

Abstract
The incidence of nonmelanoma skin cancer (NMSC) continues to rise, partly because of aging, the frequency of early childhood sunburns, and sporadic extreme recreational sun exposure. A nonsurgical approach to selected cutaneous malignancy could possibly reduce the cost as well as morbidity of surgical treatment for NMSC. There has been growing interest in isolating compounds that could suppress or reverse the biochemical changes necessary for cutaneous malignancies to progress by pharmacologic intervention. By targeting diverse pathways recognized as important in the pathogenesis of nonmelanoma skin cancers, a combination approach with multiple agents or addition of chemopreventative agents to topical sunscreens may offer the potential for novel and synergistic therapies in treating nonmelanoma skin cancer. This preliminary information will expand to include more therapeutic options for NMSC in the future.
Keywords: Newer agents, nonmelanoma skin cancers, prevention, treatment
Introduction
What was known?
The conventional treatments for these diseases were surgical procedures such as curettage and electrodessication, excision, chemotherapy, radiation, cryotherapy, or use of lasers.
However, because of the time and expense incurred for this procedure, it is indicated only in patients with aggressive tumors or those where disfigurement or functional impairment is a risk.
The common feature of all these above-mentioned treatments is that, they are quite unspecific without targeting the tumor itself or its environment. This leads to unnecessary adverse effects in the surrounding tissue such as scar formation or other cosmetically disfiguring events.
Besides, they have high rates of treatment failure, morbidity, and mortality; hence alternative treatment modalities for patients with aggressive or advanced disease are needed.
There has been a significant increase in worldwide incidence of nonmelanoma skin cancer (NMSC) by about 10% per annum and presently 2-3 million new cases of NMSC are diagnosed worldwide every year.[1,2] Basal cell carcinoma (BCC) is a slow growing, locally invasive epidermal tumor which accounts for 75% of cases. BCC has a metastatic rate of < 0.1%.[3,4] Majority of the remainder cases of NMSC are accounted by cutaneous squamous cell carcinoma (SCC) arising from dysplastic epidermal keratinocytes.[5] However, SCC has a significant rate of metastasis (0.3-3.7%), as compared to BCC.[6] Most countries do not have cancer registries for NMSC and reported figures are likely to be underestimated.[7]
Actinic keratoses (AKs) or solar keratoses, are premalignant cutaneous lesions that primarily occur in sun-exposed areas. AKs are clinically important lesions due to their ability to develop into a SCC. Furthermore, they are considered a risk factor for subsequent development of melanoma and NMSC. Older fair-skinned populations (Fitzpatrick skin phototypes I–III) are frequently afflicted by these lesions. Other important risk factors include cumulative ultraviolet (UV) radiation exposure and old age. Immunodeficiency or individuals with certain genetic syndromes, such as xeroderma pigmentosum and albinism too have a higher risk.[8,9]
Etiopathogenesis
A combination of environmental, genetic, phenotypical, immunological, and cultural factors can lead to the development of NMSC.
Environmental factors
Environmental exposure to UV light is one of the major risk factor. This is demonstrated by the surge in its incidence in sunnier climates, lesser rates in darker skin types, and the fact that a significant proportion of tumors arise over sun-exposed skin. SCC is frequently associated with chronic UV exposure, whereas BCC has been more linked to intermittent and childhood sun exposure.[10,11,12] UV light is presumed to stimulate direct DNA mutation via covalent bonding between adjacent pyrimidines (UVB) and production of reactive oxygen species (UVA) [Figure 1].[13] A 2.5-fold increase in SCC risk and 1.5-fold increase in BCC risk is seen due to the use of tanning devices.[14] Treatment of various skin diseases with phototherapy too can lead to NMSC. This association is stronger with psoralen photochemotherapy than with narrowband UVB.[12]
Figure 1.
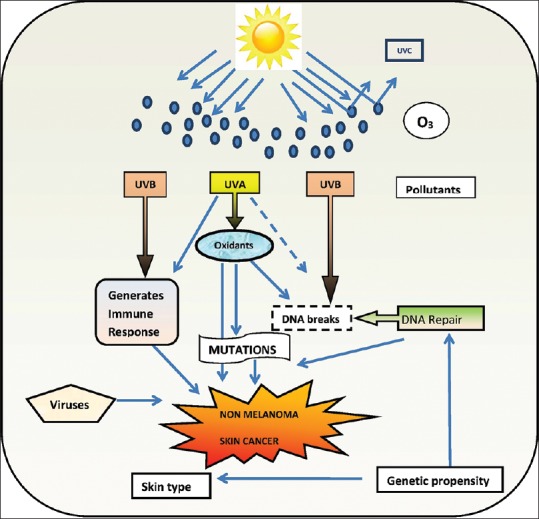
Etiological factors for nonmelanoma skin cancer
The amounts and proportion of chemical elements to which humans are exposed have been influencing human health in the past and present. Ancient Greeks and Romans knew arsenic, as a therapeutic agent. Currently, industrial and environmental exposure to arsenic impacts health more than medicinal.[15] Millions of people worldwide are currently being poisoned by inorganic arsenic, an environmental toxicant; moreover chronically exposed individuals are vulnerable to arsenicosis or arsenic poisoning.[16] Worldwide chronic arsenic toxicity has become a human health threat.[17] The “largest mass poisoning of a population in history” has been the arsenic contamination of drinking water in Bangladesh. This inorganic arsenic contamination is of natural origin, with arsenic thought to be released to the groundwater from the surrounding sediment.[18] Besides more than six million inhabitants in nine districts of West Bengal, exposed to exceedingly high concentrations of arsenic, an estimated 300,000 people demonstrated arsenic-induced skin lesions in these districts. This is regarded as the greatest arsenic calamity in the world.[19] International Agency for Research on Cancer considers arsenic as a class I human carcinogen due to its increased risk for skin cancer, besides internal cancers, such as lung and bladder cancer.[18] Numerous systems in the human body are at risk due to chronic human exposure: Dermal (exfoliative dermatitis, keratosis, vitiligo, and skin cancer), peripheral neuropathy, encephalopathy, bronchitis, pulmonary fibrosis, hepatosplenomegaly akin to noncirrhotic portal fibrosis (NCPF), portal hypertension, peripheral vascular disease and black foot disease (BFD), arteriosclerosis and cancers of lung, urinary bladder, other internal organs and diabetes.[15] The dermal tissue seems to be fairly vulnerable to the effects of arsenic. Arsenic-induced skin lesions appear to be the most common and early symptoms of arsenicosis.[17] Among these, hyperkeratoses (HKs) are frequent early manifestations of arsenicosis in humans. HKs can be precursor lesions of nonmelanoma skin cancers (NMSCs), however the dynamic forces behind their development and how they may eventually advance to NMSCs are unidentified.[20] Upregulation of inflammatory signals like cytokines and tumor necrosis factor-alpha (THF-α), oxidative stress, hypomethylation, decreased DNA repair and apoptosis, cell proliferation, angiogenesis, activation of several enzymes like methyl transferase which converts inorganic arsenic to monomethylarsonic acid (MMA) and dimethylarsonic acid (DMA), and glutathione (GSH) in in vivo and in vitro in experimental rat liver slices are some of the proposed biological mechanisms in experimental animals.[15] Especially, an arsenic-induced tumor suppressorome, a complex of 17 tumor suppressors known to be silenced in human cancers has been identified. This discovery represents a crucial indication in extrication of a probable epigenetic mode of arsenic-induced disease.[16] Even before the typical dermatological symptoms of arsenicosis starts manifesting, serum levels of catalase and myeloperoxidase may possibly be used as biomarkers of early arsenic exposure-induced disease.[21] Some of these diseases have been recurrently linked with overproduction of reactive oxygen species that leads to DNA damage and improper functioning of body's antioxidant defense mechanism. Natural polyphenols present in tea could be an excellent source of antioxidants. It has demonstrated that polyphenols and extracts of green as well as black tea modulate sodium arsenite (As III)-induced DNA damage. Furthermore, tea improved recovery of DNA damage, demonstrating repair as confirmed by unscheduled DNA synthesis and prominent expression of DNA repair enzyme poly adenosine diphosphate (ADP) ribose polymerase. Thus, it is presumed that the antioxidant potential and repair-inducing capacity of tea could assist in combating severe genotoxic effects induced by arsenic in human population.[22] Remediation by arsenic-safe drinking water can diminish dermatological expression and cytogenetic damage; however is incapable to offset the non-dermatological symptoms.[23] Treatment of arsenicosis is unsatisfactory and is mostly symptomatic.[24]
Genetic factors
Knowledge about fundamental aspects of cancer biology has increased because of advent of cytogenetic analysis. This has led to better understanding and appreciation of not only the process but progress of cancer development as well. An in depth analysis of chromosomal aberrations associated with tumors has become possible due to recent progresses in culturing techniques and new cytogenetic technologies. Various chromosomal alterations have been identified because of classical cytogenetic techniques, besides fluorescence-based techniques such as fluorescence in-situ hybridization (FISH) and comparative genomic hybridization. Even the development of newer technologies including laser capture microdissection and comparative genomic hybridization arrays will allow more refined analysis. These aberrations besides helping in defining the stages and classifications of nonmelanoma skin cancer, may also implicate chromosomal regions involved in progression and metastatic potential.[25]
Genetic mutations are an important cofactor in the development of NMSC. The p53 tumor suppressor gene and gene product are among the most diverse as well as complex. They are shown to have a direct correlation with cancer development and have been shown to occur in nearly 50% of all cancers. p53 mutations are particularly common in skin cancers and UV irradiation has been shown to be a primary cause of specific ‘signature’ mutations that can result in oncogenic transformation [Figure 2]. There are certain ‘hot spots’ in the p53 gene where mutations are commonly found that result in a mutated dipyrimidine site.[26]
Figure 2.
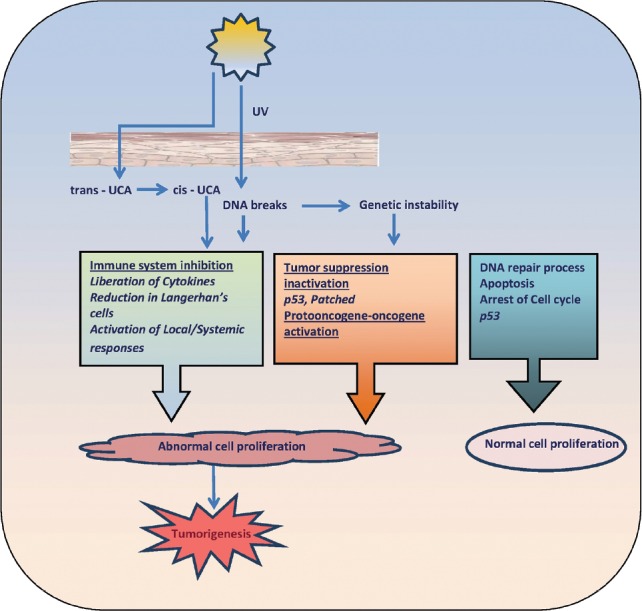
Ultraviolet light and skin carcinoma
Besides, the discovery of mutation of patched gene (PTCH) on chromosome 9q22 underlying basal cell nevus syndrome (BCNS), a genodermatosis associated with multiple BCC, has greatly forwarded the understanding of genetics underpinning BCC. Patched 1, the protein product of PTCH, is a cell surface receptor, inhibiting smoothened (SMO), a G-protein-coupled receptor. Cessation of SMO inhibition by patched 1 initiates a signal cascade that leads to the activation of glioma transcription factor-1 (GLI-1) [Figure 3]. Dysregulation of this pathway by either the loss of PTCH or uncontrolled expression of SMO results in cell proliferation and differentiation. Mutations of either PTCH or SMO have been found in 70% of sporadic human BCCs. Other reported genes associated with pathogenesis of BCC are cytochrome P450 (CYP), glutathione S-transferase (GST), and P53. CYP and GST are known to detoxify mutagens.[7] The cytochrome P450 (CYP) supergene family has more than 30 isoforms, which catalyze biotransformation of several xenobiotics, often as the first step of a two-phase detoxification. The resulting highly reactive intermediate serves as a substrate for phase-two enzymes, including members of GST supergene family. GSTs can also catalyze detoxification of products of oxidative stress (e.g., lipid and DNA hydroperoxides). Polymorphisms in GSTT1 and CYP2D6 are associated with susceptibility to NMSC.
Figure 3.
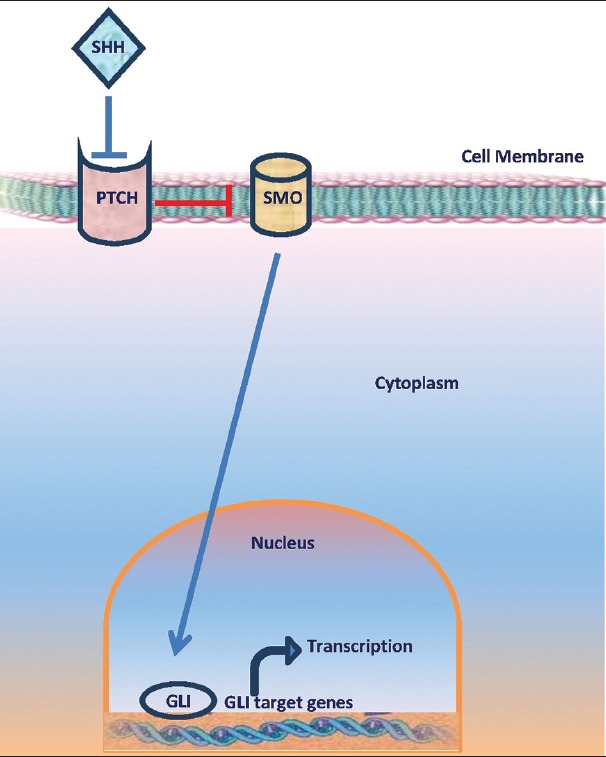
Hedgehog signaling pathway
Progression of precursor lesions of AK to SCC represents a multistep accumulation of genetic damage, whereas sporadic BCC develops de novo. Over 72% of SCC have been noted to develop within AK or actinically damaged skin, also known as background field cancerization.[27,28] Significant histological and genetic similarity is present between AK and SCC. Additionally, errors of p53 signalling have been associated in both. Mutations in P53 have been recognized in 69% to over 90% of invasive SCC.[29,30]
Human telomerase is a distinctive enzyme that uses RNA as a template to add telomere repeats at chromosome ends to compensate for telomere loss during cell division. Majority of immortal and tumor cells have extensive telomerase activity and demonstrate no net loss of telomere length during proliferation, unlike healthy cells. Consequently, telomerase reactivation in cells causes tumor immortalization. Additionally, investigators demonstrated telomerase activation to be important in pathogenesis of BCC and SCC.[31]
Phenotypical factors
Melanocortin-1 receptor (MC1R) gene variants, agouti signaling protein (ASIP) and tyrosinase (TYR), are associated with fair skin, red hair, and increased melanoma risk; and evidence suggests that they might be important independent risk factors for NMSC.[32] Blue eyes, increasing number of melanocytic nevi, and Fitzpatrick skin type are some of the major phenotypic features linked with increased risk of NMSC.[29,33] Human papillomavirus, by prolonging keratinocyte cell cycle and increasing the degradation of p53, has been implicated to be pathogenic for SCC.[34,35]
Immunological factors
Furthermore, organ transplant recipients and immunosuppressed patients too could develop NMSC. A 65-250-fold increase in the incidence of SCC and ten-fold increase in BCC have been observed in these categories of patients.[36] An increase in incidence rates of 7% for NMSC after 1 year and 45% after 11 years implies a correlation with the duration of immunosuppression. Following a primary SCC, the risk of developing a second NMSC within 5 years is 66%.[38] Furthermore, at presentation, the tumors are often more deeply invasive, with decreased histological differentiation and greater risk of metastasis.[39]
Cultural
Persistent use of a kangri by the Kashmiri people to combat the cold temperature during winter induces kangri cancer, a unique thermally-induced SCC of the skin.[40] This kangri, customary fire pot, is a resourceful method of providing warmth. The kangri is an earthenware container with an outer encasement of wickerwork that is filled with ignited coal inside for providing a source of heat in winter months. Its long-term use may result in development of erythema ab igne, a reticulate hypermelanosis with erythema, subsequently transforming into cutaneous cancer.[41] The variety of skin cancer in Kashmir valley is radically dissimilar from rest of the country. This occurrence of skin cancer was reported by Maxwell. A substantial risk of locoregional metastasis in 30-50% cases is attributed to an aggressive biological behavior. Due to the distinctive environmental distribution of kangri cancer, there is scarcity of literature concerning the natural history, locoregional, and distant metastatic pattern and treatment recommendations in these tumors.[42] kangri cancer appears on the legs and abdomen unlike classical UV-induced SCC of the skin. Tp53 is a principal target of constant hyperthermia in the progress of kangri cancer in the temperate risk Kashmiri population. The nature of the respective environmental carcinogens could attribute differences in Tp53 mutation spectrum of UV-induced SCC of the skin and kangri cancer. Tp53 may function as a potential molecular marker and prognostic tool, at least in a subset of sporadic kangri tumors.[43] Arg72Pro SNP (single-nucleotide polymorphism) is implicated in kangri cancer and that the rare, proline-related allele is connected with higher susceptibility to kangri cancer.[40] Results favor the use of postoperative radiotherapy to primary and prophylactic treatment of regional nodes on lines of head and neck tumors in these cases.[42]
Chronic inflammation
Several chronic dermatoses may possibly turn out to be malignant, such as factitial dermatitis, chronic ulcer, lichen sclerosus, lichen planus (LP), prurigo nodularis, etc., Lichen simplex chronicus (LSC) is a regular skin disorder characterized by circumscribed, lichenified, pruritic plaque secondary to local repetitive trauma, particularly rubbing and scratching. Rubbing and scratching contributes to an overload of inflammatory mediators, leading to modification in the process of keratinocyte proliferation and differentiation.[44] SCC arising within the lesion of LSC is rare. It is theorized that chronic irritation of burn cuits in addition to modified mechanisms of skin reparation perhaps could have produced carcinogenic substances. An additional assumption is that chronic cutaneous inflammatory processes, with oncogenic-like “overdrive” of growth factors persistently stimulating epithelial cells, may well lead to malignant transformation. The unremitting and recurring disturbance on inflamed tissues might have considerably contributed to progress of the neoplasia. This type of carcinoma might also be included in the group of SCC arising within chronic cutaneous conditions.[45]
The clinical manifestations of lupus erythematosus, a chronic inflammatory autoimmune disease, range from an indolent chronic cutaneous form to a severe potentially life-threatening disease. A subphenotype of chronic cutaneous lupus erythematosus, discoid lupus erythematosus, is characterized by scaly disk-shaped plaques which may be localized or widespread. Arising largely on sun-exposed skin, these might seldom advance to SCC. The pathogenesis of discoid lupus erythematosus includes intricate interactions between several susceptibility genes implicated in immune responses and clearance of apoptotic cells on one hand and environmental factors on the other.[46,47,48]
Genodermatoses, age, skin post radiotherapy, hematological diseases (e.g., leukemia, lymphoma), and chronic inflammation/ulcers are some of the other risk factors for NMSC [Figure 4].[49,50]
Figure 4.
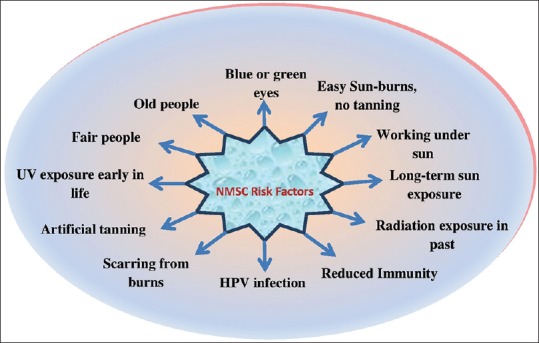
Risk factors for nonmelanoma skin cancer
Prevention
Solar ultraviolet radiation (UVR) is the main causative factor in the development of NMSC. UVR plays a variety of roles in induction of skin cancers. It can serve as a complete carcinogen or as a promoter of carcinogenesis. Tumor suppressor gene p53 is a common target of UVR-induced mutations. The immunosuppressive effect of UVR contributes to its carcinogenic activity. Finally, any one of these effects of UVR may contribute to induction of skin cancers by other agents such as X-rays, viruses, or chemical carcinogens. Primary prevention of UVR exposure is the most effective means of reducing UVR carcinogenesis.[51]
AK has been identified as a precursor of SCC, but not of BCC. AKs are far more common than SCC, making them excellent targets for chemoprevention. Cancer chemoprevention can prevent or delay the occurrence of cancer in high risk populations using dietary or chemical interventions. UVR-induced changes serve as a basis for development of novel agents.
Prevention of cutaneous sequelae of chronic arsenic exposure is multifaceted. Most important being cessation of consuming contaminated water. Person-to-person reporting of well test results, well labeling, village and individual health education, and installation of more deeply situated wells are some of the interventions that have been implemented. Rainwater harvesting, filtration, and removal of arsenic from current water supplies via individual devices or treatment plants are some of the other modalities to reduce consumption of arsenic-contaminated water. Also, necessary is treatment of surface water with pressure filtration and disinfection. To diminish the injurious consequences of arsenic on skin, certain dietary modifications as well as vitamin supplementation are beneficial. These influence methylation and subsequent detoxification of arsenic. Supplementation with vitamin E, selenium, folic acid, riboflavin, and pyridoxine are the possible additives. It is to be hoped that a blend of improvement proposals and programs with supervision by medical community for dormant sequelae and public health education in addition to counseling will alleviate the toxic brunt of chronic arsenic exposure in this population, and steer potential efforts worldwide.[18]
Kangri cancer is preventable by improving socioeconomic condition of the society, educating society regarding its use, reduce its use among children to decline its latent period; promote other means of heating such as electric/gas heaters, etc.; and general understanding regarding unfavorable effects associated with kangri. Surgery forms the mainstay of treatment in this cancer. Use of prophylactic regional nodal irradiation has proved capable of reducing both the locoregional and the distant relapse. However, as very little data is available on this aspect, further trials are required to establish its role firmly.[42]
For patients with chronic inflammatory dermatoses, regular examination and long-term follow-up are essential because these patients have a much higher propensity to develop new cutaneous malignancies. Sun protective clothing and sun avoidance during peak hours too are essential.
Goal is to develop agents for use in alone or in combination to improve chemoprevention efficacy and reduce skin cancer incidence.[52]
Secondary prevention includes a full body examination for early detection and several treatment modalities that may prevent further development and recurrence.
Treatment
The conventional treatments for these diseases were surgical procedures such as curettage and electrodesiccation, excision with so far the best outcome in terms of remission rates, and micrographic surgery. For the vast majority of patients with these tumors, surgical treatment remains the standard of care and is successful. Mohs micrographic surgery continues to be the ‘gold standard’ for treatment. Till recently, traditional methods of chemotherapy and radiation were used in the treatment of patients with metastatic or unresectable NMSC. Although radiation therapy is effective, its use is limited because of the side effects induced. Radiation therapy can be used in certain patients who are not surgical candidates. Other ablative treatment modalities are cryotherapy or use of lasers (Er:YAG, CO2). The common feature of all these above mentioned treatments is that, they are quite unspecific without targeting the tumor itself or its environment. This leads to unnecessary adverse effects in the surrounding tissue such as scar formation or other cosmetically disfiguring events. Besides, they have high rates of treatment failure, morbidity, and mortality; hence alternative treatment modalities for patients with aggressive or advanced disease are needed. Treatments should be tailored to tumor type, location, size, and histological pattern; and although surgical methods remain the most frequently used, newer noninvasive treatments can be used in select tumors and may reduce morbidity.[53]
Novel strategies are urgently needed to address millions of NMSC treated. There has been much excitement owing to the recent research elucidating molecular basis of cancer development and subsequent arrival of targeted molecular inhibitors for cancer therapy.[54,55]
Newer agents currently in development or being studied for prevention of NMSC include the following.
D, L-alpha-difluoromethylornithine
Polyamines are aliphatic cations present in all cells with important physiological roles [Figure 5]. Biosynthetic and catabolic enzymes intricately control the polyamine levels in normal cells. The biosynthetic enzymes are ornithine decarboxylase, S-adenosylmethionine decarboxylase, spermidine synthase, and spermine synthase. The catabolic enzymes include spermidine/spermine acetyltransferase, flavin containing polyamine oxidase, copper containing diamine oxidase, and possibly other amine oxidases. The increased levels of polyamines in cancer cells as compared to that of normal cells could be due to multiple abnormalities in control of polyamine metabolism and uptake.[37] Increased polyamine synthesis and inflammation have long been associated with intraepithelial neoplasia, which are risk factors for cancer development in humans. Targeting polyamine metabolism (by use of polyamine synthesis inhibitors or polyamine catabolism activators) has been studied for many cancers, including colon, prostate, and skin.[56]
Figure 5.
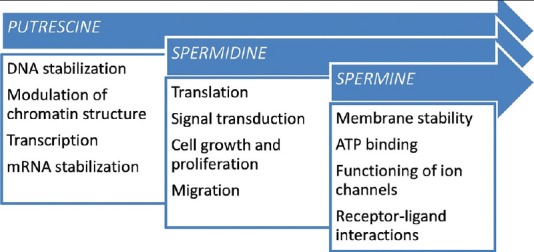
Role of polyamines
D, L-alpha-difluoromethylornithine (DFMO) was synthesized over 20 years ago. It was hoped that this enzyme-activated, irreversible inhibitor of ornithine decarboxylase, the first enzyme in polyamine synthesis, would be effective as chemotherapy for hyperproliferative diseases, including cancer and/or infectious processes. DFMO was generally found to exert cytostatic effects on mammalian cells and tissues. DFMO was shown to inhibit carcinogen-induced cancer development in a number of rodent models and interest in developing this compound as a preventive agent has increased.[57] DFMO also inhibits tumors in models of skin carcinogenesis. Thus, DFMO is a good candidate chemopreventive agent in humans at increased risk of NMSC.[58] DFMO treatment was associated with a significantly higher than background occurrence of diarrhea, hearing loss, and stomatitis and a slightly higher than background occurrence of nausea, headache, myalgia, emotional lability, and dizziness.[59]
Immunomodulators
Recently, there has been a growing interest in immunomodulators, or upregulators of the immune response, for the treatment and cure of various forms of skin cancer, including melanoma and NMSC, cutaneous T cell lymphoma, Kaposi's sarcoma, cutaneous extramammary Paget's disease, and vulvar intraepithelial carcinoma neoplasia. Numerous strategies to enhance the host's immune response against cancer cells and/or cancer cell antigenicity have been evaluated. These include recombinant cytokines, immunomodulators, dendritic cell immunization, tumor antigen vaccination, T cell-based immunotherapy, and gene therapy. Thus, immunomodulators are becoming essential in the treatment of patients who are poor surgical candidates and/or require noninvasive therapy.[60]
Imiquimod (it also functions as an immunomodulator)
There is strong evidence that ultraviolet (UV) light plays a central role in molecular pathogenesis of NMSC development. UV light results in DNA damage, loss of activity of tumor suppressor genes, overexpression of oncogenes, and other genes related to enhanced growth and survival as well as tissue invasion. Additionally, UV light impairs the cutaneous immune response, especially Langerhans cell antigen-presenting function, resulting in immune tolerance to developing tumor cells [Figure 2]. Immunotherapy of NMSC has been attempted in the form of dinitrobenzene sensitization followed by topical application on the tumor, intralesional interferon (IF/n) injections, or perilesional interleukin-2 (IL-2). These treatments, although showing promise, have not been developed because of lower efficacy compared with surgical approaches, morbidity associated with treatments, as well as the expense of using recombinant cytokine treatments. The topical immune response modifier imiquimod has developed as a novel local treatment for selected NMSCs. Studies indicate the presence of activated, natural killer (NK) cells (innate immunity), T-lymphocytes (adaptive immunity), antigen-presenting cells, and cytokines consistent with a delayed-type hypersensitivity reaction (Th1-lymphocyte cytokine pattern).[61]
Imiquimod acts as an immune modulator via its binding to the toll receptor 7 (TLR-7) present on dendritic cells, macrophages, and monocytes as demonstrated by both in vivo and in vitro studies. Proinflammatory mediators like IFN-α, TNF-α, IL-1, IL-12, IL-6, IL-8, and IL-10 are liberated following activation of these cells. Activation of adaptive immune response towards the TH-1 or cell-mediated pathway and inhibition the TH-2 pathway occurs as a result of these cytokines. These immune mediated activities including upregulation of NK-cell activity is thought to be important for control of viruses and tumors. Additionally, topical application of imiquimod increases the functional maturation and migration of Langerhan cells to regional lymph nodes. These responses hence increase antigen presentation to naive T cells and inducing a more specific immune response. Recent evidence suggests a possible direct action of imiquimod on BCC and SCC. The addition of imiquimod resulted in the induction of Fas receptors on tumor cells, and is postulated to lead to their subsequent apoptosis via binding of FasR to Fas ligand, induction by neighboring BCC cells, and infiltration of lymphocytes. There has been a high response rate with imiquimod for treatment of superficial BCC, with clearance rates ranging from 70 to 90% in a number of clinical trials. This novel immunotherapy represents a new, nonsurgical treatment option in the care of patients with NMSC.[62]
The most frequent side effects include flu-like symptoms, fatigue, diarrhea, fever, skin blistering, erosion, excoriation, flaking, edema, paresthesia, pruritus, burning, tenderness, stinging, crusting, rash, and superficial ulcer may occur. Local reactions like erythema often occur in 33-80%. Less frequent include back pain, headache, myalgia, hyperkeratosis, rhinitis, severe erythema, vitiligo-like hypopigmentation, and upper respiratory infection have been reported. Rare side effects reported are alopecia, chills, diarrhea, dizziness, dyspepsia, fatigue, fever, lymphadenopathy, sinusitis, vomiting, and angioedema.[63]
Ingenol mebutate
The diterpene ester ingenol mebutate (also referred to as PEP005, ingenol-3-angelate) is derived from the plant Euphorbia peplus. Crude euphorbia extract causes local toxicity and transient inflammation when applied topically and has been used in the treatment of warts, skin keratosis, and skin cancer.[64]
Presently topical agents for field therapy of AKs have single mechanism of action with a drawback that it must be applied for weeks. However, ingenol mebutate gel, a novel drug for field therapy of AKs, appears to have a dual mechanism of action: (1) Rapid lesion necrosis and (2) specific neutrophil-mediated, antibody-dependent cellular cytotoxicity [Figure 6]. Due to rapid destruction of AK lesions after application of ingenol mebutate gel, treatment is required only for 2 or 3 days. Any residual dysplastic epidermal cells are subsequently targeted by immune-mediated response. Thus, this dual mechanism of action provides efficacy similar to that of current topical agents but with substantially shorter treatment duration. It has been observed that cytotoxic functions of neutrophils recruited to the tumor in response to topical application of ingenol mebutate are crucial for effective ablation of the treated lesion.[65]
Figure 6.
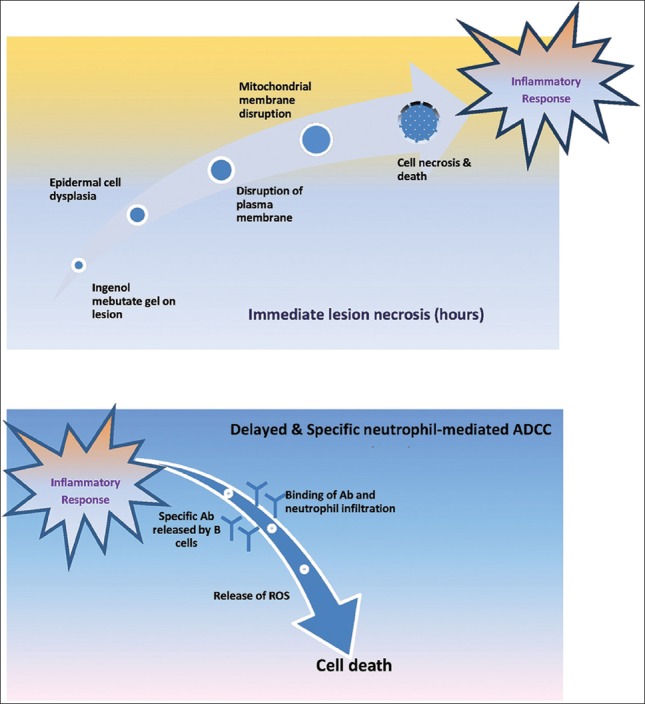
Mechanism of ingenol mebutate
Treatment with ingenol mebutate topically induces primary necrosis of tumor cells, activates protein kinase C, and demonstrates an acute T cell-independent inflammatory response characterized by a pronounced neutrophil infiltrate. Ingenol mebutate stimulated macrophage inflammatory protein-2 (MIP-2)/IL-8, TNF-alpha, and IL-1beta; all mediators of neutrophil recruitment and activation, both in vitro and in mice. Additionally, ingenol mebutate induced human endothelial cells causing neutrophil adhesion and also activated human neutrophils to generate tumoricidal reactive oxygen intermediates. There was a significant increase in the level of anticancer antibodies (Abs), resulting in promotion of neutrophil-mediated Ab-dependent cellular cytotoxicity (ADCC) in vitro, in tumors treated with ingenol mebutate. There was a > 70% increase in tumor relapse rates with ingenol mebutate treatment of tumors grown in SCID mice. Hence, these data suggest a key role for neutrophil-mediated ADCC in preventing relapse. Initial chemoablation followed by a neutrophil-dependent ADCC-mediated eradication of residual disease, demonstrates that neutrophils can be induced to mediate ingenol mebutate-mediated cure of tumors.[66]
Ingenol mebutate resulted in the production of anti-cancer CD8 T cells that could regress metastases. Additionally, ingenol mebutate-mediated cure synergized with several CD8 T cell-based immunotherapies to regress further distant metastases. Upregulation of CD80 and CD86 expression on dendritic cells in vivo, and promotion of CD8 T cell induction when co-delivered with a protein antigen, are some of the adjuvant properties of ingenol mebutate. Therefore, ingenol mebutate emerges as a novel local chemotherapeutic immunostimulatory debulking agent along with immunotherapies to promote regression of metastases.[67]
The most consistently reported side effects of ingenol mebutate are erythema, flakiness, crusting, pruritus, and pain; experienced by up to 97.5% patients, with effects peaking between days 3 and 8 and mostly resolved by day 15. A favorable safety profile has been consistently reported with no detectable systemic absorption; long-term (12 months) follow-up data showed no treatment-related serious adverse events or deaths.[68]
5-Fluorouracil (5-FU)
Topical chemotherapy with 5-FU was shown to result in selective eradication of premalignant and superficial malignant lesions of skin. It was at Roswell Park Memorial Institute that Edmund Klein developed protocol for application of a highly effective topical anticancer agent, 5-FU, for treating skin cancer. 5-FU produced complete tumor regression in a statistically significant number of lesions and is still in use today, especially for premalignant growths. This was the chief among Klein's accomplishments at Roswell Park Memorial Institute for skin cancer with topical 5-FU. Because of his pioneering and persistent efforts, he was honored with a Clinical Research Award from the Albert and Mary Lasker Foundation in 1972, the highest award in American Medicine. Topical 5-FU decreases cell proliferation and induces cellular death, particularly in cells with high mitotic rates, through inhibition of thymidylate synthetase, which interferes with DNA synthesis. Evidence suggests that 5-FU used as a topical chemotherapeutic agent in NMSC has been effective for treatment of superficial BCC (sBCC), in situ SCC, and AKs. Due to lack of penetration through the dermis, 5-FU is generally not recommended for invasive BCCs and SCCs.[69,70]
Local irritation, erythema, swelling, desquamation, and tenderness are common treatment site reactions. Topical fluorouracil is not recommended for SCC and high recurrence rates (21·4%) reported at 10-year follow-up suggest that this treatment is not appropriate for nodular BCC.[71]
Retinoids
Among these treatments, topical and systemic retinoids have demonstrated their efficacy in decreasing the risk of developing BCC and SCC. Retinoids induce apoptosis, arrest growth, stimulate differentiation of tumor cells during carcinogenesis, and downregulate the overexpression of cyclooxygenase-2 (COX-2) induced by UVR, causing a decrease in prostaglandins, which are increased in NMSC. Isotretinoin and acitretin are the most common systemic retinoids used for NMSC chemoprevention. They may decrease the morbidity and mortality seen in patients with single, high-risk, and multiple primary cancers; particularly in those with organ transplants, immunosuppression, xeroderma pigmentosum, and BCNS.[72]
COX-2 inhibitors
Besides induction of DNA damage and p53 mutations, chronic exposure to UV irradiation results in a constitutive upregulation of COX-2 expression. This in turn enhances formation of its primary product in skin, prostaglandin E2 (PGE2). In mouse, as well as human, UV-induced skin cancers and premalignant lesions, COX-2 has shown to be constitutively overexpressed. Ligand-independent activation of epidermal growth factor receptor (EGFR) occurs because of UV exposure. This subsequently activates mitogen-activated protein kinase resulting in transcriptional activation of COX-2 gene. It is evident from use of COX-2-specific inhibitors and genetic manipulation of COX-2 expression that UV induction of COX-2 in skin contributes to the induction of epidermal hyperplasia, edema, inflammation, and counters induction of apoptosis after UV exposure. Similarly, inhibition of COX-2 activity or reduced expression in COX-2 knockout mice led to a significantly reduced UV-induced tumorigenesis. However, overexpression of COX-2 in transgenic mice increased UV-induced tumor development. These studies demonstrate the crucial role of COX-2 in the development of UV-related nonmelanoma skin cancers.[73] These results suggest that COX-2 inhibitors could have potent anticarcinogenic effects in UVB-induced skin cancer.
Betulinic acid
New treatments using innovative mechanisms to stimulate tumor cell death are required with plants playing a significant role as a source for potential anticancer compounds. One highly promising class of natural compounds is the triterpenoids, with betulinic acid as the most prominent representative. BetA is a naturally occurring pentacyclic triterpene that demonstrates numerous biological activities including potent antitumor properties. Their direct effect is on mitochondria where it induces Bax/Bak-independent cytochrome-C release. This effect is responsible for its antitumor activity. The ability to directly trigger mitochondrial membrane permeabilization plays a key role in apoptotic process that seals the cell's fate. The normal tissue remains relatively resistant to BetA compared to the potent cytotoxicity of BetA against a variety of cancer types and nonmalignant cells, indicating a therapeutic window. Because agents that exert a direct action on mitochondria could trigger cell death under conditions in which conventional chemotherapeutics fail, there is growing interest to develop such compounds as experimental cancer therapeutics. Hence, mitochondrion targeted agents such as BetA holds great promise as an innovative strategy to overcome certain forms of drug resistance in human cancers.[74] Evidence from in vitro studies has demonstrated this agent to be potently effective not only against a variety of cancer cells, but also those derived from therapy resistant and refractory tumors. In vivo preclinically applied BetA exhibited some significant anticancer effects without any systemic toxicity in rodents. Coadministration of BetA with other therapies also led to tumor cell death. Additionally, several potent derivatives have been discovered.[75] BetA has shown to exhibit a variety of biological activities including inhibition of human immunodeficiency virus (HIV), antibacterial, antimalarial, anti-inflammatory, anthelmintic, and antioxidant properties.[76]
Perillyl alcohol
POH is a monoterpene isolated from essential oils of lavendin, peppermint, spearmint, cherries, celery seeds, and several other plants. POH induces apoptosis in tumor cells without affecting normal cells and can revert tumor cells back to a differentiated state. It has effects on numerous cellular substances responsible to control cell growth and differentiation. It has been shown to increase mannose-6-phosphate/insulin-like growth factor II receptors, increase tissue growth factor beta receptors, increase Bak, decrease ras protein prenylation, decrease ubiquinone synthesis, and induce phase I and phase II detoxification systems. In animal studies it has been shown to regress pancreatic, mammary, and liver tumors. It exhibits possible application as a chemopreventative agent for colon, skin, and lung cancer. Additionally, it could be a chemotherapeutic agent for neuroblastoma and prostate and colon cancer. Thus, POH might be used for chemoprevention of human skin cancer and that inhibition of activator protein-1 activity is functionally related to inhibition of skin carcinogenesis.[77,78]
The only adverse event that occurred frequently was a mild or moderate local reaction (rash, redness and/or erythema) that occurred at a rate of 56 and 54% for the low and high POH dose, respectively. Similarly, the phase 1 trial also demonstrated lack of any serious adverse events and no significant differences in mild, study-related adverse events between treatment and placebo groups.[79]
Cyclopamine
Hedgehog (hh) signaling has come a long way since its discovery by C. Nüsslein-Volhard and E. F. Wieschaus. It governs processes like cell proliferation, differentiation, and tissue patterning. Hence, it is regarded as a key regulator in embryogenesis. It is also associated in the maintenance of stem cells, tissue repair, and regeneration in adults. However, hh signaling has a second much darker face: It plays a major role in a variety of human cancers where it promotes growth and enables proliferation of tumor stem cells. The etiology of medulloblastoma and BCC is connected aberrant hh activity. Furthermore, cancers of prostate, pancreas, colon, and breasts; rhabdomyosarcoma; and leukemia too are dependent on irregular hh activity. Recent evidence has demonstrated that hh signaling can be source of a significant novel category of therapeutic agents with far-reaching implications in oncology. Thus, alteration of hh signaling by means of small molecules has evolved as an important tool against these hh-dependent cancers. Cyclopamine, a unique natural product with a fascinating history, was the first identified inhibitor of hh signaling and its story is closely linked to progress in the whole field.[80]
Cyclopamine is a steroidal alkaloid isolated from corn lily Veratrum californiucum, which is very common in subalpine meadows. It has exhibited inhibitory activity against the hedgehog family of intercellular signaling proteins. This membrane complex, particularly the sonic hedgehog (SHH) subtype, present in mammals, has recently been implicated in the development of BCC in the white population. The hedgehog signaling pathway is modulated by two transmembrane proteins, tumor suppressor PTCH, and proto-oncogene SMO [Figure 3]. The signaling is activated when SHH protein binds directly to PTCH receptor (encoded from PTCH 1 gene), causing its inhibition and further repression of expression of target genes. When SHH protein is not present, PTCH 1 inhibits the activity of SMO, which is a key activator of the pathway, thus preventing expression of target genes, such as GLI-1. Binding of SHH to the PTCH receptor eliminates that inhibition of PTCH on SMO, resulting in a dysregulated hyperactivity of the hedgehog pathway [Figure 7]. Good examples of activation and dysregulation of the hedgehog pathway represent human sporadic development of BCCs and BCNS, where germ-line mutations in the PTCH 1 gene or in any of the components of hedgehog pathway contribute with their development. Other neoplasias that have been related to mutations in the hedgehog signaling pathway include breast carcinomas, odontogenic keratocysts, medulloblastomas, sebaceous nevus, trichoepitheliomas, fibrosarcomas, and dermatofibromas. Clinical studies are needed to evaluate the efficacy and safety of topical cyclopamine for treatment of NMSC.[72]
Figure 7.
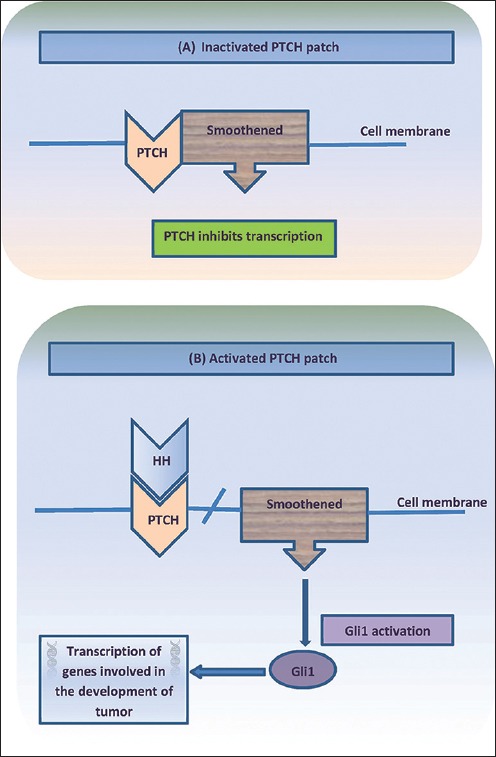
Hedgehog signaling pathway and tumorigenesis
Although no longer in clinical development due to increased potency and bioavailability of cyclopamine derivatives, cyclopamine remains an important agent in preclinical models of hh inhibition. Years of laboratory investigations have led to Phase I trials of several SMO inhibitors which appear to be relatively well-tolerated either as single agents or in combination regimens with conventional chemotherapy. Modest side effects include nausea, dysgeusia, muscle cramps, and fatigue, with rare grade 3 adverse events observed. It is unclear, however, what side effects may occur in children, as permanent defects in bone growth have been seen in mouse models of hh inhibitors which may complicate their clinical development for pediatric tumors such as medulloblastoma. The known teratogenic effects of cyclopamine, cautions for the need to prevent pregnancy in treated subjects.[81]
Photodynamic therapy
Photodynamic therapy (PDT) consists of a chemical reaction stimulated by light energy which selectively destroys tissue. The reaction needs a photosensitizer in the target tissue, a light source, and oxygen [Figure 8]. The most extensively studied photosensitizing agents for PDT are 5-aminolevulinic acid (ALA) for treatment of AK and methyl-aminolevulinate, which has been approved for treatment of AK, BCC, and Bowen's disease. When localized in the target tissue, photosensitizer is activated by light to produce oxygen intermediates that destroy target tissue cells. PDT should be considered as a therapeutic option, particularly in the case of patients with superficial, multiple, or disseminated lesions and for immunosuppressed patients. Recently, PDT has been indicated for a wide range of dermatological conditions such as photodamaged skin, acne, hidradenitis, scleroderma, psoriasis, warts, and leishmaniosis among others. During exposure to light, patients experience a burning or stinging sensation or pruritus, which is restricted to treated area. The discomfort and pain begins minutes after initiating irradiation, reflecting nervous stimulus and/or tissue damage caused by reactive oxygen. Pain, the principal side effect, remains for some hours and decreases over time. With respect to systemic photosensitizers, generalized skin photosensitivity is the principal side effect. ALA may lead to nausea and vomiting in 7-19% of cases and also to transitory changes in liver enzymes. With respect to topical PDT, erythema and edema may be treated with topical or even systemic corticoids. The normal course includes crusting, desquamation, and pruritus of varying intensity for around 2-8 weeks. Photophobia and visual discomfort may occur. Dyschromias are generally reversible within a few months. Blisters, ulcers, and necrosis are rare; suggesting a high dose of energy with phototoxicity. In addition, allergic reaction to photosensitizers or to vehicles should be considered.[82]
Figure 8.
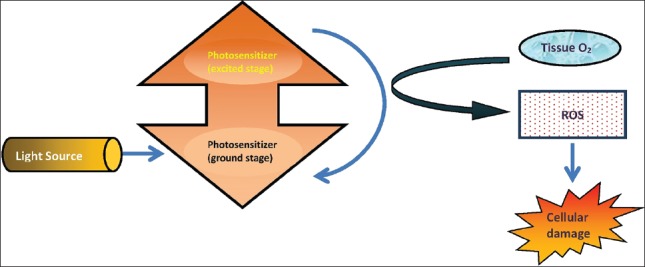
Photodynamic therapy
Afamelanotide
Alpha-melanocyte-stimulating hormone (α-MSH) is a tridecapeptide that is produced by skin itself from the precursor proopiomelanocortin. It crucially mediates UV light-induced tanning after binding to melanocortin-1 receptors (MC-1R) expressed on surface of epidermal melanocytes. The potent pigment-inducing and also cytoprotective actions of α-MSH are the rationale for performance in patients with photodermatoses such as erythropoietic protoporphyria [Figure 9]. Since α-MSH has shown promising anti-inflammatory and antifibrotic properties in numerous preclinical studies, it will be most interesting to evaluate these effects in further clinical pilot studies[83] Meanwhile, the regulated α-MSH analogue afamelanotide is showing promise for its photoprotective potential, and is undergoing phase II and III clinical trials in people with photosensitivity disorders and those prone to NMSC. The photoprotective and other biological effects of α-MSH analogues await full determination.[84] The main side-effects are burning or stinging pain during light exposure, posttreatment erythema, oedema, and temporary postinflammatory hypopigmentation or hyperpigmentation.[85]
Figure 9.
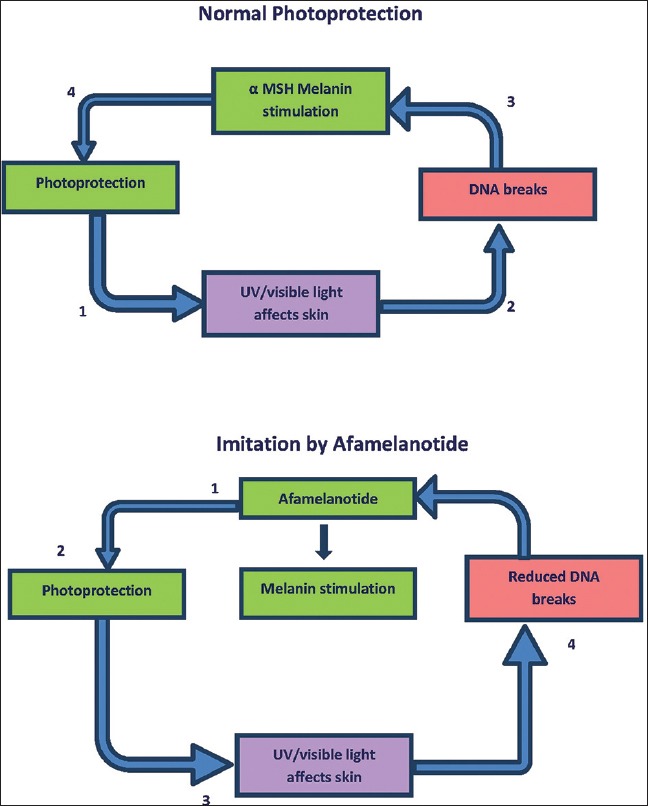
Afamelanotide mechanism
Epidermal growth factor receptor inhibitors
As mentioned earlier UV light plays a fundamental role as an initiator and promoter of carcinogenesis of SCC. This allows accumulation of genetic alterations that allows a selective growth advantage. The Tp53 (p53) gene frequently mutates and Ras is often activated, however with low frequency of mutations. It is the extracellular signals that govern whether cells move from a quiescent state into an active proliferative state. An increase in the formation of growth factors and its receptors on tumor cells can be often seen that gives rise to such an autocrine circuit facilitating cellular division. Frequent mutations in EGFR have been detected in lung cancer. These are present at EGFR tyrosine kinase domain (TK). EGFR TK mutations induce signaling pathways downstream and specifically activate antiapoptotic pathways. These mutations are correlated with clinical response of patients to tyrosine kinase inhibitors (gefinitib and erlotinib), as tumor cells are addicted to continuous activation of specific signaling pathways. Abnormalities in expression of epidermal growth factor receptor (EGFR) and/or its ligands in head and neck SCC (HNSCC) have been demonstrated.[86]
The EGFR is a member of HER family of tyrosine kinase growth factor receptors. Binding to EGFR by its natural ligands, specifically epidermal growth factor (EGF) or transforming growth factor (TGF)-alpha, causes a conformational change in the receptor. This induces homo- or heterodimerization or oligomerization with other EGFR molecules or other HER family members. Subsequently, dimerization stimulates intracellular tyrosine kinase, autophosphorylation, and activation of signal transduction molecules. This results in cell cycle progression, reduced apoptotic capacity, angiogenesis, and metastatic phenotype. EGFR is expressed on normal human cells and also across a range of malignancies. Tumor EGFR expression correlates with poor prognosis and resistance to therapy.
Cetuximab is a chimeric human-murine monoclonal antibody that binds competitively to EGFR. Binding of antibody to EGFR prevents activation of receptor by endogenous ligands. Thus, proliferation, angiogenesis, invasiveness, and metastasis is reduced, and apoptosis is enhanced. This also causes internalization and degradation of antibody-receptor complex, downregulating EGFR expression [Figure 10]. Addition of cetuximab enhances activity of radiotherapy in locoregional head and neck cancer. Its concomitant administration with platinum-based chemotherapy improves response in a proportion of patients with platinum-refractory recurrent or metastatic SCC of head and neck, as is cetuximab monotherapy. Similarly cetuximab added to cisplatin monotherapy in first-line treatment of recurrent or metastatic SCC of head and neck, the objective response rate is significantly improved. An acneiform rash that occurred in 70-80% of patients is the most commonly reported adverse event associated with cetuximab treatment. Presence of characteristic rash is significantly associated with response and/or survival. Thus, development of acneiform rash could probably become a key clinical prognostic marker. Serious cetuximab-related toxicities include hypersensitivity reactions. Cetuximab is biologically active across a range of clinical scenarios in SCC of head and neck. However, additional studies would be necessary in establishing its role in routine management of head and neck cancer.[87]
Figure 10.
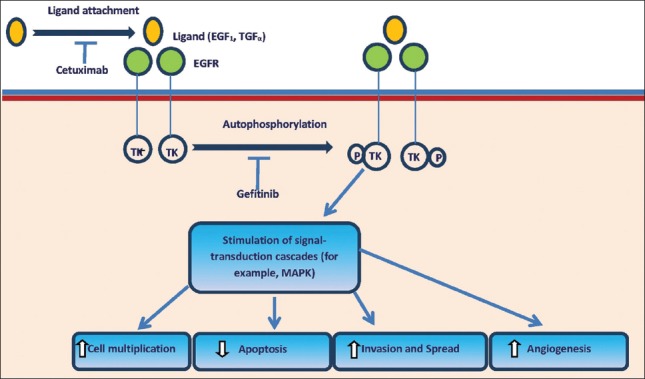
Cetuximab mechanism
The most common cutaneous side effects to EGFR inhibitors are follicular or acneiform eruptions, nail disorders, xerosis, and desquamation. Although topical and oral antibiotics with or without topical corticosteroids usually are safe and effective treatment options for acneiform eruptions due to EGFR inhibitors, they are not always successful in refractory cases. Oral isotretinoin for management of acneiform reactions to EGFR inhibitors (EGFRIs) is recommended when lesions persist or worsen despite antibiotic treatment.[88] There is a significant risk of developing nail toxicity in cancer patients receiving EGFRIs. The risk is independent of the underlying agent. Adequate monitoring and early intervention are recommended to prevent debilitating toxicity and suboptimal dosing of EGFRI.[89] Patients could present with papular-pustular eruption typically affecting the face, chest, and back; which appears in average 13.5 days after starting the drug treatment. The patients underwent oral treatment with minocycline or doxycycline and topical treatment with metronidazole, benzoyl peroxide and/or corticosteroids. All patients showed improvement of lesions. Patients also presented with periungual pyogenic granulomas, which were associated with paronychia, after an average of 8 weeks of treatment. There was improvement of the lesions with topical treatment (antibiotics, corticosteroids and antiseptics). Xerosis was observed in some patients. Other less frequent adverse side effects such as telangiectasia and angiomas, hair and eyelash alterations, and eruptive melanocytic nevi are also described. Treatment with EGFRI was maintained in most patients.[90]
Thus, to summarize, this review discusses the recent developments and future possibilities in the use of targeted molecular inhibitors for treatment of NMSC. The development of novel, more pathogenesis-based therapies are challenging new approaches [Figure 11].
Figure 11.
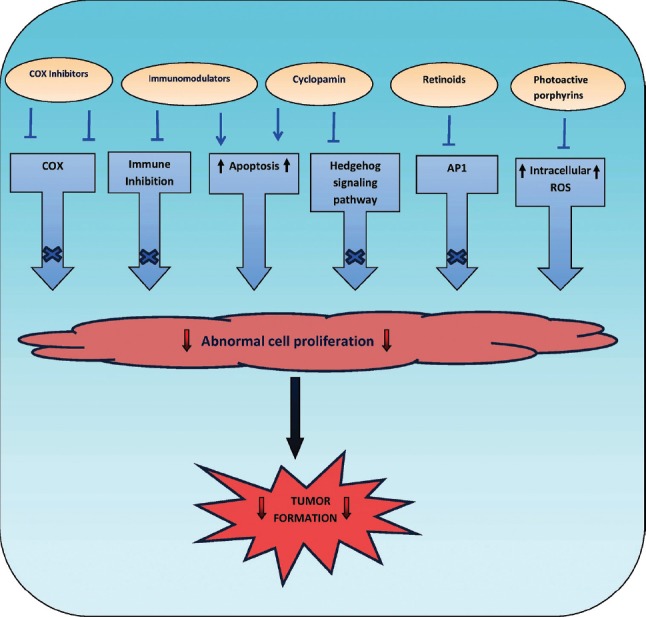
Pathogenesis-based treatment approaches for NMSC
Discussion and Conclusion
Though the incidence of skin cancers has been increasing since the last few decades worldwide, conventional wisdom has it that the incidence of all varieties of skin cancers is lower among Indians due to protective effects of melanin. Though data in India are unavailable, several smaller reports suggest that there could a rise in the incidence of NMSCs in India. Hence, it could be possible that factors other than ultraviolet radiation may be responsible in the development of these cancers, especially in the skin types prevalent in India. Additionally, SCC is predominant in the Indian population rather than BCC. The atypical forms of NMSC reported in Indian scenario could range from the one associated with discoid lupus erythematosus to the exemplary kangri cancer in Kashmir not forgetting arsenicosis. Moreover, the other uncommon occurrences of SCC on preexisting dermatological conditions include the following: LP, LP hypertrophicus, LSC, psoriasis, and chronic lymphedema.[91]
The ever-growing incidence of primary cutaneous malignancies has heralded the development of multiple treatment options. Though surgical modalities remain the mainstay of treatment of NMSC, it is imperative for the dermatologic surgeon be aware of all treatment modalities to assist the patient in making most informed decision possible, thereby achieving a most favorable outcome. Currently, numerous strategies are available to treat primary cutaneous malignancies. Although not recommended for certain aggressive tumors, many of these novel regimens involve noninvasive techniques. Thus, both the nonsurgeon and surgeon alike should be apprised of all treatment options, their indications, and their efficacy.
Many nonsurgical treatment options exist for primary cutaneous malignancies. In determining which modalities to implement, consideration must be given to careful patient selection, tumor type, and appropriate expectations. Additionally, adjunctive/combination therapy may be used before or after surgery.
Since AK is a precursor to SCC and has been linked to potential BCC progression, prevention, and early treatment of AK has been recommended to reduce the potential for progression towards malignant disease. Evidence of progression of AK to NMSC supports the concept that treating AK may serve as a rational strategy for reducing the incidence of NMSC.
Immunological and genetic research into NMSC has paved way for innovative therapeutic modalities for patients who were previously without any viable treatment alternatives. While still in preliminary stages, these agents may become integral drugs in the armamentarium of managing cutaneous carcinoma. As our understanding of the genetic foundation of these diseases improves, so too will the potential for diagnosing and managing NMSC.
What is new?
Immunological and genetic research into NMSC has paved the way for innovative therapeutic modalities for patients who were previously without any viable treatment alternatives.
While still in preliminary stages, these agents may become integral drugs in the armamentarium of managing cutaneous carcinoma.
Footnotes
Source of Support: Nil
Conflict of Interest: Nil.
References
- 1.Schulze HJ, Cribier B, Requena L, Reifenberger J, Ferrándiz C, Garcia Diez A, et al. Imiquimod 5% cream for the treatment of superficial basal cell carcinoma: Results from a randomized vehicle-controlled phase III study in Europe. Br J Dermatol. 2005;152:939–47. doi: 10.1111/j.1365-2133.2005.06486.x. [DOI] [PubMed] [Google Scholar]
- 2. [Last accessed on 2012 June 9]. Available from: http://www.who.int/uv/faq/skincancer/en/index 1.html .
- 3.Ting PT, Kasper R, Arlette JP. Metastatic basal cell carcinoma: Report of two cases and literature review. J Cutan Med Surg. 2005;9:10–5. doi: 10.1007/s10227-005-0027-1. [DOI] [PubMed] [Google Scholar]
- 4.Lo JS, Snow SN, Reizner GT, Mohs FE, Larson PO, Hruza GJ. Metastatic basal cell carcinoma: Report of twelve cases with a review of the literature. J Am Acad Dermatol. 1991;24:715–9. doi: 10.1016/0190-9622(91)70108-e. [DOI] [PubMed] [Google Scholar]
- 5.Macbeth AE, Grindlay DJ, Williams HC. What's new in skin cancer. An analysis of guidelines and systematic reviews published in 2008-2009? Clin Exp Dermatol. 2011;36:453–8. doi: 10.1111/j.1365-2230.2011.04087.x. [DOI] [PubMed] [Google Scholar]
- 6.Samarasinghe V, Madan V, Lear JT. Management of high-risk squamous cell carcinoma of the skin. Expert Rev Anticancer Ther. 2011;11:763–9. doi: 10.1586/era.11.36. [DOI] [PubMed] [Google Scholar]
- 7.Samarasinghe V, Madan V. Nonmelanoma skin cancer. J Cutan Aesthet Surg. 2012;5:3–10. doi: 10.4103/0974-2077.94323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Salasche SJ. Epidemiology of actinic keratoses and squamous cell carcinoma. J Am Acad Dermatol. 2000;42:4–7. doi: 10.1067/mjd.2000.103342. [DOI] [PubMed] [Google Scholar]
- 9.Grossman D, Leffell DJ. The molecular basis of nonmelanoma skin cancer: New understanding. Arch Dermatol. 1997;133:1263–70. [PubMed] [Google Scholar]
- 10.Kricker A, Armstrong BK, English DR, Heenan PJ. Does intermittent sun exposure cause basal cell carcinoma? A case-control study in Western Australia. Int J Cancer. 1995;60:489–94. doi: 10.1002/ijc.2910600411. [DOI] [PubMed] [Google Scholar]
- 11.Rosso S, Zanetti R, Martinez C, Tormo MJ, Schraub S, Sancho-Garnier H, et al. The multicentre south European study ‘Helios’.II: Different sun exposure patterns in the aetiology of basal cell and squamous cell carcinomas of the skin. Br J Cancer. 1996;73:1447–54. doi: 10.1038/bjc.1996.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zak-Prelich M, Narbutt J, Sysa-Jedrzejowska A. Environmental risk factors predisposing to the development of basal cell carcinoma. Dermatol Surg. 2004;30:248–52. doi: 10.1111/j.1524-4725.2004.30089.x. [DOI] [PubMed] [Google Scholar]
- 13.Samarasinghe V, Madan V, Lear JT. Focus on basal cell carcinoma. J Skin Cancer 2011. 2011:328615. doi: 10.1155/2011/328615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smeets NW, Kuijpers DI, Nelemans P, Ostertag JU, Verhaegh ME, Krekels GA, et al. Mohs’ micrographic surgery for treatment of basal cell carcinoma of the face-results of a retrospective study and review of the literature. Br J Dermatol. 2004;151:141–7. doi: 10.1111/j.1365-2133.2004.06047.x. [DOI] [PubMed] [Google Scholar]
- 15.Pimparkar BD, Bhave A. Arsenicosis: Review of recent advances. J Assoc Physicians India. 2010;58:617–24. 629. [PubMed] [Google Scholar]
- 16.Smeester L, Rager JE, Bailey KA, Guan X, Smith N, García-Vargas G, et al. Epigenetic changes in individuals with arsenicosis. Chem Res Toxicol. 2011;24:165–7. doi: 10.1021/tx1004419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rahman MM, Ng JC, Naidu R. Chronic exposure of arsenic via drinking water and its adverse health impacts on humans. Environ Geochem Health. 2009;31:189–200. doi: 10.1007/s10653-008-9235-0. [DOI] [PubMed] [Google Scholar]
- 18.Ruiz de Luzuriaga AM, Ahsan H, Shea CR. Arsenical keratoses in Bangladesh--update and prevention strategies. Dermatol Clin. 2011;29:45–51. doi: 10.1016/j.det.2010.09.003. [DOI] [PubMed] [Google Scholar]
- 19.Biswas R, Ghosh P, Banerjee N, Das JK, Sau T, Banerjee A, et al. Analysis of T-cell proliferation and cytokine secretion in the individuals exposed to arsenic. Hum Exp Toxicol. 2008;27:381–6. doi: 10.1177/0960327108094607. [DOI] [PubMed] [Google Scholar]
- 20.Bailey K, Xia Y, Ward WO, Knapp G, Mo J, Mumford JL, et al. Global gene expression profiling of hyperkeratotic skin lesions from inner Mongolians chronically exposed to arsenic. Toxicol Pathol. 2009;37:849–59. doi: 10.1177/0192623309351725. [DOI] [PubMed] [Google Scholar]
- 21.Banerjee M, Banerjee N, Ghosh P, Das JK, Basu S, Sarkar AK, et al. Evaluation of the serum catalase and myeloperoxidase activities in chronic arsenic-exposed individuals and concomitant cytogenetic damage. Toxicol Appl Pharmacol. 2010;249:47–54. doi: 10.1016/j.taap.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sinha D, Dey S, Bhattacharya RK, Roy M. In vitro mitigation of arsenic toxicity by tea polyphenols in human lymphocytes. J Environ Pathol Toxicol Oncol. 2007;26:207–20. doi: 10.1615/jenvironpatholtoxicoloncol.v26.i3.50. [DOI] [PubMed] [Google Scholar]
- 23.Paul S, Das N, Bhattacharjee P, Banerjee M, Das JK, Sarma N, et al. Arsenic-induced toxicity and carcinogenicity: A two-wave cross-sectional study in arsenicosis individuals in West Bengal, India. J Expo Sci Environ Epidemiol. 2013;23:156–62. doi: 10.1038/jes.2012.91. [DOI] [PubMed] [Google Scholar]
- 24.Guha Mazumder DN. Chronic arsenic toxicity and human health. Indian J Med Res. 2008;128:436–47. [PubMed] [Google Scholar]
- 25.Ashton KJ, Carless MA, Griffiths LR. Cytogenetic alterations in nonmelanoma skin cancer: A review. Genes Chromosomes Cancer. 2005;43:239–48. doi: 10.1002/gcc.20183. [DOI] [PubMed] [Google Scholar]
- 26.Benjamin CL, Melnikova VO, Ananthaswamy HN. p53 protein and pathogenesis of melanoma and nonmelanoma skin cancer. Adv Exp Med Biol. 2008;624:265–82. doi: 10.1007/978-0-387-77574-6_21. [DOI] [PubMed] [Google Scholar]
- 27.McGillis ST, Fein H. Topical treatment strategies for nonmelanoma skin cancer and precursor lesions. Semin Cutan Med Surg. 2004;23:174–83. doi: 10.1016/j.sder.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 28.de Lima Vazquez V, Sachetto T, Perpetuo NM, Carvalho AL. Prognostic factors for lymph node metastasis from advanced squamous cell carcinoma of the skin of the trunk and extremities. World J Surg Oncol. 2008;6:73. doi: 10.1186/1477-7819-6-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boukamp P. Non-melanoma skin cancer: What drives tumor development and progression? Carcinogenesis. 2005;26:1657–67. doi: 10.1093/carcin/bgi123. [DOI] [PubMed] [Google Scholar]
- 30.Kathpalia VP, Mussak EN, Chow SS, Lam PH, Skelley N, Time M, et al. Genome-wide transcriptional profiling in human squamous cell carcinoma of the skin identifies unique tumor-associated signatures. J Dermatol. 2006;33:309–18. doi: 10.1111/j.1346-8138.2006.00075.x. [DOI] [PubMed] [Google Scholar]
- 31.Boldrini L, Loggini B, Gisfredi S, Zucconi Y, Di Quirico D, Biondi R, et al. Evaluation of telomerase in non-melanoma skin cancer. Int J Mol Med. 2003;11:607–11. doi: 10.3892/ijmm.11.5.607. [DOI] [PubMed] [Google Scholar]
- 32.Madan V, Lear JT, Szeimies RM. Non-melanoma skin cancer. Lancet. 2010;375:673–85. doi: 10.1016/S0140-6736(09)61196-X. [DOI] [PubMed] [Google Scholar]
- 33.Naldi L, DiLandro A, D’Avanzo B, Parazzini F. Host-related and environmental risk factors for cutaneous basal cell carcinoma: Evidence from an Italian case-control study. J Am Acad Dermatol. 2000;42:446–52. doi: 10.1016/s0190-9622(00)90217-2. [DOI] [PubMed] [Google Scholar]
- 34.Michel A, Kopp-Schneider A, Zentgraf H, Gruber AD, de Villiers EM. E6/E7 expression of human papillomavirus type 20 (HPV-20) and HPV-27 influences proliferation and differentiation of the skin in UV-irradiated SKH-hr1 transgenic mice. J Virol. 2006;80:11153–64. doi: 10.1128/JVI.00954-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bouwes Bavinck JN, Neale RE, Abeni D, Euvrard S, Green AC, Harwood CA, et al. EPI-HPV-UV-CA group. Multicenter study of the association between betapapillomavirus infection and cutaneous squamous cell carcinoma. Cancer Res. 2010;70:9777–86. doi: 10.1158/0008-5472.CAN-10-0352. [DOI] [PubMed] [Google Scholar]
- 36.Euvrard S, Kanitakis J, Claudy A. Skin cancers after organ transplantation. N Eng J Med. 2003;348:1681–91. doi: 10.1056/NEJMra022137. [DOI] [PubMed] [Google Scholar]
- 37.Thomas T, Thomas TJ. Polyamine metabolism and cancer. J Cell Mol Med. 2003;7:113–26. doi: 10.1111/j.1582-4934.2003.tb00210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Euvard S, Kanitakis J, Decullier E, Butnaru AC, Lefrançois N, Boissonnat P, et al. Subsequent skin cancers in kidney and heart transplant recipients after the first squamous cell carcinoma. Transplantation. 2006;81:1093–100. doi: 10.1097/01.tp.0000209921.60305.d9. [DOI] [PubMed] [Google Scholar]
- 39.Smith KJ, Hamza S, Skelton H. Histologic features in primary cutaneous squamous cell carcinomas in immunocompromised patients focusing on organ transplant patients. Dermatol Surg. 2004;30:634–41. doi: 10.1111/j.1524-4725.2004.30149.x. [DOI] [PubMed] [Google Scholar]
- 40.Pandith AA, Khan NP, Rashid N, Azad N, Zaroo I, Hafiz A, et al. Impact of codon 72 Arg>Pro single nucleotide polymorphism in Tp53 gene in the risk of kangri cancer: A case control study in Kashmir. Tumour Biol. 2012;33:927–33. doi: 10.1007/s13277-012-0318-2. [DOI] [PubMed] [Google Scholar]
- 41.Wani I. Kangri cancer. Surgery. 2010;147:586–8. doi: 10.1016/j.surg.2009.10.025. [DOI] [PubMed] [Google Scholar]
- 42.Teli MA, Khan NA, Darzi MA, Gupta M, Tufail A. Recurrence pattern in squamous cell carcinoma of skin of lower extremities and abdominal wall (Kangri cancer) in Kashmir valley of Indian subcontinent: Impact of various treatment modalities. Indian J Dermatol. 2009;54:342–6. doi: 10.4103/0019-5154.57610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hussain I, ul Rehman S, Afroze D, Zahoor L, Abdullah S, Hafiz A, et al. Mutational spectrum of conserved regions of Tp53 and PTEN genes in Kangri cancer (of the skin) in the Kashmiri population. Mutat Res. 2009;676:5–10. doi: 10.1016/j.mrgentox.2009.02.011. [DOI] [PubMed] [Google Scholar]
- 44.Wu M, Wang Y, Bu W, Jia G, Fang F, Zhao L. Squamous cell carcinoma arising in lichen simplex chronicus. Eur J Dermatol. 2010;20:858–9. doi: 10.1684/ejd.2010.1124. [DOI] [PubMed] [Google Scholar]
- 45.Tiengo C, Deluca J, Belloni-Fortina A, Salmaso R, Galifi F, Alaibac M. Occurrence of squamous cell carcinoma in an area of lichen simplex chronicus: Case report and pathogenetic hypothesis. J Cutan Med Surg. 2012;16:350–2. doi: 10.1177/120347541201600513. [DOI] [PubMed] [Google Scholar]
- 46.Motswaledi MH, Khammissa RA, Wood NH, Meyerov R, Lemmer J, Feller L. Discoid lupus erythematosus as it relates to cutaneous squamous cell carcinoma and to photosensitivity. SADJ. 2011;66:340–3. [PubMed] [Google Scholar]
- 47.Harper JG, Pilcher MF, Szlam S, Lind DS. Squamous cell carcinoma in an African American with discoid lupus erythematosus: A case report and review of the literature. South Med J. 2010;103:256–9. doi: 10.1097/SMJ.0b013e3181c98ba9. [DOI] [PubMed] [Google Scholar]
- 48.Alsanafi S, Werth VP. Squamous cell carcinomas arising in discoid lupus erythematosus scars: Unusual occurrence in an African-American and in a sun-protected area. J Clin Rheumatol. 2011;17:35–6. doi: 10.1097/RHU.0b013e3182051928. [DOI] [PubMed] [Google Scholar]
- 49.Parren LJ, Frank J. Hereditary tumour syndromes featuring basal cell carcinomas. Br J Dermatol. 2011;165:30–4. doi: 10.1111/j.1365-2133.2011.10334.x. [DOI] [PubMed] [Google Scholar]
- 50.Telfer NR, Colver GB, Morton CA British Association of Dermatologists. Guidelines for the management of basal cell carcinoma. Br J Dermatol. 2008;159:35–48. doi: 10.1111/j.1365-2133.2008.08666.x. [DOI] [PubMed] [Google Scholar]
- 51.Molho-Pessach V, Lotem M. Ultraviolet radiation and cutaneous carcinogenesis. Curr Probl Dermatol. 2007;35:14–27. doi: 10.1159/000106407. [DOI] [PubMed] [Google Scholar]
- 52.Einspahr JG, Bowden GT, Alberts DS. Skin cancer chemoprevention: Strategies to save our skin. Recent Results Cancer Res. 2003;163:151–64. doi: 10.1007/978-3-642-55647-0_14. [DOI] [PubMed] [Google Scholar]
- 53.Neville JA, Welch E, Leffell DJ. Management of nonmelanoma skin cancer in 2007. Nat Clin Pract Oncol. 2007;4:462–9. doi: 10.1038/ncponc0883. [DOI] [PubMed] [Google Scholar]
- 54.Szeimies RM, Karrer S. Towards a more specific therapy: Targeting nonmelanoma skin cancer cells. Br J Dermatol. 2006;154:16–21. doi: 10.1111/j.1365-2133.2006.07232.x. [DOI] [PubMed] [Google Scholar]
- 55.O’Bryan KW, Ratner D. The role of targeted molecular inhibitors in the management of advanced nonmelanoma skin cancer. Semin Cutan Med Surg. 2011;30:57–61. doi: 10.1016/j.sder.2011.01.004. [DOI] [PubMed] [Google Scholar]
- 56.Babbar N, Gerner EW. Targeting polyamines and inflammation for cancer prevention. Recent Results Cancer Res. 2011;188:49–64. doi: 10.1007/978-3-642-10858-7_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Meyskens FL, Jr, Gerner EW. Development of difluoromethylornithine (DFMO) as a chemoprevention agent. Clin Cancer Res. 1999;5:945–51. [PubMed] [Google Scholar]
- 58.Einspahr JG, Nelson MA, Saboda K, Warneke J, Bowden GT, Alberts DS. Modulation of biologic endpoints by topical difluoromethylornithine (DFMO), in subjects at high-risk for nonmelanoma skin cancer. Clin Cancer Res. 2002;8:149–55. [PubMed] [Google Scholar]
- 59.Laukaitis CM, Gerner EW. DFMO: Targeted risk reduction therapy for colorectal neoplasia. Best Pract Res Clin Gastroenterol. 2011;25:495–506. doi: 10.1016/j.bpg.2011.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Berman B, Perez OA, Zell D. Immunological strategies to fight skin cancer. Skin Therapy Lett. 2006;11:1–7. [PubMed] [Google Scholar]
- 61.Gaspari AA, Sauder DN. Immunotherapy of basal cell carcinoma: Evolving approaches. Dermatol Surg. 2003;29:1027–34. doi: 10.1046/j.1524-4725.2003.29295.x. [DOI] [PubMed] [Google Scholar]
- 62.Navi D, Huntley A. Imiquimod 5 percent cream and the treatment of cutaneous malignancy. Dermatol Online J. 2004;10:4. [PubMed] [Google Scholar]
- 63.Jobanputra KS, Rajpal AV, Nagpur NG. Imiquimod. Indian J Dermatol Venereol Leprol. 2006;72:466–9. doi: 10.4103/0378-6323.29352. [DOI] [PubMed] [Google Scholar]
- 64.Ersvaer E, Kittang AO, Hampson P, Sand K, Gjertsen BT, Lord JM, et al. The protein kinase C agonist PEP005 (Ingenol 3-Angelate) in the treatment of human cancer: A balance between efficacy and toxicity. Toxins (Basel) 2010;2:174–94. doi: 10.3390/toxins2010174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rosen RH, Gupta AK, Tyring SK. Dual mechanism of action of ingenol mebutate gel for topical treatment of actinic keratoses: Rapid lesion necrosis followed by lesion-specific immune response. J Am Acad Dermatol. 2012;66:486–93. doi: 10.1016/j.jaad.2010.12.038. [DOI] [PubMed] [Google Scholar]
- 66.Challacombe JM, Suhrbier A, Parsons PG, Jones B, Hampson P, Kavanagh D, et al. Neutrophils are a key component of the antitumor efficacy of topical chemotherapy with ingenol-3-angelate. J Immunol. 2006;177:8123–32. doi: 10.4049/jimmunol.177.11.8123. [DOI] [PubMed] [Google Scholar]
- 67.Le TT, Gardner J, Hoang-Le D, Schmidt CW, MacDonald KP, Lambley E, et al. Immunostimulatory cancer chemotherapy using local ingenol-3-angelate and synergy with immunotherapies. Vaccine. 2009;27:3053–62. doi: 10.1016/j.vaccine.2009.03.025. [DOI] [PubMed] [Google Scholar]
- 68.Ali FR, Wlodek C, Lear JT. The role of ingenol mebutate in the treatment of actinic keratoses. Dermatol Ther (Heidelb) 2012;2:8. doi: 10.1007/s13555-012-0008-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Berman B, Villa AM, Ramirez CC. Mechanisms of action of new treatment modalities for actinic keratosis. J Drugs Dermatol. 2006;5:167–73. [PubMed] [Google Scholar]
- 70.Chakrabarty A, Geisse JK. Medical therapies for non-melanoma skin cancer. Clin Dermatol. 2004;22:183–8. doi: 10.1016/j.clindermatol.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 71.Reymann F. Treatment of basal cell carcinoma of the skin with 5-fluorouracil ointment. A 10-year follow-up study. Dermatologica. 1979;158:368–72. doi: 10.1159/000250782. [DOI] [PubMed] [Google Scholar]
- 72.Amini S, Viera MH, Valins W, Berman B. Nonsurgical innovations in the treatment of nonmelanoma skin cancer. J Clin Aesthet Dermatol. 2010;3:20–34. [PMC free article] [PubMed] [Google Scholar]
- 73.Rundhaug JE, Fischer SM. Cyclo-oxygenase-2 plays a critical role in UV-induced skin carcinogenesis. Photochem Photobiol. 2008;84:322–9. doi: 10.1111/j.1751-1097.2007.00261.x. [DOI] [PubMed] [Google Scholar]
- 74.Fulda S. Betulinic acid: A natural product with anticancer activity. Mol Nutr Food Res. 2009;53:140–6. doi: 10.1002/mnfr.200700491. [DOI] [PubMed] [Google Scholar]
- 75.Mullauer FB, Kessler JH, Medema JP. Betulinic acid, a natural compound with potent anticancer effects. Anticancer Drugs. 2010;21:215–27. doi: 10.1097/CAD.0b013e3283357c62. [DOI] [PubMed] [Google Scholar]
- 76.Yogeeswari P, Sriram D. Betulinic acid and its derivatives: A review on their biological properties. Curr Med Chem. 2005;12:657–66. doi: 10.2174/0929867053202214. [DOI] [PubMed] [Google Scholar]
- 77.Belanger JT. Perillyl alcohol: Applications in oncology. Altern Med Rev. 1998;3:448–57. [PubMed] [Google Scholar]
- 78.Barthelman M, Chen W, Gensler HL, Huang C, Dong Z, Bowden GT. Inhibitory effects of perillyl alcohol on UVB-induced murine skin cancer and AP-1 transactivation. Cancer Res. 1998;58:711–6. [PubMed] [Google Scholar]
- 79.Stratton SP, Alberts DS, Einspahr JG, Sagerman PM, Warneke JA, Curiel-Lewandrowski C, et al. A phase 2a study of topical perillyl alcohol cream for chemoprevention of skin cancer. Cancer Prev Res (Phila) 2010;3:160–9. doi: 10.1158/1940-6207.CAPR-09-0183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Heretsch P, Tzagkaroulaki L, Giannis A. Modulators of the hedgehog signaling pathway. Bioorg Med Chem. 2010;18:6613–24. doi: 10.1016/j.bmc.2010.07.038. [DOI] [PubMed] [Google Scholar]
- 81.Lin TL, Matsui W. Hedgehog pathway as a drug target: Smoothened inhibitors in development. Onco Targets Ther. 2012;5:47–58. doi: 10.2147/OTT.S21957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Issa MC, Manela-Azulay M. Photodynamic therapy: A review of the literature and image documentation. An Bras Dermatol. 2010;85:501–11. doi: 10.1590/s0365-05962010000400011. [DOI] [PubMed] [Google Scholar]
- 83.Böhm M, Luger TA. Alpha-melanocyte-stimulating hormone. From bench to bedside. Hautarzt. 2010;61:497–504. doi: 10.1007/s00105-009-1891-1. [DOI] [PubMed] [Google Scholar]
- 84.Langan EA, Nie Z, Rhodes LE. Melanotropic peptides: More than just ‘Barbie drugs’ and ‘sun-tan jabs’? Br J Dermatol. 2010;163:451–5. doi: 10.1111/j.1365-2133.2010.09891.x. [DOI] [PubMed] [Google Scholar]
- 85.Wilson BD, Mang TS, Stoll H, Jones C, Cooper M, Dougherty TJ. Photodynamic therapy for the treatment of basal cell carcinoma. Arch Dermatol. 1992;28:1597–601. [PubMed] [Google Scholar]
- 86.Uribe P, Gonzalez S. Epidermal growth factor receptor (EGFR) and squamous cell carcinoma of the skin: Molecular bases for EGFR-targeted therapy. Pathol Res Pract. 2011;207:337–42. doi: 10.1016/j.prp.2011.03.002. [DOI] [PubMed] [Google Scholar]
- 87.Burtness B. The role of cetuximab in the treatment of squamous cell cancer of the head and neck. Expert Opin Biol Ther. 2005;5:1085–93. doi: 10.1517/14712598.5.8.1085. [DOI] [PubMed] [Google Scholar]
- 88.Requena C, Llombart B, Sanmartín O. Acneiform eruptions induced by epidermal growth factor receptor inhibitors: Treatment with oral isotretinoin. Cutis. 2012;90:77–80. [PubMed] [Google Scholar]
- 89.Garden BC, Wu S, Lacouture ME. The risk of nail changes with epidermal growth factor receptor inhibitors: A systematic review of the literature and meta-analysis. J Am Acad Dermatol. 2012;67:400–8. doi: 10.1016/j.jaad.2011.10.009. [DOI] [PubMed] [Google Scholar]
- 90.Santiago F, Gonçalo M, Reis JP, Figueiredo A. Adverse cutaneous reactions to epidermal growth factor receptor inhibitors: A study of 14 patients. An Bras Dermatol. 2011;86:483–90. doi: 10.1590/s0365-05962011000300010. [DOI] [PubMed] [Google Scholar]
- 91.Panda S. Nonmelanoma skin cancer in India: Current scenario. Indian J Dermatol. 2010;55:373–8. doi: 10.4103/0019-5154.74551. [DOI] [PMC free article] [PubMed] [Google Scholar]