

Abstract
Objective:
In a large series of patients with cervical artery dissection (CeAD), a major cause of ischemic stroke in young and middle-aged adults, we aimed to examine frequencies and correlates of family history of CeAD and of inherited connective tissue disorders.
Methods:
We combined data from 2 large international multicenter cohorts of consecutive patients with CeAD in 23 neurologic departments participating in the CADISP-plus consortium, following a standardized protocol. Frequency of reported family history of CeAD and of inherited connective tissue disorders was assessed. Putative risk factors, baseline features, and 3-month outcome were compared between groups.
Results:
Among 1,934 consecutive patients with CeAD, 20 patients (1.0%, 95% confidence interval: 0.6%–1.5%) from 17 families (0.9%, 0.5%–1.3%) had a family history of CeAD. Family history of CeAD was significantly more frequent in patients with carotid location of the dissection and elevated cholesterol levels. Two patients without a family history of CeAD had vascular Ehlers-Danlos syndrome with a mutation in COL3A1. This diagnosis was suspected in 2 additional patients, but COL3A1 sequencing was negative. Two patients were diagnosed with classic and hypermobile Ehlers-Danlos syndrome, one patient with Marfan syndrome, and one with osteogenesis imperfecta, based on clinical criteria only.
Conclusions:
In this largest series of patients with CeAD to date, family history of symptomatic CeAD was rare and inherited connective tissue disorders seemed exceptional. This finding supports the notion that CeAD is a multifactorial disease in the vast majority of cases.
Cervical artery dissection (CeAD), a major cause of ischemic stroke in young adults, is considered a complex disease, with multiple genetic variants and environmental factors contributing to its occurrence.1,2 No consistent genetic association has been found so far.1 Given the low incidence of the disease in the general population, heritability estimates based on twin or family studies are not available, and data on family history of CeAD are scarce. Only one hospital-based study from the Mayo Clinic, published 18 years ago, systematically examined the frequency of family history of CeAD in a cohort of 200 patients with CeAD, reporting a frequency of 2.5%.3 This could be an overestimation because of a referral bias, given the single tertiary center recruitment in a highly specialized institution.
Moreover, some rare inherited connective tissue disorders (CTDs) are known to cause or to be associated with CeAD, i.e., mostly vascular Ehlers-Danlos syndrome (vEDS), and perhaps, to a lesser extent, other types of EDS, Marfan syndrome, or osteogenesis imperfecta, although the evidence for these is much weaker.2,4 However, the prevalence of these monogenic diseases has never been systematically assessed in large patient series. Evaluating the frequency of heritable forms of CeAD in a multicenter setting could be important to guide screening and genetic counseling strategies for patients with CeAD and their families.
We aimed to examine the frequencies and correlates of family history of CeAD and of the aforementioned inherited CTD in 2 large contemporary multicenter cohorts of patients with CeAD (CADISP-plus consortium).
METHODS
Study population.
The multicenter CADISP-plus consortium comprised 2,021 patients with CeAD from both the CADISP clinical study (n = 983)5 and the Paris-Lariboisière/Zürich/Bern CeAD registry (n = 1,038).6,7 Structure and methods of each of these studies have been described in detail previously.5,7–9 Briefly, we included consecutive patients evaluated in a department of neurology with a diagnosis of CeAD, following a standardized protocol (table 1, figures e-1 and e-2, and e-Methods [on the Neurology® Web site at Neurology.org] for inclusion criteria and recruiting centers). To be included, patients with CeAD had to present with a mural hematoma, aneurysmal dilation, long tapering stenosis, intimal flap, double lumen, or occlusion >2 cm above the carotid bifurcation revealing an aneurysmal dilation or a long tapering stenosis after recanalization, in a cervical artery (internal carotid or vertebral); iatrogenic dissections were excluded. Monogenic CTDs, which were an exclusion criterion for genetic analyses, were included in the CADISP clinical study. The Paris-Lariboisière/Zürich/Bern CeAD registry included patients with first-ever CeAD only.
Table 1.
Participating centers
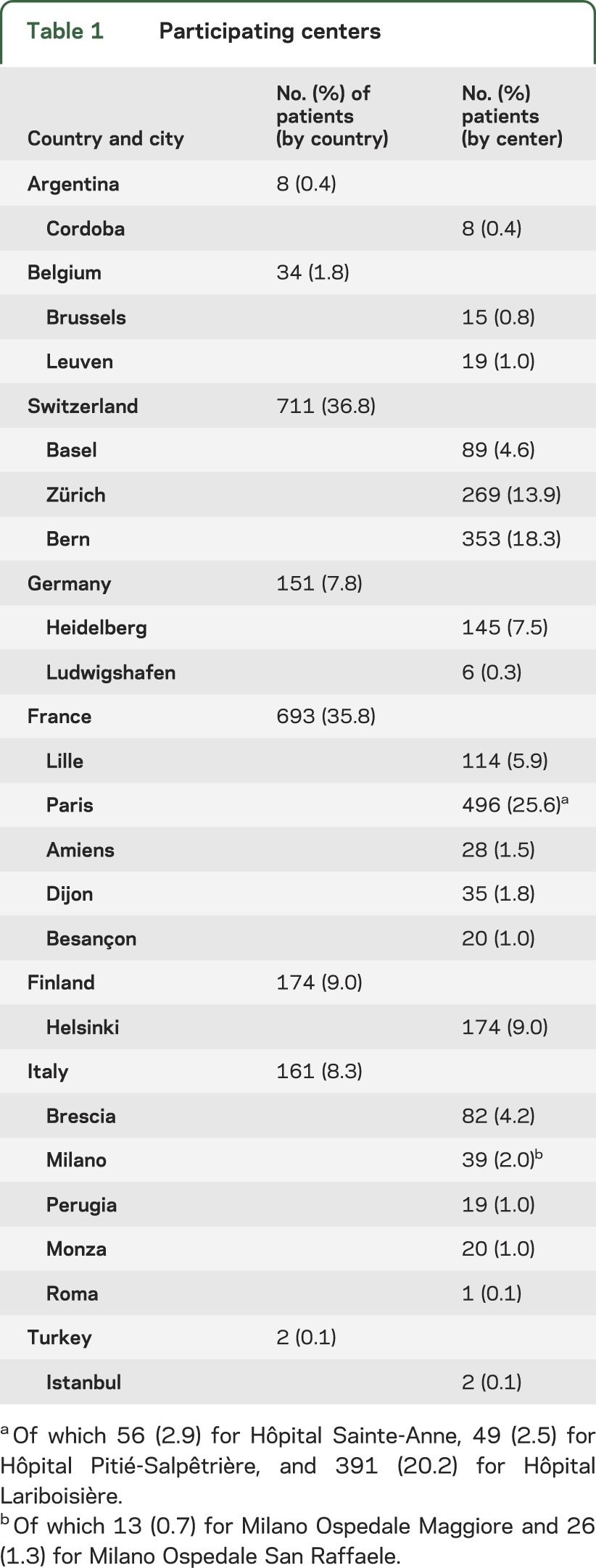
Between 2004 and 2009, the CADISP clinical study included all consecutive patients evaluated in several departments of neurology with a diagnosis of CeAD. Patients were recruited both prospectively and retrospectively. Retrospective patients are participants who had a qualifying event before the beginning of the study in each center and were identified through local registries of patients with CeAD. The vast majority of these had a qualifying event between 1999 and 2009 (<4% before 1999). In the Paris-Lariboisière/Zürich/Bern Registry on CeAD, patients were included prospectively between 1985 and 2013 (e-Methods).
After excluding patients without reliable information on family history, 1,934 patients with CeAD were available (CADISP study: n = 921; Paris-Lariboisière/Zürich/Bern registry: n = 1,013), from 8 countries and 23 centers (figure e-1).
Variable definition.
Family history of CeAD was defined by reported history of CeAD in father, mother, siblings, children, or any other defined relative of the index patient. Whenever possible, medical records were obtained for the affected relative to confirm the diagnosis and specify the age at occurrence of CeAD, the dissection site, and presence or absence of cerebral or retinal ischemia (ischemic stroke or TIA, including transient monocular blindness). We systematically searched for history of previously diagnosed vEDS, classic or hypermobile EDS,10,11 Marfan syndrome,12 or other known inherited CTD according to established criteria.4 When patients were aphasic or had cognitive deficits, information on personal and family history was collected from a close relative. Each patient's personal medical records were examined in detail. When an inherited CTD was suspected based on suggestive clinical features or family history, the patient was sent to a specialized genetics center for further diagnostic workup.
Presence of cerebral or retinal ischemia, of arterial occlusion, stenosis, aneurysmal dilation (e-Methods), or multiple dissections at admission, and 3-month CeAD recurrence rates were recorded. Putative risk factors (hypertension, hypercholesterolemia, migraine, infection, and trauma preceding the dissection) are defined in e-Methods.
Statistical analyses.
To compare clinical and radiologic characteristics as well as risk factor distribution between CeAD patients with and without a family history of CeAD, we first performed univariate analyses using Fisher exact test for categorical and Wilcoxon-Mann-Whitney test for continuous variables. Multivariable logistic regression models were then applied, adjusting for age and sex, as well as for study (CADISP clinical study, Paris-Lariboisière/Zürich/Bern CeAD registry) in a secondary analysis. Sensitivity analyses were performed after excluding one random patient from each of the 3 related patient pairs with a family history of CeAD. In addition, we verified the stability of associations after additionally adjusting for the retrospective vs prospective recruitment for CADISP clinical study participants. Analyses were performed using Statistical Analyses System software, version 9.2 (SAS Institute, Cary, NC).
Standard protocol approvals, registrations, and patient consents.
The study protocols were approved by relevant local authorities in all participating centers and conducted according to national rules concerning ethics committee approval and informed consents.5,7 The CADISP study is registered with clinicaltrials.gov (NCT00657969); see also www.cadisp.com.
RESULTS
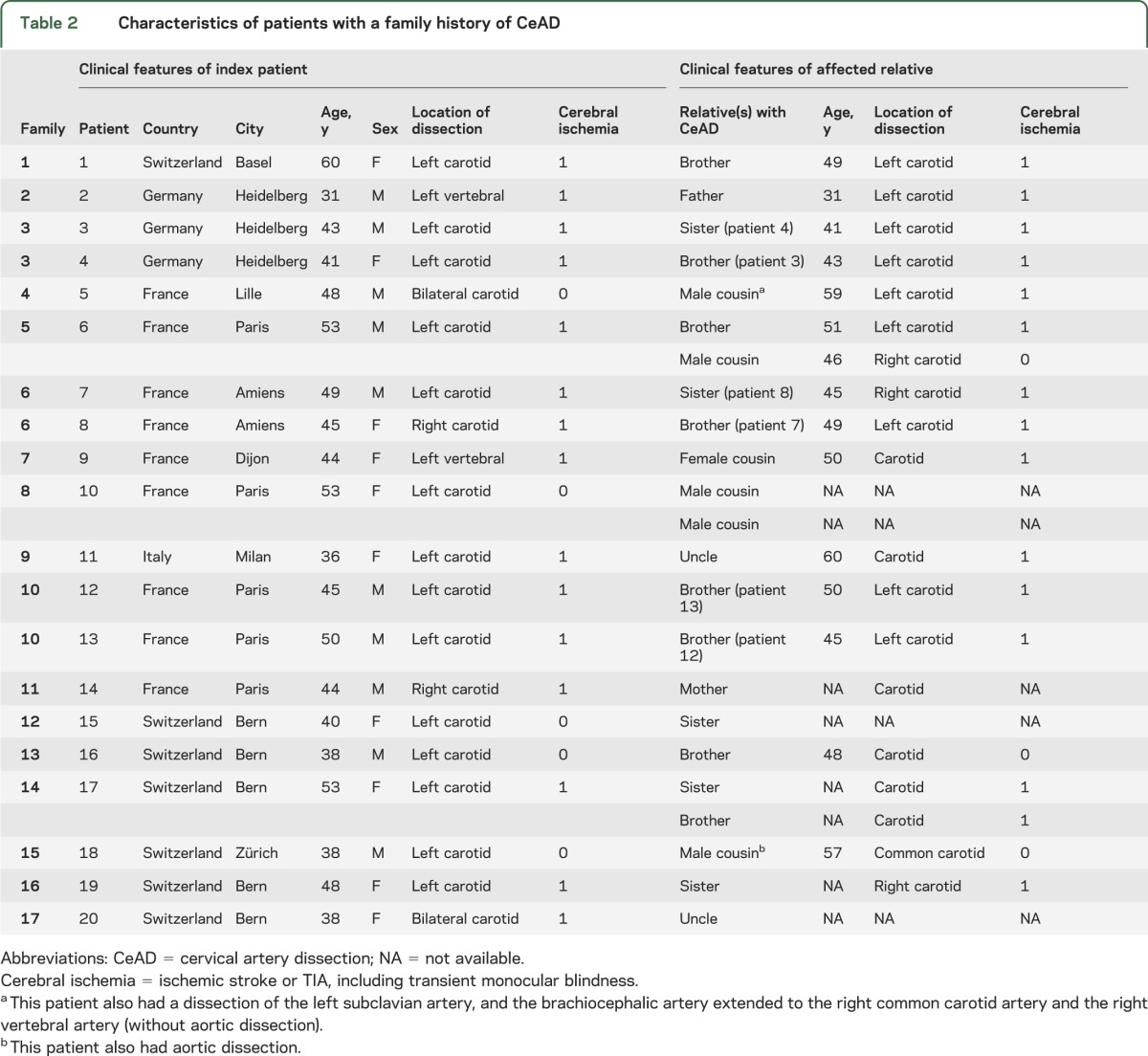
Among 1,934 patients with CeAD, 20 patients (1.0%, 95% confidence interval [CI]: 0.6%–1.5%) from 17 families (0.9%, 0.5%–1.3%) had a family history of CeAD; of these, 13 patients had a family history of CeAD in a first-degree relative (0.7%, 0.3%–1.0%). Characteristics of these patients are presented in table 2. In most cases, there were only 2 affected cases per family, including the index patient. Two families had 3 affected members. In one of these, only males were affected, including 2 brothers and 1 maternal cousin, suggesting a possible X-linked inheritance pattern. For 3 families (6 patients), both affected relatives were included in the CADISP-plus study (table 2). Of the remaining 14 patients with a positive family history, details on CeAD characteristics of the affected relative could be obtained in 11 patients. These characteristics remained undetermined for 3 patients (15%).
Table 2.
Characteristics of patients with a family history of CeAD
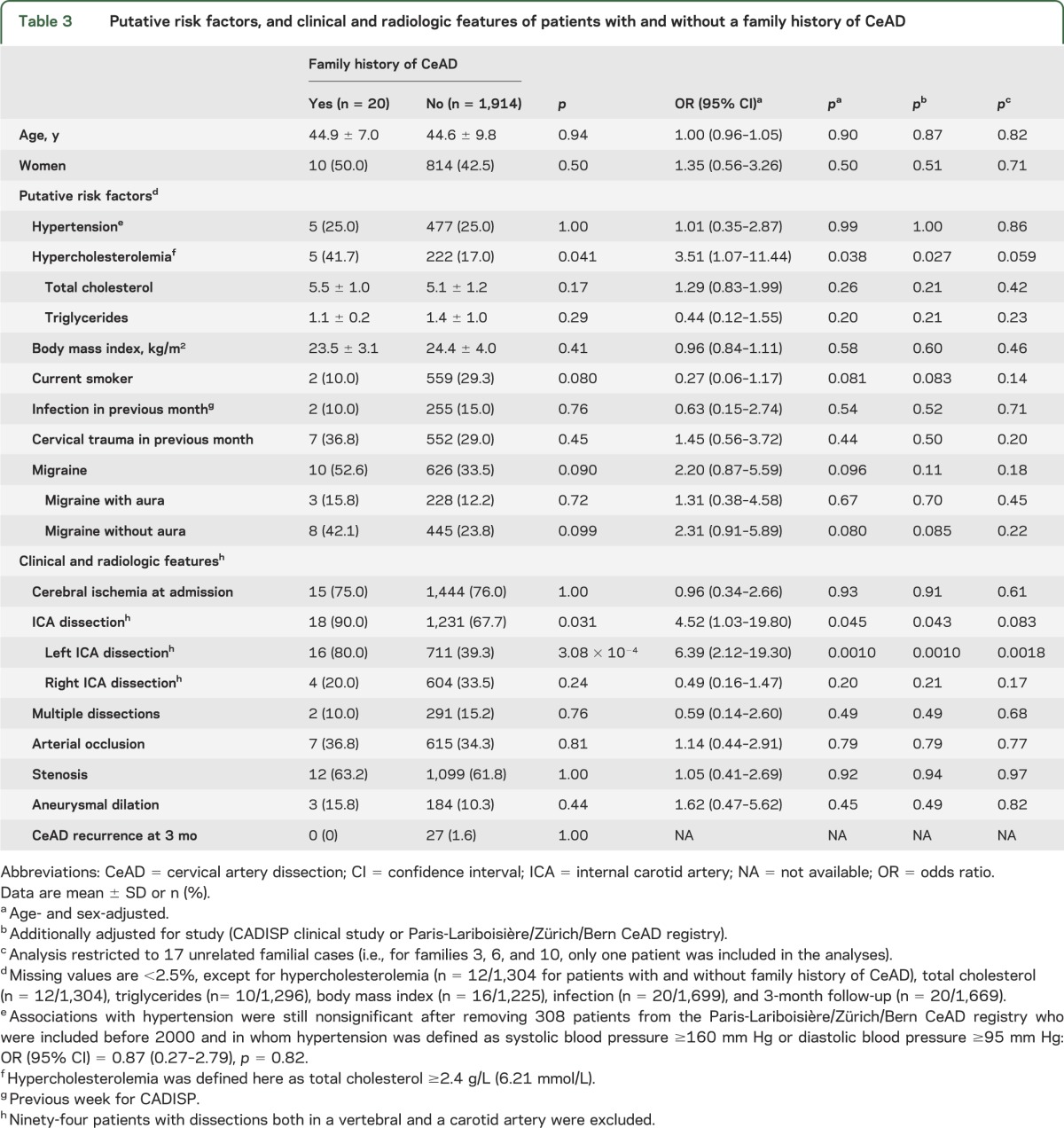
Patients with CeAD who had a family history of CeAD more often had carotid (mostly left carotid) than vertebral dissection and more frequently had elevated total cholesterol levels (≥6.20 mmol/L) at admission compared to patients with CeAD without such a family history (table 3). There was also a nonsignificant trend toward a higher prevalence of migraine, especially migraine without aura (table 3). Patients with a family history of CeAD did not differ significantly from those without such a family history regarding other putative risk factors, baseline characteristics (especially frequency of multiple dissections), and short-term (3 months) recurrence rate (table 3). Results were substantially unchanged, although no longer reaching statistical significance, when excluding one random patient from each of the 3 related patient pairs with a family history of CeAD.
Table 3.
Putative risk factors, and clinical and radiologic features of patients with and without a family history of CeAD
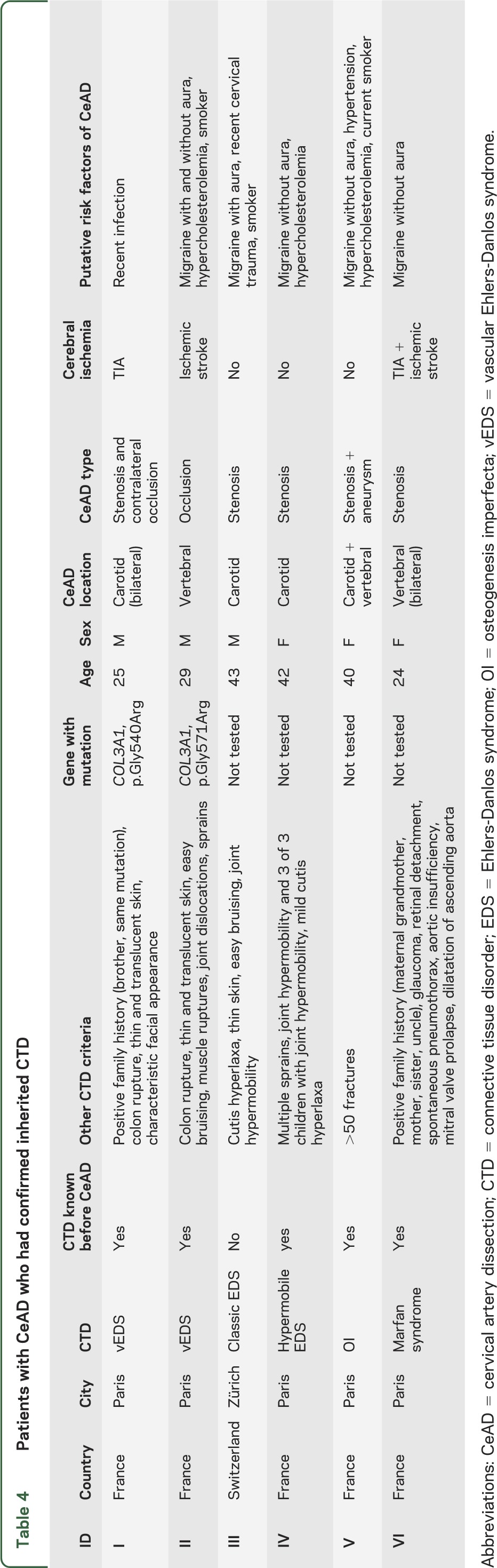
None of the patients with a family history of CeAD had been diagnosed with a known inherited CTD. Among patients with CeAD who had a family history of CeAD, 2 patients (0.1%, 0.0%–0.2%) had clinically and genetically confirmed vEDS, with mutations detected in the COL3A1 gene. In both cases, CeAD occurred before age 30 years, and the diagnosis of vEDS was already known before the dissection, with a history of colon rupture. Characteristics of the dissection and other clinical features of vEDS in these patients are described in table 4. There were 2 additional patients in whom vEDS was suspected after the CeAD, but COL3A1 DNA sequencing was negative. A detailed description of these patients is provided in table e-1. One patient had clinical diagnosis of Marfan syndrome, because he had a positive family history and fulfilled the revised Ghent criteria, despite the absence of genetic testing (table 4).12 Two additional patients had features of Marfan syndrome, but did not fulfill established clinical diagnostic criteria (table e-1).12 One patient was diagnosed with classic EDS, one with hypermobile EDS and one with osteogenesis imperfecta, all without genetic confirmation, but fulfilling clinical diagnostic criteria (table 4).10,11,13
Table 4.
Patients with CeAD who had confirmed inherited CTD
DISCUSSION
In the largest series of patients with CeAD to date, comprising nearly 2,000 patients recruited through departments of neurology (mean age 45 years, 57% men), family history of CeAD was very rare, and was associated with carotid location and elevated cholesterol levels. A trend toward an association of familial CeAD with migraine without aura was also observed. Underlying inherited CTDs, known to be associated with dissection, were exceptional, and all occurred in patients without family history of CeAD. Two patients had clinically and genetically confirmed vEDS. One patient had Marfan syndrome, one was diagnosed with classic EDS, one with hypermobile EDS, and one with osteogenesis imperfecta, based on clinical criteria.
In the only previous study to our knowledge that systematically addressed the frequency of family history of CeAD in 200 patients with CeAD,3 10 patients from 8 families had a family history of dissection. Of these, 5 patients (2.5%, 0.3%–4.7%) from 3 families (1.5%, 0%–3.2%) had a family history of dissection in the cervical arteries (the remaining being in intracranial arteries, aorta, or renal artery) and in 4 patients, the family history of CeAD was in a first-degree relative (2.0%, 0.1%–3.9%).3 The slightly higher prevalence of familial CeAD compared with our series could be explained by sampling fluctuation (CIs overlap), or selection bias, because recruitment through a single tertiary center could have led to an overrepresentation of unusual, familial cases,3 while our multicenter recruitment may be more representative of the community. Although rare, the observed frequency of family history of CeAD is higher than expected by chance given the low disease incidence. Indeed, based on an estimated CeAD prevalence of 1/1,000,14,15 the probability for a patient with CeAD with 1 sibling and 2 children of having a first-degree relative who had a CeAD by mere chance is <0.5% (e-Methods).
Although power is limited by the small number of familial cases, we describe some differences in characteristics of patients with and without a family history of CeAD, with a higher frequency of carotid dissection (90% vs 68%) and of hypercholesterolemia (42% vs 17%) in the former. These findings suggest that genetic determinants of CeAD may differ according to the dissection site. The more marked familial clustering for carotid dissection, which was suggested previously in a study comparing patients with CeAD who had a family history of CeAD with published findings from patients with sporadic CeAD,16 could point to a more important contribution of genetic factors in this vessel location. Most patients with a family history of CeAD had a dissection in the left carotid artery, suggesting that inherited anatomical factors might have a role. The higher frequency of hypercholesterolemia in patients with CeAD who had a positive family history is intriguing, because we have previously shown that patients with CeAD have a significantly lower prevalence of hypercholesterolemia than healthy controls or patients with an ischemic stroke of another etiology matched on age and sex.9 This may reflect that familial CeAD has different underlying mechanisms from sporadic CeAD, and may also point to shared pathways or shared genetic variation between CeAD and lipid metabolism, with allelic heterogeneity. Of note, for patients in whom detailed information was available, most relatives had a dissection in the same artery type as the index patients (17/19); age at CeAD occurrence was often similar within families, as previously described.15
Only 2 patients with CeAD (without family history of CeAD) had a genetically confirmed diagnosis of vEDS, which was known before the CeAD. From a clinical perspective, our findings suggest that clinically obvious inherited CTDs are extremely rare in patients with CeAD, because only 0.1% of our patients had a diagnosis of vEDS that was clinically evident and genetically confirmed. vEDS is a very rare condition, with a prevalence estimated at 0.2–1/100,000,17 and although CeAD is one of its classic complications, it was described in only 2% of the patients in the largest, partly retrospective series of patients with genetically confirmed vEDS.18,19 The first major complication of vEDS was shown to occur before age 40 in 80% of 220 patients,19 and the reported mean age at occurrence of the first cerebrovascular complication was 28 years.18 In agreement with these reports, CeAD occurred before age 30 in both patients with vEDS in our series. One of 1,934 patients was previously known to have Marfan syndrome, which is close to the prevalence of this disease in the general population, estimated between 1/3,000 and 1/5,000.20 Three patients, who had their dissection between 2001 and 2006, had been diagnosed beforehand with classic or hypermobile EDS and with osteogenesis imperfecta (prevalence for these diseases range between approximately 1/100,000 and 1/20,000), without molecular confirmation, but the genetic investigation of these disorders was challenging before the advent of next-generation sequencing, and not thought to be a requirement for diagnosis. Clinical implications are less dramatic because these diseases do not expose to an increased risk of life-threatening complications, such as vascular or hollow organ rupture, as is the case for vEDS.4 Overall, inherited CTDs, including EDS, Marfan syndrome, and osteogenesis imperfecta, appeared very rare in this large multicenter series of patients with CeAD, and CeAD was typically not the first symptom revealing the inherited disorder.
Our study has limitations. Only short-term clinical follow-up was available at this stage, so that we could not verify the previously reported association of CeAD family history with long-term CeAD recurrence.3 The number of patients with a family history of CeAD being small, association estimates have wide CIs and require confirmation in independent samples. Family members with a history of CeAD were not all included in CADISP-plus, therefore precluding detailed clinical description, although minimal information on CeAD characteristics could be obtained in the majority of cases. The diagnosis of CeAD could not formally be verified in 17% of the affected relatives. An underestimation of familial CeAD cannot be excluded, because (1) many CeADs causing strokes among family members remained undiagnosed in the last century because of lack of knowledge and proper imaging equipment, (2) CeAD with minor or no clinical symptoms, or leading to very severe rapidly fatal stroke, is probably still underdiagnosed, (3) some patients may have limited knowledge of their relatives' health. However, history of symptomatic CeAD is clinically more relevant and, while a systematic screening of CeAD in all first-degree relatives of patients with CeAD with arterial imaging might increase the diagnostic yield by potentially capturing some cases of asymptomatic CeAD, it would be unrealistic and ethically challenging to perform on such a large sample. Patients with CeAD who had inherited CTDs were diagnosed only in centers from Paris (1.0% of all Paris patients) and Zürich (0.4% of all Zürich patients). Although these were among the cities with the largest recruitment, it could point to an underestimation of such disorders in other centers. In France, many national referral centers for rare vascular disorders are located in Paris, which could lead to a recruitment and ascertainment bias. However, the proportion of patients with CeAD with inherited CTD remains small (≤1.0%), even in the cities with the largest number of diagnosed cases. Because patients with severe inherited CTDs, such as vEDS, often benefit from close multidisciplinary follow-up in highly specialized referral centers, they may also be hospitalized there when they have a CeAD, and may therefore not have been included in our study. However, given the low prevalence of these disorders, this is unlikely to have led to a major underestimation of their frequency among patients with CeAD at population level. Finally, we cannot formally exclude mild forms of inherited CTD with incomplete penetrance or CeAD as the first isolated symptom,15,21 because patients were not systematically screened for mutations in genes such as COL3A1, FBN1, TGFBR1, or TGFRB2, responsible for vEDS,19 Marfan syndrome,12 and Loeys-Dietz syndrome.22 In earlier studies, systematic search for mutations in COL3A1 among 53 patients with CeAD,23–26 and in TGFBR1 and TGFBR2 among 56 consecutive patients with CeAD,21 identified potentially deleterious mutations in COL3A1 among 2 cousins with CeAD,23 and in TGFBR2 in 2 unrelated patients with CeAD,21 but none of them fulfilled the clinical diagnostic criteria of vEDS or Loeys-Dietz syndrome, possibly pointing to mutations with incomplete penetrance.10,11,22 Whether identifying such mutations is clinically relevant, especially regarding risk of CeAD recurrence, other vascular complications, and familial risk, is unclear to date. Noteworthy, skin biopsies performed in series of 7 to 65 patients with CeAD have shown that about half of these patients have ultrastructural skin connective tissue abnormalities on electron microscopy, the most common pattern being described as composite fibrils within collagen bundles and elastic fiber abnormalities with minicalcifications and fragmentation.2,27–30 Moreover, skin biopsies performed in healthy relatives of 3 index patients with CeAD (from 3 distinct families) have suggested that these ultrastructural skin connective tissue changes may be inherited according to an autosomal dominant pattern, but linkage studies were inconclusive.31 Similar pathologic changes in the wall of temporal arteries were also described in discordant identical twin pairs, both in the affected and unaffected twin.32 These findings could suggest underlying connective tissue fragility in patients with CeAD, which may in some cases be inherited, perhaps as part of uncharacterized inherited CTD with incomplete penetrance.
In 2 large multicenter cohorts of patients with CeAD, both family history of symptomatic CeAD and known inherited CTDs were very rare. In clinical practice, given the psychological distress and anxiety that is often generated by the occurrence of CeAD,33 which usually affects young individuals with few or no vascular risk factors,9 this is important information for patients and their families. Based on current knowledge, we believe that systematic screening for mutations in genes responsible for inherited CTDs is not clinically justified in monosymptomatic cases.4 For research purposes, our findings justify ongoing efforts to identify genetic susceptibility factors for CeAD by genetic association studies,4 or next-generation sequencing, rather than a family-based linkage approach, except for the very rare families with multiple cases.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the staff and participants of all centers from the CADISP study and the Paris-Lariboisière/Zürich/Bern CeAD registry for their important contributions: Mrs. Marja Metso (RN), Department of Neurology, Helsinki University Central Hospital, Helsinki, Finland; Ms. Laurence Bellengier (MS), Pr. Christian Libersa (MD, PhD), Pr. Dominique Deplanque (MD, PhD), Centre d'Investigation Clinique, University Hospital of Lille, France; Dr. Nathalie Fievet (PhD), INSERM U744, Pasteur Institute, Lille, France; Dr. Jean-Christophe Corvol (MD, PhD), Mrs. Sylvie Montel (RN), and Mrs. Christine Rémy (RN), Centre d'Investigation Clinique, Pitié-Salpêtrière University Hospital, Paris, France; Ana Lopes Da Cruz (PhD), Laboratory of Experimental Neurology, ULB, Brussels, Belgium; Annet Tiemessen (MS), Stroke Team, University Hospital Basel, Switzerland; Dr. Raffaele Palmirotta (MD, PhD), Department of Laboratory Medicine and Advanced Biotechnologies, IRCCS San Raffaele, Rome, Italy; and Mrs. Marianne Kormann (RN) and Mrs. Andrea Surtmann Huguenin (RN), Department of Neurology, University Hospital Inselspital, Bern, Switzerland. The authors also thank Dr. William R. Davidson, Jr., at Penn State Hershey Heart and Vascular Institute, PA, for the clinical information he kindly provided.
GLOSSARY
- CADISP
Cervical Artery Dissection and Ischemic Stroke Patients
- CeAD
cervical artery dissection
- CI
confidence interval
- CTD
connective tissue disorder
- EDS
Ehlers-Danlos syndrome
- vEDS
vascular Ehlers-Danlos syndrome
Footnotes
Supplemental data at Neurology.org
AUTHOR AFFILIATIONS
From the Department of Neurology (S.D., D.H., H.C., C.S., M.-G.B.), Lariboisière Hospital, DHU Neurovasc Paris Sorbonne, Paris; INSERM Unit UMR-S 1161 (S.D., S.S., D.H., H.C., E.T.-L.), Paris; University Paris 7–Denis-Diderot (S.D., S.S., D.H., H.C., C.S., E.T.-L., M.-G.B.), DHU Neurovasc Sorbonne Paris Cité; Department of Neurology (S.D., D.L.), EA1046, Lille University Hospital; Department of Epidemiology and Public Health (S.D., J.C.), INSERM U744, Pasteur Institute, Lille, France; INSERM U897 (S.D.), Bordeaux University, Bordeaux, France; Department of Neurology (B.G.S., H.S., S.J., U.F., M.A.), University Hospital Inselspital, Bern, Switzerland; Department of Neurology (J.J.M.), Sanatorio Allende, Cordoba, Argentina; Department of Neurology (M.K., C.L., C.G.-G.), Heidelberg University Hospital, Germany; Department of Neurology (H.S.), Zürich University Hospital, Switzerland; Electron Microscopy Laboratory (I.H.), Institute of Pathology University Hospital, Germany; Department of Neurology and Stroke Center (S.E., P.A.L.), Basel University Hospital; Neurorehabilitation Unit (S.E.), University and University Center for Medicine of Aging and Rehabilitation Basel, Felix Platter Hospital, Basel, Switzerland; Department of Neurology (T.M.M., A.J.M., T.T.), Helsinki University Central Hospital, Finland; Department of Clinical and Experimental Sciences (A.P., P.C.), Neurology Clinic, University of Brescia, Italy; KU Leuven–University of Leuven (V.T.), Department of Neurosciences, VIB–Vesalius Research Center, Experimental Neurology–Laboratory of Neurobiology, Leuven; Vesalius Research Center (V.T.), VIB, Leuven, Belgium; Paris Descartes University (E.T.), INSERM UMR S894, Department of Neurology, Sainte-Anne Hospital, Paris; Department of Neurology (E.T.), University Hospital of Caen, France; Department of Rehabilitation (S.P.), Santa Lucia Hospital, Rome; Department of Neurology (M.S.), San Raffaele University Hospital, Milano, Italy; Department of Neurology (Y.S.), Pitié-Salpêtrière University Hospital, Paris; Department of Neurology (Y.B.), Dijon University Hospital, France; Istanbul University (A.A.), Cerrahpasa Medical School, Neurology Department, Turkey; Department of Neurology (C. Lichy), Memmingen Hospital, Germany; Department of Neurology (C. Lamy), Amiens University Hospital, France; Department of Neurology (A.G.), Ludwigshafen Hospital, Germany; Stroke Unit (V.C.), Perugia University Hospital, Italy; Department of Rehabilitation (T.B.), Schmieder-Klinik, Heidelberg, Germany; Department of Molecular Genetics (E.T.-L.), Lariboisière Hospital, Paris; Division of Medical Genetics (D.P.G.), Referral Center for Fabry Disease and Inherited Disorders of Connective Tissue, CHU Raymond Poincaré (AP-HP), Garches; University of Versailles–St Quentin en Yvelines (D.P.G.), EA 2493, Montigny; National Referral Center for Rare Vascular Diseases (M.F.), Hôpital Européen Georges-Pompidou HEGP, Paris, France; NeuroCenter (R.W.B.), Clinic Hirslanden Zürich, Switzerland; and Cerebrovascular Disease Unit (A.B.), IRCCS Foundation C. Besta Neurological Institute, Milan Italy.
AUTHOR CONTRIBUTIONS
Dr. Stéphanie Debette: study concept and design, analysis and interpretation, statistical analysis, study supervision or coordination, drafting/revising the manuscript, obtaining funding, acquisition of data. Dr. Barbara Goeggel Simonetti: analysis and interpretation, drafting/revising the manuscript, acquisition of data. Mrs. Sabrina Schilling: statistical analysis, analysis and interpretation. Dr. Juan José Martin: acquisition of data, drafting/revising the manuscript. Dr. Manja Kloss: acquisition of data, drafting/revising the manuscript. Dr. Hakan Sarikaya: acquisition of data, drafting/revising the manuscript. Dr. Ingrid Hausser: drafting/revising the manuscript. Dr. Stefan Engelter: acquisition of data, drafting/revising the manuscript. Dr. Tiina M. Metso: acquisition of data, drafting/revising the manuscript. Dr. Alessandro Pezzini: acquisition of data, drafting/revising the manuscript. Dr. Vincent Thijs: acquisition of data, drafting/revising the manuscript. Dr. Emmanuel Touzé: acquisition of data, drafting/revising the manuscript. Dr. Stefano Paolucci: acquisition of data, drafting/revising the manuscript. Dr. Paolo Costa: acquisition of data. Dr. Maria Sessa: acquisition of data, drafting/revising the manuscript. Dr. Yves Samson: acquisition of data, drafting/revising the manuscript. Dr. Yannick Béjot: acquisition of data, drafting/revising the manuscript. Dr. Ayse Altintas: acquisition of data, drafting/revising the manuscript. Dr. Antti J. Metso: acquisition of data, drafting/revising the manuscript. Dr. Dominique Hervé: acquisition of data. Dr. Christoph Lichy: acquisition of data. Dr. Simon Jung: acquisition of data, drafting/revising the manuscript. Dr. Urs Fischer: acquisition of data. Dr. Chantal Lamy: acquisition of data, drafting/revising the manuscript. Dr. Armin Grau: acquisition of data, drafting/revising the manuscript. Dr. Hugues Chabriat: acquisition of data, drafting/revising the manuscript. Dr. Valeria Caso: acquisition of data, drafting/revising the manuscript. Dr. Philippe A. Lyrer: acquisition of data, drafting/revising the manuscript. Dr. Christian Stapf: acquisition of data. Dr. Turgut Tatlisumak: acquisition of data, drafting/revising the manuscript. Dr. Tobias Brandt: acquisition of data, drafting/revising the manuscript. Dr. Elisabeth Tournier-Lasserve: drafting/revising the manuscript. Dr. Dominique P. Germain: acquisition of data, drafting/revising the manuscript. Dr. Michael Frank: acquisition of data, drafting/revising the manuscript. Dr. Ralf W. Baumgartner: acquisition of data, drafting/revising the manuscript. Dr. Caspar Grond-Ginsbach: acquisition of data, drafting/revising the manuscript. Dr. Marie-Germaine Bousser: analysis and interpretation, acquisition of data, drafting/revising the manuscript, obtaining funding. Dr. Didier Leys: analysis and interpretation, acquisition of data, drafting/revising the manuscript, obtaining funding. Dr. Jean Dallongeville: analysis and interpretation, acquisition of data, drafting/revising the manuscript, obtaining funding. Dr. Anna Bersano: study concept and design, analysis and interpretation, study supervision or coordination, drafting/revising the manuscript, obtaining funding, acquisition of data. Dr. Marcel Arnold: study concept and design, analysis and interpretation, study supervision or coordination, drafting/revising the manuscript, acquisition of data.
STUDY FUNDING
Contrat de Projet Etat-Region; Projet Hospitalier de Recherche Clinique Régional; Fondation de France; Adrinord-EA2691; Institut Pasteur de Lille; INSERM U744; Emil Aaltonen, Paavo Ilmari Ahvenainen, Päivikki and Sakari Sohlberg, Aarne Koskelo, Maire Taponen, Aarne and Aili Turunen, Biomedicum Helsinki Foundation, Finnish Brain Foundation, Lilly, Alfred Kordelin, Orion-Farmos and Maud Kuistila Foundations; Finnish Medical Foundation; Helsinki University Central Hospital Research Fund; Academy of Finland; Helsinki University Medical Foundation; Basel Stroke-Funds; Käthe-Zingg-Schwichtenberg Fonds (Swiss Academy of Medical Sciences); Swiss Heart Foundation; Swiss National Science Foundation. Stéphanie Debette is supported by a Chair of Excellence grant from the French National Research Agency (Agence Nationale de la Recherche) and from the Leducq Foundation. Vincent Thijs is supported by a Clinical Investigatorship from FWO Flanders.
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Debette S, Markus HS. The genetics of cervical artery dissection: a systematic review. Stroke 2009;40:e459–e466. [DOI] [PubMed] [Google Scholar]
- 2.Grond-Ginsbach C, Debette S. The association of connective tissue disorders with cervical artery dissections. Curr Mol Med 2009;9:210–214. [DOI] [PubMed] [Google Scholar]
- 3.Schievink WI, Mokri B, Piepgras DG, Kuiper JD. Recurrent spontaneous arterial dissections: risk in familial versus nonfamilial disease. Stroke 1996;27:622–624. [DOI] [PubMed] [Google Scholar]
- 4.Debette S, Germain DP. Neurologic manifestations of inherited disorders of connective tissue. Handb Clin Neurol 2014;119:565–576. [DOI] [PubMed] [Google Scholar]
- 5.Debette S, Grond-Ginsbach C, Bodenant M, et al. Differential features of carotid and vertebral artery dissections: the CADISP study. Neurology 2011;77:1174–1181. [DOI] [PubMed] [Google Scholar]
- 6.von Babo M, De Marchis GM, Sarikaya H, et al. Differences and similarities between spontaneous dissections of the internal carotid artery and the vertebral artery. Stroke 2013;44:1537–1542. [DOI] [PubMed] [Google Scholar]
- 7.Arnold M, Kappeler L, Georgiadis D, et al. Gender differences in spontaneous cervical artery dissection. Neurology 2006;67:1050–1052. [DOI] [PubMed] [Google Scholar]
- 8.Debette S, Metso TM, Pezzini A, et al. CADISP-genetics: an international project searching for genetic risk factors of cervical artery dissections. Int J Stroke 2009;4:224–230. [DOI] [PubMed] [Google Scholar]
- 9.Debette S, Metso T, Pezzini A, et al. Association of vascular risk factors with cervical artery dissection and ischemic stroke in young adults. Circulation 2011;123:1537–1544. [DOI] [PubMed] [Google Scholar]
- 10.De Paepe A, Malfait F. The Ehlers-Danlos syndrome: a disorder with many faces. Clin Genet 2012;82:1–11. [DOI] [PubMed] [Google Scholar]
- 11.Beighton P, De Paepe A, Steinmann B, Tsipouras P, Wenstrup RJ. Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK). Am J Med Genet 1998;77:31–37. [DOI] [PubMed] [Google Scholar]
- 12.Loeys BL, Dietz HC, Braverman AC, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet 2010;47:476–485. [DOI] [PubMed] [Google Scholar]
- 13.Sillence DO, Senn A, Danks DM. Genetic heterogeneity in osteogenesis imperfecta. J Med Genet 1979;16:101–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee VH, Brown RD, Jr, Mandrekar JN, Mokri B. Incidence and outcome of cervical artery dissection: a population-based study. Neurology 2006;67:1809–1812. [DOI] [PubMed] [Google Scholar]
- 15.Debette S, Leys D. Cervical-artery dissections: predisposing factors, diagnosis, and outcome. Lancet Neurol 2009;8:668–678. [DOI] [PubMed] [Google Scholar]
- 16.Grond-Ginsbach C, de Freitas GR, Campos CR, et al. Familial occurrence of cervical artery dissection: coincidence or sign of familial predisposition? Cerebrovasc Dis 2012;33:466–470. [DOI] [PubMed] [Google Scholar]
- 17.Germain DP. Ehlers-Danlos syndrome type IV. Orphanet J Rare Dis 2007;2:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.North KN, Whiteman DA, Pepin MG, Byers PH. Cerebrovascular complications in Ehlers-Danlos syndrome type IV. Ann Neurol 1995;38:960–964. [DOI] [PubMed] [Google Scholar]
- 19.Pepin M, Schwarze U, Superti-Furga A, Byers PH. Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. N Engl J Med 2000;342:673–680. [DOI] [PubMed] [Google Scholar]
- 20.Keane MG, Pyeritz RE. Medical management of Marfan syndrome. Circulation 2008;117:2802–2813. [DOI] [PubMed] [Google Scholar]
- 21.Pezzini A, Drera B, Del Zotto E, et al. Mutations in TGFBR2 gene cause spontaneous cervical artery dissection. J Neurol Neurosurg Psychiatry 2011;82:1372–1374. [DOI] [PubMed] [Google Scholar]
- 22.Loeys BL, Dietz HC. Loeys-Dietz syndrome. In: Pagon RA, Bird TC, Dolan CR, Stephens K, editors. GeneReviews [Internet]. Seattle: University of Washington; 2008:1993–2014. [PubMed] [Google Scholar]
- 23.Martin JJ, Hausser I, Lyrer P, et al. Familial cervical artery dissections: clinical, morphologic, and genetic studies. Stroke 2006;37:2924–2929. [DOI] [PubMed] [Google Scholar]
- 24.Kuivaniemi H, Prockop DJ, Wu Y, et al. Exclusion of mutations in the gene for type III collagen (COL3A1) as a common cause of intracranial aneurysms or cervical artery dissections: results from sequence analysis of the coding sequences of type III collagen from 55 unrelated patients. Neurology 1993;43:2652–2658. [DOI] [PubMed] [Google Scholar]
- 25.van den Berg JS, Limburg M, Kappelle LJ, Pals G, Arwert F, Westerveld A. The role of type III collagen in spontaneous cervical arterial dissections. Ann Neurol 1998;43:494–498. [DOI] [PubMed] [Google Scholar]
- 26.von Pein F, Valkkila M, Schwarz R, et al. Analysis of the COL3A1 gene in patients with spontaneous cervical artery dissections. J Neurol 2002;249:862–866. [DOI] [PubMed] [Google Scholar]
- 27.Ulbricht D, Diederich NJ, Hermanns-Le T, Metz RJ, Macian F, Pierard GE. Cervical artery dissection: an atypical presentation with Ehlers-Danlos-like collagen pathology? Neurology 2004;63:1708–1710. [DOI] [PubMed] [Google Scholar]
- 28.Uhlig P, Bruckner P, Dittrich R, Ringelstein EB, Kuhlenbaumer G, Hansen U. Aberrations of dermal connective tissue in patients with cervical artery dissection (sCAD). J Neurol 2008;255:340–346. [DOI] [PubMed] [Google Scholar]
- 29.Brandt T, Hausser I, Orberk E, et al. Ultrastructural connective tissue abnormalities in patients with spontaneous cervicocerebral artery dissections. Ann Neurol 1998;44:281–285. [DOI] [PubMed] [Google Scholar]
- 30.Brandt T, Orberk E, Weber R, et al. Pathogenesis of cervical artery dissections: association with connective tissue abnormalities. Neurology 2001;57:24–30. [DOI] [PubMed] [Google Scholar]
- 31.Wiest T, Hyrenbach S, Bambul P, et al. Genetic analysis of familial connective tissue alterations associated with cervical artery dissections suggests locus heterogeneity. Stroke 2006;37:1697–1702. [DOI] [PubMed] [Google Scholar]
- 32.Volker W, Kuhlenbaumer G, Dittrich R, et al. Two sets of identical twins with cervical artery dissection concordant for temporal artery pathology. Neurology 2008;71:1035–1037. [DOI] [PubMed] [Google Scholar]
- 33.Fischer U, Ledermann I, Nedeltchev K, et al. Quality of life in survivors after cervical artery dissection. J Neurol 2009;256:443–449. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.