

Abstract
We report an approach to preventing bacterial biofilm formation that is based on the surface-mediated release of 5,6-dimethyl-2-aminobenzimidazole (DMABI), a potent and non-bactericidal small-molecule inhibitor of bacterial biofilm growth. Our results demonstrate that DMABI can be encapsulated in thin films of a model biocompatible polymer [poly(lactide-co-glycolide), PLG] and be released in quantities that inhibit the formation of Pseudomonas aeruginosa biofilms by up to 75–90% on surfaces that otherwise support robust biofilm growth. This approach enables the release of this new anti-biofilm agent for over one month, and it can be used to inhibit biofilm growth on both film-coated surfaces and other adjacent surfaces (e.g., on other uncoated surfaces and at air/water interfaces). Our results demonstrate a non-bactericidal approach to the prevention of biofilm growth and provide proof of concept using a clinically relevant human pathogen. In contrast to coatings designed to kill bacteria on contact, this release-based approach should also permit the design of strategically placed depots that disseminate DMABI more broadly and exert inhibitory effects over larger areas, which could prove useful in applications where the design or function of a surface prohibits the application of a uniform coating. This approach could also be used to design new polymer-coated surfaces useful for fundamental studies of bacterial biofilm growth. In a broader context, the non-bactericidal nature of DMABI could also provide opportunities to address concerns related to evolved resistance that currently face approaches based on the release of traditional microbicidal agents (e.g., antibiotics). Finally, the results of initial in vitro mammalian cell culture studies indicate that DMABI is not toxic to cells at concentrations required for strong anti-biofilm activity, suggesting that this new agent is well suited for further investigation in biomedical and personal care contexts. The surface-mediated approach reported here provides new tools useful for fundamental studies of biofilm formation and could, with further development, prove useful in a range of other industrial and commercial contexts in which bacterial biofilm growth is endemic.
Keywords: Biofilms, Thin Films, Drug Delivery, Polymers, Surfaces and Interfaces
Introduction
Many species of bacteria colonize surfaces and form complex, structured communities called ‘biofilms’ that protect against external threats, aid in the capture and trafficking of nutrients, and otherwise promote survival in hostile environments.[1–2] Biofilms are ubiquitous and often harmless in natural environments, but they can pose persistent and costly challenges in many industrial, commercial, and health-related contexts.[3–4] The financial and societal burdens incurred by biofilm-associated bacterial infections alone are enormous.[5–6] As just one example, biofilms formed by the Gram-negative bacterium Pseudomonas aeruginosa – a species identified as the primary cause of infection in cystic fibrosis patients,[7–10] a leading source of infection in burn victims,[10] and one of the most common bacteria found on the surfaces of indwelling medical devices[11–13] – can increase treatment costs substantially and impact the quality of life in a broad spectrum of patients.[14] Biofouling and other issues associated with the formation of biofilms on the surfaces of pipes, ship hulls, and other objects also present substantial challenges in many other contexts.[15–17]
Biofilms can be removed from surfaces using physical methods (e.g., scraping) or by treatment with chemical agents that either kill the bacteria (e.g., antibiotics)[6, 18] or disrupt the matrix of excreted proteins, DNA, and polysaccharides that provide structural support to the biofilm (e.g., dispersants).[6, 19–21] The usefulness of these approaches depends upon the location of the biofilm and the context in which it has formed. For example, high concentrations of dispersants can often be applied readily to clear biofilms in an industrial setting, but may not be appropriate in the context of implanted medical devices. Approaches based on the administration of antibiotics are also generally limited by the fact that bacteria growing in biofilms can be 10–1000 times more resistant to treatment with these conventional drugs than non-biofilm-associated bacteria.[22–23] For these and other reasons, approaches to preventing the initial formation of biofilms (as opposed to approaches designed to remove them after they have formed) would be broadly useful and have been the target of intensive study.[6, 24–25] The work reported here takes a step toward addressing these important issues by developing a materials-based and non-bactericidal approach to preventing biofilm growth at surfaces and interfaces, the primary locations at which biofilms form.
Many different materials-based approaches have been developed in efforts to prevent or reduce biofilm growth. These typically fall into one of two general categories: (i) non-leaching surface treatments that either prevent the initial attachment of bacteria or kill them on contact,[24–28] or (ii) coatings designed to control or sustain the release of antimicrobial agents (e.g., antibiotics).[25, 29–31] These approaches can be used alone or in combination, and each has advantages or limitations that can render them more or less useful depending on the needs or restrictions of a particular application. For example, whereas coatings that prevent attachment or kill bacteria on contact have the potential to prevent the growth of bacteria directly on the surfaces to which they are applied, approaches that release antimicrobial compounds also have the potential to prevent growth in adjacent areas (e.g., in solution or on other nearby surfaces, etc.). On the other hand, while many of these microbicidal approaches have the potential to prevent biofilm formation, they also, unfortunately, have the potential to contribute further to the emerging problem of evolved bacterial resistance.[32] To address this potential issue, we[33] and others[34–36] have reported approaches to the release of non-bactericidal small-molecules that can inhibit the growth of biofilms through mechanisms that do not result in cell death, including the disruption of bacterial cell-cell communication, or “quorum sensing”.[37–38] These and other non-bactericidal agents represent an exciting area of fundamental research and provide new opportunities to clear bacterial infections and control other important aspects of bacterial behavior (e.g., virulence, motility, etc.).[39–40]
The work reported here was motivated by the recent discovery of a new class of small molecule anti-biofilm agents based on the 2-aminobenzimidazole (ABI) scaffold[41] and a recent study from our group demonstrating that certain ABI derivatives are highly active in P. aeruginosa.[42] These ABI-based agents are hydrolytically stable, straightforward to synthesize, and several of these derivatives are among the most potent anti-biofilm agents reported to date against this pathogen. Although the mechanisms through which these compounds exert their anti-biofilm activities are not completely understood, our recent results demonstrate (i) that ABI derivatives can modulate quorum sensing pathways in P. aeruginosa and (ii) that they inhibit biofilm growth through mechanisms that are non-bactericidal.[42] In this current investigation, we sought to develop methods for the immobilization and surface-mediated release of our most potent ABI-based anti-biofilm agent (5,6-dimethyl-2-aminobenzimidazole, DMABI) and design new polymer-based coatings that could be used to prevent or reduce the growth of bacterial biofilms at surfaces and interfaces. Our results suggest the basis of a new non-bactericidal and surface-based approach to biofilm inhibition that could prove useful as a tool for fundamental studies of bacterial biofilm growth and in a range of different biomedical, personal care, and industrial contexts in which biofilm growth is endemic.
Materials and Methods
Materials
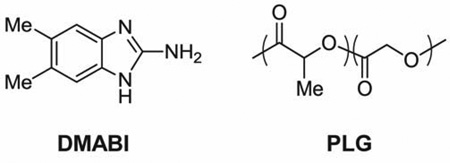
Reagent grade solvents, standard salts and media, and crystal violet were purchased from Sigma Aldrich (Milwaukee, WI), Fisher Scientific (Pittsburgh, PA), or other commercial sources and used without further purification unless otherwise noted. Poly(D,L-lactide-co-glycolide) (PLG, 50L:50G, MW = 12,000–16,000) and poly(L-lysine) hydrobromide (MW = 100,000–140,000) were purchased from Polysciences (Warrington, PA). Dimethyl-2-aminobenzimidazole (DMABI) was synthesized using a previously reported procedure.[42] The P. aeruginosa strain PAO1 used in biofilm assays was provided by Professor Barbara Iglewski (University of Rochester).
General Considerations
All assays involving the use of P. aeruginosa were performed in a 95:5 mixture of M9+ media:LB media using M9+ medium with the following previously reported composition: 47.7 mM Na2HPO4, 21.7 mM KH2PO4, 8.6 mM NaCl, 18.7 mM NH4Cl, 1 mM MgSO4, 0.1 mM CaCl2, 0.4% (w/v) L-Arg, 0.5% (w/v) casamino acids, 0.2% (w/v) anhydrous α-D(+)-glucose, 0.2% (w/v) sodium succinate dibasic hexahydrate, 0.2% (w/v) citric acid monohydrate, and 0.2% (w/v) L-glutamic acid monopotassium salt monohydrate.[42] All experiments that did not involve bacteria were performed in M9 buffer (47.7 mM Na2HPO4, 21.7 mM KH2PO4, 8.6 mM NaCl, and 18.7 mM NH4Cl) to prevent contaminating growth and avoid interfering absorbance by other components of M9+ and LB. Phosphate-buffered saline (PBS) used for washing steps in microtiter plate biofilm assays was prepared with the following composition: 10 mM Na2HPO4, 1.76 mM KH2PO4, 137 mM NaCl, and 2.7 mM KCl. All solutions and buffers were prepared using 18 MΩ water to minimize trace metal contamination, and were used at pH ~7.35. All media solutions were sterilized by passing them through a 0.22 µm PES (PolyEtherSulfone) membrane filter into autoclaved glassware. All plate-based assays were performed in flat-bottomed 96-well microtiter plates made of either untreated polystyrene (Costar 3370), untreated polypropylene (Costar 3364), or glass-coated polypropylene (SUN-SRi 400 062). For assays performed in non-polystyrene plates, samples were later transferred to flat-bottomed polystyrene or quartz (Hellma) 96-well plates to permit characterization of absorbance using a plate reader. For release studies, microtiter plates were sealed using plate-sealing mats (Costar 3080) to prevent evaporation; plates were covered with standard lids for biofilm studies. Film-coated glass substrates used in these studies were fabricated on glass chips (e.g., 5 mm × 7 mm) cut from larger microscope slides (Fisher Scientific) and, in the case of biofilm studies, were attached to the lids of microtiter plates (see text) using epoxy. All film-coated substrates and microtiter plates were sterilized directly before use by UV-C germicidal irradiation for 30 min in a Baker SterilGARD III Advance Biological Safety Cabinet. Absorbance measurements were acquired using a BioTek Synergy 2 plate reader running Gen 5 1.05 software. The wavelength of maximum absorption (λmax) of DMABI in M9 buffer was determined to be 284 nm, at which the molar extinction coefficient (ε) of DMABI was 8880 A284M−1cm−1. Data were analyzed using Microsoft Office Excel 2007. Biofilm assay data are shown as a percentage of the positive control (i.e., growth on PLG films that did not contain DMABI) and represent the average and standard error (STE) of three replicate wells. For the characterization of surface morphology by scanning electron microscopy (SEM), an accelerating voltage of 3 kV was used to obtain images on a LEO DSM 1530 scanning electron microscope. Samples were coated with a thin layer of gold using a sputterer (45 s at 45 mA, 50 mTorr) prior to analysis. Digital pictures were acquired using a Canon Power Shot S5IS digital camera.
Fabrication of DMABI-Containing Films
PLG films containing DMABI were fabricated on the surfaces of glass chips or the bottoms of the wells of solvent-resistant, 96-well plates using a solvent casting approach. Prior to fabrication, all surfaces were cleaned by rinsing with acetone, exposed to oxygen plasma for 5 min, soaked in a solution of poly(L-lysine) hydrobromide (1 mg/mL in 18 MΩ water) for 1 hr, and then dried in an oven at 60 °C for 1 hr. DMABI-loaded PLG films containing different specified amounts of DMABI were then fabricated by (1) preparing a series of different stock solutions containing DMABI and PLG (prepared in acetone, with each solution containing a constant concentration of PLG, but varying amounts of DMABI), and (2) depositing equal aliquots of these solutions onto both sides of glass chips or directly into the wells of 96-well plates via pipette. Evaporation of acetone from these films was achieved by either allowing film-coated substrates to stand at room temperature (for glass chips) or by heating them to ~30 °C on a hot plate (for microtiter plates) for 30 min. Film-coated substrates were then placed in a vacuum desiccator at room temperature for at least 12 h to remove any residual solvent prior to use in subsequent experiments. This procedure permitted the fabrication of a series of substrates coated with polymer films containing the same amount of PLG but different known concentrations of DMABI. A list of the volumes and concentrations of stock solutions used, as well as the volumes of the final casting solutions deposited for each of the experiments described below, can be found in Table S1 of the Supporting Information.
Characterization of DMABI Release Profiles
The release of DMABI from film-coated substrates was characterized by incubating the films in M9 buffer and measuring changes in solution absorbance at 284 nm (the absorbance maximum of DMABI) as a function of time. For these experiments, 200 µL of buffer was added directly to the wells of a microtiter plate containing either film-coated bottoms or film-coated glass chips. The plates were sealed to prevent evaporation and incubated without shaking at 37 °C. At pre-determined times, 100-µL aliquots were removed from 3–4 replicate wells and transferred to the wells of a 96-well quartz plate. Absorbance measurements were made (with path length correction to normalize to a 1 cm path length) in each well and corrected by solvent blank subtraction, and the molar extinction coefficient of DMABI (ε = 8880 A284 M−1cm−1) was used to convert these values to molar concentrations of DMABI released per well.
Bacteriological Methods and Characterization of Biofilm Inhibition
The ability of film-coated substrates to inhibit the formation of bacterial biofilms was characterized using (i) films deposited directly on the bottoms of the wells of microtiter plates, and (ii) films deposited on glass chips and suspended from microtiter plate lids (see schematic illustrations in Figures 2A and 3A). For suspended-chip experiments, DMABI-loaded films were prepared in a manner identical to that reported above for chip-based release experiments, except that (i) films were cast on the bottom 7 mm of glass chips (5 mm × 10 mm in size), and (ii) the uncoated ends of these slides were fixed to the undersides of standard-sized 96-well plate lids (see text for additional discussion). The bioactivity of released DMABI was characterized using P. aeruginosa and a previously described crystal violet (CV) static biofilm assay protocol.[42] A bacterial inoculating culture was prepared by centrifuging an aliquot of an overnight culture (OD600 of ~1.0; 0.5 cm path length) and re-suspending the pellet in 95:5 M9+:LB medium to an OD600 of ~0.1–0.2. This culture was added to the wells of microtiter plates in 200-µL aliquots. As a no-bacteria negative control, an identical series of film-coated substrates was incubated in M9 buffer in the absence of P. aeruginosa. Plates were covered with a standard microtiter plate lid (for experiments using coated well bottoms) or a lid containing attached glass chips (for suspended-film experiments) and incubated statically at 37 °C for 24 h. After incubation, the bacterial suspension was removed by inverting the plate, and each well was washed twice with 225 µL of PBS to remove loosely bound material not associated with attached biofilm. For experiments in which suspended substrates were used, the substrates were washed twice by soaking in fresh PBS-filled wells for 30 seconds. Biofilms were fixed by placing uncovered plates and lids with suspended chips in a 37 °C oven and thermally dehydrating overnight. Substrates were then treated for 15 min at room temperature with a CV staining solution (200 µL per well; 0.1% (w/v) CV in 95:5 water to ethanol). The staining solution was then removed by inverting the plate and substrates were washed once with 200 µL of PBS and twice with 200 µL of 18 MΩ water. The plates and suspended chips were dried at 37 °C and then imaged on a white light trans-illuminator using a digital camera. The amount of CV retained by the biofilm was quantified by re-solubilizing (or “de-staining”) the CV in 30% acetic acid and measuring the absorbance at 590 nm using a two-step process. To characterize the amount of biofilm at the well bottoms, 100 µL of 30% acetic acid was added to each well, gently pipetted to re-solubilize the CV, and then characterized by UV/visible spectrophotometry. After removal of the first aliquot of acetic acid, wells were then refilled with 225 µL of 30% acetic acid to characterize the amount of biofilm originally present at the air/water interface, as previously described.[42] Experiments to characterize the ability of film-coated substrates to inhibit biofilm over longer periods (see text) were performed in the following manner. After an initial 24-hr challenge in the presence of bacteria, suspended chips were washed twice as described above. Suspended chips were then subjected to two additional and successive 24-hr challenges by transferring them to the wells of new plates containing 200 µL of a fresh 1-in-10 inoculating culture of P. aeruginosa. These additional challenges, and procedures used to characterize extents of biofilm formation resulting from them, were performed using the methods described above.
Figure 2.

(A) Schematic showing experimental setup for characterization of biofilm growth in PLG-coated wells. DMABI was loaded in PLG films solvent-cast on the bottom surface of each well of a microtiter plate and incubated for 24 hours in the presence of a suspension of bacteria. (B) Plot of biofilm growth (as a percentage of growth measured in control wells containing PLG films that did not contain DMABI) as a function of initial DMABI loading. Biofilm growth was characterized and quantified on the bottoms of film-coated wells (closed circles) and at the air/water interface in each well (open circles). (C) Digital pictures of film-coated wells fabricated to contain 18 different loadings ranging from 0.001 to 0.40 µmol DMABI after 24 hours of incubation in the presence of bacteria. Key loading values are labeled; a complete list of all loadings used is included in Table S1. Images shown are of wells after staining of biomass with crystal violet (Row I) and after subsequent de-staining to remove crystal violet (Row II; see text). Results arising from these staining and de-staining procedures were used to quantify amounts of biofilm growth and calculate the results shown in (B). Pictures of wells used as controls containing no polymer coating (bare wells, *) or a polymer coating that did not contain DMABI (PLG only, **) are included for comparison. To aid in interpretation of references to color made in the main text, a color version of this Figure is included as Figure S4 of the Supporting Information.
Figure 3.

(A) Schematic showing experimental setup for characterization of biofilm growth in uncoated wells using suspended film-coated substrates. DMABI-containing PLG films cast on the surfaces of glass chips were suspended in suspensions of bacteria contained in the uncoated wells of a microtiter plate (see text). (B) Plot of biofilm growth (as a percentage of growth measured in control wells containing PLG films that did not contain DMABI) as a function of initial DMABI loading. Films were fabricated to contain 18 different loadings of DMABI ranging from 0.001 to 0.40 µmol; a complete list of all loadings used is included in Table S1. Biofilm growth was characterized and quantified on the bottoms of film-coated wells after three successive 24-hour challenges in the presence of bacteria (see text). Data correspond to Challenge I (0–24 hours, closed triangles), Challenge II (24–48 hours, closed squares), and Challenge III (48–72 hours, closed diamonds). (C) Digital pictures of crystal violet-stained biomass present in each well after each 24-hour challenge. Characterization of the amount of crystal violet in each of these wells (determined using a de-staining procedure) was used to quantify amounts of biofilm growth and calculate the results shown in (B). Pictures of control wells that contained chips coated with PLG only (no DMABI, **) are included for comparison. To aid in interpretation of references to color made in the main text, a color version of this Figure is included as Figure S5 of the Supporting Information.
Results and Discussion
Film Fabrication and Characterization of DMABI Release Profiles
We selected 5,6-dimethyl-2-aminobenzimidazole (DMABI) as an anti-biofilm agent for use in this investigation for several reasons. First, this small molecule is among the most potent small-molecule anti-biofilm agents reported against P. aeruginosa (IC50 value of 4.0 µM when administered to bacterial suspensions).[42] Second, as described above, DMABI inhibits biofilm formation through a mechanism that is non-bactericidal. Third, the stability of this compound against chemical hydrolysis (relative to other small molecule modulators of biofilm growth)[42–43] renders it well suited for evaluation in long-term release and biofilm inhibition experiments. In addition, and with respect to potential biomedical applications of this approach, initial in vitro assays conducted as part of this study indicated that DMABI is not toxic to mammalian cells at concentrations required to promote strong biofilm inhibition (see Figure S1 of the Supporting Information). Lastly, DMABI is soluble in a broad range of organic solvents, which provides flexibility with respect to the range of materials and fabrication procedures that can be used to encapsulate and release it.
We used thin solvent-cast films of poly(lactide-co-glycolide) (PLG) as a model matrix for these proof-of-concept studies. PLG is a well-known biocompatible, biodegradable, and FDA-approved polymer that has been used for the encapsulation and release of many other small molecule agents,[30, 44–47] and numerous past studies demonstrate that the structure of this polymer can be varied to exert broad control over the rates at which small molecules can be released (e.g., over periods ranging from several days to weeks or months by changing the ratio of lactide to glycolide in the backbone of the polymer).[48] All experiments described herein were conducted using films fabricated from PLG having a 50:50 ratio of lactide to glycolide repeat units. As described in further detail below, this polymer provides for the extended release of DMABI, but it can also be formulated to release concentrations of DMABI above the IC50 value of this agent within the first several hours of incubation in physiological media. This polymer was thus well suited for many of the short-term (e.g., 24 – 72 hour) bacteriological experiments reported here.
We performed a series of experiments to characterize the loading and release of DMABI from PLG films that were solvent-cast in the wells of 96-well microtiter plates (see Materials and Methods for additional details). This microtiter plate platform was designed to facilitate the subsequent evaluation of these materials in bacterial biofilm assays described below. The use of solvent-casting methods (as opposed to dip-coating or spin-coating protocols) also provided precise and convenient control over the total amounts of DMABI loaded into each film. PLG films fabricated using this approach were ~6.8 ± 3.5 µm thick and were generally smooth, uniform, and devoid of large-scale surface defects, as determined by scanning electron microscopy (SEM; see Figure S2 of the Supporting Information).
Figure 1 shows representative release profiles of PLG films (fabricated in the wells of a glass-coated microtiter plate) containing either 0.1 µmol (16.3 µg; closed diamonds) or 0.025 µmol (4.1 µg; closed triangles) of DMABI upon incubation in M9 buffer at 37 °C. Inspection of these data reveals that DMABI was released from these films over a period of at least 40 days, at which point ~80% of the compound initially loaded into the films had been released. Further inspection reveals release to proceed without substantial burst release or, more importantly, the presence of an initial lag phase. Characterization of DMABI-loaded films solvent-cast in the wells of polypropylene microtiter plates and on the surfaces of planar glass substrates (used in bacteria-based experiments described below) exhibited similar extended release profiles (see Figure S3 of the Supporting Information). While many different approaches could be used to vary or tune the rates at which DMABI can be released from these films (or from films fabricated using a range of other degradable or non-degradable polymers), the release profiles shown in Figure 1 were well suited for the in vitro evaluation of surface-mediated biofilm inhibition in the experiments described below.
Figure 1.

Plot of release vs. time for two solvent-cast PLG films loaded with DMABI [initial loadings of 4.1 µg (triangles) and 16.3 µg (diamonds)] incubated in M9 buffer (pH 7.35) at 37 °C. Each data point represents the average of 4 replicate wells; error bars are STE.
Release of DMABI from Film-Coated Surfaces Inhibits Formation of P. aeruginosa Biofilms
We next sought to determine whether surfaces coated with DMABI-loaded films could prevent the formation of biofilms in the clinically relevant pathogen P. aeruginosa using a standard static biofilm growth assay. Although the IC50 value of DMABI for biofilm inhibition when added exogenously to solution is known (4.0 µM),[42] the range of DMABI loadings required to inhibit initial or longer-term biofilm growth in these release-based experiments was not known at the outset of these studies. We therefore fabricated a series of films with 18 different initial loadings of DMABI ranging from 0.001 to 0.4 µmol per well in the bottoms of polypropylene microtiter plates (as described above and shown schematically in Figure 2A). These experiments were conducted using two identical sets of films: one set was incubated in the presence of P. aeruginosa in a modified M9 media and the other was incubated in M9 buffer in the absence of bacteria (as a negative control, and to facilitate characterization of the amount of DMABI released at relevant time points, as described above). The amounts of biofilm growth on the bottoms of film-coated wells and at the air/water interface in each well (Figure 2A), two measures used frequently to characterize biofilm growth in static culture, were quantified after 24 hours by staining the resulting biomass with crystal violet.
Figure 2B shows a plot of percent biofilm growth (as a function of initial DMABI loading, and relative to growth in wells containing control films that did not contain DMABI) on the bottoms of film-coated wells (closed circles) and at the air/water interface in each well (open circles). Inspection of these results reveals that films with DMABI loadings at or below 0.025 µmol per well did not substantially inhibit the growth of biofilms (i.e., less than 50% inhibition) in either of these interfaces over this 24-hour incubation period. Further inspection, however, reveals that films containing higher loadings (0.035 to 0.1 µmol DMABI; Region D) inhibited biofilm formation at both of these interfaces substantially (i.e., greater than 50% inhibition, with a maximum inhibition of up to 85%). Differences in biofilm growth on film-coated well bottoms were also evident by visual inspection of crystal violet-stained wells, as shown in Figure 2C. The presence and intensity of blue stain in Row I indicates the presence and relative amount of biofilm in each well. Row 2 shows images of the same wells in Row I after the de-staining procedure used to extract crystal violet and obtain the quantitative results shown in Figure 2B.
We draw several conclusions on the basis of these results. First, the results shown in Figure 2 demonstrate that DMABI is released in a form that is bioactive and at levels that are sufficient to strongly inhibit the formation of P. aeruginosa biofilms on surfaces coated with a polymer that otherwise supports robust biofilm growth. Second, these results demonstrate that this approach can also be used to inhibit biofilm growth in other environments and locations (e.g., at air/water interfaces) that are adjacent to film-coated surfaces but that are not themselves coated. The similarity of the loading/response profiles for biofilm inhibition at these two different locations (Figure 2B) suggests that DMABI prevents the formation of biofilm on polymer-coated surfaces through a mechanism that involves the interaction of bacteria with released (i.e., soluble) DMABI, as opposed to alternate mechanisms that, for example, could depend upon surface-based interactions between bacteria and DMABI adsorbed to or presented locally at film-coated surfaces.
Third, we note that while the DMABI-loaded films used here do not result in substantial burst release (Figure 1), formulations containing loadings ranging from 0.035 to 0.1 µmol per well (Figure 2C, Region D) release amounts of DMABI that are sufficient to strongly inhibit biofilm formation over the first 24 hours of incubation. Characterization of DMABI release from films incubated in the absence of bacteria showed that films with loadings at or above 0.05 µmol yielded in-well molar solution concentrations of DMABI of at least 15.0 µM within the first 6 hours of incubation, a concentration that is over three times the IC50 value of 4.0 µM reported for this agent. This is an important design consideration from a practical point of view, because a substantial lag phase (or the release of concentrations of DMABI below its IC50 value during this initial period) would provide bacteria a ‘head start’ with which to initiate significant levels of biofilm growth (e.g., as shown clearly in Figures 2B and 2C for films with loadings below 0.035 µmol). We return to a consideration of the time-dependent release of DMABI and its influence on the ability of these films to inhibit biofilm formation over longer periods again in the discussion below.
Finally, we note that wells containing the four highest concentrations of DMABI used here (i.e., loadings ranging from 0.14 to 0.4 µmol per well; Region E of Row I, Figure 2C) adsorbed significantly more crystal violet than films of intermediate loading (e.g., from 0.035 to 0.1 µmol; Region D, as discussed above). Inspection of the corresponding wells shown in Row II of Figure 2C, however, reveals that a significant amount of crystal violet also remained in these samples after the de-staining procedure – in stark contrast to films at lower DMABI loadings, for which de-staining resulted in the removal of almost all visible stain. The presence of this residual stain prevented quantification of biofilm growth on the polymer films and at the air/water interfaces of these wells at these four highest loadings. The reasons for this residual staining at these higher DMABI loadings are not completely understood. In this context, however, we note that past studies by our group demonstrate that DMABI remains an inhibitor (and is not an activator) of biofilm formation in P. aeruginosa at concentrations up to ~3.2 mM,[42] a concentration that is significantly above those generated by the films used in this current investigation.
The fact that this residual staining was observed only at the four highest mass fractions of DMABI used here (i.e., loadings ranging from 0.14 to 0.4 µmol; Region E) suggested that this could result from physicochemical changes in those higher-loading films (e.g., changes in porosity) that could enable more effective penetration of biofilm and/or lead to more effective retention of stain by the polymer matrix itself (i.e., such that crystal violet itself is more difficult to remove quantitatively). SEM images of DMABI-loaded films before and after incubation revealed that while the surfaces of low-loading films (e.g., 0.01 µmol) remained smooth and uniform after incubation for 24 hours, films at higher loadings (0.14 µmol) exhibited a porous morphology (see Figure S2 of the Supporting Information). We note that we also observed residual, non-specific staining in otherwise identical PLG films loaded with high mass fractions of DMABI that were incubated in the absence of bacteria (data not shown). The intensity and uniformity of non-specific staining in these no-bacteria controls varied from experiment to experiment and staining in these cases was generally less intense that that observed when films were incubated in the presence of bacteria (Figure 2). Additional studies will be required to characterize changes in the structures of these films more completely. However, we conclude that higher loaded films are able to retain crystal violet through mechanisms that are at least partially independent of biofilm formation and, more importantly, that a useful window of lower DMABI concentrations exists (e.g., Region D; loadings ranging from 0.035 to 0.1 µmol) over which these films can be formulated to strongly inhibit biofilm formation on PLG-coated surfaces.
Inhibition on Adjacent Uncoated Surfaces and Over Extended Periods
The ability to use anti-biofilm surface coatings to inhibit biofilm growth at other locations (e.g., on other nearby or immediately adjacent surfaces that are not themselves coated) would significantly expand the utility of this approach and could be useful in a range of fundamental and applied contexts. We therefore conducted an additional series of experiments to characterize the ability of DMABI-loaded films to inhibit the formation of P. aeruginosa biofilms on adjacent uncoated surfaces. We also investigated the ability of these films to inhibit biofilm formation over periods of time longer than those used in the initial studies described above. These studies were difficult to conduct using the coated well-bottom approach described above for several reasons, including the fact that, because DMABI prevents biofilm growth through a mechanism that is non-bactericidal, prolonged incubation and additional growth of bacteria results in depletion of nutrients in the media. To permit periodic characterization of changes in biofilm growth over time, we designed experiments using DMABI-loaded PLG films solvent-cast onto the surfaces of planar glass substrates (cut to fit into the wells of a microtiter plate and attached to a plate lid to facilitate suspension in media during culture; see schematic in Figure 3A and Materials and Methods for more details). This approach allowed for the iterative insertion and removal of arrays of substrates coated with films containing different loadings of DMABI into multiple cultures of bacteria, as well as a practical and straightforward format for characterizing the inhibition of biofilm growth on the bottoms of uncoated plate wells. This approach was used to screen and characterize the ability of DMABI-loaded films (at 18 different loadings, ranging from 0.001 µmol to 0.4 µmol as used in the experiments described above) to inhibit biofilm formation over three discrete and successive 24-hour periods by periodically removing the film-coated substrates and then incubating them further in fresh cultures of bacteria.
Figure 3B shows a plot of percent P. aeruginosa biofilm growth as function of DMABI loading for film-coated substrates subjected to three different 24-hour challenges in the presence of bacteria: Challenge I (over 0–24 hours, closed triangles), Challenge II (over 24–48 hours, closed squares), and Challenge III (over 48–72 hours, closed diamonds). Qualitative differences in biofilm growth during each of these challenges are also shown in the pictures of crystal violet-stained wells shown in Figure 2C. The loading/response profiles for biofilm inhibition on the uncoated well bottoms during Challenge I were generally similar to those described above for data obtained using polymer-coated wells [e.g., an onset of biofilm inhibition at loadings at or above 0.025 µmol (see Regions D of Figures 3B and 3C) and up to ~90% inhibition of biofilm growth on uncoated well bottoms at the highest DMABI loadings (Region E)]. Importantly, these results also demonstrate clearly that films capable of strongly inhibiting biofilm growth during Challenge I were also able to strongly inhibit biofilm growth upon subsequent introduction to new cultures of bacteria in two additional 24-hour trials (Challenges II and III). For example, films containing loadings of DMABI ranging from 0.14 to 0.40 µmol (Region E) continued to inhibit biofilm growth on these uncoated surfaces at levels up to ~90% during these two additional challenges (i.e., over a total of three days). Visual inspection of the crystal violet-stained and de-stained suspended release substrates used in these experiments revealed staining and residual staining trends that were analogous to those discussed above and shown in Figure 2C for films on plate bottoms containing higher mass fractions of DMABI (data not shown).
Our data demonstrate that these suspended films retain their anti-biofilm activities upon repeated introduction to fresh cultures of bacteria. These results are important because they underscore the ability of these films to promote the gradual and sustained release of useful amounts of DMABI over time. We note that closer inspection of the results in Regions D of Figures 3B and 3C, corresponding to films with intermediate DMABI loadings (ranging from 0.025 to 0.10 µmol), reveals the onset and extent of inhibition in this loading range to shift and deteriorate from challenge to challenge. This trend suggests that while these films are able to strongly inhibit biofilm growth over an initial 24-hour period, these intermediate formulations may be less appropriate for applications that require longer-term inhibition.
The results above provide several interesting avenues for future investigation. In contrast to surfaces and coatings that are designed to kill bacteria on contact, the release-based methods described here should permit the placement of strategically-placed depots or inserts, or the design of partial, patterned, and non-uniform coatings that disseminate DMABI more broadly and exert inhibitory effects over larger areas. These methods could be particularly useful in cases where the design, function, scale, or surface properties of a device, assay plate, or other object prohibit the direct application of a uniform polymer coating. In addition, this polymer-based approach could prove useful for the design of new surface-based tools that could facilitate other fundamental and mechanistic studies of bacterial biofilm growth. Although the molecular mechanisms through which ABI-based agents inhibit biofilm growth are not yet completely understood, the non-bactericidal nature of DMABI could also potentially lead to methods that address concerns associated with evolved resistance currently faced by approaches based on the use of conventional microbicidal agents (e.g., antibiotics). Approaches that exploit orthogonal, non-bactericidal mechanisms of action should also make possible the development of polymer-based formulations containing combinations of DMABI and other bioactive agents that could act synergistically to inhibit biofilm formation or attenuate other bacterial behaviors (e.g., virulence, motility, etc.) more effectively. Although additional studies will be required to investigate potential mammalian cell toxicity more extensively, the results of our initial in vitro cell culture studies (Figure S1) suggest that DMABI is well suited for further investigation in biomedical contexts (that is, DMABI does not appear to be substantially toxic to cells at concentrations required to act as a strong inhibitor of P. aeruginosa biofilm formation).
Summary and Conclusions
We have reported an approach to the encapsulation and surface-mediated release of a new and potent small-molecule inhibitor of bacterial biofilm growth. Our results demonstrate that the anti-biofilm agent DMABI can be encapsulated in films of a hydrolytically degradable polymer, and that this approach can be tuned to release quantities of DMABI that are sufficient to prevent the growth of P. aeruginosa biofilms. This approach provides a mechanism for the extended and surface-mediated release of this new anti-biofilm agent (e.g., for over one month) and can be used to coat surfaces and design objects that can be used to inhibit the growth of bacterial biofilms on both film-coated surfaces and other adjacent surfaces and interfaces (e.g., on nearby, uncoated solid surfaces and at air/water interfaces, two locations where biofilms grow and are problematic). The results of this investigation provide a new polymer-based and non-bactericidal approach to inhibiting the growth of biofilms on surfaces and demonstrate proof of concept using a notorious and clinically relevant human pathogen. More broadly, the approach reported here provides new surface-based tools that could prove useful for fundamental studies of bacterial biofilm formation and methods that could, with further development, prove valuable in a range of other industrial and commercial contexts.
Acknowledgments
Financial support to H.E.B and D.M.L. was provided by the Office of Naval Research (N00014-07-1-0255), the National Institutes of Health (AI063326 for H.E.B), the Burroughs Wellcome Fund, and the UW Vilas Trust. This work made use of shared facilities supported, in part, by the National Science Foundation through a grant to the Materials Research Science and Engineering Center (MRSEC) at the University of Wisconsin (DMR 1121288). A.H.B. is a NSF Graduate Research Fellow. A.S.B. was funded in part by an NIH Chemistry-Biology Interface Training Grant (NIGMS T32 GM008505). We gratefully acknowledge Prof. Barbara Iglewski (University of Rochester) for donation of the P. aeruginosa PAO1 strain.
Footnotes
Supporting Information. Supporting information including results of mammalian cell toxicity assays, SEM images of PLG films, additional DMABI release profiles, and details of film fabrication procedures can be found online at doi:
References
- 1.Stoodley P, Sauer K, Davies DG, Costerton JW. Annu. Rev. Microbiol. 2002;56:187–209. doi: 10.1146/annurev.micro.56.012302.160705. [DOI] [PubMed] [Google Scholar]
- 2.Flemming HC, Wingender J. Nat. Rev. Microbio. 2010;8:623–633. doi: 10.1038/nrmicro2415. [DOI] [PubMed] [Google Scholar]
- 3.Hall-Stoodley L, Costerton JW, Stoodley P. Nat. Rev. Microbio. 2004;2:95–108. doi: 10.1038/nrmicro821. [DOI] [PubMed] [Google Scholar]
- 4.Donlan RM, Costerton JW. Clin. Microbiol. Rev. 2002;15:167–193. doi: 10.1128/CMR.15.2.167-193.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Costerton JW, Stewart PS, Greenberg EP. Science. 1999;284:1318–1322. doi: 10.1126/science.284.5418.1318. [DOI] [PubMed] [Google Scholar]
- 6.Francolini I, Donelli G. Fems Immunol Med Mic. 2010;59:227–238. doi: 10.1111/j.1574-695X.2010.00665.x. [DOI] [PubMed] [Google Scholar]
- 7.Wagner VE, Iglewski BH. Clin Rev Allergy Immunol. 2008;35:124–134. doi: 10.1007/s12016-008-8079-9. [DOI] [PubMed] [Google Scholar]
- 8.Koch C, Høiby N. Lancet. 1993;341:1065–1069. doi: 10.1016/0140-6736(93)92422-p. [DOI] [PubMed] [Google Scholar]
- 9.Govan JRW, Deretic V. Microbiol. Rev. 1996;60:539–574. doi: 10.1128/mr.60.3.539-574.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lyczak JB, Cannon CL, Pier GB. Microbes Infect. 2000;2:1051–1060. doi: 10.1016/s1286-4579(00)01259-4. [DOI] [PubMed] [Google Scholar]
- 11.Donlan RM. Emerging Infect. Dis. 2001;7:277–281. doi: 10.3201/eid0702.010226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Passerini L, Lam K, Costerton JW, King EG. Crit. Care Med. 1992;20:665–673. doi: 10.1097/00003246-199205000-00020. [DOI] [PubMed] [Google Scholar]
- 13.Mittal R, Sharma S, Chhibber S, Aggarwal S, Gupta V, Harjai K. FEMS Immunol. Med. Microbiol. 2010;58:237–243. doi: 10.1111/j.1574-695X.2009.00627.x. [DOI] [PubMed] [Google Scholar]
- 14.Hancock REW, Speert DP. Drug Resist. Updat. 2000;3:247–255. doi: 10.1054/drup.2000.0152. [DOI] [PubMed] [Google Scholar]
- 15.Beech IB, Sunner J. Curr. Opin. Biotechnol. 2004;15:181–186. doi: 10.1016/j.copbio.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 16.Coetser SE, Cloete TE. Crit. Rev. Microbiol. 2005;31:213–232. doi: 10.1080/10408410500304074. [DOI] [PubMed] [Google Scholar]
- 17.Huq A, Whitehouse CA, Grim CJ, Alam M, Colwell RR. Curr. Opin. Biotechnol. 2008;19:244–247. doi: 10.1016/j.copbio.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 18.Simoes M. Curr. Med. Chem. 2011;18:2129–2145. doi: 10.2174/092986711795656216. [DOI] [PubMed] [Google Scholar]
- 19.Chen X, Stewart PS. Water Res. 2000;34:4229–4233. [Google Scholar]
- 20.Kaplan JB. Int J Artif Organs. 2009;32:545–554. doi: 10.1177/039139880903200903. [DOI] [PubMed] [Google Scholar]
- 21.Xiong YH, Liu Y. Appl. Microbiol. Biotechnol. 2010;86:825–837. doi: 10.1007/s00253-010-2463-0. [DOI] [PubMed] [Google Scholar]
- 22.Davies D. Nat Rev Drug Discov. 2003;2:114–122. doi: 10.1038/nrd1008. [DOI] [PubMed] [Google Scholar]
- 23.Harmsen M, Yang LA, Pamp SJ, Tolker-Nielsen T. Fems Immunol Med Mic. 2010;59:253–268. doi: 10.1111/j.1574-695X.2010.00690.x. [DOI] [PubMed] [Google Scholar]
- 24.Bruellhoff K, Fiedler J, Moller M, Groll J, Brenner RE. Int J Artif Organs. 2010;33:646–653. doi: 10.1177/039139881003300910. [DOI] [PubMed] [Google Scholar]
- 25.Banerjee I, Pangule RC, Kane RS. Adv Mater. 2011;23:690–718. doi: 10.1002/adma.201001215. [DOI] [PubMed] [Google Scholar]
- 26.Kingshott P, Griesser HJ. Curr Opin Solid State Mater Sci. 1999;4:403–412. [Google Scholar]
- 27.Rana D, Matsuura T. Chem. Rev. 2010;110:2448–2471. doi: 10.1021/cr800208y. [DOI] [PubMed] [Google Scholar]
- 28.Glinel K, Thebault P, Humblot V, Pradier CM, Jouenne T. Acta Biomater. 2012;8:1670–1684. doi: 10.1016/j.actbio.2012.01.011. [DOI] [PubMed] [Google Scholar]
- 29.Hetrick EM, Schoenfisch MH. Chem. Soc. Rev. 2006;35:780–789. doi: 10.1039/b515219b. [DOI] [PubMed] [Google Scholar]
- 30.Wu P, Grainger DW. Biomaterials. 2006;27:2450–2467. doi: 10.1016/j.biomaterials.2005.11.031. [DOI] [PubMed] [Google Scholar]
- 31.Zilberman M, Elsner JJ. J Control Release. 2008;130:202–215. doi: 10.1016/j.jconrel.2008.05.020. [DOI] [PubMed] [Google Scholar]
- 32.Campoccia D, Montanaro L, Speziale P, Arciola CR. Biomaterials. 2010;31:6363–6377. doi: 10.1016/j.biomaterials.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 33.Breitbach AS, Broderick AH, Jewell CM, Gunasekaran S, Lin Q, Lynn DM, Blackwell HE. Chem. Commun. 2011;47:370–372. doi: 10.1039/c0cc02316g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Farber BF, Wolff AG. J Biomed Mater Res. 1993;27:599–602. doi: 10.1002/jbm.820270506. [DOI] [PubMed] [Google Scholar]
- 35.Baveja JK, Willcox MDP, Hume EBH, Kumar N, Odell R, Poole-Warren LA. Biomaterials. 2004;25:5003–5012. doi: 10.1016/j.biomaterials.2004.02.051. [DOI] [PubMed] [Google Scholar]
- 36.Melander C, Moeller PDR, Ballard TE, Richards JJ, Huigens RW, Cavanagh J. Int. Biodeterior. Biodegradation. 2009;63:529–532. doi: 10.1016/j.ibiod.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Njoroge J, Sperandio V. EMBO Mol Med. 2009;1:201–210. doi: 10.1002/emmm.200900032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kalia VC, Purohit HJ. Crit Rev Microbiol. 2011;37:121–140. doi: 10.3109/1040841X.2010.532479. [DOI] [PubMed] [Google Scholar]
- 39.Rasko DA, Sperandio V. Nat Rev Drug Discov. 2010;9:117–128. doi: 10.1038/nrd3013. [DOI] [PubMed] [Google Scholar]
- 40.Sintim HO, Smith JAI, Wang J, Nakayama S, Yan L. Future Med Chem. 2010;2:1005–1035. doi: 10.4155/fmc.10.185. [DOI] [PubMed] [Google Scholar]
- 41.Rogers SA, Huigens RW, Melander C. J. Am. Chem. Soc. 2009;131:9868–9869. doi: 10.1021/ja9024676. [DOI] [PubMed] [Google Scholar]
- 42.Frei R, Breitbach AS, Blackwell HE. Angew. Chem. Int. Ed. 2012;51:5226–5229. doi: 10.1002/anie.201109258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Geske GD, Wezeman RJ, Siegel AP, Blackwell HE. J. Am. Chem. Soc. 2005;127:12762–12763. doi: 10.1021/ja0530321. [DOI] [PubMed] [Google Scholar]
- 44.Anderson JM, Shive MS. Adv. Drug Delivery Rev. 1997;28:5–24. doi: 10.1016/s0169-409x(97)00048-3. [DOI] [PubMed] [Google Scholar]
- 45.Jain RA. Biomaterials. 2000;21:2475–2490. doi: 10.1016/s0142-9612(00)00115-0. [DOI] [PubMed] [Google Scholar]
- 46.Chen G, Ushida T, Tateishi T. Macromol Biosci. 2002;2:67–77. [Google Scholar]
- 47.Panyam J, Labhasetwar V. Adv. Drug Delivery Rev. 2003;55:329–347. doi: 10.1016/s0169-409x(02)00228-4. [DOI] [PubMed] [Google Scholar]
- 48.Fredenberg S, Wahlgren M, Reslow M, Axelsson A. Int J Pharm. 2011;415:34–52. doi: 10.1016/j.ijpharm.2011.05.049. [DOI] [PubMed] [Google Scholar]