region followed by DNA sequencing and BLAST analysis.
Results:
Among the 75 HIV positive patients, P. jirovecii was detected in 19 (25.33%) patients by the staining techniques, and in 23 (30.65%) patients each by PCR targeting mtLSU region and by RT- PCR targeting kex1 gene of P. jirovecii. PCR based DNA sequencing targeting mtLSU region revealed 97-100 per cent sequence homology with P. jirovecii sequences in GenBank.
Interpretation & conclusions:
Of the three techniques for detection of P. jirovecii evaluated in this study, false negativity was found to be more in staining technique and it also required high technical expertise to interpret the result. Both nested PCR and RT-PCR were reliable and equally sensitive, in rapid detection of P. jirovecii, but RT-PCR technique also generated the copy numbers for knowing the severity of infection.
Keywords: DNA sequencing, GMS, HIV, kex 1, KOHcalcoflour, mtLSU, Pneumocystis jirovecii
Pneumocystis jirovecii a fungus belonging to Pneumocystidaceae family, is an opportunistic pathogen causing Pneumocystis pneumonia (PNP), in immunocompromised patients, specially in about 20 per cent of AIDS patients. The timely diagnosis and treatment of P. jirovecii infection remain a challenge to the clinicians and mycology laboratories, where the gold standard has been visualization of characteristic P. jirovecii cysts and/or trophozoites in lung tissue biopsy specimens1 due to non availability of in vitro culture techniques for isolation and identification of P. jirovecii from clinical specimens. The application of PCR in the diagnosis of the infection has improved the laboratory diagnosis due to its high sensitivity and specificity. The first report using molecular amplification methods of detection of P. jirovecii was published by Wakefield et al in 19902. Since then many different Pneumocystis genes have been proposed as targets for detection of P. jirovecii in clinical samples2,3,4,5.
The commonly targeted genes for the detection of Pneumocystis are internal transcriber spacer region (ITS)5, major surface glycoprotein5 (MSG), rRNA region5, 18s RNA5, 5s rRNA5, dihydrofolate reductase6, mtLSU7, thymidylate synthase5,8. Of all these, mtLSU7 region plays an important role and is commonly used for detection of P. jirovecii. Another target gene is the Kex1 gene coding for serine endopeptidase of P. jirovecii in processing proteins that maintain cell surface integrity6. Real time (RT)-PCR allows accurate quantification of DNA and has the potential to discriminate between asymptomatic carriage of P. jirovecii and clinical disease based on pathogen load. There are several RT-PCR assays using a variety of gene targets for detection of P. jirovecii in respiratory samples and a high inter-laboratory agreement among RT-PCR assays has been described9.
The present study was aimed to evaluate and compare three different detection techniques - KOH/Calcoflour and Grocott methanamine silver staining (GMS), with the commercially available RT-PCR targeting kex1 gene of P. jirovecii, and nested PCR targeting mtLSU region followed by DNA sequencing and BLAST analysis to deduce the sequence homology. These moelcular techniques were applied directly to all the clinical specimens and compared for sensitivity, specificity, and ease and rapidity of preparation and interpretation to detect the incidence of Pneumocystis pneumonia infection in HIV positive patients.
Material & Methods
A total of 150 sputum samples collected from HIV positive (N = 75) and HIV negative (N = 75) patients were included in the study. Among the 75 HIV positive patients, 66 were males (88%) and nine were females (12%). The sample size was calculated based on the prevalence rate of Pneumocystis pneumonia in HIV positive patients reported in a study conducted at the Government Hospital of Thoracic Medicine, Tambaram Sanitorium Chennai10. The required sample size based on Chennai population was 111 and 150 clinical samples were included in the present study. The power of the study was 85 per cent and the level of significance was 5 per cent. The samples were collected in sterile Uricol containers, transported in coolant box to the laboratory and refrigerated at 4°C until further processing to maintain the structural integrity of the cellular components. Induced sputum samples from HIV positive patients with clinical suspicion of PCP (n = 75) were collected from patients attending Government Hospital of Thoracic Medicine, Chennai, India. Clinical suspicion of the infection was made based on the radiological picture and symptoms strongly suggestive of PCP infection like persistent non-productive cough, dyspnoea, history of protracted fever of many weeks, duration with radiological findings (i.e. bilateral perihilar reticulonodular involvement and ground glass appearance) based on the inclusion and exclusion criteria of Lederberger et al11. Consecutive samples collected during December 2009 to March 2011 were included as the test specimens. The HIV negative (n = 75) sputum samples collected from patients suffering from respiratory infections other than PCP, attending Vision Research Foundation Referral Laboratory, Chennai, during the same period were included as the control samples. The patients were divided into five age groups 1-20, >20-30, >30-40, >40-50, and >50-60 yr of age. The details of the patients enrolled in the study are given in Table I.
Table I.
Details of patients (n=150, 75 HIV positive and 75 HIV negative) included in the study
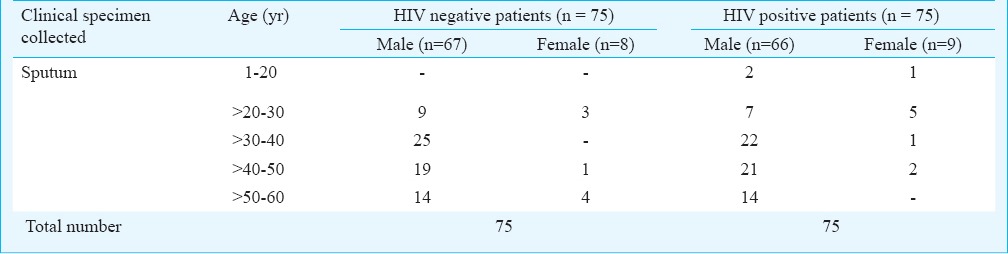
The study protocol was approved by both the Institutions’ Research Committee and Ethics Committees (Vision Research Foundation and Government Hospital of Thoracic Medicine, Chennai). PCR targeting mtLSU and RT-PCR targeting kex1 gene were optimized using the standard ATCC strain P. carinii PRA 159, obtained through LGC Promochem, Bangalore, India. The study was conducted at the Department of Microbiology, L & T Microbiology Research Centre, Vision Research Foundation, Chennai.
Microscopic investigations: Sputum smears were subjected to KOH/Calcoflour and Grocott methanamine silver (GMS) staining.
KOH calcoflour staining - One drop of calcoflour and one drop of 10 per cent potassium hydroxide was added to each slide, the coverslip was applied and the preparation was observed under fluorescence microscope (Nikon, India).
GMS staining - The sputum smear was flooded with 5 per cent chromic acid for five minutes, washed with water, microwaved (medium temperature) for 50 sec (till the smear turned yellowish brown) in a Coplin jar containing 50 ml of methenamine working solution. The slides were rinsed again with distilled water, treated with 1 per cent gold chloride for 2 to 5 sec, rinsed with distilled water, exposed to 5 per cent sodium thiosulphate for one minute, counterstained with a light green working solution, cleared in xylene, and examined under 100X of light microscope (Nikon, India).
DNA extraction for semi nested PCR targeting mtLSU region: Extraction of DNA from the standard ATCC strain of Pneumocystis (P. carinii PRA 159) and from the induced sputum specimens was carried out using fungal DNA extraction kit (Golecha's DNA extraction kit, Chennai, Tamil Nadu), according to the manufacturer's instruction.
Semi nested PCR targeting mtLSU region5: Standardization of PCR targeting the mtLSU region was optimized by using the Hot start Taq polymerase enzyme (Bio Source Surgicals, India). Hot start Taq DNA polymerase is a mixture of Taq DNA polymerase and an aptamer based inhibitor. The inhibitor binds reversibly to the enzyme, inhibiting polymerase activity at temperatures below 45°C, but releases the enzyme during normal cycling conditions allowing reactions to be set up at room temperature.
Briefly, 50 µl PCR reaction was set with, 200 µM (8 µl) of each dNTPs [2µl each of idATP, dTTP, dGTP, dCTP (Bangalore genei, India)]; 5µl of 10X buffer, 10 pico mole of each of the primers (pAZ102-E:5’-GATGGCTGTTTCCAAGCCCA-3’, pAZ102-H:5’-GTGTACGTTGCAAAGTACTC-3’ for the first round and 10 pm of pAZ102-E; 5’-GATGGCTGTTTCCAAGCCCA-3’, pAZ102-L2:5’-ATAAGGTAGATAGTCGAAAG-3’ for the second round)5; 0.5 µl of Hot Start Taq DNA polymerase, 25 µl of milliQ water, and 10 µl of the template DNA for the I round of PCR and for the second round of seminested PCR 30 µl of milliQ water and 5µl of I round amplified product were added. The Hot Start Taq DNA polymerase was added after the initial denaturation of 94°C for 5 min. The optimization was carried out initially with the standard ATCC PRA P. carinii 159 strain. The first round consisted of denaturation at 94°C for 20 sec, annealing at 56 °C for 20 sec, extension at 72 °C for 20 sec for 35 cycles followed by final extension at 72 °C for 5 min. The second round consisted of denaturation at 94°C for 20 sec, annealing at 50 °C for 20 sec, extension at 72 °C for 20 sec for 35 cycles followed by final extension at 72 °C for 5 min5. PCR using primers targeting mtLSU region generated 346 base pair product size for the first round and 120bp product size for the second round of amplification.
Real time PCR targeting kex1 gene: The DNA extraction was carried out with the Qiagen (Hilden, Germany) kit method by adding 4μl (per sample) of internal extraction control DNA provided in the real time kit along with the lysis buffer and 200 µl of induced sputum samples and proceeded further according to the manufacturers instructions. (The internal extraction control DNA should not be added directly to the unprocessed biological sample as this leads to degradation and a loss in signal.
Amplification of the Kex1 gene: The standardization of RT-PCR targeting Kex1 gene of P. jirovecii was carried out initially using Taq enzyme and by using various concentrations of magnesium chloride (ranging from 1-3mM). But the amplification was not observed. The amplification of the Kex1 gene of P. jirovecii was carried out using Primer Design™ (Southampton, United Kingdom) genesig kit following the manufacturer's instruction.
Briefly, 15µl PCR reaction was set with, 200µM (4 µl) of each dNTPs [dATP, dTTP, dGTP, dCTP (Bangalore genei)]; 2µl of 10X buffer, 1µl of pathogen primer/probe mix, 1µl internal extraction control primer, 7µl of RNAse/DNAse free water and 0.5 µl of Hot Start Taq DNA polymerase. The enzyme was added after the initial denaturation of 94°C for 5 min followed by denaturation at 95°C for 10 sec with combined annealing and extension at 60°C for 60 sec for 50 cycles.
PCR based DNA sequencing targeting mtLSU gene: The amplified products of the samples positive by the mtLSU PCR method were subjected to DNA sequencing. Sequencing of the amplified products were performed in a 10 µl reaction volume, containing 1.0 µl of RR mix, 3.0 µl of sequencing buffer, 1.0 µl of each primer (1:25 diluted), 3.0 µl MilliQ water and 1.0 µl of amplified product for the mtLSU.
Amplification was carried out in Perkin-Elmer thermocycler USA using 25 cycles of 96°C for 10 sec, 50°C for five sec, 60°C for four minutes with initial denaturation at 96°C for one minute.
The cycle-sequenced products were then purified using 125mM EDTA and 3M sodium acetate. The purified products were denatured with 10 µl formamide and loaded onto ABI Prism 3130 AVANT (Applied Biosystems, USA) genetic analyzer. The sequences were analysed by Bio Edit sequence alignment software, USA and BLAST analysis [www.ncbi.nlm.nih.gov/BLAST] was done to confirm the sequenced data with the standard strains and to determine the percentage homology.
Statistical analysis: The results of the semi nested PCR targeting mtLSU region were statistically analysed in comparison with the results of control samples using Chi square test.
Results
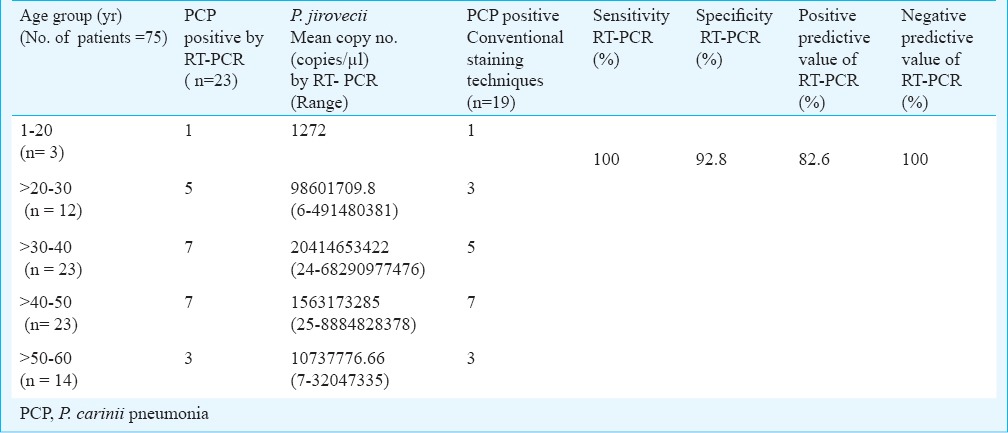
Of the 75 sputum samples collected from HIV positive and clinically suspected PCP patients, 19 (25.33%) were positive by staining (GMS and KOH staining). The mtLSU PCR revealed a specific 346 bp at the end of first round of amplification followed by 120 bp after the second round amplification. Twenty three (30.65%) samples were positive by semi nested PCR targeting mtLSU region when compared with control group P<0.0001. Among the 23 HIV positive patients in whom P. jirovecii was detected, 18 (78.2%) were male and five (21.7 %) were female patients (Table II).
Table II.
Evaluation of RT-PCR and nested PCR against conventional staining techniques for detection of P. jirovecii from clinically suspected Pneumocystis pneumonia (PCP) patients
DNA sequencing performed on the mtLSU amplicons revealed 97-100 per cent sequence similarity and the RT-PCR targeting kex-1 gene revealed concordant results for the same 23 sputum samples, which were positive by PCR targeting the mtLSU region.
PCR based DNA sequencing of P. jirovecii ATCC targeting mtLSU region revealed 99 per cent BLAST score with the nucleotide sequences of P. jirovecii strains deposited in Genbank. To further analyse the genetic homology existing among P. jirovecii detected in HIV positive patients, a dendrogram was constructed using DAMBE Software (Figure).
Fig.
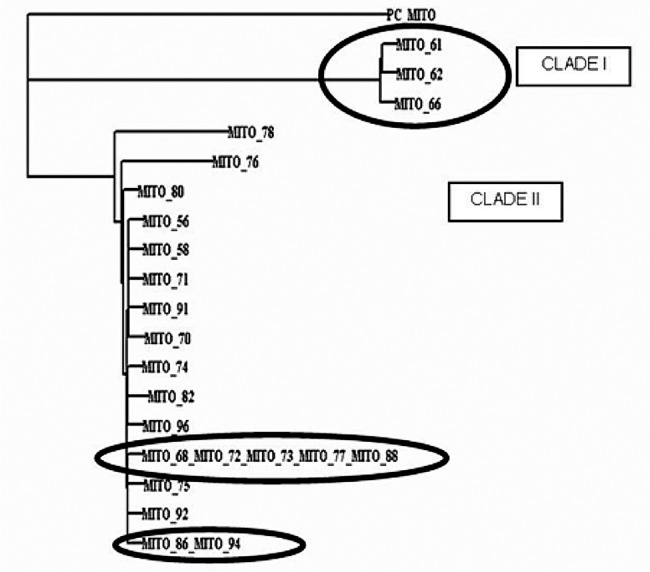
Dendrogram constructed using mtLSU sequences with DAMBE(data analysis in molecular biology and evolution) software. MITO denotes mitochondrial gene (mtr gene) which has been targeted for PCR. The numbers denote the clinical specimen number. The dendrogram revealed two separate clades - Clade I (3 amplified products of mitochondrial gene) together and Clade II. There were 2 sets of identical P. jirovecii strains which clustered in the same line (Clade II amplified products of mitochondrial gene) and two belonging to one subdivision in the clade II. The similar patterns in the clades are highlighted.
The dendrogram revealed two separate clades – Clade I and Clade II. There were two sets of identical P. jirovecii isolates which clustered in the same line (Clade II). One set comprised five isolates belonging together, of which one isolate belonged to the age group >20 - 30 yr (491480381 copies/µl), three belonged to the age group of >30 - 40 yr (8342, 929354, and 427 copies/µl), and one isolate belonged to the age group >40 - 50 yr (8884828 copies/µl). The other set belonged to the age group >40 - 50 yr with high copy numbers 491480380852, 8884828378 copies/µl, respectively. Two sputum samples (76, 78) in clade II were clustered away from the rest of the specimens. [specimen 76 (age- 48 yr/male), having copy number 42726, and specimen 78 (age - 40 yr/male) having copy number 7447721].
On comparing the results of staining and mtLSU PCR, the four samples negative by staining (KOH and GMS) technique were positive for the first round of mtLSU PCR. Comparing RT-PCR and mtLSU PCR, sputum specimens which revealed copy numbers as low as 6, 7, 24, 25 copies/µl were observed to be positive in the first round of PCR targeting mtLSU region of P. jirovecii. These findings indicate the high sensitivity of mtLSU primers. For further analysis, the 23 positive sputum samples were divided into five age groups based on the P. jirovecii copy numbers (Table II). From the results, the mean of copy number >1,00,000 copies/ µl of P. jirovecii was found high in the age group >30-40 yr. The sensitivity, specificity, positive predictive value and negative predictive value of RT-PCR in comparison with the conventional staining techniques employed in the present study were 100, 92.5, 82.6 and 100 per cent (Table II).
The remaining 52 sputum specimens collected from HIV positive and clinically suspected Pneumocystis pneumonia patients were negative by staining and by both the PCR targeting mtLSU region and also by the RT- PCR targeting the kex1 gene of P. jirovecii. The 75 samples collected from HIV negative patients suffering from respiratory infections other than PCP were found negative by both the detection techniques.
Discussion
The different laboratory microscopic techniques for detecting the PCP infection are reported to have varying sensitivities, and the applicability of PCR assay in the laboratory diagnosis of PCP has not been extensively studied. In the present study, staining technique, the real time PCR technique targeting Kex1 of P. jirovecii and the semi nested PCR technique targeting the mtLSU region for the rapid detection of P. jirovecii were applied on the clinical samples. GMS staining was considered as the traditional “gold standard”.
Gupta et al12, compared the PCR techniques targeting the MSG gene, mtLSU region and the ITS region and observed that MSG was relatively more sensitive when single round PCR assay was used. However, on comparing the two nested PCR assays, targeting mtLSU and the ITS region, both assays were found to be equally sensitive with results more or less comparable12. Lu et al5, developed a PCR method was developed targeting portions of 18S rRNA, mitochondrial (mt) rRNA, 5S rRNA, thymidylate synthase (TS), and dihydrofolate reductase (DHFR) genes. Thirty bronchoalveolar lavage (BAL) specimens were examined and all were positive by both the ITS-PCR and the 18S rRNA gene PCR, whereas only 26 (87%), 18 (60%), 10 (33%), and 7 (23%) of were positive by mt rRNA gene PCR, TS gene PCR, 5S rRNA gene PCR, and DHFR gene PCR, respectively5. There are several studies on RT- PCR targeting various genes of P. jirovecii14,15,16,17. The commonly targeted regions are heat shock protein 70 (HSP70) gene, the MSG gene, and beta-tubulin gene13 of P. jirovecii.
Rohner et al6 compared the results obtained by two staining methods (toluidine blue and calcofluor white) and two quantitative (q) RT-PCR assays for Kex-1 primer pair and β-tubulin for the detection of P. jirovecii in BAL specimens. Of the 186 BAL samples, 21 (11.3%) were microscopically positive with both stains as well as by qPCR. Of the remaining 165 samples, 153 (82%) were negative by both microscopy and PCR (PCR with the two sets of primers); the remaining 12 samples (7%) were Kex-1-based PCR positive but microscopically negative. Of these latter samples, 10 (6%) were also positive with the primers targeting the β-tubulin gene6.
The major modification employed in the present study for both the semi nested PCR targeting the mtLSU region of P. jirovecii, and for RT- PCR targeting the Kex1 region of P. jirovecii, was the use of Hot start Taq polymerase enzyme, which aided in improving the sensitivity of mtLSU PCR. Taking microscopy as a reference, the sensitivity of qPCR targeting the Kex-1 gene was 100 per cent and the specificity was 92.4 per cent.
The primers described by Lu et al5 were chosen for the PCR targeting the mtLSU region. The novel thermal profile along with the use of Hot start Taq polymerase enzyme increased the sensitivity of PCR targeting the mtLSU region of P. jirovecii in such a way that even the copy numbers as low as 6 copies/µl were detected at the end of single round of amplification. Detection of P. jirovecii organism by the RT-PCR detection method aided in generating the results in about three hours including the time taken for DNA extraction, whereas the time taken by the semi-nested PCR targeting the mtLSU region was five hours. Though the semi-nested PCR targeting mtLSU region was cost effective, the copy numbers could be obtained by the real-time PCR which was not possible with the semi-nested PCR.
The target sequence within the Kex1 gene has previously been shown to be a good genetic marker for P. jirovecii in clinical real-time PCR based studies. This gene has no known homology with other species in the Pneumocystis at the primer binding sites14,15. The primers and probe sequences in the kit used in the present study had 100 per cent homology with a broad range of clinically relevant reference sequences based on a comprehensive bioinformatics analysis.
The three techniques were evaluated on cost-effectiveness as well as sensitivity. Of the three, the RT- PCR and PCR was the simplest, taking approximately 3 h. In case of PCR targeting mtLSU region, DNA extraction followed by PCR and sequence analysis was time consuming. In case of conventional staining techniques, only small amounts of reagents were required to stain a slide. Though the cost of reagents for the conventional staining techniques are comparatively less, the reading and interpretation of the stained slides requires expertise in interpretation of the results to generate reliable reports.
Fillaux et al16 have reported that the direct immunofluorescence assay can be replaced by a real-time PCR assay given its feasibility, sensitivity, and specificity, for the detection of P. jirovecii. The two molecular techniques included in the present study were reliable and rapid for the detection of P. jirovecii but RT-PCR technique was more useful in giving the reliable results with copy numbers of P. jirovecii. Further studies on more number of patients will help in molecular typing of different clades of P. jirovecii by targeting mtLSU region.
The main limitation of the study was that the follow up of the patients after treatment could not be done and a future study has to be planned including the follow up to utilize RT-PCR as a prognostic marker for treatment outcome based on the reduction of copy numbers by RT-PCR. The laboratories must aim at balancing the cost and ease of a testing method against the sensitivity and the goal of preventing unnecessary and invasive clinical procedures to reduce the risk involved for the patients.
Acknowledgment
The technical assistance provided by Shri A. Kamalavinayagam is acknowledged. Authors acknowledge the funding provided by the Indian Council of Medical Research (ICMR), New Delhi, India, for part of the research work included in this study.
References
- 1.Hauser PM, Bille J, Lass-Flor C, Geltner C, Feldmesser M, Levi M, et al. Multicenter, prospective clinical evaluation of respiratory samples from subjects at risk for Pneumocystis jirovecii infection by use of a commercial real-time PCR assay. J Clin Microbiol. 2011;49:1872–8. doi: 10.1128/JCM.02390-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wakefield AE, Pixley FJ, Banerji S, Sinclair K, Miller RF, Moxon ER, et al. Detection of Pneumocystis carinii with DNA amplification. Lancet. 1990;336:451–3. doi: 10.1016/0140-6736(90)92008-6. [DOI] [PubMed] [Google Scholar]
- 3.Lu JJ, Bartlett MS, Shaw MM, Queener SF, Smith JW, Ortiz-Rivera M, et al. Typing of Pneumocystis carinii strains that infect humans based on nucleotide sequence variations of internal transcribed spacers of rRNA genes. J Clin Microbiol. 1994;32:2904–12. doi: 10.1128/jcm.32.12.2904-2912.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kovacs JA, Hiemenz JW, Macher AM, Stover D, Murray HW, Shelhamer J, et al. Pneumocystis carinii pneumonia: a comparison between patients with the acquired immunodeficiency syndrome and patients with other immunodeficiencies. Ann Intern Med. 1984;100:663–71. doi: 10.7326/0003-4819-100-5-663. [DOI] [PubMed] [Google Scholar]
- 5.Lu JJ, Chen CH, Bartlett MS, Smith JW, Lee CH. Comparison of six different PCR methods for detection of Pneumocystis carinii. J Clin Microbiol. 1995;33:2785–8. doi: 10.1128/jcm.33.10.2785-2788.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rohner P, Jacomo V, Studer R, Schrenzel J, Graf JD. Detection of Pneumocystis jirovecii by two staining methods and two quantitative PCR assays. Infection. 2009;37:261–5. doi: 10.1007/s15010-008-8027-x. [DOI] [PubMed] [Google Scholar]
- 7.Edman JC, Edman U, Cao M, Lundgren B, Kovacs JA, Santi DV. Isolation and expression of the Pneumocystis carinii dihydrofolate reductase gene. Proc Natl Acad Sci USA. 1989;86:8625–9. doi: 10.1073/pnas.86.22.8625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Edman U, Edman JC, Lundgren B, Santi DV. Isolation and expression of the Pneumocystis carinii thymidylate synthase gene. Proc Natl Acad Sci USA. 1989;86:6503–7. doi: 10.1073/pnas.86.17.6503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Linssen CF, Jacobs JA, Beckers P, Templeton KE, Bakkers J, Kuijper EJ, et al. Inter-laboratory comparison of three different real-time PCR assays for the detection of Pneumocystis jirovecii in bronchoalveolar lavage fluid samples. J Med Microbiol. 2006;55:1229–35. doi: 10.1099/jmm.0.46552-0. [DOI] [PubMed] [Google Scholar]
- 10.Deivanayagam CN, Rajasekaran S, Senthilnathan V, Krishnarajasekhar OR, Raja K, Chandrasekar C, et al. Clinico-radiological spectrum of tuberculosis among HIV sero-positive-a tambaram study. Indian J Tuberc. 2001;48:123–7. [Google Scholar]
- 11.Ledergerber B, Mocroft A, Reiss P, Furrer H, Kirk O, Bickel M, et al. Eight European Study Group. Discontinuation of secondary prophylaxis against Pneumocystis carinii pneumonia in patients with HIV infection who have a response to antiretroviral therapy. N Engl J Med. 2001;344:168–74. doi: 10.1056/NEJM200101183440302. [DOI] [PubMed] [Google Scholar]
- 12.Gupta R, Mirdha BR, Guleria R, Mohan A, Kabra SK, Kumar L, et al. Use of different primer directed sequence amplification by polymerase chain reaction for identification of Pneumocystis jirovecii in clinical samples. Indian J Chest Dis Allied Sci. 2008;50:321–7. [PubMed] [Google Scholar]
- 13.Banerji S, Wakefield AE, Allen AG, Maskell DJ, Peters SE, Hopkin JM. The cloning and characterization of the arom gene of Pneumocystis carinii. J Gen Microbiol. 1993;139:2901–14. doi: 10.1099/00221287-139-12-2901. [DOI] [PubMed] [Google Scholar]
- 14.Dyer M, Volpe F, Delves CJ, Somia N, Burns S, Scaife JG. Cloning and sequence of a beta-tubulin cDNA from Pneumocystis carinii: possible implications for drug therapy. Mol Microbiol. 1992;6:991–1001. doi: 10.1111/j.1365-2958.1992.tb02165.x. [DOI] [PubMed] [Google Scholar]
- 15.Huggett JF, Taylor MS, Kocjan G, Evans HE, Morris-Jones S, Gant V, et al. Development and evaluation of a real-time PCR assay for detection of Pneumocystis jirovecii DNA in bronchoalveolar lavage fluid of HIV-infected patients. Thorax. 2008;63:154–9. doi: 10.1136/thx.2007.081687. [DOI] [PubMed] [Google Scholar]
- 16.Fillaux J, Berry A. Real-time PCR assay for the diagnosis of Pneumocystis jirovecii pneumonia. Methods Mol biol. 2013;943:159–70. doi: 10.1007/978-1-60327-353-4_11. [DOI] [PubMed] [Google Scholar]
- 17.Harris JR, Marston BJ, Sangrujee N, DuPlessis D, Park B. Cost-effectiveness analysis of diagnostic options for Pneumocystis pneumonia (PCP) PLoS One. 2011;6:e23158. doi: 10.1371/journal.pone.0023158. [DOI] [PMC free article] [PubMed] [Google Scholar]