

Abstract
Candida albicans is a commensal in heathy people but has the potential to become an opportunistic pathogen and is responsible for half of all clinical infections in immunocompromised patients. Central to understanding C. albicans behavior is the white-opaque phenotypic switch, in which cells can undergo an epigenetic transition between the white state and the opaque state. The phenotypic switch regulates multiple properties, including biofilm formation, virulence, mating, and fungus-host interactions. Switching between the white and opaque states is associated with many external stimuli, such as oxidative stress, pH, and N-acetylglucosamine, and is directly regulated by the Wor1 transcriptional circuit. The Hog1 stress-activated protein kinase (SAPK) pathway is recognized as the main pathway for adapting to environmental stress in C. albicans. In this work, we first show that loss of the HOG1 gene in a/a and α/α cells, but not a/α cells, results in 100% white-to-opaque switching when cells are grown on synthetic medium, indicating that switching is repressed by the a1/α2 heterodimer that represses WOR1 gene expression. Indeed, switching in the hog1Δ strain was dependent on the presence of WOR1, as a hog1Δ wor1Δ strain did not show switching to the opaque state. Deletion of PBS2 and SSK2 also resulted in C. albicans cells switching from white to opaque with 100% efficiency, indicating that the entire Hog1 SAPK pathway is involved in regulating this unique phenotypic transition. Interestingly, all Hog1 pathway mutants also caused defects in shmoo formation and mating efficiencies. Overall, this work reveals a novel role for the Hog1 SAPK pathway in regulating white-opaque switching and sexual behavior in C. albicans.
INTRODUCTION
Candida albicans is a diploid hemiascomycete yeast that is commonly found in the human gastrointestinal tract (1, 2). However, C. albicans can become a pathogen that attacks multiple organs in the body, including kidney and brain, resulting in life-threatening systemic infections with high mortality rates (3). Phenotypic transitions represent an important mechanism for the striking ability of Candida cells to colonize a wide variety of habitats with diverse properties. For example, under conditions of various environmental signals, the yeast forms of C. albicans can switch to filamentous cells (4–8). The reversible yeast-hypha transition regulated by a core set of transcription factors that mediate its developmental program is complex (7, 9–13). Hyphal formation also plays a major role in biofilm formation, which is closely linked with the propensity to cause infection and is associated with antifungal resistance (14–16).
In addition to the yeast-hypha transition, an important morphological change in C. albicans is the white-opaque switch (17). This epigenetic transition between white and opaque states regulates many aspects of C. albicans biology. In particular, opaque cells are mating competent (18, 19), whereas white cells do not mate but can generate biofilms in response to pheromones (20, 21). The regulation of mating between C. albicans a/a and α/α opaque cells occurs via pheromone signaling between cells of opposite mating types (18, 19, 22, 23). Pheromone responses induce a mitogen-activated protein kinase (MAPK) signal transduction cascade leading to the formation of mating projections (shmoos) and subsequent cell-cell conjugation (21, 24). Furthermore, opaque cells are less susceptible to phagocytosis than white cells due to the lack of secretion of a chemoattractant for leukocytes (25–27), indicating that switching plays a role in host-fungus interactions. Thus, the phenotypic plasticity of C. albicans in response to different host niches is important for its ability to colonize and infect the host.
C. albicans cells typically exist in the default white state, where they are round and form bright, shiny colonies, but can stochastically switch to the opaque state, where cells are more elongated and form darker, flatter colonies (17). The white-opaque switch is regulated by the master regulator Wor1, which acts in concert with several other transcription factors as part of a transcriptional network (28, 29). This network is also controlled by the a1/α2 complex which is produced in a/α heterozygous cells and which represses opaque-cell formation (30), although a recent study showed that some clinical a/α cells have the ability to switch to opaque under certain culture conditions (31). Several environmental factors, including CO2, temperature, oxidative stress, N-acetylglucosamine (NAG), and growth rate, also regulate white-opaque switching (32–34), suggesting that the transition between white and opaque cells is extremely sensitive to external signals and dependent on the expression levels of the master regulator Wor1.
In eukaryotic cells, MAP kinases are responsible for transducing a variety of extracellular signals for growth and differentiation (35, 36). One of them, the high-osmolarity glycerol (Hog1) pathway, is not only responsible for the cellular response to osmotic stress but is also required for responses to UV radiation, temperature, and oxidative stress in the budding yeast (37–39). This pathway is known as the Hog1 stress-activated protein kinase (SAPK) pathway or the Hog1 mitogen-activated protein kinase (MAPK) pathway. Osmosensing triggers Hog1 activation through the stimulation of a MAPK kinase kinase (MAPKKK), Ssk2, and a MAPK kinase (MAPKK), Pbs2 (35, 39, 40).
In regard to white-opaque switching, several stresses have been found to promote the switch from white to opaque (32–34). We therefore hypothesized that the Hog1 SAPK pathway of C. albicans could be involved in regulating this phenotypic transition. In this study, the results of deletion of HOG1, PBS2, and SSK2 genes in C. albicans MTLa/a, MTLα/α, and MTLa/α strains were compared. Mutants in all three strain backgrounds showed high sensitivity to osmotic and oxidative stress, indicating that the roles of the Hog1 SAPK pathway are similar in different cell types. Interestingly, homozygous MTLa/a and MTLα/α Hog1 pathway mutants exhibit 100% switching from white to opaque but also display shorter shmoos and have reduced mating efficiencies. Overall, our data therefore reveal a novel role for the Hog1 SAPK pathway in white-opaque switching and indicate a further route for the integration of environmental signals into the white-opaque regulatory circuit.
MATERIALS AND METHODS
Media and reagents.
Yeast extract-peptone-dextrose (YPD), Spider, and synthetic complete dextrose (SCD) media used in experiments were prepared as described previously with slight modifications (38, 41). YPD medium, composed of 2% (wt/vol) peptone, 1% (wt/vol) yeast extract, and 2% (wt/vol) glucose, was used for cell growth experiments. Nourseothricin-resistant strains were selected on YPD medium supplemented with 0.2 mg/ml norseothricin (Werner BioAgents). SCD consisted of 0.7% (wt/vol) yeast nitrogen base without amino acids, 0.17% complete amino acids powder, and 2% (wt/vol) glucose. Synthetic complete (SC) medium was formulated as SCD medium without the addition of glucose. Preparation of Lee's N-acetylglucosamine (Lee's NAG), which contained 1.25% N-acetylglucosamine (Alfa Aesar), followed an established protocol (34). SCD, SC, and Lee's NAG media were used for white-opaque phenotypic transition assays. Spider medium (pH 7.2), containing 1% (wt/vol) mannitol, 1% (wt/vol) nutrient broth, and 0.4% (wt/vol) dipotassium phosphate (Showa Chemical Industry Co.), was used for opaque cell maintenance. Leu−, Arg−, and Arg− Leu− media were used to perform a quantitative mating assay. Agar was added to reach a final concentration of 2% (wt/vol) to make solid plates. All chemicals were purchased from Sigma-Aldrich Chemical Co., unless otherwise stated.
Plasmid and strain constructions.
Strains and primers used in this study are listed in Table 1 and Table 2, respectively.
TABLE 1.
Strains used in this studya
| Strain | MTL type | Genotype | White or opaque | Reference or source |
|---|---|---|---|---|
| SC5314 | a/α | Wild type | White | 22 |
| RBY717 | a/a | ura3::imm434/URA3 iro1::imm434/IRO1 | White | 22 |
| RBY722 | α/α | ura3::imm434/URA3 iro1::imm434/IRO1 | White | 22 |
| YL8 | a/a | hog1/hog1 | White | This study |
| YL9 | a/a | hog1/hog1 | White | This study |
| YL11 | a/a | hog1/hog1::HOG1 | White | This study |
| YL12 | a/a | hog1/hog1::HOG1 | White | This study |
| YL26 | α/α | hog1/hog1 | White | This study |
| YL27 | α/α | hog1/hog1 | White | This study |
| YL31 | a/α | hog1/hog1 | White | This study |
| YL34 | a/α | hog1/hog1 | White | This study |
| YL35 | α/α | hog1/hog1::HOG1 | White | This study |
| YL36 | α/α | hog1/hog1::HOG1 | White | This study |
| YL38 | a/α | hog1/hog1::HOG1 | White | This study |
| YL39 | a/α | hog1/hog1::HOG1 | White | This study |
| YL45 | a/a | ura3::imm434/URA3 iro1::imm434/IRO1 | Opaque | This study |
| YL46 | a/a | ura3::imm434/URA3 iro1::imm434/IRO1 | Opaque | This study |
| YL47 | α/α | ura3::imm434/URA3 iro1::imm434/IRO1 | Opaque | This study |
| YL48 | α/α | ura3::imm434/URA3 iro1::imm434/IRO1 | Opaque | This study |
| YL97 | a/a | arg4/arg4 | White | This study |
| YL98 | a/a | arg4/arg4 | White | This study |
| YL133 | α/α | leu2/leu2 | White | This study |
| YL134 | α/α | leu2/leu2 | White | This study |
| YL135 | a/a | wor1/wor1 | White | This study |
| YL136 | a/a | hog1/hog1 wor1/wor1 | White | This study |
| YL191 | a/a | hog1/hog1 leu2/leu2 | White | This study |
| YL192 | a/a | hog1/hog1 leu2/leu2 | White | This study |
| YL193 | a/a | ssk2/ssk2 | White | This study |
| YL194 | a/a | ssk2/ssk2 | White | This study |
| YL195 | a/a | pbs2/pbs2 | White | This study |
| YL196 | a/a | pbs2/pbs2 | White | This study |
| YL226 | a/a | hog1/hog1::HOG1 leu2/leu2 | White | This study |
| YL227 | a/a | hog1/hog1::HOG1 leu2/leu2 | White | This study |
| YL234 | a/a | hog1/hog1 leu2/leu2 | Opaque | This study |
| YL235 | a/a | hog1/hog1 leu2/leu2 | Opaque | This study |
| YL238 | a/a | arg4/arg4 | Opaque | This study |
| YL239 | a/a | arg4/arg4 | Opaque | This study |
| YL259 | a/a | hog1/hog1 | Opaque | This study |
| YL260 | a/a | hog1/hog1 | Opaque | This study |
| YL267 | α/α | hog1/hog1 | Opaque | This study |
| YL268 | α/α | hog1/hog1 | Opaque | This study |
| YL271 | a/a | hog1/hog1::HOG1 leu2/leu2 | Opaque | This study |
| YL272 | a/a | hog1/hog1::HOG1 leu2/leu2 | Opaque | This study |
| YL273 | α/α | leu2/leu2 | Opaque | This study |
| YL274 | α/α | leu2/leu2 | Opaque | This study |
| YL275 | a/a | OP4/OP4::OP4p-GFP | White | This study |
| YL276 | a/a | OP4/OP4::OP4p-GFP | White | This study |
| YL279 | a/a | hog1/hog1 OP4/OP4::OP4p-GFP | White | This study |
| YL280 | a/a | hog1/hog1 OP4/OP4::OP4p-GFP | White | This study |
| YL298 | a/a | hog1/hog1 OP4/OP4::OP4p-GFP | Opaque | This study |
| YL299 | a/a | hog1/hog1 OP4/OP4::OP4p-GFP | Opaque | This study |
| YL304 | α/α | hog1/hog1 arg4/arg4 | White | This study |
| YL305 | α/α | hog1/hog1 arg4/arg4 | White | This study |
| YL328 | α/α | hog1/hog1::HOG1 arg4/arg4 | White | This study |
| YL329 | α/α | hog1/hog1::HOG1 arg4/arg4 | White | This study |
| YL330 | α/α | hog1/hog1 arg4/arg4 | Opaque | This study |
| YL331 | α/α | hog1/hog1 arg4/arg4 | Opaque | This study |
| YL332 | α/α | hog1/hog1::HOG1 arg4/arg4 | Opaque | This study |
| YL333 | α/α | hog1/hog1::HOG1 arg4/arg4 | Opaque | This study |
| YL400 | α/α | pbs2/pbs2 | White | This study |
| YL401 | α/α | pbs2/pbs2 | White | This study |
| YL402 | α/α | ssk2/ssk2 | White | This study |
| YL403 | α/α | ssk2/ssk2 | White | This study |
| YL404 | a/α | pbs2/pbs2 | White | This study |
| YL405 | a/α | pbs2/pbs2 | White | This study |
| YL442 | a/α | ssk2/ssk2 | White | This study |
| YL443 | a/α | ssk2/ssk2 | White | This study |
| YL458 | α/α | ssk2/ssk2 leu2/leu2 | Opaque | This study |
| YL459 | α/α | ssk2/ssk2 leu2/leu2 | Opaque | This study |
| YL469 | a/a | pbs2/pbs2 arg4/arg4 | Opaque | This study |
| YL470 | a/a | pbs2/pbs2 arg4/arg4 | Opaque | This study |
| YL484 | α/α | pbs2/pbs2 leu2/leu2 | Opaque | This study |
| YL485 | α/α | pbs2/pbs2 leu2/leu2 | Opaque | This study |
| YL498 | a/a | ssk2/ssk2 arg4/arg4 | Opaque | This study |
| YL499 | a/a | ssk2/ssk2 arg4/arg4 | Opaque | This study |
| YL524 | a/a | pbs2/pbs2 arg4/arg4 | White | This study |
| YL525 | a/a | pbs2/pbs2 arg4/arg4 | White | This study |
| YL526 | α/α | pbs2/pbs2 leu2/leu2 | White | This study |
| YL527 | α/α | pbs2/pbs2 leu2/leu2 | White | This study |
| YL528 | a/a | ssk2/ssk2 arg4/arg4 | White | This study |
| YL529 | a/a | ssk2/ssk2 arg4/arg4 | White | This study |
| YL530 | α/α | ssk2/ssk2 leu2/leu2 | White | This study |
| YL531 | α/α | ssk2/ssk2 leu2/leu2 | White | This study |
| YL532 | a/a | ssk2/ssk2 | Opaque | This study |
| YL533 | a/a | ssk2/ssk2 | Opaque | This study |
| YL534 | a/a | pbs2/pbs2 | Opaque | This study |
| YL535 | a/a | pbs2/pbs2 | Opaque | This study |
| YL536 | α/α | pbs2/pbs2 | Opaque | This study |
| YL537 | α/α | pbs2/pbs2 | Opaque | This study |
| YL538 | α/α | ssk2/ssk2 | Opaque | This study |
| YL539 | α/α | ssk2/ssk2 | Opaque | This study |
| YL899 | a/a | OP4/OP4::OP4p-GFP | Opaque | This study |
| YL900 | a/a | OP4/OP4::OP4p-GFP | Opaque | This study |
All of the strains were SC5314 derived.
TABLE 2.
Oligonucleotides used in this study
| Oligonucleotide | Sequence (5′–3′) |
|---|---|
| CH1 | GGAGCGGGGCCCCGAGCCGAGAACAGACTTTGAAC |
| CH2 | GGAGCGCTCGAGTGTTGTTGTTATTATTCCACGGTG |
| CH3 | GGAGCGCCGCGGTCTTCAAAAATACAAGCTAGCAATTATAG |
| CH4 | GGAGCGGAGCTCTTGCCCACCAAAGTGTTTGG |
| 2 | CCTTCAATTGCATCGTAAGTACC |
| 3 | GAAGATGACTCAGGTCATGC |
| 4 | GTGGTCAATGGAGCTGATAC |
| 5 | ACATGTGGTCGCCCAACTCC |
| 10 | GGAGCGGGGCCCCACACATGTTTGATTCGATTCTTG |
| 11 | GGAGCGCTCGAGAGCTAATGTTGCTGGAGACGAC |
| 70 | GGAGCGGGTACCCCCTTCCCCCTGTTAACTTTG |
| 71 | GGAGCGCTCGAGGGGCCCTTTTCCAATTGAAC |
| 72 | GGAGCGCCGCGGGAGGGAAACCAGAGCTTATTCC |
| 73 | GGAGCGGAGCTCAAGACTCCTAAACCGGAATTACAATT |
| 78 | GGAGCGGGGCCCTGGTCCAATCTGGTTGGGAG |
| 79 | GGAGCGCTCGAGCGCTGTCCCTCTCTCTCGC |
| 80 | GGAGCGCCGCGGGCCCAGGTTATTGGGCTGTA |
| 81 | GGAGCGGAGCTCCTATGCAAATGGAGGTTGGGAA |
| 114 | GGAGCGGGGCCCGACAGGAATCACGTCATCGTTG |
| 115 | GGAGCGCTCGAGTGTTGGGATGGCCAGCG |
| 116 | GGAGCGGCGGCCGCCTTCCATCACAACACACAACACG |
| 117 | GGAGCGCCGCGGGGAATCCATGAACTACCTGCTTC |
| 122 | GGAGCGGGGCCCGTTGCTGGGTTTGGTGAAGC |
| 123 | GGAGCGCTCGAGTGAAAAAAGGGCACCCCAG |
| 124 | GGAGCGCCGCGGGCCTGGTAGATGTAATGGTGGC |
| 125 | GGAGCGGAGCTCGAACAAACGAACCACCTCGC |
| 191 | ATGAGTAAGGGAGAAGAACTTTTCACT |
| 192 | GGAGCGCTCGAGTTATTTGTATAGTTCATCCATGCCAT |
| 193 | GGAGCGGGGCCCGATAATGCAAGAGCTCGCCACCAC |
| 194 | AGTGAAAAGTTCTTCTCCCTTACTCATTGTAAATTATTTATATTTGTATGTGTGTAGGA |
To generate hog1Δ strains, 5′ and 3′ flanking regions of HOG1 were amplified using primer pair CH1 and CH2 and primer pair CH3 and CH4. The 5′ and 3′ PCR products were digested with ApaI and XhoI and with SacII and SacI and cloned into plasmid pSFS2A (42) to generate pSFS-HOG1 knockout (KO). This plasmid was linearized with ApaI and SacI and then transformed into SC5314 (a/α), RBY717 (a/a), and RBY722 (α/α) to generate strains YL31, YL34, YL8, YL9, YL26, and YL27. To construct the HOG1 complementary plasmid, primers 10 and 11 were used to amplify the promoter and open reading frame (ORF) of the HOG1 gene. The PCR product was digested with ApaI and XhoI and cloned into pSFS2A to construct pSFS-HOG1 AB. This construct was linearized with BsaBI and transformed into YL31, YL34, YL8, YL9, YL26, and YL27 to create the YL38, YL39, YL11, YL12, YL35, and YL36 strains.
For the wor1Δ mutant and hog1Δ wor1Δ double mutant, the pSFS-WOR1 KO plasmid (32) was cut with KpnI and SacI and transformed into RBY717 and YL8 to create strains YL135 and YL136.
To create the ssk2Δ mutants, 5′ and 3′ flanking regions of the SSK2 gene were amplified using primer pair 70 and 71 and primer pair 72 and 73. The 5′ and 3′ PCR products were digested with KpnI and ApaI and with SacII and SacI and cloned into pSFS2A to generate pSFS-SSK2 KO. KpnI and SacI were used to linearize these constructs. Linearized pSFS-SSK2 KO plasmid was transformed into SC5314, RBY717, and RBY722 to generate the YL442, YL443, YL193, YL194, YL402, and YL403 strains.
For pbs2Δ strains, 5′ and 3′ flanking regions of the PBS2 gene were amplified using primer pair 78 and 79 and primer pair 80 and 81. The 5′ and 3′ PCR products were digested with ApaI and XhoI and with SacII and SacI. The digested PCR products were cloned into plasmid pSFS2A to generate the pSFS-PBS2 KO plasmid. The constructed plasmid was linearized with NheI and SacI and transformed into SC5314, RBY717, and RBY722 to generate the YL404, YL405, YL195, YL196, YL400, and YL401 strains.
To generate green fluorescent protein (GFP) reporter strains of OP4/OP4::OP4p-GFP in RBY717 and the hog1Δ mutant, the OP4 promoter and GFP gene were amplified with primer pair 191 and 192 and primer pair 193 and 194, respectively. The PCR products were mixed and amplified again with primer pair 191 and 194 to generate an OP4-GFP gene fragment. The fused fragment was digested with ApaI and SacI and cloned into pSFS2A to construct pSFS-OP4-GFP. The construct was digested with BsgI and transformed into RBY717, YL8, and YL9 to create strains YL275, YL276, YL279, and YL280.
To delete the ARG4 gene in C. albicans, the hog1Δ, hog1Δ::HOG1, pbs2Δ, and ssk2Δ mutants, primer pair 114 and 115, and primer pair 116 and 117 were used to amplify the 5′ and 3′ flanking regions of the ARG4 gene, respectively. The PCR products were digested with ApaI and XhoI and NotI and SacII and cloned into plasmid pSFS2A to construct pSFS-ARG4 KO. The construct was digested with ApaI and SacII and transformed into RBY717, YL26, YL27, YL35, YL36, YL193, YL194, YL195, and YL196 to generate the YL97, YL98, YL304, YL305, YL328, YL329, YL528, YL529, YL524, and YL525 strains.
To delete the LEU2 gene, the hog1Δ, hog1Δ::HOG1, pbs2Δ, and ssk2Δ mutants, primer pair 122 and 123, and primer pair 124 and 125 were used to amplify the 5′ and 3′ flanking regions of LEU2 gene, respectively. The PCR products were digested with ApaI and XhoI and with SacII and SacI and cloned into plasmid pSFS2A to construct pSFS-LEU2 KO. The construct was digested with ApaI and SacI and transformed into RBY722, YL8, YL9, YL11, YL12, YL402, YL403, YL400, and YL401 to generate the YL133, YL134, YL191, YL192, YL226, YL227, YL530, YL531, YL526, and YL527 strains.
Opaque cells of each strain were obtained and purified after treatment with Lee's NAG (Table 1).
Quantitation of opaque-cell formation.
Overnight cultures of C. albicans were plated onto SC, SCD, or Lee's NAG media supplemented with phloxine B and incubated at 25°C for 4 to 7 days. Opaque colonies were stained red. The white-to-opaque switching efficiency was expressed as follows: (number of opaque colonies)/(total number of colonies) × 100%.
Sensitivity tests.
Hydrogen peroxide and sodium chloride were purchased from Sigma-Aldrich Co., St. Louis, MO, USA, and MDBio, Inc., Shandong, China, respectively. Determinations of susceptibilities to hydrogen peroxide and sodium chloride were performed by spotting serial 4-fold dilutions of 105 cells (total volume of 3 μl) from overnight cultures of C. albicans onto YPD plates supplemented with 10 mM H2O2 or 1 M NaCl. Plates were incubated at 30°C and examined after 48 h.
Western blotting.
To examine the level of Hog1 phosphorylation, 200 μl of overnight cultures was diluted into 10 ml of YPD broth and grown for 5 h at 30°C. H2O2 was added to each culture to reach a final concentration of 10 mM, and the reaction mixture was incubated for 15 min after the addition. Samples were collected by centrifugation, suspended in 10 mM Tris-HCl (pH 8.0), and then incubated at 100°C for 10 min to lyse cells. Equal amounts of proteins were analyzed by SDS-polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride (PVDF) membrane (Bio-Rad Laboratories, Inc., Hercules, CA, USA). ScHog1 polyclonal antibody (Santa Cruz Biotechnology, Inc., Dallas, TX, USA) and phospho-p38 MAPK (Thr180/Tyr182) antibody (Cell Signaling Technology, Inc., Danvers, MA, USA) were used to detect Hog1 and phosphorylated Hog1 proteins, respectively. Blots were developed with Clarity Western ECL substrate (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Pheromone response assays.
To test formation of mating projections, opaque cells of MTLa/a strains were grown in Spider medium at 25°C overnight. Overnight cultures (500 μl) were diluted into 3 ml of Spider broth, and α-pheromone peptide (GFRLTNFGYFEPG; synthesized by GMbiolab Co., Ltd.) was added to reach a final concentration of 10 μg/ml as previously described (43). After 8 h of incubation at 25°C, an Eclipse Ti inverted microscope (Nikon Instruments Inc., Melville, NY, USA) was used to examine the mating projections.
Quantitative mating assays.
To generate auxotrophic strains, ARG4 and LEU2 genes were deleted in MTLa/a and MTLα/α strains, respectively. Opaque cells were grown in Spider medium at 25°C overnight. Cells (2 × 107) of MTLa/a and MTLα/α strains were mixed and placed on Spider plates at 25°C for 48 h. The mixture was collected, diluted, and plated onto selective medium, Arg−, Leu−, and Arg− Leu− plates. Mating efficiencies were calculated by the number of tetraploid colonies on Arg− Leu− plates divided by the total colony number on Arg− or Leu− plates.
Fluorescence-activated cell sorter (FACS) analysis.
Tetraploid cells from mating progeny were incubated with 95% ethanol for at least 1 h at 4°C and washed with Tris-EDTA (TE) buffer (50 mM Tris-HCl, 5 mM EDTA, pH 8.0). Samples were incubated with RNase A solution (2 mg/ml RNase A, 5 mM EDTA, 50 mM Tris-HCl, pH 8.0) for at least 2 h at 37°C and then with pepsin solution (5 mg/ml pepsin, 55 mM HCl) for 30 min at 37°C and washed with TE buffer (50 mM Tris-HCl, 5 mM EDTA, pH 7.5). Samples were incubated with Sytox solution (1 mM Sytox green, 5 mM EDTA, 50 mM Tris-HCl, pH 7.5) for 1 h at 4°C. A BD FACSCanto II system (Becton, Dickinson and Company, Franklin Lakes, New Jersey, USA) was used to examine the fluorescence of samples.
Fluorescence microscopy.
White cells and opaque cells of OP4 promoter-driven GFP reporter strains of the wild-type (WT) strain or hog1Δ mutants were mixed, and the expression of fluorescence was examined by the use of an Eclipse Ti inverted microscope (Nikon Instruments Inc., Melville, NY, USA).
RESULTS
Deletion of the HOG1 gene promotes opaque-cell formation.
To understand the role of Hog1 in regulation of white-opaque switching, hog1Δ mutants of MTLa/a, MTLα/α, and MTLa/α white cells were plated on synthetic complete (SC), synthetic complete dextrose (SCD), and N-acetylglucosamine (NAG) media supplemented with phloxine B to test their switching frequencies. Figure 1A shows that both a/a and α/α colonies of hog1Δ mutants switched to the opaque state (100% switching) when grown on SC medium, whereas a/α cells remained in the white state. Reintegration of the HOG1 gene in each mutant restored the WT phenotype on SC medium, with colonies remaining in the white state.
FIG 1.

Deletion of the HOG1 gene induces white-to-opaque switching in MTLa/a and MTLα/α strains of C. albicans on SC plates. White cells from YPD medium were plated onto SC, SCD, or Lee's N-acetylglucosamine plates containing phloxine B and incubated at 25°C for 4 to 7 days. Opaque cells were stained red. At least 1,000 colonies were examined for each strain. White-to-opaque switching efficiency (percentage of average ± standard deviation [SD]) is indicated below each image. “<” indicates that no opaque-cell formation was observed. (A) MTLa/a and MTLα/α of hog1Δ mutants were able to undergo white-to-opaque switching on SC plates. (B) C. albicans strains were not able to undergo white-to-opaque switching on SCD plates. (C) N-acetylglucosamine induced 100% of white-to-opaque switching in homozygous hog1Δ mutants. (D) Only opaque cells expressed the GFP fluorescence in strains of the WT strain and the hog1Δ mutant. BF, bright field. Scale bar, 20 μm.
Interestingly, the high rates of switching observed with hog1 mutants were not observed on medium supplemented with glucose (SCD) (Fig. 1B), suggesting that glucose plays a negative role in opaque-cell formation. Upon NAG treatment, hog1Δ mutants from both homozygous strain backgrounds also showed 100% opaque switching rates, whereas the WT and complemented mutant strains showed intermediate (63% to 85%) levels of opaque-cell formation (Fig. 1C). To test whether opaque-looking cells were expressing opaque state-specific genes, we expressed the OP4 promoter fused to GFP in both wild-type and hog1Δ strains. As shown in Fig. 1D, only the opaque cells exhibited strong GFP expression, whereas white cells showed no GFP expression. Together, these results demonstrate that Hog1 in C. albicans possesses a novel role in the regulation of white-opaque switching.
Induction of opaque-cell formation in hog1Δ cells requires Wor1.
We observed that hog1Δ a/α cells remain locked in the white phenotype (Fig. 1). This is presumably due to the role of the a1/α2 heterodimer in inhibiting WOR1 expression (30), thereby impeding opaque-cell formation. To determine whether WOR1 is necessary for opaque-cell formation in hog1Δ mutants, hog1Δ wor1Δ double-knockout strains were created. In contrast to the hog1Δ mutant, the hog1Δ wor1Δ mutant lacked the ability to switch to opaque when plated on SC medium (Fig. 2).
FIG 2.

The lack of WOR1 gene strongly inhibits Hog1-mediated white-to-opaque switching. White cells of the MTLa/a WT strain, hog1Δ mutants, wor1Δ mutants, and hog1Δ wor1Δ double mutants were plated onto SC plates supplemented with phloxine B. Images were taken after a 7-day incubation at 25°C. Opaque cells were stained red. The total numbers of cells examined for each strain were 1,000 to 1,200. Switching frequency was expressed as a percentage of the mean ± SD and is shown below each image. “<” indicated that no opaque or opaque-sectored colony was observed.
Both Psb2 and Ssk2 are involved in white-opaque switching.
The SAPK signaling cascade consists of three serine/threonine protein kinases: MAPKKK (Ssk2), MAPKK (Pbs2), and MAPK (Hog1) (39, 40, 44). Ssk2 phosphorylates Pbs2, which in turn phosphorylates and activates Hog1. To determine whether Pbs2 and Ssk2 also regulate the white-opaque transition, mutants were constructed and plated on SC medium. Similarly to the hog1Δ mutant (Fig. 1), the pbs2Δ and ssk2Δ mutants exhibited 100% opaque-cell formation on SC medium supplemented with phloxine B (Fig. 3). Once again, switching occurred only in MTL homozygous strains and not in the MTL heterozygous strain.
FIG 3.

MAPKKK Ssk2 and MAPKK Pbs2 are involved in the regulation of white-opaque switching. White cells of each mutant were plated onto the SC medium supplemented with phloxine B and incubated at 25°C for 7 days. White-to-opaque switching efficiencies were expressed as percentages of averages ± standard deviations (SD) and are shown below each image. At least 1,000 colonies were examined. “<” indicates that no opaque-cell formation was observed.
Functions of the SAPK components are well conserved in both homozygous and heterozygous strains.
The Hog1 SAPK pathway is necessary for resistance to osmotic and oxidative stresses. However, previous studies were performed in C. albicans a/α heterozygous cells (45, 46). We therefore compared and tested the roles of SAPK pathway components in both the heterozygous and homozygous strain backgrounds. As shown in Fig. 4A, mutants of each SAPK component were highly sensitive to both osmotic and oxidative stress in a/α, a/a, and α/α cells. Western blotting also revealed that the function of SAPK in C. albicans is conserved and that Hog1 activation is regulated by Ssk2 and Pbs2 in MTL homozygous strains, as it is in heterozygous strains (Fig. 4B).
FIG 4.
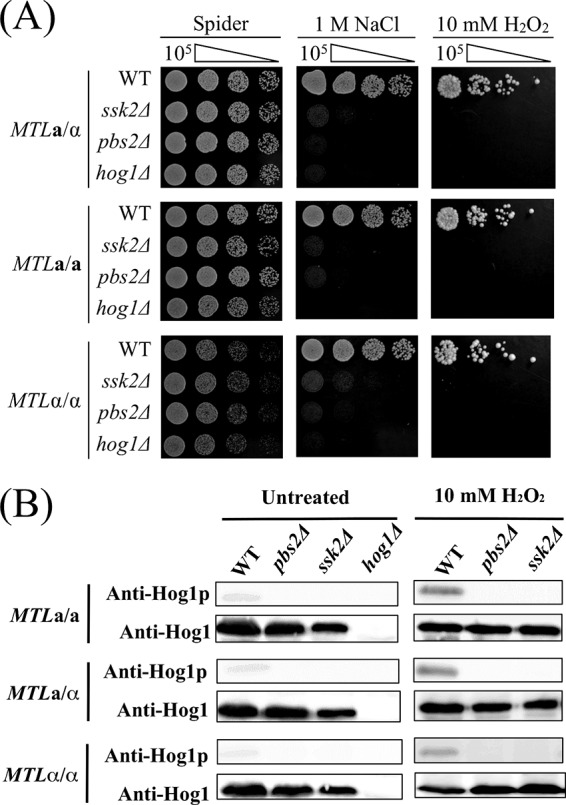
The roles of SSK2, PBS2, and HOG1 in regard to osmotic and oxidative stresses are indistinguishable between homozygous and heterozygous strains of C. albicans. (A) Serially diluted cells of MTLa/a, MTLa/α, and MTLα/α strains of the wild-type, ssk2Δ, pbs2Δ, and hog1Δ mutant strains were spotted onto Spider medium supplemented with 1 M NaCl or 10 mM hydrogen peroxide. (B) Phosphorylation levels of Hog1p were analyzed in MTLa/α, MTLa/a, and MTLα/α strains of ssk2Δ and pbs2Δ mutants. H2O2 (10 mM) was added to exponentially growing cultures, and samples were collected after a 15-min treatment. Anti-Hog1 antibody and phospho-p38 (Thr180/Tyr182) antibody were used to detect Hog1 and phospho-Hog1 (Hog1p), respectively.
Deletion of HOG1, PBS2, and SSK2 genes results in shorter mating projections.
C. albicans strains must undergo several developmental steps before having sex (47). Most isolates are diploid a/α strains and have to undergo homozygosis to generate a/a and α/α cells. These homozygous cells must then switch from white to opaque to become mating competent (18, 19). Pheromone treatment of C. albicans opaque cells activates mating responses and leads to the formation of mating projections (21–23, 43). We further examined the mating characteristics of opaque cells obtained from hog1Δ, pbs2Δ, and ssk2Δ mutants. Surprisingly, mutants of each SAPK component exhibited marked reductions in the length of their mating projections (10 to 12 μm) relative to the length seen with the wild-type strain (24 μm) (Fig. 5). These results indicate that although deletion of HOG1, PBS2, and SSK2 promotes opaque-cell formation, these mutants are less capable of forming mating projections of normal lengths.
FIG 5.
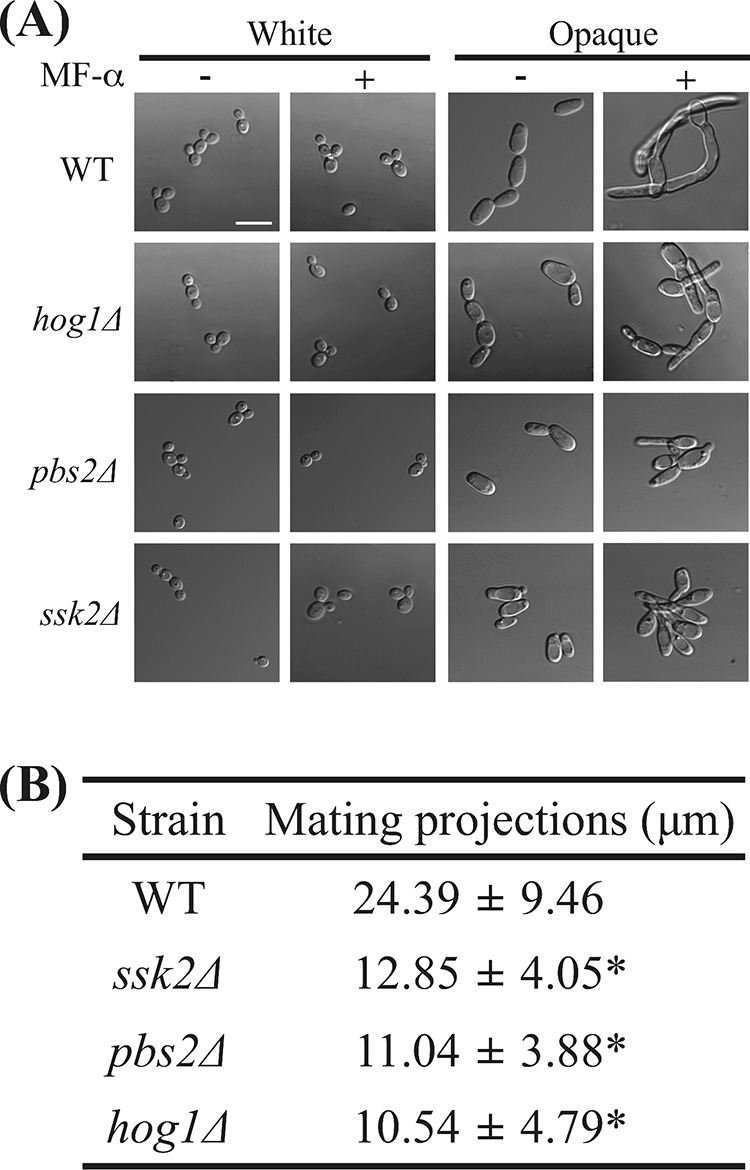
ssk2Δ, pbs2Δ, and hog1Δ mutants exhibited shorter mating projections. (A) White cells and opaque cells of the hog1Δ, ssk2Δ, pbs2Δ, and WT strains were challenged with or without α-factor for 8 h at 25°C. The cells were then imaged with a Nikon Eclipse Ti microscope. (B) At least 50 cells were analyzed to measure the projection length after an 8-h pheromone treatment. The mating projection length of each strain is expressed as the mean ± SD. MF-α, C. albicans pheromone peptide. *, P < 0.001.
ssk2Δ, pbs2Δ, and hog1Δ mutants exhibit reduced mating efficiencies.
Given that ssk2Δ, pbs2Δ, and hog1Δ strains exhibited shorter mating projections (Fig. 5), it is possible that these mutants would also have defects in mating. Opaque MTLa/a and MTLα/α cells containing different selection markers were constructed as described in Materials and Methods. Quantitative mating assays were performed, and the results are shown in Fig. 6A. As expected on the basis of the defective mating responses, the mating efficiencies of ssk2Δ, pbs2Δ, and hog1Δ mutants were 0.6%, 1.6%, and 2.7%, respectively (Fig. 6A). In contrast, the wild-type strain showed a mating efficiency of 16.7% (Fig. 6A). All the mating progeny were confirmed by PCR and FACS analysis to be tetraploid MTLa/α cells (Fig. 6B and C).
FIG 6.
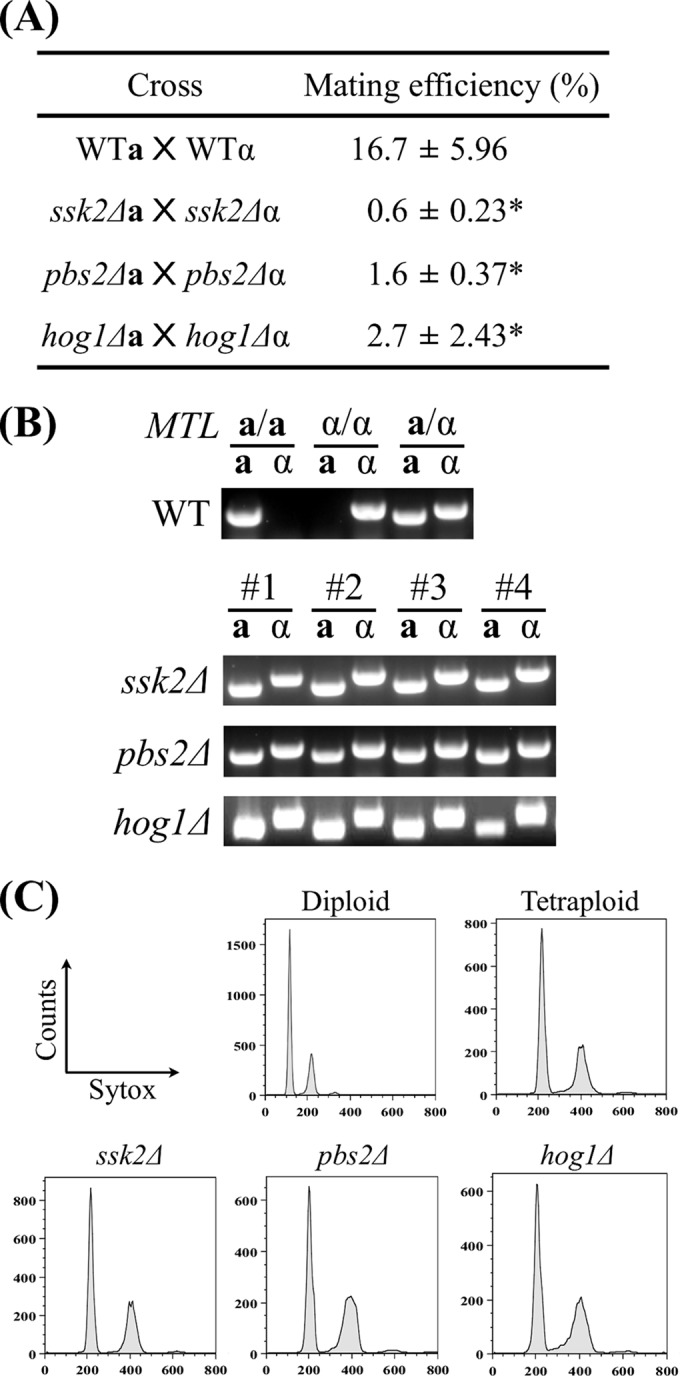
Mating efficiencies of ssk2Δ, pbs2Δ, and hog1Δ mutants were significantly reduced. (A) Auxotrophic strains were coincubated on Spider medium for 48 h, and cells were subsequently plated onto selective medium to determine the mating efficiencies. Mating efficiencies of each cross are expressed as means ± SD. Experiments were repeated three times at least. (B) The OPBa gene and OPBα gene (primer pair 2 and 3 and primer pair 4 and 5) were used to determine the mating type. (C) Progeny ploidies were determined by the use of a BD FACSCanto II system. *, P < 0.05.
DISCUSSION
C. albicans is the most important fungal pathogen in humans, where many infections arise and the difficulties for treatment are due to its inherent flexible lifestyle and ability to avoid the immune system. In addition to the yeast-hypha transition, epigenetic variation via white-opaque switching is perhaps the best example of morphological plasticity. In this work, we first demonstrated that the Hog1 SAPK signaling cascade is involved in regulating the white-opaque epigenetic transition. Inactivation of SSK2, PBS2, and HOG1 genes in MTLa/a and MTLα/α strains led to high sensitivity to osmotic and oxidative stresses and also promoted switching en masse to the opaque cell state under certain culture conditions. These results indicate that although the SAPK in C. albicans is evolutionarily well conserved among eukaryotes, the biological functions of each component kinase are highly dependent on the lifestyles of the species and their environment.
Given that HOG1 can impact white/opaque phenotypic states, we were curious whether the postmodification states of Hog1 differ between white and opaque cells. However, cells of the two types showed similar levels of phosphorylation of the Hog1 protein (data not shown). Consistent with previous results, expression levels of Hog1 were also comparable between white and opaque cells (48). Nevertheless, we cannot rule out the possibility that changes in gene expression levels or postmodification states of Hog1 occur during the white-to-opaque switching process.
C. albicans Hog1 SAPK is activated by many environmental cues, including osmotic and oxidative stimuli, heavy metal, glucose, and cationic peptides (45, 49–51). Two of them, oxidative-stress levels and glucose concentrations, have been reported to be involved in opaque-cell formation and its stability (32, 34, 52). Our studies highlighted that deletion of HOG1 led to switching of white cells to the opaque state on SC and NAG media. These observations could be because removal of HOG1 causes cells to be more sensitive to internal and external oxidative microenvironments that drive cells to the opaque state. Furthermore, it is possible that a transcription factor regulated by Hog1 impacts WOR1 expression and switching frequencies (Fig. 7). Indeed, our studies demonstrated that Wor1 plays a crucial role in the morphological change in hog1Δ mutants, as no white-to-opaque switching was observed in MTLa/α cells or in hog1Δ wor1Δ mutants. Whether genetic control (induction of Wor1 expression) or environmental control (increased sensitivity to stress conditions) or a combination of these factors induces switching remains an open question. Despite high switching rates of hog1Δ mutants on SC and NAG media, we have not observed opaque-colony formation on SCD plates. Similar observations have been reported to show that the presence of glucose (dextrose) inhibited opaque-cell formation and its stability (34, 52). However, the exact mechanism by which glucose or its metabolism influences phenotypic switching or the stability of opaque cells remains obscure (Fig. 7).
FIG 7.
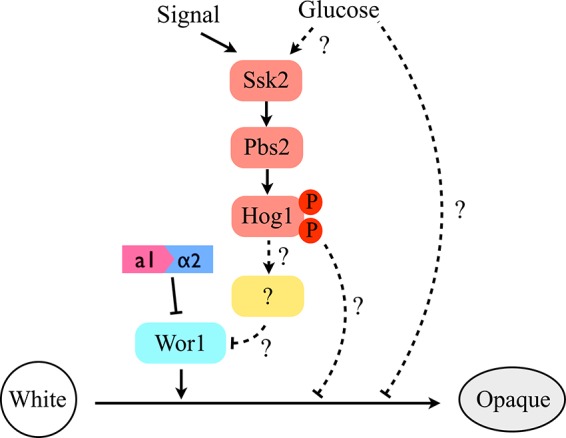
A proposed model of Hog1 MAPK pathway-mediated white-opaque switching in C. albicans. The master regulator of Wor1 and the a1/α2 heterodimer still control the white-opaque switching. The presence of glucose could be a negative factor for opaque-cell formation. The dashed lines represent unknown mechanisms or unidentified regulators for Hog1 SAPK in regulating this phenotypic transition.
The pheromone response for C. albicans is mediated by Cph1, a downstream target of Cek1/Cek2 MAP kinases (21). Deletion of the CPH1 gene resulted in loss of pheromone-stimulated cell adhesion in white cells and mating-projection formation in opaque cells (21). Despite loss of Hog1 SAPK components promoting opaque-cell formation, upon pheromone challenge, hog1Δ, pbs2Δ, and ssk2Δ mutants exhibited projections that were shorter than those of the wild-type strain. Apparently, the pheromone response in these mutant cells is decreased relative to the level in the wild type. These mutants also exhibited a significant decrease in the level of sexual mating. It is clear that C. albicans cells have to switch to the opaque state before having sex and that the mechanisms of the switch and mating response are disparate (22, 53). For example, when the pheromone receptor Ste2 or Cph1 is removed, cells are still able to switch to the opaque state but cannot respond to pheromones and cannot undergo a-α mating (21).
Regulatory interactions between the Hog1 and mating pathways in Saccharomyces cerevisiae have been reported (54–60). Analyses of the antagonistic manners of the potential interactions of two MAPKs were conducted via examination of the insulation mechanism, a physical separation of the shared components of two cascades (54, 59, 61). Since the Hog1 and mating pathways share several components, including Cdc2, Ste20, Ste50, and Ste11, it is therefore possible that Hog1-mediated phosphorylation might cause the conformational changes of shared components to control cell fate (59, 61). The Ste5 and Pbs2 scaffold proteins exclusively required for the mating and hyperosmotic responses, respectively, also appear to regulate and maintain the response specificity (61). Additionally, pathway cross-inhibition has been previously addressed (61). When Hog1 is deleted or inhibited, mating is promoted under conditions of hyperosmotic stress (54, 57). A recent study further indicated that activation of Hog1 can (i) phosphorylate Ste50, leading to the downregulation of mating signal, and (ii) repress the Fus3 response shown by Rck2, a protein kinase of the cell cycle checkpoint (60). It was previously noted that S. cerevisiae contains two osmosensors (Sho1 and Sln1) for Hog1 activation (39). The shared components of Cdc2, Ste20, Ste50, and Ste11 in S. cerevisiae cooperate only with the Sho1-mediated branch (39, 60). Interestingly, although a conserved mechanism by which external signals are sensed and relayed to the SAPK pathway has been observed in C. albicans, mechanisms for activation of the SAPK pathways in the budding yeast and C. albicans have diverged. Indeed, Hog1 SAPK signaling in C. albicans is entirely dependent on a sole osmosensor, Sln1, whereas the Sho1 branch in C. albicans plays no role in Hog1 activation (39, 46). These results therefore suggest a more interesting issue: the potential mechanisms by which the Hog1 SAPK pathway impacts mating in C. albicans.
Taking the data together, white and opaque cells differ in many aspects, as they exhibit different colony morphologies and different mating efficiencies, colonize different niches in the host, and have different interactions with host immune cells. Our work has identified a novel role for the Hog1 SAPK pathway in mediating white-opaque switching. Whether there is a niche in the human body that inhibits Hog1 signaling, leading to increased opaque-cell formation and potential escape from the immune system, is therefore an interesting issue. Future studies will investigate the roles of several key upstream regulatory factors and downstream targets of the Hog1 SAPK pathway to determine if these factors function in regulating this unique phenotypic transition in the human fungal pathogen C. albicans.
ACKNOWLEDGMENTS
We thank Richard Bennett for stains and plasmids. We also thank Richard Bennett and members of his laboratory for critical reading of and comments on the manuscript. We are grateful to the staff of Technology Commons, College of Life Science, National Taiwan University, for the help with the BD FACSCanto II system.
This research was supported by NSC102-2320-B-002-027-MY3, MOST 103-2628-B-002-003-MY3, and NTU 103R7787 from the National Science Council, the Ministry of Science and Technology, and National Taiwan University, respectively.
Footnotes
Published ahead of print 24 October 2014
REFERENCES
- 1.Gargas A, Taylor JW. 1995. Phylogeny of discomycetes and early radiations of the apothecial Ascomycotina inferred from SSU rDNA sequence data. Exp. Mycol. 19:7–15. 10.1006/emyc.1995.1002. [DOI] [PubMed] [Google Scholar]
- 2.Hedges SB. 2002. The origin and evolution of model organisms. Nat. Rev. Genet. 3:838–849. 10.1038/nrg929. [DOI] [PubMed] [Google Scholar]
- 3.Ruhnke M, Maschmeyer G. 2002. Management of mycoses in patients with hematologic disease and cancer – review of the literature. Eur. J. Med. Res. 7:227–235. [PubMed] [Google Scholar]
- 4.Buffo J, Herman MA, Soll DR. 1984. A characterization of pH-regulated dimorphism in Candida albicans. Mycopathologia 85:21–30. 10.1007/BF00436698. [DOI] [PubMed] [Google Scholar]
- 5.Mardon D, Balish E, Phillips AW. 1969. Control of dimorphism in a biochemical variant of Candida albicans. J. Bacteriol. 100:701–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Simonetti N, Strippoli V, Cassone A. 1974. Yeast-mycelial conversion induced by N-acetyl-D-glucosamine in Candida albicans. Nature 250:344–346. 10.1038/250344a0. [DOI] [PubMed] [Google Scholar]
- 7.Sudbery PE. 2011. Growth of Candida albicans hyphae. Nat. Rev. Microbiol. 9:737–748. 10.1038/nrmicro2636. [DOI] [PubMed] [Google Scholar]
- 8.Taschdjian CL, Burchall JJ, Kozinn PJ. 1960. Rapid identification of Candida albicans by filamentation on serum and serum substitutes. AMA J. Dis. Child. 99:212–215. 10.1001/archpedi.1960.02070030214011. [DOI] [PubMed] [Google Scholar]
- 9.Sudbery P, Gow N, Berman J. 2004. The distinct morphogenic states of Candida albicans. Trends Microbiol. 12:317–324. 10.1016/j.tim.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 10.Stoldt VR, Sonneborn A, Leuker CE, Ernst JF. 1997. Efg1p, an essential regulator of morphogenesis of the human pathogen Candida albicans, is a member of a conserved class of bHLH proteins regulating morphogenetic processes in fungi. EMBO J. 16:1982–1991. 10.1093/emboj/16.8.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Si H, Hernday AD, Hirakawa MP, Johnson AD, Bennett RJ. 2013. Candida albicans white and opaque cells undergo distinct programs of filamentous growth. PLoS Pathog. 9:e1003210. 10.1371/journal.ppat.1003210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shapiro RS, Uppuluri P, Zaas AK, Collins C, Senn H, Perfect JR, Heitman J, Cowen LE. 2009. Hsp90 orchestrates temperature-dependent Candida albicans morphogenesis via Ras1-PKA signaling. Curr. Biol. 19:621–629. 10.1016/j.cub.2009.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shapiro RS, Robbins N, Cowen LE. 2011. Regulatory circuitry governing fungal development, drug resistance, and disease. Microbiol. Mol. Biol. Rev. 75:213–267. 10.1128/MMBR.00045-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nobile CJ, Fox EP, Nett JE, Sorrells TR, Mitrovich QM, Hernday AD, Tuch BB, Andes DR, Johnson AD. 2012. A recently evolved transcriptional network controls biofilm development in Candida albicans. Cell 148:126–138. 10.1016/j.cell.2011.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nobile CJ, Mitchell AP. 2005. Regulation of cell-surface genes and biofilm formation by the Candida albicans transcription factor Bcr1p. Curr. Biol. 15:1150–1155. 10.1016/j.cub.2005.05.047. [DOI] [PubMed] [Google Scholar]
- 16.Nobile CJ, Mitchell AP. 2007. Microbial biofilms: e pluribus unum. Curr. Biol. 17:R349–R353. 10.1016/j.cub.2007.02.035. [DOI] [PubMed] [Google Scholar]
- 17.Slutsky B, Staebell M, Anderson J, Risen L, Pfaller M, Soll DR. 1987. “White-opaque transition”: a second high-frequency switching system in Candida albicans. J. Bacteriol. 169:189–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lockhart SR, Pujol C, Daniels KJ, Miller MG, Johnson AD, Pfaller MA, Soll DR. 2002. In Candida albicans, white-opaque switchers are homozygous for mating type. Genetics 162:737–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Magee BB, Magee PT. 2000. Induction of mating in Candida albicans by construction of MTLa and MTLalpha strains. Science 289:310–313. 10.1126/science.289.5477.310. [DOI] [PubMed] [Google Scholar]
- 20.Daniels KJ, Park YN, Srikantha T, Pujol C, Soll DR. 2013. Impact of environmental conditions on the form and function of Candida albicans biofilms. Eukaryot. Cell 12:1389–1402. 10.1128/EC.00127-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin CH, Kabrawala S, Fox EP, Nobile CJ, Johnson AD, Bennett RJ. 2013. Genetic control of conventional and pheromone-stimulated biofilm formation in Candida albicans. PLoS Pathog. 9:e1003305. 10.1371/journal.ppat.1003305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bennett RJ, Johnson AD. 2006. The role of nutrient regulation and the Gpa2 protein in the mating pheromone response of C. albicans. Mol. Microbiol. 62:100–119. 10.1111/j.1365-2958.2006.05367.x. [DOI] [PubMed] [Google Scholar]
- 23.Bennett RJ, Uhl MA, Miller MG, Johnson AD. 2003. Identification and characterization of a Candida albicans mating pheromone. Mol. Cell. Biol. 23:8189–8201. 10.1128/MCB.23.22.8189-8201.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Magee BB, Legrand M, Alarco AM, Raymond M, Magee RT. 2002. Many of the genes required for mating in Saccharomyces cerevisiae are also required for mating in Candida albicans. Mol. Microbiol. 46:1345–1351. 10.1046/j.1365-2958.2002.03263.x. [DOI] [PubMed] [Google Scholar]
- 25.Geiger J, Wessels D, Lockhart SR, Soll DR. 2004. Release of a potent polymorphonuclear leukocyte chemoattractant is regulated by white-opaque switching in Candida albicans. Infect. Immun. 72:667–677. 10.1128/IAI.72.2.667-677.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lohse MB, Johnson AD. 2008. Differential phagocytosis of white versus opaque Candida albicans by Drosophila and mouse phagocytes. PLoS One 3:e1473. 10.1371/journal.pone.0001473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sasse C, Hasenberg M, Weyler M, Gunzer M, Morschhauser J. 2013. White-opaque switching of Candida albicans allows immune evasion in an environment-dependent fashion. Eukaryot. Cell 12:50–58. 10.1128/EC.00266-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morschhäuser J. 2010. Regulation of white-opaque switching in Candida albicans. Med. Microbiol. Immunol. 199:165–172. 10.1007/s00430-010-0147-0. [DOI] [PubMed] [Google Scholar]
- 29.Zordan RE, Miller MG, Galgoczy DJ, Tuch BB, Johnson AD. 2007. Interlocking transcriptional feedback loops control white-opaque switching in Candida albicans. PLoS Biol. 5:e256. 10.1371/journal.pbio.0050256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang G, Wang H, Chou S, Nie X, Chen J, Liu H. 2006. Bistable expression of WOR1, a master regulator of white-opaque switching in Candida albicans. Proc. Natl. Acad. Sci. U. S. A. 103:12813–12818. 10.1073/pnas.0605270103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xie J, Tao L, Nobile CJ, Tong Y, Guan G, Sun Y, Cao C, Hernday AD, Johnson AD, Zhang L, Bai FY, Huang G. 2013. White-opaque switching in natural MTLa/α isolates of Candida albicans: evolutionary implications for roles in host adaptation, pathogenesis, and sex. PLoS Biol. 11:e1001525. 10.1371/journal.pbio.1001525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alby K, Bennett RJ. 2009. Stress-induced phenotypic switching in Candida albicans. Mol. Biol. Cell 20:3178–3191. 10.1091/mbc.E09-01-0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang G, Srikantha T, Sahni N, Yi S, Soll DR. 2009. CO(2) regulates white-to-opaque switching in Candida albicans. Curr. Biol. 19:330–334. 10.1016/j.cub.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang G, Yi S, Sahni N, Daniels KJ, Srikantha T, Soll DR. 2010. N-acetylglucosamine induces white to opaque switching, a mating prerequisite in Candida albicans. PLoS Pathog. 6:e1000806. 10.1371/journal.ppat.1000806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Correia I, Alonso-Monge R, Pla J. 2010. MAPK cell-cycle regulation in Saccharomyces cerevisiae and Candida albicans. Future Microbiol. 5:1125–1141. 10.2217/fmb.10.72. [DOI] [PubMed] [Google Scholar]
- 36.Xu JR. 2000. MAP kinases in fungal pathogens. Fungal Genet. Biol. 31:137–152. 10.1006/fgbi.2000.1237. [DOI] [PubMed] [Google Scholar]
- 37.Folch-Mallol JL, Garay-Arroyo A, Lledias F, Covarrubias Robles AA. 2004. The stress response in the yeast Saccharomyces cerevisiae. Rev. Latinoam. Microbiol. 46:24–46. [PubMed] [Google Scholar]
- 38.Liu H, Kohler J, Fink GR. 1994. Suppression of hyphal formation in Candida albicans by mutation of a STE12 homolog. Science 266:1723–1726. 10.1126/science.7992058. [DOI] [PubMed] [Google Scholar]
- 39.Smith DA, Morgan BA, Quinn J. 2010. Stress signalling to fungal stress-activated protein kinase pathways. FEMS Microbiol. Lett. 306:1–8. 10.1111/j.1574-6968.2010.01937.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Monge RA, Roman E, Nombela C, Pla J. 2006. The MAP kinase signal transduction network in Candida albicans. Microbiology 152:905–912. 10.1099/mic.0.28616-0. [DOI] [PubMed] [Google Scholar]
- 41.Guthrie C, Fink GR. (ed). 1991. Methods in enzymology, vol 194 Guide to yeast genetics and molecular biology. Academic Press, Inc, San Diego, CA. [PubMed] [Google Scholar]
- 42.Reuss O, Vik A, Kolter R, Morschhauser J. 2004. The SAT1 flipper, an optimized tool for gene disruption in Candida albicans. Gene 341:119–127. 10.1016/j.gene.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 43.Lin CH, Choi A, Bennett RJ. 2011. Defining pheromone-receptor signaling in Candida albicans and related asexual Candida species. Mol. Biol. Cell 22:4918–4930. 10.1091/mbc.E11-09-0749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.de Dios CH, Roman E, Monge RA, Pla J. 2010. The role of MAPK signal transduction pathways in the response to oxidative stress in the fungal pathogen Candida albicans: implications in virulence. Curr. Protein Pept. Sci. 11:693–703. 10.2174/138920310794557655. [DOI] [PubMed] [Google Scholar]
- 45.Alonso-Monge R, Navarro-Garcia F, Roman E, Negredo AI, Eisman B, Nombela C, Pla J. 2003. The Hog1 mitogen-activated protein kinase is essential in the oxidative stress response and chlamydospore formation in Candida albicans. Eukaryot. Cell 2:351–361. 10.1128/EC.2.2.351-361.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cheetham J, Smith DA, da Silva Dantas A, Doris KS, Patterson MJ, Bruce CR, Quinn J. 2007. A single MAPKKK regulates the Hog1 MAPK pathway in the pathogenic fungus Candida albicans. Mol. Biol. Cell 18:4603–4614. 10.1091/mbc.E07-06-0581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ni M, Feretzaki M, Sun S, Wang X, Heitman J. 2011. Sex in fungi. Annu. Rev. Genet. 45:405–430. 10.1146/annurev-genet-110410-132536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tuch BB, Mitrovich QM, Homann OR, Hernday AD, Monighetti CK, De la Vega FM, Johnson AD. 2010. The transcriptomes of two heritable cell types illuminate the circuit governing their differentiation. PLoS Genet. 6:e1001070. 10.1371/journal.pgen.1001070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rodaki A, Bohovych IM, Enjalbert B, Young T, Odds FC, Gow NA, Brown AJ. 2009. Glucose promotes stress resistance in the fungal pathogen Candida albicans. Mol. Biol. Cell 20:4845–4855. 10.1091/mbc.E09-01-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Smith DA, Nicholls S, Morgan BA, Brown AJ, Quinn J. 2004. A conserved stress-activated protein kinase regulates a core stress response in the human pathogen Candida albicans. Mol. Biol. Cell 15:4179–4190. 10.1091/mbc.E04-03-0181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vylkova S, Jang WS, Li W, Nayyar N, Edgerton M. 2007. Histatin 5 initiates osmotic stress response in Candida albicans via activation of the Hog1 mitogen-activated protein kinase pathway. Eukaryot. Cell 6:1876–1888. 10.1128/EC.00039-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lohse MB, Hernday AD, Fordyce PM, Noiman L, Sorrells TR, Hanson-Smith V, Nobile CJ, DeRisi JL, Johnson AD. 2013. Identification and characterization of a previously undescribed family of sequence-specific DNA-binding domains. Proc. Natl. Acad. Sci. U. S. A. 110:7660–7665. 10.1073/pnas.1221734110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Alby K, Bennett RJ. 2010. Sexual reproduction in the Candida clade: cryptic cycles, diverse mechanisms, and alternative functions. Cell. Mol. Life Sci. 67:3275–3285. 10.1007/s00018-010-0421-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.O'Rourke SM, Herskowitz I. 1998. The Hog1 MAPK prevents cross talk between the HOG and pheromone response MAPK pathways in Saccharomyces cerevisiae. Genes Dev. 12:2874–2886. 10.1101/gad.12.18.2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Harris K, Lamson RE, Nelson B, Hughes TR, Marton MJ, Roberts CJ, Boone C, Pryciak PM. 2001. Role of scaffolds in MAP kinase pathway specificity revealed by custom design of pathway-dedicated signaling proteins. Curr. Biol. 11:1815–1824. 10.1016/S0960-9822(01)00567-X. [DOI] [PubMed] [Google Scholar]
- 56.Nelson B, Parsons AB, Evangelista M, Schaefer K, Kennedy K, Ritchie S, Petryshen TL, Boone C. 2004. Fus1p interacts with components of the Hog1p mitogen-activated protein kinase and Cdc42p morphogenesis signaling pathways to control cell fusion during yeast mating. Genetics 166:67–77. 10.1534/genetics.166.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Westfall PJ, Thorner J. 2006. Analysis of mitogen-activated protein kinase signaling specificity in response to hyperosmotic stress: use of an analog-sensitive HOG1 allele. Eukaryot. Cell 5:1215–1228. 10.1128/EC.00037-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hao N, Zeng Y, Elston TC, Dohlman HG. 2008. Control of MAPK specificity by feedback phosphorylation of shared adaptor protein Ste50. J. Biol. Chem. 283:33798–33802. 10.1074/jbc.C800179200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Patterson JC, Klimenko ES, Thorner J. 2010. Single-cell analysis reveals that insulation maintains signaling specificity between two yeast MAPK pathways with common components. Sci. Signal. 3:ra75. 10.1126/scisignal.2001275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nagiec MJ, Dohlman HG. 2012. Checkpoints in a yeast differentiation pathway coordinate signaling during hyperosmotic stress. PLoS Genet. 8:e1002437. 10.1371/journal.pgen.1002437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schwartz MA, Madhani HD. 2004. Principles of MAP kinase signaling specificity in Saccharomyces cerevisiae. Annu. Rev. Genet. 38:725–748. 10.1146/annurev.genet.39.073003.112634. [DOI] [PubMed] [Google Scholar]