

Abstract
Background
High-throughput expression data, such as gene expression and metabolomics data, exhibit modular structures. Groups of features in each module follow a latent factor model, while between modules, the latent factors are quasi-independent. Recovering the latent factors can shed light on the hidden regulation patterns of the expression. The difficulty in detecting such modules and recovering the latent factors lies in the high dimensionality of the data, and the lack of knowledge in module membership.
Methods
Here we describe a method based on community detection in the co-expression network. It consists of inference-based network construction, module detection, and interacting latent factor detection from modules.
Results
In simulations, the method outperformed projection-based modular latent factor discovery when the input signals were not Gaussian. We also demonstrate the method's value in real data analysis.
Conclusions
The new method nMLSA (network-based modular latent structure analysis) is effective in detecting latent structures, and is easy to extend to non-linear cases. The method is available as R code at http://web1.sph.emory.edu/users/tyu8/nMLSA/.
Keywords: matrix decomposition, modularity, latent factors, network, community detection
Background
Modularity is a common characteristic of high-throughput biological data [1]. In a large system, the biological units, i.e. features (genes, proteins, or metabolites) are organized into quasi-autonomous modules. In expression data, each expression module can be modeled reasonably well using the latent factor approach [2,3]. Given the involvement of thousands of features, an unknown number of modules, and unknown module membership of the features, it is difficult to faithfully detect the modules and recover the underlying latent factors controlling the modules.
Dimension reduction methods at the global level, such as Principal Component Analysis (PCA), Independent Component Analysis (ICA) [4], sparse PCA [5,6], and Bayesian decomposition [7] are not effective in detecting localized signals. Clustering methods group co-expressed features together [8], which may help identify modules that are controlled by a single underlying signal [9,10]. However in real data, the features involved in the same module may not co-express when more than one latent factors control the module. We previously proposed the projection-based Modular Latent Structure Analysis (MLSA) [11], which detects modules using iteratively re-weighted singular value decomposition (SVD). So far there are no other modular decomposition methods. In this study, we seek to improve the method using a totally different approach. Our goal is to develop a method that is more intuitive, flexible, and involves less ad hoc parameter choices.
Using networks constructed from expression data can provide a flexible framework for module detection [12-14]. Here we present a method to identify modules and the underlying latent signals in three steps: (1) constructing a co-expression network based on statistical inference and local false discovery rate (lfdr); (2) detecting communities in the network; and (3) recovering interacting latent factors from the modules.
The goal of the algorithm is to achieve modular matrix decomposition. We attempt to solve the problem by assembling tools from some well-established fields. The first is the reverse engineering of genome-scale networks. There are a number of methods available in this area, which were designed with different objectives, including Gaussian Graphical Models where the absence of an edge signifies conditional independence [15,16], co-expression network where edges signify marginal dependence [13], information theory-based networks [17], and Bayesian networks [18]. In this study, we designed our own method to estimate an inference-based co-expression network using the local false discovery rate (lfdr) concept [19-21]. The use of local fdr makes the procedure adaptive to shifts of baseline correlation levels and avoids constructing overly dense networks when there are pervasive low-level correlations between genes. Once the network is constructed, we borrow a method from the mature field of community detection in large networks [22-25]. This is followed by latent factor extraction and rotation using factor analysis methods [26]. Added together, the assembled tools make a very good heuristic solution to the modular decomposition problem.
We demonstrate the superiority of the new method against existing modular and global decomposition methods using simulations, and apply the method to a real dataset to show it detects biologically meaningful modules that are controlled by multiple latent factors.
Methods
The objective
Given a data matrix Gp×n with p features measured in n conditions, we seek to assign subgroups of the features into modules, such that within each module, the expression levels of the features can be modeled by a linear factor model
where q is the size of the module, r is the number of latent factors controlling the module, L is the regulation strength (loading) matrix, and E is the residual matrix. Our interest is estimating (1) the number of modules, (2) the module membership of the features, (3) the activities of the latent factors controlling each module (F matrix), and (4) the regulation strength of each factor on each feature (L matrix).
The estimation procedure
Figure 1 illustrates the procedure using a toy dataset with two modules. Generally, three steps are involved.
Figure 1.
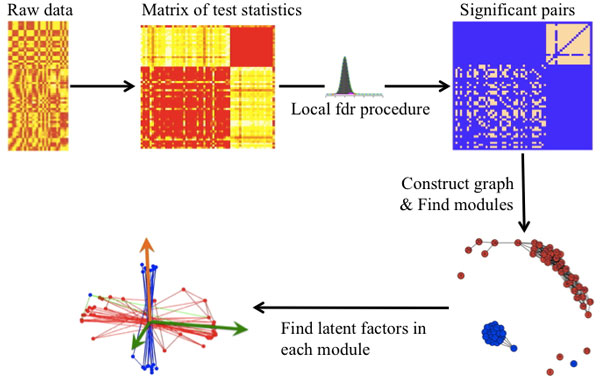
An illustration of the procedure using a toy example dataset. The features belong to two modules. One module is controlled by a single factor, and the other controlled by two factors.
Step 1. Constructing co-expression network based on local fdr. We use the concept of local false discovery rate (lfdr) to establish links between features [19]. First, we compute the correlation coefficients between all pairs of features. Secondly, we transform the correlation coefficients by
so that the distribution of the resulting statistic is close to normal under the null hypothesis that the pair of features are independent [27]. Thirdly, we compute the local false discovery rate using Efron's procedure [19]. The local fdr is a statistical statement of how likely two features are independent given we observe the statistics from all pairs of features. Fourth, if the local fdr value for a pair of features is smaller than a threshold, e.g. 0.2, an edge is established between the two features.
Step 2. Module detection in the co-expression network. We first use a well-established method that detects dense sub-graphs from a sparse graph by short random walks [25]. To fine-tune the results, we conduct an additional community-merging step. For a pair of communities that contains features and within-community connections, and that contains features and within-community connections, we divide the number of between-community connections by the expected number of connections if the communities were indeed one
We then pool all the values computed from all pairs of communities and examine the distribution. Any outlier , defined by a value higher than the median plus four times the difference between the 75th percentile and the median, signifies a community pair that should be merged into a single community.
Step 3. Detecting latent factors from each module. For each module, we first conduct an eigenvalue decomposition of the covariance matrix, and select all eigenvectors that account for at least 5% of the data variance. We then find the projection length of each feature onto each eigenvector , where i denotes the feature and j denotes the eigenvector. The value is the number of features in the module, and is the number of eigenvectors under consideration.
Two eigenvectors are considered "interactive" if the correlation of the projection length of the features onto these two vectors is statistically significant. We initiate a selected vector set with only the first eigenvector. Then from the second eigenvector on, if the eigenvector is interactive with any vector in the selected set, it is added to the selected set. Otherwise we stop the iteration and return the selected vector set as the latent variables of the module. If more than one eigenvector is selected, we rotate them using oblique rotation [26].
Step 4. The overall factor model. After finding a collection of F matrices, we can combine them into an overall factor model with a sparse loading matrix to interpret the gene expression. Let K be the total number of latent factors found, B be the combined factor activity matrix of all the factor scores, L be the loading matrix, and E be the unexplained expression, we have a factor model,
The values in L can be filled in two ways. The first is by performing linear regression of each gene against only the factors of the modules the gene is assigned to. Alternatively, we can perform regularized regression of each gene against all the factors using lasso [28] with BIC (Bayesian information criterion) model selection.
Simulation study
We refer to our method as "Network-based Modular Latent Factor Analysis (nMLSA)". We compared the method with MLSA [11], PCA, ICA [29], factor analysis with oblique rotation [26], gene shaving [9], and sparse principal component analysis (SPCA) [5]. In each simulation, we generated a gene expression dataset with 10 modules. Every module consisted of 100 simulated genes. The number of latent factors controlling the module was randomly selected between 1 and 3. An additional 1000 pure noise genes were generated from the standard Gaussian distribution. We vary the following parameters in the simulations:
(1) The latent factor scores were either independent Gaussian, or randomly chosen from a mixture of four types: Gaussian, sine wave, square wave, and sawtooth wave (Additional file 1 Figure S1). The setting stayed the same for every module in each simulated dataset.
(2) Different levels of within-module loading sparsity, i.e. proportion of zero loadings, were tested. The sparsity of the loading matrix was achieved by drawing samples from the binomial distribution. After the non-zero positions in the loading matrix was determined, for every simulated gene, if there were m controlling factors, we divided [0, 1]into m regions by drawing (m-1) samples from the uniform distribution between 0 and 1. We then used the sizes of the regions as the loadings for the gene. Half of the loadings were then multiplied by -1 to generate negative loadings. The sparsity levels tested were 0%, 30% and 60%. The setting stayed the same for every module in each simulated dataset.
(3) After multiplying the loading matrix and the factor score matrix to generate the simulated expression matrix, Gaussian random noise was added to achieve different signal to noise ratios (values used: 1, 2). The setting stayed the same for every module in each simulated dataset.
The number of samples was set at 100. All possible combinations of the parameters were tested, each repeated 100 times.
To judge the performance of the methods, we used the information of the true hidden factors to group the identified factors. Let K be the combined hidden factor count from all modules in the simulated dataset. We first performed linear regression of every identified factor against each hidden factor group (those controlled the same module), and recorded the multiple R2. The identified factor was assigned to the hidden factor group with which it had the largest R2 value. The K identified factors with the largest R2 values were retained for the next step. Second, we performed linear regression of every true hidden factor against the identified factors assigned to its group, and recorded the multiple R2 as the level of recovery of the true hidden factor. The ideal method should yield multiple R2 values close to one. After repeating the simulation from every parameter setting 100 times, we compared the methods by the distribution of the multiple R2 values.
Results
Simulation results
The simulation results are summarized in Figure 2. Each sub-plot represents a parameter setting. The relative frequencies (10 equal-sized bins between 0 and 1, equivalent to the histogram) of the R2 values are plotted in Figure 1. Different colors represent different methods. The curves are effectively histograms of the multiple R2 values. The curve of a better method should show higher frequency in larger R2 values. In all the scenarios, clearly nMLSA (red) and MLSA (blue) outperformed the other methods.
Figure 2.
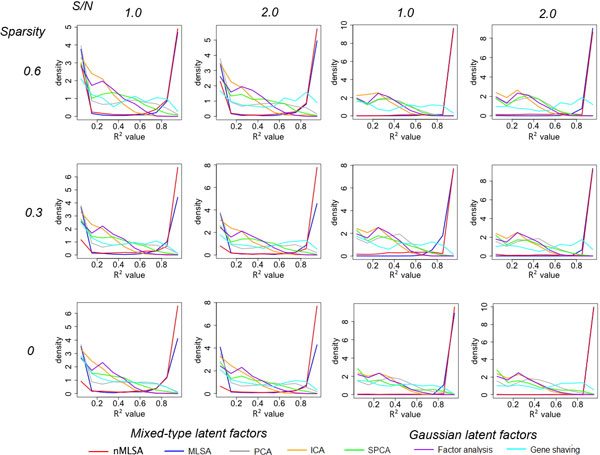
Simulation results from modular latent structure models. In every simulation, 10 modules, each consisting of 100 simulated genes, were generated. The number of latent factors per module was randomly selected between 1 and 3. The latent factors were either independent Gaussian (two right columns), or randomly chosen from a mixture of four types (two left columns). Gaussian random noise was added to achieve different signal to noise ratios (columns), and different levels of within-module sparsity (proportion of zero loadings) were tested (rows). An additional 1000 pure noise genes were generated from the standard Gaussian distribution. Each simulation setting was repeated 100 times. The success of latent factor recovery was evaluated by the R2 values obtained by the regression of each latent factor against the identified factors assigned to the module to which the latent factor belongs. The relative frequencies (10 equal-sized bins between 0 and 1, equivalent to the histogram) of the R2 values are plotted.
When the true signals were Gaussian (Figure 2; two right columns), nMLSA and MLSA yielded similar results. Both methods recovered the hidden factors almost perfectly in all sparsity (rows) and noise (columns) settings. When the true signals were randomly drawn from four different types (Figure 2; two left columns), nMLSA outperformed MLSA. Both methods tend to either fully recover or totally miss a hidden factor, as indicated by spikes at R2 = 1 and R2 = 0. However when the within-module sparsity was moderate to low (30% and 0%), nMLSA showed a roughly 3-fold reduction in the chance to miss hidden factors, and accordingly a much higher chance to faithfully recover the hidden factors.
Real data analysis
The Spellman cell cycle data consists of four time-series, each covering roughly two cell cycles [30]. The array data consists of 73 conditions and 6178 genes. Because of phase differences, the cell cycle-related genes cannot be easily summarized by clusters although many of them exhibit periodic patterns [31]. We applied nMLSA to the cell cycle data as a whole, in order to discover common patterns across the four time series. Our method identified 7 modules containing 10 latent factors in total. The two largest modules each contained two latent factors (Figure 3).
Figure 3.
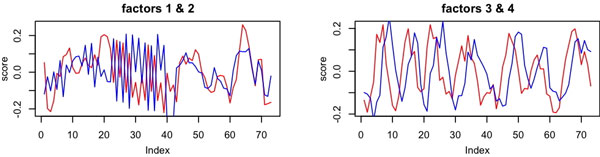
The top four factors from the Spellman dataset form two modules. On the x-axis the four time series are displayed in sequential order.
While MLSA also detected the second module, it failed to detect module 1 found by nMLSA (Figure 3, left panel). Functional analyses using Gene Ontology [32] indicate the module is highly biologically meaningful. Based on hypergeometric tests using the GOStats package [33], genes associated with the first factor of the module strongly over-represent biological processes related to RNA processing and the ribosome, which is central to protein biosynthesis (Table 1). Genes associated with the second factor over-represent biological processes related to protein degradation, transport and localization (Table 2). Protein transport and localization processes are naturally coordinated with protein biosynthesis. Evidences also point to the co-regulation of protein biosynthesis and protein degradation, under normal circumstances and experimental interference [34-36].
Table 1.
Top 25 GO terms overrepresented by genes associated with factor 1.
| GOBPID | Pvalue | Term |
|---|---|---|
| GO:0042254 | 3.27E-75 | ribosome biogenesis |
| GO:0022613 | 2.04E-68 | ribonucleoprotein complex biogenesis |
| GO:0034470 | 6.20E-67 | ncRNA processing |
| GO:0034660 | 1.31E-65 | ncRNA metabolic process |
| GO:0006364 | 1.55E-60 | rRNA processing |
| GO:0016072 | 1.47E-59 | rRNA metabolic process |
| GO:0006396 | 3.00E-51 | RNA processing |
| GO:0071843 | 1.48E-50 | cellular component biogenesis at cellular level |
| GO:0042273 | 5.74E-33 | ribosomal large subunit biogenesis |
| GO:0000460 | 4.04E-30 | maturation of 5.8S rRNA |
| GO:0000466 | 2.05E-29 | maturation of 5.8S rRNA from tricistronic rRNA transcript (SSU-rRNA, 5.8S rRNA, LSU-rRNA) |
| GO:0016070 | 1.33E-26 | RNA metabolic process |
| GO:0042274 | 4.46E-26 | ribosomal small subunit biogenesis |
| GO:0044085 | 3.25E-25 | cellular component biogenesis |
| GO:0030490 | 1.57E-21 | maturation of SSU-rRNA |
| GO:0000462 | 3.11E-21 | maturation of SSU-rRNA from tricistronic rRNA transcript (SSU-rRNA, 5.8S rRNA, LSU-rRNA) |
| GO:0000469 | 3.59E-21 | cleavage involved in rRNA processing |
| GO:0010467 | 7.58E-21 | gene expression |
| GO:0090304 | 2.29E-16 | nucleic acid metabolic process |
| GO:0009451 | 2.58E-16 | RNA modification |
| GO:0000478 | 8.00E-16 | endonucleolytic cleavage involved in rRNA processing |
| GO:0000479 | 8.00E-16 | endonucleolytic cleavage of tricistronic rRNA transcript (SSU-rRNA, 5.8S rRNA, LSU-rRNA) |
| GO:0006139 | 1.09E-15 | nucleobase-containing compound metabolic process |
| GO:0000447 | 1.37E-15 | endonucleolytic cleavage in ITS1 to separate SSU-rRNA from 5.8S rRNA and LSU-rRNA from tricistronic rRNA transcript (SSU-rRNA, 5.8S rRNA, LSU-rRNA) |
| GO:0000472 | 1.42E-14 | endonucleolytic cleavage to generate mature 5'-end of SSU-rRNA from (SSU-rRNA, 5.8S rRNA, LSU-rRNA) |
Table 2.
Top 25 GO terms overrepresented by genes associated with factor 2.
| GOBPID | Pvalue | Term |
|---|---|---|
| GO:0006511 | 1.99E-09 | ubiquitin-dependent protein catabolic process |
| GO:0019941 | 2.44E-09 | modification-dependent protein catabolic process |
| GO:0044257 | 2.65E-09 | cellular protein catabolic process |
| GO:0051603 | 3.79E-09 | proteolysis involved in cellular protein catabolic process |
| GO:0043632 | 4.21E-09 | modification-dependent macromolecule catabolic process |
| GO:0030163 | 7.26E-09 | protein catabolic process |
| GO:0010499 | 2.24E-08 | proteasomal ubiquitin-independent protein catabolic process |
| GO:0044265 | 9.94E-08 | cellular macromolecule catabolic process |
| GO:0007005 | 1.38E-07 | mitochondrion organization |
| GO:0043623 | 2.59E-07 | cellular protein complex assembly |
| GO:0043248 | 3.56E-07 | proteasome assembly |
| GO:0009057 | 5.19E-07 | macromolecule catabolic process |
| GO:0006508 | 5.25E-07 | proteolysis |
| GO:0071842 | 3.02E-06 | cellular component organization at cellular level |
| GO:0015031 | 4.89E-06 | protein transport |
| GO:0008104 | 6.28E-06 | protein localization |
| GO:0043161 | 6.81E-06 | proteasomal ubiquitin-dependent protein catabolic process |
| GO:0045184 | 7.86E-06 | establishment of protein localization |
| GO:0010498 | 1.01E-05 | proteasomal protein catabolic process |
| GO:0006461 | 1.35E-05 | protein complex assembly |
| GO:0034613 | 1.41E-05 | cellular protein localization |
| GO:0070271 | 2.51E-05 | protein complex biogenesis |
| GO:0070727 | 2.56E-05 | cellular macromolecule localization |
| GO:0034621 | 3.00E-05 | cellular macromolecular complex subunit organization |
| GO:0009987 | 3.93E-05 | cellular process |
The second module is even more intuitive biologically. The factor scores showed that the second module was governed by two periodic latent factors with similar periodicity but different phases (Figure 3, right). Genes of this module showed clear periodic behavior with different phase shifts (Figure 4), which is consistent with the biological knowledge that cell-cycle genes are activated at different phases of the cell cycle [30]. We analyzed the functionalities of the genes associated with each factor using gene ontology (GO). It was clear that cell cycle-related biological processes dominated the list of top processes overrepresented by genes associated with either latent factors (Tables 3 &4). Other methods used in the simulations, except MLSA, could not group cell cycle genes with different phase shift into a single module.
Figure 4.
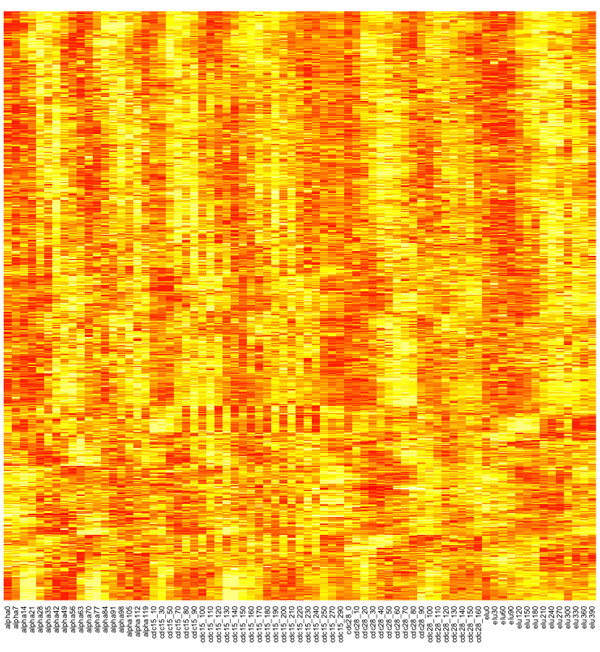
Expression levels of genes involved in module 2. Genes are hierarchically clustered. The four time series are displayed in sequential order.
Table 3.
Top 25 GO terms overrepresented by genes associated with factor 3.
| GOBPID | Pvalue | Term |
|---|---|---|
| GO:0000278 | 4.02E-19 | mitotic cell cycle |
| GO:0022402 | 4.08E-17 | cell cycle process |
| GO:0007049 | 6.05E-17 | cell cycle |
| GO:0022403 | 5.72E-15 | cell cycle phase |
| GO:0007017 | 8.75E-15 | microtubule-based process |
| GO:0048285 | 2.31E-13 | organelle fission |
| GO:0000280 | 5.08E-13 | nuclear division |
| GO:0051301 | 8.45E-13 | cell division |
| GO:0000226 | 8.50E-13 | microtubule cytoskeleton organization |
| GO:0000087 | 9.22E-13 | M phase of mitotic cell cycle |
| GO:0007067 | 1.39E-12 | mitosis |
| GO:0000279 | 2.71E-09 | M phase |
| GO:0007010 | 1.75E-08 | cytoskeleton organization |
| GO:0007059 | 2.33E-08 | chromosome segregation |
| GO:0030472 | 3.11E-08 | mitotic spindle organization in nucleus |
| GO:0032886 | 3.46E-08 | regulation of microtubule-based process |
| GO:0070507 | 3.46E-08 | regulation of microtubule cytoskeleton organization |
| GO:0016043 | 5.99E-08 | cellular component organization |
| GO:0007051 | 7.48E-08 | spindle organization |
| GO:0051329 | 7.69E-08 | interphase of mitotic cell cycle |
| GO:0007052 | 1.09E-07 | mitotic spindle organization |
| GO:0051325 | 1.12E-07 | interphase |
| GO:0006928 | 1.15E-07 | cellular component movement |
| GO:0007018 | 2.33E-07 | microtubule-based movement |
| GO:0010564 | 6.27E-07 | regulation of cell cycle process |
Table 4.
Top 25 GO terms overrepresented by genes associated with factor 4.
| GOBPID | Pvalue | Term |
|---|---|---|
| GO:0007049 | 5.54E-08 | cell cycle |
| GO:0051301 | 8.21E-08 | cell division |
| GO:0000278 | 7.99E-07 | mitotic cell cycle |
| GO:0000087 | 1.12E-06 | M phase of mitotic cell cycle |
| GO:0022402 | 1.57E-06 | cell cycle process |
| GO:0022403 | 6.92E-06 | cell cycle phase |
| GO:0048285 | 2.16E-05 | organelle fission |
| GO:0000280 | 3.03E-05 | nuclear division |
| GO:0010458 | 3.55E-05 | exit from mitosis |
| GO:0000910 | 3.70E-05 | Cytokinesis |
| GO:0000279 | 4.43E-05 | M phase |
| GO:0007067 | 7.91E-05 | Mitosis |
| GO:0033205 | 0.000523865 | cell cycle cytokinesis |
| GO:0032506 | 0.000572574 | cytokinetic process |
| GO:0010970 | 0.001012686 | microtubule-based transport |
| GO:0030473 | 0.001012686 | nuclear migration along microtubule |
| GO:0030705 | 0.001012686 | cytoskeleton-dependent intracellular transport |
| GO:0072384 | 0.001012686 | organelle transport along microtubule |
| GO:0000114 | 0.00177365 | regulation of transcription involved in G1 phase of mitotic cell cycle |
| GO:0046459 | 0.00185316 | short-chain fatty acid metabolic process |
| GO:0007018 | 0.002062712 | microtubule-based movement |
| GO:0016575 | 0.002062712 | histone deacetylation |
| GO:0032392 | 0.002399826 | DNA geometric change |
| GO:0010696 | 0.002438214 | positive regulation of spindle pole body separation |
| GO:0007097 | 0.002544351 | nuclear migration |
Discussions
In this study, we developed the network-based modular latent structure analysis (nMLSA). It is aimed at detecting expression modules and latent factors controlling the modules, the same goal as the original MLSA [11]. Compared to MLSA, the new method is based on a totally different setup, and is substantially advantageous. Firstly, the number of tuning parameters and heuristic choices is substantially less compared to MLSA. Secondly, the method is much more intuitive to understand. Thirdly, it is more flexible. As an example, one can easily limit the gene relations to positive correlations and ignore negative correlations using nMLSA, while MLSA has to take both positive and negative correlations. Fourth, nMLSA can be adapted for nonlinearly associated modules if a nonlinear association measure is used in the co-expression network building, while MLSA is limited to linear relations. In the nonlinear case, it is difficult to define latent factors. The challenge is subject to our future studies.
Instead of using hard cutoffs, nMLSA utilizes the concept of local false discovery rate (lfdr). As different datasets exhibit different levels of baseline correlation [37], using hard cutoffs on correlations may result in unsatisfactory results. Using local false discovery rate procedures that are flexible in the null distribution estimation, nMLSA is naturally adaptive to the characteristics of the data. Given the nMLSA procedure relies on existing network community detection algorithms, it is admitted that the performance of the method relies on the choice of the community detection algorithm. The research field of community detection is mature and a number of good methods are available. Thus it is not difficult to tune the method to achieve good performance.
Conclusions
In summary, the new network-based method nMLSA is more effective than existing methods in recovering biologically meaningful latent variables and latent variable groups. The method can potentially be extended to detect nonlinearly associated modules if a nonlinear association measure is used to build the network.
Competing interests
None.
Authors' contributions
TY developed the computational method, conducted simulations. TY and YB conducted real data analyses, interpreted the results, and drafted the manuscript.
Supplementary Material
Figure S1. The four types of input signal from which the data were simulated.
Contributor Information
Tianwei Yu, Email: tianwei.yu@emory.edu.
Yun Bai, Email: yunba@pcom.edu.
Acknowledgements
This work was partially supported by NIH grants P20HL113451, P30AI50409 and U19AI090023. The funding source to publish the publication cost is NIH grant U19AI090023.
This article has been published as part of BMC Bioinformatics Volume 15 Supplement 13, 2014: Selected articles from the 9th International Symposium on Bioinformatics Research and Applications (ISBRA'13): Bioinformatics. The full contents of the supplement are available online at http://www.biomedcentral.com/bmcbioinformatics/supplements/15/S13.
References
- Wagner GP, Pavlicev M, Cheverud JM. The road to modularity. Nat Rev Genet. 2007;8:(12):921–931. doi: 10.1038/nrg2267. [DOI] [PubMed] [Google Scholar]
- Yu T, Li KC. Inference of transcriptional regulatory network by two-stage constrained space factor analysis. Bioinformatics. 2005;21(21):4033–4038. doi: 10.1093/bioinformatics/bti656. [DOI] [PubMed] [Google Scholar]
- Liao JC, Boscolo R, Yang YL, Tran LM, Sabatti C, Roychowdhury VP. Network component analysis: reconstruction of regulatory signals in biological systems. Proc Natl Acad Sci USA. 2003;100(26):15522–15527. doi: 10.1073/pnas.2136632100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TW. Independent component analysis : theory and applications. Boston: Kluwer Academic Publishers; 1998. [Google Scholar]
- Zou H, Hastie T, Tibshirani R. Sparse principal component analysis. Journal of Computational and Graphical Statistics. 2006;15(2):265–286. doi: 10.1198/106186006X113430. [DOI] [Google Scholar]
- Carvalho CM, Chang J, Lucas JE, Nevins JR, Wang Q, West M. High-Dimensional Sparse Factor Modeling: Applications in Gene Expression Genomics. Journal of the American Statistical Association. 2008;103(484):1438–1456. doi: 10.1198/016214508000000869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moloshok TD, Klevecz RR, Grant JD, Manion FJ, Speier WFt, Ochs MF. Application of Bayesian decomposition for analysing microarray data. Bioinformatics. 2002;18(4):566–575. doi: 10.1093/bioinformatics/18.4.566. [DOI] [PubMed] [Google Scholar]
- Gan G, Ma C, Wu J. Data clustering : theory, algorithms, and applications. Philadelphia, Pa. Alexandria, Va.: SIAM. American Statistical Association; 2007. [Google Scholar]
- Hastie T, Tibshirani R, Eisen MB, Alizadeh A, Levy R, Staudt L, Chan WC, Botstein D, Brown P. 'Gene shaving' as a method for identifying distinct sets of genes with similar expression patterns. Genome Biol. 2000;1(2):RESEARCH0003. doi: 10.1186/gb-2000-1-2-research0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan S, Li KC. Context-dependent clustering for dynamic cellular state modeling of microarray gene expression. Bioinformatics. 2007;23(22):3039–3047. doi: 10.1093/bioinformatics/btm457. [DOI] [PubMed] [Google Scholar]
- Yu T. An exploratory data analysis method to reveal modular latent structures in high-throughput data. BMC Bioinformatics. 2010;11:440. doi: 10.1186/1471-2105-11-440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Q, Lemmens K, Sanchez-Rodreiquez A, Thijs IM, Meysman P, Sun H, Fierro AC, Engelen K, Marchal K. Directed module detection in a large-scale expression compendium. Methods Mol Biol. 2012;804:131–165. doi: 10.1007/978-1-61779-361-5_8. [DOI] [PubMed] [Google Scholar]
- Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A, Horvath S. Network module detection: Affinity search technique with the multi-node topological overlap measure. BMC Res Notes. 2009;2:142. doi: 10.1186/1756-0500-2-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J, Wang P, Zhou N, Zhu J. Partial Correlation Estimation by Joint Sparse Regression Models. J Am Stat Assoc. 2009;104(486):735–746. doi: 10.1198/jasa.2009.0126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer J, Strimmer K. An empirical Bayes approach to inferring large-scale gene association networks. Bioinformatics. 2005;21(6):754–764. doi: 10.1093/bioinformatics/bti062. [DOI] [PubMed] [Google Scholar]
- Margolin AA, Nemenman I, Basso K, Wiggins C, Stolovitzky G, Dalla Favera R, Califano A. ARACNE: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinformatics. 2006;7(Suppl 1):S7. doi: 10.1186/1471-2105-7-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis B, Wong W. Learning causal bayesian network structures from experimental data. Journal of the American Statistical Association. 2008;103:778–789. doi: 10.1198/016214508000000193. [DOI] [Google Scholar]
- Efron B, Tibshirani R. Empirical Bayes methods and false discovery rates for microarrays. Genet Epidemiol. 2002;23(1):70–86. doi: 10.1002/gepi.1124. [DOI] [PubMed] [Google Scholar]
- Strimmer K. A unified approach to false discovery rate estimation. BMC Bioinformatics. 2008;9:303. doi: 10.1186/1471-2105-9-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guedj M, Robin S, Celisse A, Nuel G. Kerfdr: a semi-parametric kernel-based approach to local false discovery rate estimation. BMC Bioinformatics. 2009;10:84. doi: 10.1186/1471-2105-10-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball B, Karrer B, Newman ME. Efficient and principled method for detecting communities in networks. Physical review E, Statistical, nonlinear, and soft matter physics. 2011;84(3 Pt 2):036103. doi: 10.1103/PhysRevE.84.036103. [DOI] [PubMed] [Google Scholar]
- Hofman JM, Wiggins CH. Bayesian approach to network modularity. Physical review letters. 2008;100(25):258701. doi: 10.1103/PhysRevLett.100.258701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman ME. Spectral methods for community detection and graph partitioning. Physical review E, Statistical, nonlinear, and soft matter physics. 2013;88(4):042822. doi: 10.1103/PhysRevE.88.042822. [DOI] [PubMed] [Google Scholar]
- Pons P, Latapy M. Computing Communities in Large Networks Using Random Walks. Lecture Notes in Computer Science. 2005;3733:284–293. doi: 10.1007/11569596_31. [DOI] [Google Scholar]
- Bernaards CA, Jennrich RI. Gradient Projection Algorithms and Software for Arbitrary Rotation Criteria in Factor Analysis. Educational and Psychological Measurement. 2005;65:676–696. doi: 10.1177/0013164404272507. [DOI] [Google Scholar]
- Rahman NA. A course in theoretical statistics for sixth forms, technical colleges, colleges of education, universities. London,: Griffin; 1968. [Google Scholar]
- Efron B, Hastie T, Johnstone I, Tibshirani R. Least Angle Regression. Annals of Statistics. 2003;32(2):407–499. [Google Scholar]
- Hastie T, Tibshirani R, Friedman JH. The elements of statistical learning : data mining, inference, and prediction. 2. New York, NY: Springer; 2009. [Google Scholar]
- Spellman PT, Sherlock G, Zhang MQ, Iyer VR, Anders K, Eisen MB, Brown PO, Botstein D, Futcher B. Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol Biol Cell. 1998;9(12):3273–3297. doi: 10.1091/mbc.9.12.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li KC, Yan M, Yuan SS. A simple statistical model for depicting the cdc15-synchronized yeast cell-cycle regulated gene expression data. Stat Sinica. 2002;12(1):141–158. [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25(1):25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcon S, Gentleman R. Using GOstats to test gene lists for GO term association. Bioinformatics. 2007;23(2):257–258. doi: 10.1093/bioinformatics/btl567. [DOI] [PubMed] [Google Scholar]
- Dai CL, Shi J, Chen Y, Iqbal K, Liu F, Gong CX. Inhibition of protein synthesis alters protein degradation through activation of protein kinase B (AKT) J Biol Chem. 2013. [DOI] [PMC free article] [PubMed]
- Hemmerlin A. Post-translational events and modifications regulating plant enzymes involved in isoprenoid precursor biosynthesis. Plant Sci. 2013;203-204:41–54. doi: 10.1016/j.plantsci.2012.12.008. [DOI] [PubMed] [Google Scholar]
- Stein H, Honig A, Miller G, Erster O, Eilenberg H, Csonka LN, Szabados L, Koncz C, Zilberstein A. Elevation of free proline and proline-rich protein levels by simultaneous manipulations of proline biosynthesis and degradation in plants. Plant Sci. 2011;181(2):140–150. doi: 10.1016/j.plantsci.2011.04.013. [DOI] [PubMed] [Google Scholar]
- Yu T, Zhao Y, Shen S. AAPL: Assessing Association between P-value Lists. Statistical analysis and data mining. 2013;6(2):144–155. doi: 10.1002/sam.11180. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. The four types of input signal from which the data were simulated.