

Abstract
The clustered regularly interspaced short palindromic repeat (CRISPR)/Cas9 system has become a powerful and precise tool for targeted gene modification (e.g., gene knockout and gene replacement) in numerous eukaryotic organisms. Initial attempts to apply this technology to a model, the single-cell alga, Chlamydomonas reinhardtii, failed to yield cells containing edited genes. To determine if the Cas9 and single guide RNA (sgRNA) genes were functional in C. reinhardtii, we tested the ability of a codon-optimized Cas9 gene along with one of four different sgRNAs to cause targeted gene disruption during a 24-h period immediately following transformation. All three exogenously supplied gene targets as well as the endogenous FKB12 (rapamycin sensitivity) gene of C. reinhardtii displayed distinct Cas9/sgRNA-mediated target site modifications as determined by DNA sequencing of cloned PCR amplicons of the target site region. Success in transient expression of Cas9 and sgRNA genes contrasted with the recovery of only a single rapamycin-resistant colony bearing an appropriately modified FKB12 target site in 16 independent transformation experiments involving >109 cells. Failure to recover transformants with intact or expressed Cas9 genes following transformation with the Cas9 gene alone (or even with a gene encoding a Cas9 lacking nuclease activity) provided strong suggestive evidence for Cas9 toxicity when Cas9 is produced constitutively in C. reinhardtii. The present results provide compelling evidence that Cas9 and sgRNA genes function properly in C. reinhardtii to cause targeted gene modifications and point to the need for a focus on development of methods to properly stem Cas9 production and/or activity following gene editing.
INTRODUCTION
The single-celled green alga Chlamydomonas reinhardtii has proven to be an outstanding model organism for studies of mechanisms of photosynthesis, cilium/flagellum-based motility, and, more recently, the biochemical pathway for production of lipids and other materials of interest to the renewal biofuel community and specialty chemical companies. C. reinhardtii (referred to here as Chlamydomonas) grows rapidly, has a fully sequenced and annotated genome, is easily genetically transformed, is amenable to classical genetic analyses, and has associated with it both a wealth of biochemical, molecular, and cellular information and numerous experimental tools to expedite research. Because it is a haploid organism, mutant phenotypes are easily created and identified through classical screening techniques or through new, high-throughput DNA analysis approaches (1).
The advent of clustered regularly interspaced short palindromic repeat (CRISPR)/Cas9 technology for facile gene editing in numerous organism has heralded rapid progress in creating targeted gene knockouts and gene replacement by homologous recombination (2, 3). Indeed, our initial success in transient expression of the Cas9/single guide RNA (sgRNA) system in plant protoplasts and leaves (4–7) has been followed by reports of stably transformed T2 plants containing genes with permanent modifications to specifically targeted genes (see, e.g., references 8, 9, and 10). Although the Cas9 gene used in our Arabidopsis and tobacco studies was codon optimized for expression in Chlamydomonas and worked efficiently in supporting gene editing in plants (4, 9), we were unable to recover Chlamydomonas cells carrying Cas9/sgRNA-mediated targeted gene modifications in numerous experiments using the codon-optimized Cas9 gene and a variety of sgRNA genes. Because earlier experiments using transcription activator-like effector nuclease (TALEN) constructs for targeted gene modifications in our laboratory pointed to the possibility of toxic effects of continuous TALEN production in transformed Chlamydomonas cells, we sought to determine if continuous expression of Cas9 could likewise be toxic to Chlamydomonas cells. Given the success of transient expression of Cas9 and sgRNA genes in plant cells and plant leaves (4–7), we designed experiments in which Cas9 and sgRNA genes were delivered via electroporation into Chlamydomonas cells that were then harvested 24 h later and examined for DNA sequence modifications in Cas9/sgRNA target site DNA sequences in four different sgRNA-targeted genes. Here we report success in obtaining transient expression of Cas9 and sgRNA genes in Chlamydomonas resulting in precise targeting of the four independent target genes by the Cas9/sgRNA system—as verified by detailed characterization of the modified target sites.
MATERIALS AND METHODS
Construction of expression vectors.
Design and construction of the Chlamydomonas codon-optimized Cas9 gene from Streptococcus pyogenes (11), the sgRNA gene (11, 12), and the nonfunctional, out-of-frame green fluorescent protein (GFP) gene have been described earlier (4). Figure 1B displays the design of the full-length Chlamydomonas codon-optimized Cas9 gene driven by the cauliflower mosaic virus (CaMV) 35S promoter and terminated by the nopaline synthetase gene termination region (Tnos), the sgRNA gene targeting the nonfunctional, mutant GFP (mGFP) gene driven by the Arabidopsis U6 gene promoter, and the mGFP gene driven by the 35S promoter and terminated with Tnos. The 20-nucleotide (nt) mGFP target sequence is shown in Table S1 in the supplemental material, and the complete DNA sequences of all three genes are provided in Fig. S1 in the supplemental material. Following the same approach, the 20-nt target sequence used to produce the mutant GFP was inserted at the 5′ coding region of the Gaussia luciferase (Gluc) gene (GenBank accession no. EU372000.1) to create an out-of-reading frame Gluc gene (Fig. 1C). The relevant DNA sequences are provided in Fig. S2. Similarly, the 20-nt target sequence was inserted into a hygromycin resistance (hygror) gene (GenBank accession no. AF234296.1; protein identifier [ID], AAF65337.1) (Fig. 1A). The relevant DNA sequences are provided in Fig. S3 in the supplemental material. For targeting the C. reinhardtii peptidyl-prolyl cis-trans isomerase gene (i.e., the FKB12 gene; NCBI reference sequence XM 001693563.1; phytozome Cre13.g586300.t1.2) coding region, an appropriate 20-nt target sequence in the 5′ coding region of the gene was selected for sgRNA binding (Fig. 1D; the relevant DNA sequences are provided in Fig. S4 in the supplemental material).
FIG 1.

Verification of Cas9/sgRNA activity in C. reinhardtii. Designs of constructs for testing expression of the Cas9/sgRNA system in Chlamydomonas reinhardtii are shown. The genes targeted for mutagenesis were the exogenous hygromycin resistance (Hygro) gene (A), the exogenous mutant GFP (mGFP) gene (B), the exogenous mutant luciferase (Gluc) gene (C), and the endogenous FKB12 gene (D). Target sequence restriction sites DraIII (A), ApaLI (B and C), and StyI (D) are underlined. The PAM sequence is highlighted in red. PsaDP, promoter of the photosystem I reaction center subunit II (PsaD) gene of C. reinhardtii; 35S and 35ST, cauliflower mosaic virus (CAMV) 35S gene promoter and terminator, respectively; U6P, Arabidopsis thaliana U6 gene promoter; Tnos, termination region of the nopalene synthetase gene.
Chlamydomonas culture growth and transformation.
A mutant strain of Chlamydomonas lacking an intact cell wall (CC503) was maintained on solid Tris-acetate-phosphate (TAP) media (13) containing 20 g/liter agar and maintained at a light intensity of 50 μmol m−2 s−1. For use in transformation experiments, cells were inoculated into liquid cultures of TAP medium and maintained with shaking at 100 RPM under light at 200 μmol m−2 s−1 until cells reached a density of 3 × 106/ml as determined with a hemocytometer. Plasmid DNAs used in transformation experiments were isolated from freshly transformed bacterial cultures picked from plates and grown in liquid culture for 8 h at 37°C. Plasmid DNA was purified using a GeneJET Plasmid Miniprep kit (Thermo Scientific). Restriction enzyme analysis and DNA sequencing were used to verify the identity and intactness of each construct. Chlamydomonas cell cultures were grown to a density of 3 × 106/ml and then chilled on ice for 10 min prior to harvesting by centrifugation at 2,000 × g for 5 min at 4°C. Cells were resuspended with TAP–60 mM sucrose to a density of 4 × 108 cells/ml. For each transformation reaction, a 250-μl volume of cells was mixed with 2 μg plasmids in an electroporation cuvette (4-mm gap) and chilled for 5 min in a 16°C water bath prior to electroporation. Electroporation was performed using a Bio-Rad Gene Pulser II apparatus with one pulse at 0.75 kV and 25 μF without resistance. Cells were diluted into 10 ml of TAP–60 mM sucrose and transferred to a 50-ml sterile disposable tube. Cells were incubated with very gentle shaking in low light for 24 h. To extract genomic DNA, transformed cells were harvested by centrifugation and resuspended in 200 μl of DNA extraction buffer (100 mM Tris-Cl [pH 8.0], 20 mM EDTA, 500 mM NaCl, 2% Sarkosyl). DNA was extracted using 200 μl of buffer-equilibrated phenol and vigorous vortex mixing for 1.5 h. Following centrifugation, the aqueous layer was transferred to a new 1.5-ml Eppendorf tube and an equal volume of chloroform was added. After vortex mixing for 5 min, the tube was centrifuged and the aqueous layer was transferred to a new 1.5-ml Eppendorf tube. To precipitate DNA, a 2/3 volume of isopropanol was added and mixed by inverting the tube several times. DNA was harvested by centrifugation at full speed in a microcentrifuge for 20 min at room temperature. The DNA pellet was washed with 70% ethanol and dried in a 37°C oven for 5 min. DNA was resuspended with TE buffer (10 mM Tris-Cl [pH 8.0], 1 mM EDTA) containing 50 μg/ml RNase A and incubated at 37°C for 1.5 h prior to treatment with an appropriate restriction enzyme and PCR amplification as described below.
Enrichment of mutagenized target sites using restriction enzyme digestion of PCR-amplified sgRNA target site regions present in mutagenized genes.
Design of the PCR amplification of the sgRNA target sites in hygromycin, mGFP, Gluc, and FKB12 genes from Chlamydomonas cells transformed with Cas9 and sgRNA genes followed previously described procedures for PCR amplification/restriction enzyme analyses of the sgRNA target sites in mGFP genes contained in land plants expressing Cas9/sgRNA (4). To eliminate PCR amplification of the vast majority of hygromycin, GFP, Gluc, and FKB12 genes not modified by Cas9/sgRNA exposure, the extracted DNA was cleaved at the resident ApaLI cut site for the mGFP and Gluc genes, at DraIII for the hygromycin resistance gene, and at StyI for the FKB12 gene, which are located within the 20-bp sgRNA target site in close proximity to the protospacer adjacent motif (PAM) site. In this way, subsequent PCR amplifications favored production of mutagenized DNAs. For PCR amplification, primers upstream and downstream of the restriction site of each gene were employed (see Table S2 in the supplemental material). PCR-amplified DNA resistant to restriction enzyme digestion was separated from digested DNA fragments by agarose gel electrophoresis. DNA bands of the appropriate, full-length size were cut from the agarose gel and cloned into a pBlueScript vector. DNA sequencing of the cloned fragments was used to determine the nature of Cas9/sgRNA/NHEJ-mediated mutations obtained.
RESULTS AND DISCUSSION
Our experiments were designed to test for transient expression of Cas9 and sgRNA genes and the production of mutagenized target DNA sequences in exogenously supplied genes (a hygromycin resistance gene, a mutant GFP gene employed in earlier experiments to demonstrate successful expression of the Cas9/sgRNA system in Arabidopsis and tobacco [4, 9], and a mutant Gaussia luciferase [Gluc] gene) as well as in the endogenous Chlamydomonas FKB12 gene that confers sensitivity to the antibiotic rapamycin. The targeted exogenous genes were incorporated into the same vector that carried the Cas9 and sgRNA genes to allow simultaneous delivery of the DNA modifying genes and the targeted gene itself (Fig. 1A to C). The endogenous FKB12 gene was targeted using a vector containing only the Cas9 gene and an appropriately designed sgRNA gene (Fig. 1D). Following the scheme outlined in Fig. 2, Chlamydomonas cells were transformed with each of the vectors using electroporation and allowed to recover overnight. The next day, all cells in each treatment were harvested and DNA was extracted from each pool of cells. An appropriate set of primers (see Table S2 in the supplemental material) was used to perform independent PCR amplifications of DNA sequences in the target region of the three exogenous genes and the endogenous FKB12 gene (Fig. 3, lanes 1 and 2). Each DNA sequence within each 20-bp target site that was the sequence most likely to be cleaved by Cas9 nucleases (i.e., nucleotide sequences immediately upstream of the NGG PAM site) contained a restriction enzyme recognition site (DraIII for the hygror gene, ApaLI for the mGFP and mGluc genes, and StyI for the FKB12 gene). If Cas9/sgRNA-directed DNA cleavage occurred within these restriction sites and if nucleotides were deleted from or inserted into the cleavage site as a result of error-prone DNA repair by the nonhomologous end-joining mechanism, then in some cases the restriction site would be destroyed and the DNA fragments amplified by PCR would no longer be susceptible to restriction enzyme cleavage. Examination of PCR-amplified DNA fragments from all four experimental samples treated in two sequential digestions (see Materials and Methods) with the appropriate restriction enzyme (Fig. 3, lanes 3) showed that a portion of each DNA fragment was resistant to restriction enzyme digestion, indicating the presence of mutagenized target sites. If the vectors used for transformation lacked the sgRNA gene (Fig. 3A and D, lanes 4) or contained an sgRNA with a randomly chosen 20-bp DNA sequence (Fig. 3B, lane 4), complete cleavage of the PCR-amplified DNA was observed—indicating no mutagenesis. The complete contrast in results between these control experiments and experiments in which bona fide sgRNAs were supplied clearly demonstrated both that sgRNA genes were active in Chlamydomonas and that they were essential for targeted gene editing by Cas9.
FIG 2.
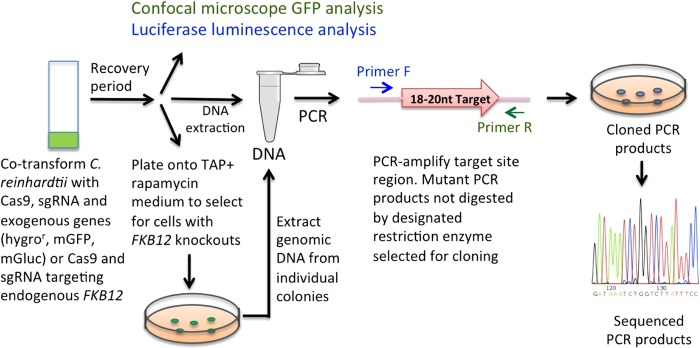
Scheme for use of the Cas9/sgRNA system in C. reinhardtii for mutagenesis of three exogenous genes (the hygromycin resistance gene, the mutant GFP gene [mGFP], the mutant luciferase gene [mGluc]) or the endogenous FKB12 gene.
FIG 3.

PCR/restriction enzyme (PCR/RE) analysis to determine rates of Cas9/sgRNA-directed mutagenesis of a target gene. (A) PCR was used to amplify target regions of hygromycin resistance genes isolated from pooled C. reinhardtii cells cotransformed with the hygromycin resistance gene and with the Cas9 gene in the absence of a sgRNA gene (lanes 2 and 4) or in the presence of a sgRNA gene (lanes 1 and 3) targeting the hygromycin resistance gene. The PCR products were then treated with DraIII to determine whether or not the DraIII restriction enzyme site in the genes had been mutagenized. The top arrow indicates the expected ∼235-bp size of PCR products from hygromycin gene target regions not cleaved by DraIII; the bottom arrow indicates the two expected ∼118-bp DNA fragments resulting from DraIII cleavage of the ∼235-bp PCR product amplified from the hygromycin resistance genes when the DraIII site had not been mutagenized. (B) Same conditions as those described for panel A but with a mutant GFP (mGFP) gene as the target gene that contained an ApaLI restriction enzyme site at the expected cleavage site for a Cas9/sgRNA complex targeted to a 20-bp sequence in the 5′ end of the mGFP gene. Lane 3 contains ApaI-digested DNA from pools of Chlamydomonas cells transformed with a Cas9 gene and a sgRNA gene targeted to the mGFP gene, while lane 4 contains similar ApaI-digested DNA but from cells transformed with a Cas9 gene and a sgRNA gene containing a 20-bp guide sequence composed of randomly selected nucleotides. The uncleaved PCR product is 280 bp, and the two products of ApaLI cleavage are ∼140 bp each. (C) Same conditions as those described for panel A but with a luciferase (Gluc) gene as the target gene that contained an ApaLI restriction enzyme site at the expected cleavage site for a Cas9/sgRNA complex targeted to a 20-bp sequence in the 5′ end of the Gluc gene. The uncleaved PCR product is 347 bp, and the two products of ApaLI cleavage are 208 bp and 139 bp, respectively. (D) Same conditions as those described for panel A but with a FKB12 gene as the target gene that contained an StyI restriction enzyme site at the expected cleavage site for a Cas9/sgRNA complex targeted to a 20-bp sequence in the coding region of the FKB12 gene. The uncleaved PCR product is 255 bp, and the two products of StyI cleavage are 127 bp and 128 bp, respectively.
As a demonstration of the specificity of mutagenesis by all four sets of Cas9/sgRNA genes at the expected cleavage site within each of the four target genes, the PCR-amplified, restriction enzyme-resistant DNA fragments obtained in each experiment were cloned and sequenced. Results showed that there were at least two independent types of mutations within each of the groups of 14 or 15 cloned genes analyzed in each of the four experiments (Fig. 4A to D). In all cases, the inserted or deleted nucleotides were in close proximity to the expected Cas9 cleavage site 3 to 4 bp upstream of the NGG PAM DNA sequence. (In each experiment, there was a portion of clones lacking an altered DNA sequence, suggesting incomplete digestion of the PCR-amplified DNA fragments during incubation with the respective restriction enzymes.) It should be noted that only mutations that change the restriction enzyme recognition DNA sequence are detected in the assay employed and that a portion of the mutagenized target genes produced in each experiment is therefore likely to be missed. Regardless, the present results provide compelling evidence that Chlamydomonas can utilize exogenously supplied Cas9 and sgRNA genes to produce Cas9 molecules and sgRNAs that are fully functional in causing targeted gene cleavage and fostering subsequent gene mutagenesis.
FIG 4.

Confirmation of Cas9/sgRNA-mediated mutagenesis of sgRNA target sites within the target genes by DNA sequencing. (A) Hygromycin resistance gene. Fourteen cloned DNA fragments of 235 bp containing DNA from PCR-amplified sgRNA target regions of hygromycin resistance genes mutated by Cas9/sgRNA treatment were subjected to DNA sequencing. DNA sequences of a segment surrounding the sgRNA target site are shown for each clone, with the sequence of the nonmutagenized DNA region shown as the top line. The 20-nucleotide target sequence for the Cas9/sgRNA complex is depicted in blue, the PAM site is depicted in red, and the DraIII recognition site is underlined in blue. For the Cas9/sgRNA-mutagenized DNA sequences, inserted nucleotides are shown in green. The net increase or decrease in fragment length and the length of insertions and/or deletions (In/Del) along with the frequency of the mutant sequence within the total of sequenced clones are provided in the column to the right. (B) Same conditions as those described for panel A but with the target mGFP gene containing an ApaLI restriction site as the sgRNA target within the 5′ target region of the gene. Deleted nucleotides are denoted by red dashes. (C) Same conditions as those described for panel A but with the target Gluc gene containing an ApaLI restriction site as the sgRNA target within the 5′ target region of the gene. (D) Same conditions as those described for panel A but with the target FKB12 gene containing an StyI restriction site as the sgRNA target within the coding region of the gene. (E) DNA sequence of the region of an endogenous FKB12 gene target for gene modification by Cas9/sgRNA (top line) and the DNA sequence of the same region of a mutagenized FKB12 gene isolated from an extremely rare Cas9/sgRNA-treated, rapamycin-resistant Chlamydomonas transformant (bottom line).
Following the successful demonstration of transient Cas9 and sgRNA gene expression reported here, the next step will be to obtain several genetically transformed cell lines in which at least one gene has been successfully targeted for gene disruption or editing. Initial experiments indicate that, in the case of Chlamydomonas, this goal will not be achieved with the same apparent ease as with several other organisms. Use of a Cas9 gene codon optimized for Chlamydomonas that has been shown to be functional in supporting targeted gene modification in Arabidopsis and tobacco (4, 9) along with sgRNA genes targeting several different endogenous Chlamydomonas genes (e.g., FKB12, with a selectable null phenotype) has, with a single exception discussed below, failed to yield viable cells with targeted gene modifications. Toxicity due to continuous expression of Cas9 in transformed cells was one potential explanation for the lack of recovery of cells containing intact Cas9 genes. To test this hypothesis, a Cas9 gene in which both nuclease domains were inactivated by mutation (14) was constructed to determine if cells transformed with this deactivated (or “dead”) Cas9 (dCas9) gene (containing a 3× Flag-tag coding sequence) and an antibiotic resistance gene (to allow selection of transformed cells) could be recovered. Of the 33 antibiotic-resistant transformants examined by PCR analysis, only 6 contained potentially intact dCas9 genes. However, protein blot analysis of the six transformants using highly sensitive anti-Flag antibodies failed to detect even weak expression of dCas9 (data not shown)—suggesting that even dCas9 may interfere with the ability of Chlamydomonas cells to survive. In an extensive set of experiments involving 16 transformations and more than 1.6 × 109 cells, we used the same codon-optimized Cas9 gene and the same sgRNA gene used successfully in the experiments described above for FKB12 gene cleavage (Fig. 3D and Fig. 4D) to target the Chlamydomonas FKB12 gene for coding region disruption. Inactivation of the FKB12 gene produces rapamycin-resistant cells (15). These experiments yielded only one rapamycin-resistant Chlamydomonas transformant with signature signs of Cas9/sgRNA gene modification. DNA sequencing of the PCR-amplified target region of the FKB12 gene from this transformant demonstrated a 2-bp deletion 3 bp upstream of the GGG PAM site (Fig. 4E)—the expected location of Cas9-directed DNA cleavage. Although it is likely that this represents a true Cas9/sgRNA-directed gene knockout (versus a random mutagenic event), the rare occurrence of this single putative positive event supports the hypothesis that Cas9 gene expression may be toxic to Chlamydomonas.
The results of the present studies provide clear evidence that Cas9 and sgRNA genes are functional in promoting targeted gene modifications in Chlamydomonas. However, compared with the success in gene editing obtained in the transient assays used in these studies, evidence for recovery of gene editing in stably transformed cells has been achieved in only one instance. The inferred toxicity of both active Cas9 and inactivated Cas9 suggests that a method that avoids or limits accumulation of active Cas9 in transformed cells will be an important future experimental goal to pursue—and will perhaps be critical to development of a practical Cas9/sgRNA system for targeted gene modification in Chlamydomonas. Use of conditional promoters to drive Cas9 gene expression and transformation with likely short-lived Cas9 mRNAs are but two of the more obvious tactics that might be tested.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the U.S. National Science Foundation (MCB-0952533 and EPSCoR-1004094) and the U.S. Department of Energy (DOE DE-EE0001052 and DOE CAB-COMM DOE DE-EE0003373).
We declare that we have no conflicts of interest.
Footnotes
Published ahead of print 19 September 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/EC.00213-14.
REFERENCES
- 1.Zhang R, Patena W, Armbruster U, Gang SS, Blum SR, Jonikas MC. 2014. High-throughput genotyping of green algal mutants reveals random distribution of mutagenic insertion sites and endonucleolytic cleavage of transforming DNA. Plant Cell 26:1398–1409. 10.1105/tpc.114.124099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mali P, Esvelt KM, Church GM. 2013. Cas9 as a versatile tool for engineering biology. Nat. Methods 10:957–963. 10.1038/nmeth.2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hsu PD, Lander ES, Zhang F. 2014. Development and applications of CRISPR-Cas9 for genome engineering. Cell 157:1262–1278. 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jiang W, Zhou H, Bi H, Fromm M, Yang B, Weeks DP. 2013. Demonstration of CRISPR/Cas9/sgRNA-mediated targeted gene modification in Arabidopsis, tobacco, sorghum and rice. Nucleic Acids Res. 41:e188. 10.1093/nar/gkt780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li JF, Norville JE, Aach J, McCormack M, Zhang D, Bush J, Church GM, Sheen J. 2013. Multiplex and homologous recombination-mediated genome editing in Arabidopsis and Nicotiana benthamiana using guide RNA and Cas9. Nat. Biotechnol. 31:688–691. 10.1038/nbt.2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nekrasov V, Staskawicz B, Weigel D, Jones JD, Kamoun S. 2013. Targeted mutagenesis in the model plant Nicotiana benthamiana using Cas9 RNA-guided endonuclease. Nat. Biotechnol. 31:691–693. 10.1038/nbt.2655. [DOI] [PubMed] [Google Scholar]
- 7.Shan Q, Wang Y, Li J, Zhang Y, Chen K, Liang Z, Zhang K, Liu J, Xi JJ, Qiu JL, Gao C. 2013. Targeted genome modification of crop plants using a CRISPR-Cas system. Nat. Biotechnol. 31:686–688. 10.1038/nbt.2650. [DOI] [PubMed] [Google Scholar]
- 8.Feng Z, Mao Y, Xu N, Zhang B, Wei P, Yang DL, Wang Z, Zhang Z, Zheng R, Yang L, Zeng L, Liu X, Zhu JK. 2014. Multigeneration analysis reveals the inheritance, specificity, and patterns of CRISPR/Cas-induced gene modifications in Arabidopsis. Proc. Natl. Acad. Sci. U. S. A. 111:4632–4637. 10.1073/pnas.1400822111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiang W, Yang B, Weeks DP. 2014. Efficient CRISPR/Cas9-mediated gene editing in Arabidopsis thaliana and inheritance of modified genes in the T2 and T3 generations. PLoS One 9:e99225. 10.1371/journal.pone.0099225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou H, Liu B, Weeks DP, Spalding MH, Yang B. 8 September 2014. Large chromosomal deletions and heritable small genetic changes induced by CRISPR/Cas9 in rice. Nucleic Acids Res. pii:gku806. 10.1093/nar/gku806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. 2012. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821. 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. 2013. RNA-guided human genome engineering via Cas9. Science 339:823–826. 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harris EH. 1989. The Chlamydomonas sourcebook: a comprehensive guide to biology and laboratory use. Academic Press, San Diego, CA. [DOI] [PubMed] [Google Scholar]
- 14.Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, Stern-Ginossar N, Brandman O, Whitehead EH, Doudna JA, Lim WA, Weissman JS, Qi LS. 2013. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154:442–451. 10.1016/j.cell.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Crespo JL, Díaz-Troya S, Florencio FJ. 2005. Inhibition of target of rapamycin signaling by rapamycin in the unicellular green alga Chlamydomonas reinhardtii. Plant Physiol. 139:1736–1749. 10.1104/pp.105.070847. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.