

Abstract
The peptide transporter Ptr2 plays a central role in di- or tripeptide import in Saccharomyces cerevisiae. Although PTR2 transcription has been extensively analyzed in terms of upregulation by the Ubr1-Cup9 circuit, the structural and functional information for this transporter is limited. Here we identified 14 amino acid residues required for peptide import through Ptr2 based on the crystallographic information of Streptococcus thermophilus peptide transporter PepTst and based on the conservation of primary sequences among the proton-dependent oligopeptide transporters (POTs). Expression of Ptr2 carrying one of the 14 mutations of which the corresponding residues of PepTst are involved in peptide recognition, salt bridge interaction, or peptide translocation failed to enable ptr2Δtrp1 cell growth in alanyl-tryptophan (Ala-Trp) medium. We observed that Ptr2 underwent rapid degradation after cycloheximide treatment (half-life, approximately 1 h), and this degradation depended on Rsp5 ubiquitin ligase. The ubiquitination of Ptr2 most likely occurs at the N-terminal lysines 16, 27, and 34. Simultaneous substitution of arginine for the three lysines fully prevented Ptr2 degradation. Ptr2 mutants of the presumed peptide-binding site (E92Q, R93K, K205R, W362L, and E480D) exhibited severe defects in peptide import and were subjected to Rsp5-dependent degradation when cells were moved to Ala-Trp medium, whereas, similar to what occurs in the wild-type Ptr2, mutant proteins of the intracellular gate were upregulated. These results suggest that Ptr2 undergoes quality control and the defects in peptide binding and the concomitant conformational change render Ptr2 subject to efficient ubiquitination and subsequent degradation.
INTRODUCTION
Nitrogen is imported into the cell in the form of an ammonium ion, amino acid, or peptide through transporter proteins in the plasma membrane. Peptide transporters typically comprise 12 transmembrane domains (TMDs) that are highly conserved among bacteria, animals, and plants. These transporters belong to the proton-coupled oligopeptide transporter (POT) family, which is also referred to as the peptide transporter (PTR) family (1–3). The POT family belongs to the major facilitator superfamily (MFS), members of which contain substrate/H+ symporters that are driven by electrochemical gradient across the plasma membrane (4, 5). In addition to being a source of nitrogen, peptides play beneficial roles in physiological activity. For example, soy peptides, the proteolytic products of soy proteins, can reduce serum lipid levels, scavenge free radicals, and reduce oxidative stress in mammalian cells (6–8). In yeast, soy peptides improve tolerance to freeze-thaw stress, repress the formation of lipid bodies (9), and accelerate ethanol fermentation (10, 11). hPepT1 and hPepT2 are human peptide transporters expressed in the small intestine and the kidney, respectively (12). hPepT1 operates as a high-capacity, low-affinity peptide transporter, whereas hPepT2 operates as a low-capacity, high-affinity transporter. In addition to peptide uptake, these transporters mediate absorption of drugs that have a steric resemblance to peptides, such as β-lactam antibiotics, antihypertensive drugs, anticancer agents, and antivirals (e.g., valacyclovir) (13–17). Hence, investigation of POT family proteins should facilitate fermented food production and drug discovery as well as contribute to our basic understanding of nutrient transport.
In the yeast Saccharomyces cerevisiae, the POT/PTR family protein Ptr2 is the major peptide transporter for importing di- and tripeptides (1, 18, 19). External amino acids regulate PTR2 transcription through the Ssy1-Ptr3-Ssy5 (SPS) signal transduction pathway. In the SPS complex, Ssy1 is a plasma membrane protein that senses external amino acids and induces transcription of PTR2 and several amino acid permease genes (20–23). Dipeptides containing N-terminal basic (Arg, Lys, and His) or bulky hydrophobic (Phe, Leu, Tyr, Trp, and Ile) amino acids, the N-end rule amino acids, also positively regulate PTR2 transcription (24, 25). These dipeptides bind directly to Ubr1 (also known as Ptr1), which leads to Ubr1-dependent degradation of Cup9, a repressor of PTR2 transcription, and consequently, PTR2 upregulation (24, 25). Although PTR2 transcription has been extensively investigated, the mechanism by which the Ptr2 protein recognizes and imports peptides is still unclear. To the best of our knowledge, the only relevant study is a mutational analysis on TMD5 of Ptr2 (26). The FVxxINxG sequence in TMD5 is highly conserved among the POT/PTR family proteins, from yeast to humans. Although replacement of F244, F247, Y248, I251, or G254 by alanine abolished the uptake of Leu-Leu and Met-Met-Met, N252A substitution exhibited differential substrate preference for Met-Met-Met over Leu-Leu (26). Thus, at least TMD5 plays a role in Ptr2 substrate recognition.
Recently, the crystal structures of bacterial homologs for mammalian peptide transporters were solved in PepTso from Shewanella oneidensis (27), PepTst from Streptococcus thermophilus (28), and GkPOT from Geobacillus kaustophilus (29). These crystal structures share a fold in the MFS proteins consisting of two 6-transmembrane bundles facing each other in the membrane with a central cavity between the two bundles. The PepTso structure formed a substrate-bound occluded state (27), whereas the PepTst structure formed an inward-open conformation (28). The GkPOT structures had both an inward-open H+-bound and an inward-open substrate/H+-bound conformation (29). These structures revealed the key mechanistic and functional roles for conserved glutamate (or aspartate) and arginine residues in the peptide-binding site, as well as a number of amino acid residues involved in peptide recognition. Interestingly, both glutamate and arginine residues are not conserved in fungal Ptr2, including S. cerevisiae and Candida albicans, in which glycine and serine residues are positioned, respectively. This suggests an unidentified mechanism of recognition or translocation of substrates in fungal Ptr2 proteins, which might account for the unsolved effects of soy peptides in yeast physiology.
Homology modeling that uses the crystal structure of a closely related protein as a template allows us to determine the functional roles of corresponding amino acid residues in structurally unknown proteins. In this study, we introduced mutations into Ptr2 to verify the significance of 19 amino acid residues based on the structural information of the POT family proteins. Yeast genetics has an advantage in that a functional analysis of a transporter protein can be performed in vivo in combination with an analysis of the expression and subcellular localization of the protein. We also show that Ptr2 undergoes an Rsp5-dependent ubiquitination that likely occurs at N-terminal lysine positions 16, 27, and 34.
MATERIALS AND METHODS
Yeast strains and culture conditions.
The wild-type strain YPH499 (MATa his3-Δ200 leu2-Δ1 lys2-801 trp1-Δ1 ade2-101 ura3-52), the ptr2Δ mutant (ptr2Δ::KanMX in YPH499 [see below]), and the ptr2ΔTRP1 mutant (ptr2Δ::KanMX TRP1 in YPH499 [see below]) were used in this study. Unless otherwise specified, ptr2Δ denotes the ptr2Δ (trp1) mutant. Cells were grown in synthetic dextrose (SD) medium or l-alanyl-l-tryptophan (Ala-Trp) medium at 25°C with shaking. Ala-Trp medium was supplemented with Ala-Trp to complement tryptophan auxotrophy (trp1) instead of l-tryptophan in SD medium and was used for functional analysis of ptr2 mutants. Ala-Trp medium was composed of a yeast nitrogen base without amino acids (6.7 g · liter−1), d-glucose (20 g · liter−1), adenine (20 μg · ml−1), histidine (20 μg · ml−1), lysine (30 μg · ml−1), leucine (90 μg · ml−1), and Ala-Trp (20 μg · ml−1). In addition, l-leucyl-l-tryptophan (Leu-Trp) was used to complement tryptophan auxotrophy. Ala-Trp, Leu-Trp, l-alanyl-l-leucine (Ala-Leu), and l-alanyl-l-alanine (Ala-Ala) were purchased from Kokusan Chemical Co. Ltd. (Tokyo, Japan).
Construction of the ptr2Δ mutant and plasmids.
PCR-based gene disruption was employed in YPH499 to create the ptr2Δ::KanMX mutant using the genome DNA from the ptr2Δ mutant (ptr2Δ::KanMX in BY4742) as a template, and the primers 5′-GGTTTGTAAAATATATAAGTGATGTACA-3′ and 5′-CATTTGAGATAATGGTATGCAAATACGC-3′. The transformant was selected on YPD agar plates containing 200 μg · ml−1 Geneticin. pJJ280 (TRP1) (30) was digested with EcoRI to obtain a TRP1 DNA fragment, which was subsequently introduced into the ptr2Δ mutant to generate the ptr2Δ TRP1 mutant.
The plasmids used in this study are listed in Table 1. pUA35 (TDH3 promoter [pTDH3]-3HA [hemagglutinin]-TDH3 terminator [tTDH3], URA3 CEN4) was used to generate a PTR2-3HA plasmid driven by its own promoter. pUA35 was digested with XhoI and SmaI to remove pTDH3. The PTR2 open reading frame (ORF), with its own promoter region (800 bp upstream of the ORF), was amplified by PCR using the genomic DNA from strain YPH499 as a template and 5′-CGGGCCCCCCCTCGAGGAAGGCAGCGGCGAACTGACTT-3′ and 5′-ACTCATGGTTCCCCCGGGATATTTGGTGGTGGATCTTAGACTTTCCATTG-3′ as the primers. The digested plasmid YCplac33 (URA3 CEN4) (31) and the PCR product were fused by the In-Fusion HD cloning kit (TaKaRa Bio Inc., Otsu, Shiga, Japan) to yield pPTR2-3HA (URA3 CEN4). PCR-based site-directed mutagenesis was performed to create each of the 19 amino acid substitutions in Ptr2 TMDs or at least one lysine-to-arginine (K-to-R) substitution in the Ptr2 N-terminal domain using pPTR2-3HA as a template with the corresponding mutant primers. To generate a PTR2-GFP (where GFP is green fluorescent protein) plasmid, pPTR2-3HA was digested with XhoI and SmaI to obtain a DNA fragment containing the PTR2 ORF with its own promoter region. The resulting fragment was cloned into pKK1 (GFP URA3 CEN4) to yield pPTR2-GFP.
TABLE 1.
Plasmids used in this study
| Plasmid | Description | Source or reference |
|---|---|---|
| pJJ280 | TRP1 | 30 |
| YCplac33 | URA3 CEN4 | 31 |
| pUA35 | pTDH3-3HA-tTDH3, URA3 CEN4 | This study |
| pKK1 | GFP URA3 CEN4 | This study |
| pPTR2–3HA | PTR2-3HA driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2-E92Q-3HA | PTR2-E92Q-3HA driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2-R93K-3HA | PTR2-R93K-3HA driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2-Y96A-3HA | PTR2-Y96A-3HA driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2-Y138A-3HA | PTR2-Y138A-3HA driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2-D149E-3HA | PTR2-D149E-3HA driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2-G168A-3HA | PTR2-G168A-3HA driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2-L196A-3HA | PTR2-L196A-3HA driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2-K205R-3HA | PTR2-K205R-3HA driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2-S209A-3HA | PTR2-S209A-3HA driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2-F244L-3HA | PTR2-F244L-3HA driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2-F247L-3HA | PTR2-F247L-3HA driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2-W362L-3HA | PTR2-W362L-3HA driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2-E480D-3HA | PTR2-E480D-3HA driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2-G487A-3HA | PTR2-G487A-3HA driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2-L488A-3HA | PTR2-L488A-3HA driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2-M504L-3HA | PTR2-M504L-3HA driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2-F507W-3HA | PTR2-F507W-3HA driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2-T510V-3HA | PTR2-T510V-3HA driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2-G514A-3HA | PTR2-G514A-3HA driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2-K16R-3HA | PTR2-K16R-3HA driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2-K27R-3HA | PTR2-K27R-3HA driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2-K34R-3HA | PTR2-K34R-3HA driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2-K77R-3HA | PTR2-K77R-3HA driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2–3K>R-3HAa | PTR2-K16,27,34R-3HA driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2–4K>R-3HA | PTR2-K16,27,34,77R-3HA driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2-E92Q-3K>R-3HA | PTR2-E92Q-K16,27,34R-3HA driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2-R93K-3K>R-3HA | PTR2-R93K-K16,27,34R-3HA driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2-K205R-3K>R-3HA | PTR2-K205R-K16,27,34R-3HA driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2-W362L-3K>R-3HA | PTR2-W362L-K16,27,34R-3HA driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2-E480D-3K>R-3HA | PTR2-E480D-K16,27,34R-3HA driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2-GFP | PTR2-GFP driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2-E92Q-GFP | PTR2-E92Q-GFP driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2-R93K-GFP | PTR2-R93K-GFP driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2-Y96A-GFP | PTR2-Y96A-GFP driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2-Y138A-GFP | PTR2-Y138A-GFP driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2-D149E-GFP | PTR2-D149E-GFP driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2-G168A-GFP | PTR2-G168A-GFP driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2-L196A-GFP | PTR2-L196A-GFP driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2-K205R-GFP | PTR2-K205R-GFP driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2-S209A-GFP | PTR2-S209A-GFP driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2-F244L-GFP | PTR2-F244L-GFP driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2-F247L-GFP | PTR2-F247L-GFP driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2-W362L-GFP | PTR2-W362L-GFP driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2-E480D-GFP | PTR2-E480D-GFP driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2-G487A-GFP | PTR2-G487A-GFP driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2-L488A-GFP | PTR2-L488A-GFP driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2-M504L-GFP | PTR2-M504L-GFP driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2-F507W-GFP | PTR2-F507W-GFP driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2-T510V-GFP | PTR2-T510V-GFP driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2-G514A-GFP | PTR2-G514A-GFP driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2–3K>R-GFP | PTR2-K16,27,34R-GFP driven by PTR2 promoter, URA3 CEN4 | This study |
| pPTR2–4K>R-GFP | PTR2-K16,27,34,77R-GFP driven by PTR2 promoter, URA3 CEN4 | This study |
K>R, K-to-R substitution.
Functional analysis of Ptr2 mutants.
The wild-type or ptr2 mutant cells were grown in SD medium at an optical density at 600 nm (OD600) value of up to ∼1.0 (1.65 × 107 cells · ml−1). The cells were collected by centrifugation, washed twice with distilled water, and resuspended in SD or Ala-Trp medium to obtain cultures at an OD600 value of 0.04. The OD600 values of cultures were measured after incubation for 20 h at 25°C.
Western blot analysis.
Whole-cell extracts were prepared as previously described (32). In brief, 108 cells were collected by centrifugation, washed once with 10 mM NaN3–10 mM NaF, and washed once in lysis buffer A (50 mM Tris-HCl, 5 mM EDTA, 10 mM NaN3, 1 mM phenylmethylsulfonyl fluoride, and one tablet of EDTA-free Complete [Roche, Mannheim, Germany] for 50 ml [pH 7.5]). The cells were broken with glass beads. Unbroken cells and debris were removed by centrifugation at 900 × g for 5 min, and the cleared lysates were treated with 5% SDS and 5% 2-mercaptoethanol at 37°C for 10 min to denature proteins. Western blot analysis was performed as previously described (32) using antibodies for HA (16B12; BabCO, Richmond, CA, USA), GFP (anti-chicken IgY; Aves Labs, Inc., Tigard, OR, USA), and Adh1 (Rockland antibodies and assays, Gilbersville, PA, USA). Adh1 was used as a loading control for the whole-cell extracts. An image analyzer (LAS4000 mini; GE Healthcare Life Sciences, Piscataway, NJ, US) was used for signal detection.
Fluorescence microscopy.
Living cells expressing GFP-tagged Ptr2 proteins were placed on a glass slide and were imaged using a fluorescence microscope model IX70 with magnification of ×100 of the objective lens (Olympus, Co. Ltd., Tokyo, Japan).
Peptide import assay.
Peptide import was analyzed using the procedure for tryptophan import by Tat2, but with modifications (33). A commercially available radiolabeled peptide, [3H]l-alanyl-l-alanine (Ala-Ala, MT-945, 14.8 GBq · mmol−1; Moravek Biochemicals, Inc., Brea, CA, USA), was used for the peptide import assay. Cells were grown in SD medium at a cell density of up to 1.5 × 107 cells · ml−1. The cells were collected by centrifugation, washed twice, and resuspended in SD medium at a cell density of about 5.0 × 107 cells · ml−1. The import assay was performed in the presence of 11.6 μg · ml−1 of nonlabeled Ala-Ala with a 1/2,000 volume of the 3H-labeled Ala-Ala. The molar concentration of nonlabeled Ala-Ala (72.6 μM) was equal to that for the 20-μg · ml−1 Ala-Trp used in the functional analysis. After incubation, the cells were trapped with a vacuum aspirator on GF/C glass filters that were stacked in pairs. The filters were washed with 10 ml cold distilled water. The quantity of imported peptides was measured using a liquid scintillation counter. Under our experimental conditions, 1 dpm of [3H]Ala-Ala can be converted to 42.4 fmol Ala-Ala. Data were expressed as mean values of Ala-Ala incorporated (pmol · 107 cells−1 · min−1) with standard deviations obtained from three independent experiments.
Structural modeling.
The ESyPred3D web server 1.0 software (http://www.fundp.ac.be/sciences/biologie/urbm/bioinfo/esypred/), an automated homology modeling program with an increased alignment performance that is based on the modeling package Modeller (34, 35), was used for modeling. The profile query for Ptr2 yielded the S. thermophilus PepTst (PDB 4APS) as a structural template (28). An alignment was created for the template, and the Ptr2 model was constructed using the PyMOL molecular graphics system, version 1.5.0.4 schrödinger, LLC (http://www.pymol.org/) (36).
RESULTS
Rationale for mutation design and homology modeling of Ptr2.
Figure 1 shows a multiple-sequence alignment of TMDs in some peptide transporters. The ExxERFxYY motif in TMD1 is highly conserved across the POT family proteins and contributes to forming the central cavity within the molecule (3). In PepTst, E25 and R26 (E92 and R93 in Ptr2, respectively) interact through a salt bridge, which is positioned close to E22 in TMD1 (E89 in Ptr2). E22Q, E25Q, and R26K mutations completely inactivate proton-driven peptide uptake but allow the retention of some activity of counterflow in PepTst (28). Y29 (TMD1) and Y68 (TMD2) (Y96 and Y138 in Ptr2, respectively) are thought to help determine peptide specificity. K126 in TMD4 (K205 in Ptr2) is conserved throughout the POT family proteins and is positioned close to the ExxERFxYY motif. K126 and E400 (TMD10) are positioned to form the peptide-binding site. The K126A mutation abolishes only proton-driven peptide import and retains counterflow activity, whereas the E400D mutation abolishes both functions (28). In human hPepT1, the equivalent glutamate (E595) is assumed to bind the N terminus of peptides. W294 in hPepT1 (W362 in Ptr2) plays a role in peptide import (37). Hence, we created E92Q, R93K, Y96A, Y138A, K205R, W362L, and E480D mutations in Ptr2. S130 (TMD4), F148 (TMD5), G407 (TMD10), L408 (TMD10), M424 (TMD11), and W427 (TMD11) in PepTst (S209, F244, G487, L488, M504, and F507 in Ptr2, respectively) are positioned at the intracellular gate, which shows hingelike movement. Hence, we created S209A, F244L, G487A, L488A, M504L, and F507W mutations in Ptr2. In addition to the potentially important 13 amino acid residues, we mutated the following 6 sites according to the pronounced conservation: D149E (TMD2), G168A (TMD3), L196A (TMD4), F247L (TMD5), T510V (TMD11), and G514A (TMD11). R33 and E300 in PepTst are highly conserved across the POT family, and the salt bridge between R33 and E300 is assumed to facilitate close packing in the extracellular gate between TMD1 and -2 and TMD7 and -8 (28). In total, 19 Ptr2 mutants were subjected to the subsequent functional analysis.
FIG 1.

Multiple-sequence alignments of TMDs for some peptide transporters. The alignments were constructed for ScPtr2 (S. cerevisiae), CaPtr2 (C. albicans), hPepT1 and hPepT2 (H. sapiens), PepTso (S. oneidensis), PepTst (S. thermophilus), and GkPOT (G. kaustophilus) using the Praline multiple-sequence alignment program (54). TMD α-helices were assigned based on the crystallographic structure of PepTst (PDB 4APS) (28). Amino acid residues that mutated in this study are marked with an asterisk (*) above the residue, and those that reside in the extracellular gate in bacterial peptide transporters are marked with a dot (•).
For the structural modeling of Ptr2, the ESyPred3D web server 1.0 software proposed an inward-open crystal structure of PepTst (PDB 4APS) as the closest template (28). In the overall modeled structure, the N- and C-terminal 6-helix bundles TMD1 and -6 and TMD7 and -12 face each other to form a pseudo-2-fold-symmetry structure (Fig. 2). There seems to be a putative trajectory for a substrate peptide in the top view of the modeled Ptr2. The 19 amino acid residues mutated in this study are mapped in the structure (Fig. 2).
FIG 2.
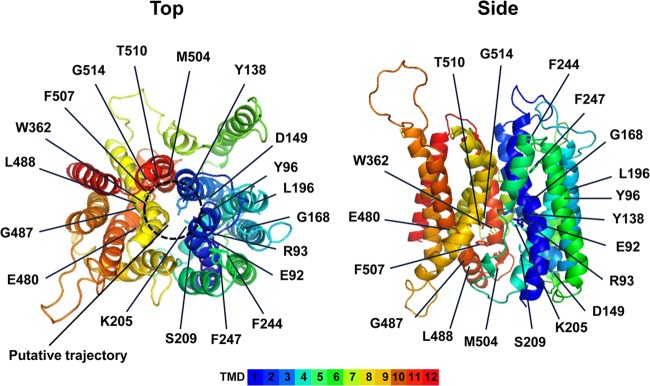
Structural model of Ptr2. The structure of Ptr2 was based on the crystal structure of PepTst inward-open conformation (PDB 4APS) as a template (28). Amino acid residues that were mutated in this study are indicated.
Functional analysis of Ptr2 mutant proteins.
To evaluate the activity of Ptr2 mutant proteins in vivo, we performed a growth assay using nutrient auxotrophy for tryptophan and leucine. First, we examined whether Ala-Trp or Ala-Leu supported growth of the wild-type or ptr2Δ mutant in the absence of tryptophan or leucine, respectively, in SD medium. We confirmed that the wild-type strain grew on 20 μg · ml−1 Ala-Trp and 40 μg · ml−1 tryptophan with equal efficiency, but ptr2Δ mutant growth was defective in 20 μg · ml−1 Ala-Trp (Fig. 3A). Ala-Leu did not support wild-type growth, even at the very high concentration of 100 μg · ml−1, suggesting that the peptide form of leucine cannot meet its high demand in protein synthesis (data not shown). Therefore, Ala-Trp was used for further experiments. Here, the wild-type strain denotes the ptr2Δ cell harboring pPTR2-3HA, and the ptr2Δ mutant denotes the cell harboring an empty vector alone.
FIG 3.
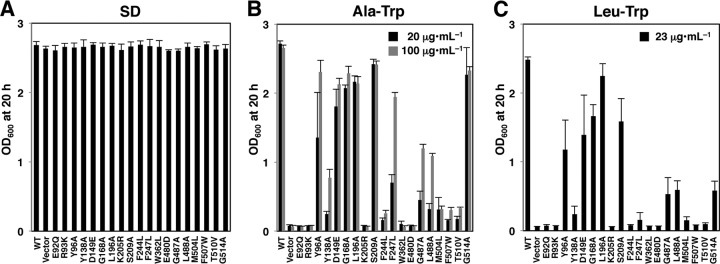
Effects of Ptr2 mutations on cell growth. 3HA-tagged wild-type or Ptr2 mutant proteins were expressed in ptr2Δ cells. The cells were incubated at 25°C for 20 h in SD (40 μg · ml−1 tryptophan) (A), Ala-Trp medium containing 20 or 100 μg · ml−1 Ala-Trp in the absence of free tryptophan (B), or Leu-Trp medium containing 23 μg · ml−1 Leu-Trp in the absence of free tryptophan (C). The starting OD600 value was 0.04.
Cells were grown in SD medium and diluted with Ala-Trp medium to obtain cultures with an OD600 of 0.04. After incubation for 20 h at 25°C, the OD600 values were measured to quantify Ptr2 mutant growth. The 19 Ptr2 mutant cells exhibited growth in the SD medium that was equal to that of the wild-type cells, whereas the mutants showed various degrees of deficient growth in Ala-Trp medium (Fig. 3B). E92Q, R93K, Y138A, K205R, F244L, W362L, E480D, M504L, F507W, and T510V mutants showed severe growth defects. Y96A, F247L, G487A, and L488A mutants showed moderate growth defects, which were partially repressed by a higher concentration of Ala-Trp (100 μg · ml−1). In contrast, D149E, G168A, L196A, S209A, and G514A mutations conferred no obvious growth defect in Ala-Trp (20 μg · ml−1) medium (Fig. 3B). A similar result was obtained with a medium containing an N-end rule dipeptide Leu-Trp (23 μg · ml−1), which was preferred by Ptr2 (25), except for the marked growth defect in the G514A mutant (Fig. 3C).
Expression and localization of Ptr2 mutant proteins.
Next, we examined whether the mutations affected Ptr2 expression. Cells were grown in SD medium and then moved to Ala-Trp (20 μg · ml−1) medium to continue incubating for 5 h. Whole-cell lysates were prepared for Western blot analysis to detect Ptr2-3HA proteins. Consistent with previous reports (25), Ptr2 was upregulated after cells were moved to Ala-Trp medium (Fig. 4A). The Ptr2 mutant proteins were also expressed in SD medium, though the expression levels varied among the strains. We found that Ptr2 carrying the E92Q, R93K, K205R, W362L, or E480D mutation was markedly downregulated after moving the cells to Ala-Trp medium. In contrast, the remaining Ptr2 mutant proteins were upregulated, as they were in the wild-type Ptr2 (Fig. 4A). It should be noted that E92, R93, K205, W362, and E480 are assumed to form the substrate-binding pocket in the Ptr2 structural model (Table 2 and Fig. 2), thereby raising the possibility that the 5 substrate-free forms of Ptr2 may have been efficiently degraded when cells were cultured in Ala-Trp medium. When Ptr2 proteins were expressed in the wild-type PTR2 trp1 strain, the 5 Ptr2 mutant proteins were upregulated when moved to Ala-Trp medium, similar to what was observed in the wild-type Ptr2 (Fig. 4B). Therefore, intracellular Ala-Trp may have prevented the 5 Ptr2 mutant proteins from degradation in the wild-type cells. Further, we examined whether conferring tryptophan prototrophy in the ptr2Δ strain, which enabled growth in Ala-Trp medium, affected the degradation of the 5 Ptr2 mutant proteins. We found that the 5 Ptr2 mutant proteins were degraded when the ptr2Δ TRP1 cells were moved to Ala-Trp medium; however, the degradation of Ptr2 was delayed (Fig. 4C). These results suggest that the substrate-binding defect of Ptr2 and the shortage of intracellular tryptophan promoted Ptr2 degradation (see Discussion).
FIG 4.

Expression of Ptr2 mutant proteins. 3HA-tagged wild-type or Ptr2 mutant proteins were expressed in ptr2Δ trp1 cells (A), PTR2 trp1 cells (B), or ptr2Δ TRP1 cells (C). Exponentially growing cells in SD medium were moved to Ala-Trp (AW) medium (20 μg · ml−1) and incubated for an additional 5 h. Whole-cell extracts were subjected to Western blot analysis to detect Ptr2-3HA and Adh1 using specific antibodies. Adh1 was used as a loading control.
TABLE 2.
Mutated amino acid residues in Ptr2 and corresponding PepTst residues
| Ptr2 mutation | TMDa | Ptr2 functionb | Corresponding PepTst residue | Description |
|---|---|---|---|---|
| None | ++ | |||
| E92Q | 1 | − | E25 | ExxERFxYY motif |
| R93K | 1 | − | R26 | ExxERFxYY motif |
| Y96A | 1 | + | Y29 | ExxERFxYY motif |
| Y138A | 2 | +/− | Y68 | Recognition of C terminus of substrates |
| D149E | 2 | ++ | D79 | Highly conserved residue |
| G168A | 3 | ++ | G98 | Highly conserved residue |
| L196A | 4 | ++ | L117 | Highly conserved residue |
| K205R | 4 | − | K124 | Interaction with the ExxERFxYY motif |
| S209A | 4 | ++ | S130 | Intracellular gate |
| F244L | 5 | − | F148 | Highly conserved residue |
| F247L | 5 | +/− | F151 | Intracellular gate |
| W362L | 7 | − | W296 | Highly conserved residue |
| E480D | 10 | − | E400 | Recognition of N terminus of substrates |
| G487A | 10 | +/− | G407 | Intracellular gate |
| L488A | 10 | +/− | L408 | Intracellular gate |
| M504L | 11 | − | M424 | Intracellular gate |
| F507W | 11 | − | W427 | Intracellular gate |
| T510V | 11 | − | S430 | Interaction with W362 |
| G514A | 11 | ++ | G434 | Highly conserved residue |
TMDs were assigned based on the crystallographic structure of PepTst (PDB 4APS).
Ptr2 function was evaluated by the complementation test for growth of ptr2Δ cells in Ala-Trp medium. Symbols: −, no growth or impaired growth in 20 μg · ml−1 Ala-Trp medium (<10% of the OD600 value with respect to the wild-type cells); +/−, impaired growth in 20 μg · ml−1 Ala-Trp medium (10 to 50% of the OD600 value with respect to the wild-type cells) or growth defect is partially rescued in 100 μg · ml−1 Ala-Trp medium; +, moderately impaired growth in 20 μg · ml−1 Ala-Trp medium (50 to 70% of the OD600 value with respect to the wild-type cells); ++, growth similar to that of the wild-type cells in 20 μg · ml−1 Ala-Trp medium (>70% of the OD600 value with respect to the wild-type cells).
GFP-tagged mutant proteins were expressed in ptr2Δ cells to determine the extent to which the mutations affected Ptr2 localization. The wild-type Ptr2-GFP was localized to the plasma membrane when cells were incubated in both SD and Ala-Trp medium (Fig. 5A, arrowheads). Intensive GFP fluorescence in the vacuole suggests that Ptr2 is subjected to degradation through the endocytic pathway. We confirmed that all mutant Ptr2-GFP proteins were localized to the plasma membrane in cells grown in SD medium, and the vacuoles were intensively stained with GFP (Fig. 5A, lower panels in R93K and W362L). In fact, partially digested products of Ptr2-GFP, which were attributed to the vacuolar signal, were observed in Western blotting using an anti-GFP antibody (Fig. 5B). Therefore, the mutant Ptr2 proteins can be delivered to the cell surface as efficiently as the wild-type protein and are delivered to the vacuole for degradation. Consistent with the Western blotting results in Fig. 4, Ptr2-GFP protein carrying the E92Q, K205R, or E480D mutation was absent from the plasma membrane when moved to Ala-Trp medium (Fig. 5A). Ptr2-GFP proteins carrying the R93K or W362L mutation were predominantly localized in the vacuole when moved to Ala-Trp medium, although these remained in the plasma membrane in some of the cells (∼25%) (Fig. 5A). In addition, Ptr2R93K-GFP and Ptr2W362L-GFP were not efficiently degraded when moved to Ala-Trp medium (Fig. 5B); however, Ptr2R93K-3HA and Ptr2W362L-3HA were rapidly degraded (Fig. 4A). These findings suggest that the C-terminal fused GFP partially stabilizes Ptr2R93K-GFP and Ptr2W362L-GFP in the plasma membrane. In addition, we expressed GFP-tagged mutant Ptr2 in the wild-type PTR2 cells and observed that they were localized to the plasma membrane, similar to the wild-type Ptr2 protein (data not shown). For the subsequent peptide import assay to be valid, we used SD medium rather than Ala-Trp medium for cell culture to ensure plasma membrane localization of Ptr2.
FIG 5.

Localization of Ptr2 mutant proteins in ptr2Δ cells. (A) GFP-tagged wild-type or Ptr2 mutant proteins were expressed in ptr2Δ cells. Exponentially growing cells in SD medium were moved to Ala-Trp (AW) medium (20 μg · ml−1) and incubated for an additional 5 h. The cells were imaged via a fluorescence microscope. Ptr2 in the plasma membrane is shown by arrowheads. Ptr2R93K-GFP and Ptr2W362L-GFP were predominantly localized in the vacuole, although they remained in the plasma membrane in some of the cells (∼25%, lower panels in R93K and W362L). DIC, differential interference contrast. (B) Whole-cell extracts were subjected to Western blot analysis to detect Ptr2-GFP and Adh1 using specific antibodies. Adh1 was used as a loading control.
Peptide uptake by Ptr2 mutant proteins.
To evaluate the effect of Ptr2 mutations quantitatively, we employed peptide import assay using a commercially available [3H]Ala-Ala. First, we performed a competitive assay to examine whether the use of Ala-Ala was valid and consistent with the functional analysis with Ala-Trp. Exponentially growing cells in SD medium were collected, washed, and transferred to [3H]Ala-Ala medium containing 11.6 μg · ml−1 nonlabeled Ala-Ala with a molar concentration that was equal to that for 20 μg · ml−1 Ala-Trp (72.6 μM). The assay was performed in the presence or absence of nonlabeled Ala-Ala or Ala-Trp in various concentrations as competitive substrates. We found that [3H]Ala-Ala uptake decreased when nonlabeled Ala-Ala increased but was more effective with nonlabeled Ala-Trp (Fig. 6A). This finding suggests that both peptides can be transported by Ptr2 but Ala-Trp is preferred over Ala-Ala. Such a preference is consistent with a recent report of a systematic competitive assay that used the fluorescence tracer substrate β-Ala-Lys (AMCA) (38).
FIG 6.

Effects of Ptr2 mutations on Ala-Ala uptake. 3HA-tagged wild-type or Ptr2 mutant proteins were expressed in ptr2Δ cells. (A) Competition import assay for 3H-labeled Ala-Ala with nonlabeled Ala-Ala or Ala-Trp. The wild-type cells were incubated for 10 min with 3H-labeled Ala-Ala in the presence of 72.6 μM nonlabeled Ala-Ala (1×) or additional nonlabeled Ala-Ala or Ala-Trp. (B) Effects of Ptr2 mutations on Ala-Ala uptake. Cells were incubated in 3H-labeled Ala-Ala in the presence of 72.6 μM nonlabeled Ala-Ala. Data are expressed as mean values of amino acid incorporated (pmol · 107 cells−1 min−1) with standard deviations obtained from three independent experiments. Inset, time dependence of Ala-Ala uptake in the wild-type cells.
Eight Ptr2 mutants, E92Q, R93K, Y138A, K205R, W362L, E480D, F507W, and T510V, were selected for peptide import assays, according to the extent of their growth defects. Peptide uptake by the wild-type cells was time dependent and exhibited a linear dependence on incubation time up to 5 min (Fig. 6B, inset). Due to the technical difficulties involved in analyzing 10 samples simultaneously, subsequent measurements were performed at 7.5 min. The wild-type cells imported Ala-Ala at 61 pmol · 107 cells−1 · min−1, whereas the ptr2Δ cells did not import it, confirming the primary role of Ptr2 in peptide import (Fig. 6B). Although the experimental conditions differed, the degree of Ptr2-mediated Ala-Ala uptake was 20-fold greater than Tat1-mediated tyrosine uptake and 40-fold greater than Tat2-mediated tryptophan uptake, the import assays being performed with 19.6 μM tyrosine and tryptophan, respectively (33). At 15-min incubation, a trace amount of Ala-Ala uptake (0.5 pmol · 107 cells−1 · min−1) was observed in the ptr2Δ mutant. This could be mediated by another peptide transporter, which is likely to be the allantoin transporter Dal5 because Opt1 and Opt2 cannot import di- or tripeptides (39). We found that all 8 Ptr2 mutants showed defective peptide uptake, with rates of up to 4.2 pmol · 107 cells−1 · min−1 (Fig. 6B). Accordingly, the 8 amino acid residues are important for peptide recognition and translocation through the Ptr2 molecule.
Ptr2 undergoes ubiquitin-dependent degradation.
To the best of our knowledge, Ptr2 has not been examined for ubiquitin-dependent degradation. Therefore, we examined whether degradation of Ptr2 and its mutant forms were dependent on Rsp5 ubiquitin ligase. We found that the wild-type Ptr2 was degraded rapidly in cells cultured in both SD and Ala-Trp media with the addition of cycloheximide (half-life, approximately 1 h), suggesting a high basal turnover for Ptr2 (Fig. 7A). The HPG1-1 mutant is defective in Rsp5 ubiquitin ligase activity due to an amino acid substitution (P514T) in the catalytic HECT domain of Rsp5 (32). Double deletion of genes encoding Rsp5-binding proteins Bul1 and Bul2 also compromises Rsp5 activity upon ubiquitination (32). We found that degradation of Ptr2 was significantly delayed in the HPG1-1 and the bul1Δbul2Δ mutants compared with that in the wild-type strain, suggesting that Ptr2 undergoes Rsp5-mediated ubiquitination (Fig. 7B). Covalent binding of ubiquitin to lysine residues likely occurs in the cytoplasmic N-terminal domain in amino acid permeases, including Gap1 (40), Tat1 (41), Tat2 (42), and Bap2 (43). Based on homology modeling by PepTst (PDB entry 4APS) as a structural template (Fig. 2) (28), the TMD1 of Ptr2 starts with isoleucine at position 78, and therefore the cytoplasmic N-terminal domain is likely composed of 77 amino acids in which 4 lysine residues are located. We first created single K-to-R substitutions in the N-terminal domain to determine whether lysine is required for Ptr2 degradation. Whole-cell extracts were prepared from ptr2Δ cells expressing 3HA-tagged Ptr2 variant proteins in SD medium. We found that K34R mutation resulted in retarded Ptr2 degradation after 2 h of cycloheximide treatment (Fig. 8A). This stabilizing effect by the K34R mutation was more prominent after 1 h of cycloheximide treatment with respect to degradation of the wild-type Ptr2 (Fig. 8A). These results suggest that K34 is primarily ubiquitinated by Rsp5 for degradation. Similar results were obtained with cell culture in Ala-Trp medium (data not shown). The 4 single K-to-R substitutions conferred normal growth on ptr2Δ cells in Ala-Trp medium, which suggests that the K-to-R variants are functional (Fig. 8B). To examine whether the remaining lysines are required for Ptr2 degradation at its maximum rate, we created multiple K-to-R substitutions in combination with K34R. Compared with the single K34R mutation, double mutations K16R-K34R and K27R-K34R further stabilized Ptr2, which was more pronounced in triple (K16R-K27R-K34R [3K-to-R]) and quadruple (K16R-K27R-K34R-K77R [4K-to-R]) mutations (Fig. 8A). This result suggests that multiple N-terminal lysines are potential targets for Rsp5-mediated ubiquitination. As observed in the ubiquitination-deficient plasma membrane proteins, Ptr23K>R-GFP and Ptr24K>R-GFP were predominantly localized to the plasma membrane in cells in both SD and Ala-Trp media in the absence of vacuolar GFP signals (Fig. 8C). Interestingly, ptr2Δ cells expressing the 3K-to-R or 4K-to-R mutant Ptr2 grew in Ala-Trp medium less efficiently than cells expressing the wild-type Ptr2 or double K-to-R mutants (Fig. 8B). We speculate that the cells consumed Ala-Trp in excess as a result of Ptr23K>R or Ptr24K>R accumulation, and hence, Ala-Trp became insufficient for supporting full growth of the Trp− strain. Otherwise, excess cytoplasmic Ala-Trp might abrogate amino acid homeostasis in the cell.
FIG 7.
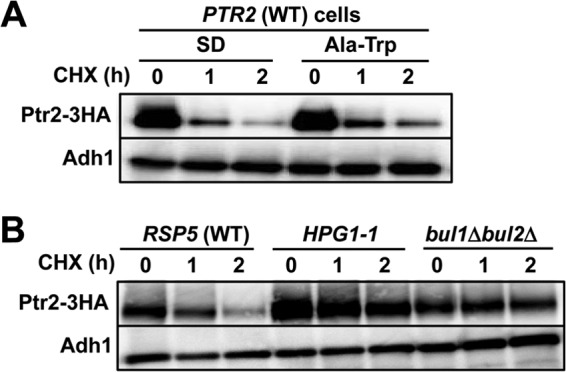
Ubiquitin-dependent degradation of Ptr2 proteins. (A) Rapid degradation of Ptr2 in the wild-type cells. (B) Stabilization of Ptr2 in the HPG1-1 (Rsp5P514T) and the bul1Δbul2Δ mutants cultured in SD medium. Whole-cell extracts were prepared after adding cycloheximide (CHX). Western blot analysis was performed to detect Ptr2-3HA and Adh1 using specific antibodies. Adh1 was used as a loading control.
FIG 8.

Determination of lysine residues required for ubiquitin-dependent Ptr2 degradation. (A) Whole-cell extracts were prepared from ptr2Δ cells expressing Ptr2-3HA with single (top) or multiple (bottom) K-to-R (K>R) substitutions. SD medium was used for culture. Western blot analysis was performed to detect Ptr2-3HA and Adh1 using specific antibodies. Adh1 was used as a loading control. (B) The OD600 values were measured after individual strains were grown in Ala-Trp medium (20 μg · ml−1) at 25°C for 20 h. The starting OD600 value was 0.04. (C) GFP-tagged Ptr2 proteins with multiple K-to-R substitutions were expressed in ptr2Δ cells. Exponentially growing cells in SD medium were moved to Ala-Trp medium (20 μg · ml−1) and incubated for an additional 5 h. The cells were imaged using a fluorescence microscope. DIC, differential interference contrast.
Next, we examined whether peptide-induced degradation of the 5 dysfunctional Ptr2 mutants E92Q, R93K, K205R, W362L, and E480D (Fig. 4A) was also ubiquitin dependent. Initially, there was no measurable difference in steady-state turnover between the wild-type Ptr2 and the dysfunctional Ptr2 mutants on treatment with cycloheximide (Fig. 9A). The HPG1-1 cells expressing Ptr2 with each mutation were grown in SD medium and then moved to Ala-Trp medium to culture for 5 h. We found that the 5 Ptr2 mutant proteins were markedly stabilized in the PTR2 HPG1-1 mutant and the ptr2Δ HPG1-1 mutant in Ala-Trp medium, suggesting that Rsp5 mediates degradation of dysfunctional Ptr2 (Fig. 9B and C). In support of this finding, the introduction of the 3K-to-R mutation to the 5 Ptr2 mutants markedly stabilized the proteins (Fig. 9D). We assume that Ptr2 is subjected to Rsp5-dependent quality control when the peptide import activity is severely impaired. Because the 5 amino acid residues are assumed to reside in the peptide-binding pocket, these Ptr2 mutants might fail to bind peptides, thus preventing concomitant conformational change. Therefore, a peptide-free outward-facing conformation of Ptr2 might be the substrate for Rsp5 ubiquitin ligase.
FIG 9.
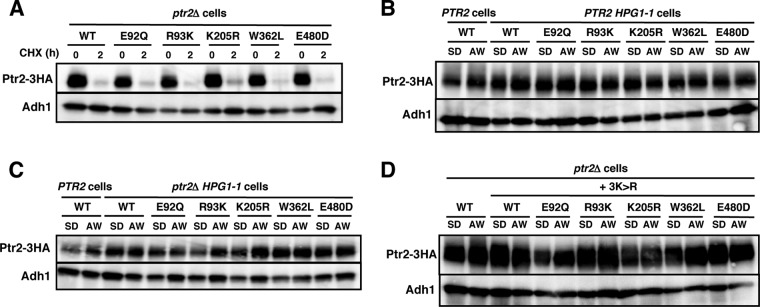
Ubiquitin-dependent degradation of dysfunctional Ptr2 proteins. (A) 3HA-tagged wild-type or Ptr2 mutant proteins were expressed in ptr2Δ cells. Cycloheximide was added to the cells in SD medium for 2 h. (B and C) 3HA-tagged wild-type or Ptr2 mutant proteins were expressed in PTR2 or PTR2 HPG1-1 cells (B) and PTR2 or ptr2Δ HPG1-1 cells (C). (D) 3HA-tagged wild-type or Ptr2 mutant proteins combined with the 3K-to-R (3K>R) substitution were expressed in ptr2Δ cells. Exponentially growing cells in SD medium were moved to Ala-Trp (AW) medium (20 μg · ml−1) and incubated for an additional 5 h. Whole-cell extracts were subjected to Western blot analysis to detect Ptr2-3HA and Adh1 using specific antibodies. Adh1 was used as a loading control.
DISCUSSION
Structural and functional implications of Ptr2.
In this study, we identified amino acid residues required for Ptr2-mediated peptide import using site-directed mutagenesis with the crystal structure of the S. thermophilus PepTst. The growth assay of Ptr2 mutants with 20 μg · ml−1 Ala-Trp as a tryptophan source allowed us to determine dysfunctional Ptr2 mutations when combined with the ptr2Δ trp1 genetic background. Of the 19 amino acid residues tested, 14 contributed to peptide import to various degrees; however, the remaining 5 residues did not, although they are highly conserved across the POT family proteins. The 14 mutations are likely to cause defects in peptide recognition or translocation through the Ptr2 molecule but not defects in structural stability or delivery to the plasma membrane of this protein. Inevitably, amino acid residues assumed to participate in proton-driven peptide uptake (E92 and R93), determination of peptide specificity (Y96 and Y138), peptide binding (K205, E480, and W362), and hingelike movement at the intracellular gate (F244, G487, L488, M504, and F507) play important roles in Ptr2 function. This suggests that the mechanisms for peptide recognition and translocation are similar for Ptr2 and other POT family proteins. However, Ptr2 and PepTst showed different salt bridge interactions at the extracellular gate. In PepTst, two salt bridge interactions facilitated close packing of the extracellular gate helices, which were R53 with E312 and R33 with E300 (28). These amino acid residues are not conserved in fungal peptide transporters (S123 with M378 and S100 with G366, respectively). Mutational analysis in PepTst showed that the R53-E312 salt bridge plays a supportive role whereas the R33-E300 salt bridge is essential during peptide transport. E300A mutation abolishes transport in both proton-driven and peptide-driven counterflow assay of PepTst, whereas R33A mutation abolishes transport only in proton-driven assays (28). These interactions are considered important for orchestrating alternating access with the POT family proteins. Therefore, salt bridges other than those corresponding to PepTst R33-E300 might play a role at the putative extracellular gate in Ptr2. Otherwise, the extracellular gate might be unnecessary in fungal peptide transporters. Currently, Ptr2 has not been sufficiently studied for proton/peptide coupling transport and counterflow activity. Such an analysis in combination with mutational mapping is crucial for describing the undefined features of Ptr2 in peptide import.
One of the central issues to be solved through peptide transporter research is how a single transporter can recognize many di-/tripeptides for transport. The sequence FYxxINxG in TMD5 is highly conserved in the POT family proteins. Alanine-scanning mutagenesis performed on the 22 amino acid residues in TMD5 revealed that the FYxxINxG sequence plays a role in substrate preference: the wild-type Ptr2 prefers Leu-Leu to Met-Met-Met for uptake at a ratio of 0.6. Five mutations, T239A, Q241A, N242A, M245A, and A260G, resulted in an increased preference for Leu-Leu over Met-Met-Met relative to the wild-type preference, whereas 4 mutations, L240A, M250A, N252A, and L258A, conferred a preference for Met-Met-Met rather than Leu-Leu, particularly N252A and L258A (19). It is rationalized in the modeled structure of Ptr2 that most of the 9 residues are facing toward the putative peptide-binding site. The substrate multispecificity of Ptr2 was examined recently using a dipeptide library in which dipeptides composed of hydrophobic or bulky amino acids (Trp, Phe, His, Tyr, Met, or Leu) were preferred by Ptr2, whereas negatively charged and/or small amino acids (Asp, Glu, Gly, or Pro) were not (38). Ala-Trp was categorized into the high-affinity preferable substrate. Transcription of PTR2 is regulated by the N-end rule pathway, through which dipeptide binding with a certain N terminus to Ubr1 promotes degradation of Cup9, a repressor of PTR2 (24, 25). Consistently, the N terminus of peptides with a high-affinity for Ptr2 mostly comprises the N-end rule amino acids (Trp, Phe, His, Tyr, Leu, Ile, and Lys) (38). In a Ptr2 structural model based on the crystal structure of S. oneidensis PepTso (PDB 2XUT), a dipeptide is situated in a peptide-binding pocket that is surrounded by multiple amino acid residues, including R93, Y138, K205, and E480. Most recently, there was one substantive publication on structural issues with the substrate multispecificity of PepTst (44). The study presented crystal structures of di- and tripeptide-bound complexes of PepTst, which revealed that Ala-Phe was oriented laterally in the binding site, whereas Ala-Ala-Ala showed an alternative vertical binding mode. Furthermore, the peptide-biding site can co-opt the same or similar pockets to accommodate the side chains of different ligands, linking their formation and dissolution with the different states in the transport cycle to facilitate peptide recognition and release.
Ubiquitin-dependent degradation of Ptr2.
PTR2 has been analyzed extensively with respect to transcriptional regulation in S. cerevisiae (45–47) and Schizosaccharomyces pombe (48). Peptide import is upregulated by growth media containing poor nitrogen sources such as allantoin, isoleucine, or proline due to upregulation of PTR2 transcription (49). In media containing rich nitrogen sources such as ammonium, PTR2 expression is downregulated via catabolite repression (50). Addition of leucine and tryptophan promotes expression of PTR2. Thus, in the present study, Ptr2 proteins were substantially expressed in SD medium that contained 90 μg · ml−1 leucine and 40 μg · ml−1 tryptophan, even though the medium contained rich nitrogen sources. Induction of PTR2 by extracellular amino acids requires the SPS (Ssy1-Ptr3-Ssy5) sensor signal transduction system (21). PTR2 transcription is also induced by dipeptides containing N-end rule amino acids (24, 25). The dipeptides bind to Ubr1 followed by degradation of the Cup9 repressor. Despite a considerable amount of transcriptional analyses, posttranslational regulation of Ptr2 has yet to be characterized. We showed that Ptr2 underwent rapid degradation when cells were cultured in either SD or Ala-Trp medium, suggesting that the steady-state turnover rate of Ptr2 is rapid but unaffected by external nitrogen species. Rapid degradation of Ptr2 depends on Rsp5 ubiquitin ligase and its binding proteins Bul1 and Bul2 in a manner similar to that of amino acid permeases Gap1 and Tat2 or uracil permease Fur4. This ubiquitination occurs most probably at K16, K27, and K34 in the N-terminal cytoplasmic domain of Ptr2; however, whether ubiquitin covalently binds to the lysines has not been tested. Ptr2 carrying the E92Q, R93K, K205R, W362L, or E480D mutation was dysfunctional and eventually was degraded through ubiquitination when cells were cultured in Ala-Trp medium. There are at least two possibilities to account for the peptide-induced degradation of dysfunctional Ptr2. First, E92, R93, K205, W362, and E480 are thought to reside in the substrate-binding pocket according to the Ptr2 structural model. Therefore, the 5 mutations could abrogate Ptr2 to form a substrate-binding occluded conformation, and hence, the 5 Ptr2 mutant proteins (here termed binding mutants) might remain in the substrate-free outward-facing conformation. Eventually, Ptr2 in this conformation is regarded as a preferential target for Rsp5-dependent ubiquitination. On the other hand, the remaining 9 Ptr2 mutant proteins, where most mutation sites are located within the intracellular gate (here termed gate mutants), could form the substrate-binding occluded conformation that may not be a preferential target for ubiquitination. Second, the gate mutants retain marginal peptide importing activity because addition of 100 μg · ml−1 Ala-Trp partially restored growth defects in ptr2Δ cells, which was not the case in the binding mutants (Fig. 3B). In the gate mutant cells, internal tryptophan might be sufficient to continue protein synthesis and prevent efficient ubiquitination of Ptr2. In contrast, the binding mutant cells are depleted for tryptophan and eventually downregulate Ptr2 for efficient degradation. This is possible because degradation of the 5 binding mutants was delayed in tryptophan prototrophic cells when moved to Ala-Trp medium (Fig. 4C). Although these models do not explain why Ptr2 is stably expressed in peptide-free SD medium, we speculate that a certain mechanism maintains Ptr2 expression insomuch as cell growth is supported by free amino acids.
Addition of uracil induces rapid downregulation of the transporter Fur4 (51). The Fur4 structure was modeled by crystal structures of Mhp1, a bacterial homolog of Fur4. The N-terminal domain of Mhp1, which is composed of 20 amino acid residues, runs parallel to the membrane along a groove between the cytoplasmic loops. In the outward-facing or ground state of Mhp1, the N-terminal domain is kept in position by hydrogen bond interactions with the loops and the C-terminal domain; however, the interactions are attenuated in the inward-facing state (51). A mechanism that is based on a conformation-sensing domain, LID (loop interaction domain), which regulates substrate-induced downregulation and quality control in Fur4, has been proposed. The LID is located within the N-terminal cytoplasmic tail that interacts with intermembrane loop regions, thereby stabilizing the outward facing or ground state of Fur4. In the proposed model, conformational change to the inward-facing state caused by substrate binding or increased import activity in Fur4 transmits to the LID, which becomes accessible to Rsp5 ubiquitin ligase (51). Although Ptr2 does not contain the LID consensus motif, it might have a similar domain in the cytoplasmic tail that senses the conformational status of this transporter.
Gap1 is the best-characterized amino acid permease for nitrogen-induced degradation. When cells are grown on poor nitrogen sources, such as urea or proline, GAP1 transcription increases and Gap1 accumulates in the plasma membrane. Upon addition of rich nitrogen sources, such as ammonium ions or glutamate, Gap1 is ubiquitinated by Rsp5 on the N-terminal K9 or K16, which is followed by delivery to the multivesicular body and then to the vacuole for degradation (40). As a new and distinct mechanism of amino acid-dependent downregulation of Gap1 activity, Gap1 itself is likely to be the sensor for high amino acid levels. It was shown that an exogenously added rich mixture of amino acids (0.25% Casamino Acids) inactivated amino acid import through Gap1K9R-K16R, which resides at the plasma membrane (52). A mutant Gap1A297V imports glycine as efficiently as wild-type Gap1 does, but it is unable to import basic amino acids such as citrulline, lysine, or arginine. Based on a competitive import assay, basic amino acids appear to bind to the active site of Gap1A297V with the same affinity as the wild-type Gap1, but after substrate binding, Gap1A297V cannot undergo the conformational changes necessary to complete the transport cycle (53). Although Casamino Acids inactivate glycine import through Gap1K9R-K16R-A297V, basic amino acids have no effect on glycine import through the mutant protein (53). Therefore, the amino acid-dependent inactivation of Gap1 at the plasma membrane requires active transport of amino acids through the permease. To validate the concept in Ptr2, we will examine whether exogenously added peptides bind to the Ptr2 gate mutants but not to the binding mutants and whether the presumed conformational changes upon peptide binding could prevent Ptr2 ubiquitination. Peptides other than Ala-Trp and Ala-Ala that can be imported through the Ptr2 binding mutants may not induce ubiquitin-dependent degradation of the transporter proteins.
ACKNOWLEDGMENTS
We thank Masahiro Sugiyama and Mayu Miyachi for technical assistance and Keisuke Ito for valuable discussions.
This work was supported by grants from the Japan Society for the Promotion of Science (No. 24580122 to F. Abe) and the Program for the Strategic Research Foundation at Private Universities by the Ministry of Education, Culture, Sports, Science, and Technology (No. 2013-2017).
Footnotes
Published ahead of print 29 August 2014
REFERENCES
- 1.Steiner HY, Naider F, Becker JM. 1995. The PTR family: a new group of peptide transporters. Mol. Microbiol. 16:825–834. 10.1111/j.1365-2958.1995.tb02310.x. [DOI] [PubMed] [Google Scholar]
- 2.Daniel H, Spanier B, Kottra G, Weitz D. 2006. From bacteria to man: archaic proton-dependent peptide transporters at work. Physiology (Bethesda) 21:93–102. 10.1152/physiol.00054.2005. [DOI] [PubMed] [Google Scholar]
- 3.Newstead S. 2011. Towards a structural understanding of drug and peptide transport within the proton-dependent oligopeptide transporter (POT) family. Biochem. Soc. Trans. 39:1353–1358. 10.1042/BST0391353. [DOI] [PubMed] [Google Scholar]
- 4.Pao SS, Paulsen IT, Saier MH., Jr 1998. Major facilitator superfamily. Microbiol. Mol. Biol. Rev. 62:1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yan N. 2013. Structural advances for the major facilitator superfamily (MFS) transporters. Trends Biochem. Sci. 38:151–159. 10.1016/j.tibs.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 6.Hori G, Wang MF, Chan YC, Komatsu T, Wong Y, Chen TH, Yamamoto K, Nagaoka S, Yamamoto S. 2001. Soy protein hydrolyzate with bound phospholipids reduces serum cholesterol levels in hypercholesterolemic adult male volunteers. Biosci. Biotechnol. Biochem. 65:72–78. 10.1271/bbb.65.72. [DOI] [PubMed] [Google Scholar]
- 7.Takenaka A, Annaka H, Kimura Y, Aoki H, Igarashi K. 2003. Reduction of paraquat-induced oxidative stress in rats by dietary soy peptide. Biosci. Biotechnol. Biochem. 67:278–283. 10.1271/bbb.67.278. [DOI] [PubMed] [Google Scholar]
- 8.Tsuruki T, Kishi K, Takahashi M, Tanaka M, Matsukawa T, Yoshikawa M. 2003. Soymetide, an immunostimulating peptide derived from soybean beta-conglycinin, is an fMLP agonist. FEBS Lett. 540:206–210. 10.1016/S0014-5793(03)00265-5. [DOI] [PubMed] [Google Scholar]
- 9.Kitagawa S, Mukai N, Furukawa Y, Adachi K, Mizuno A, Iefuji H. 2008. Effect of soy peptide on brewing beer. J. Biosci. Bioeng. 105:360–366. 10.1263/jbb.105.360. [DOI] [PubMed] [Google Scholar]
- 10.Izawa S, Ikeda K, Takahashi N, Inoue Y. 2007. Improvement of tolerance to freeze-thaw stress of baker's yeast by cultivation with soy peptides. Appl. Microbiol. Biotechnol. 75:533–537. 10.1007/s00253-007-0855-6. [DOI] [PubMed] [Google Scholar]
- 11.Ikeda K, Kitagawa S, Tada T, Iefuji H, Inoue Y, Izawa S. 2011. Modification of yeast characteristics by soy peptides: cultivation with soy peptides represses the formation of lipid bodies. Appl. Microbiol. Biotechnol. 89:1971–1977. 10.1007/s00253-010-3001-9. [DOI] [PubMed] [Google Scholar]
- 12.Nielsen CU, Brodin B. 2003. Di/tri-peptide transporters as drug delivery targets: regulation of transport under physiological and patho-physiological conditions. Curr. Drug Targets 4:373–388. 10.2174/1389450033491028. [DOI] [PubMed] [Google Scholar]
- 13.Wenzel U, Thwaites DT, Daniel H. 1995. Stereoselective uptake of beta-lactam antibiotics by the intestinal peptide transporter. Br. J. Pharmacol. 116:3021–3027. 10.1111/j.1476-5381.1995.tb15958.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tamai I, Nakanishi T, Hayashi K, Terao T, Sai Y, Shiraga T, Miyamoto K, Takeda E, Higashida H, Tsuji A. 1997. The predominant contribution of oligopeptide transporter PepT1 to intestinal absorption of beta-lactam antibiotics in the rat small intestine. J. Pharm. Pharmacol. 49:796–801. 10.1111/j.2042-7158.1997.tb06115.x. [DOI] [PubMed] [Google Scholar]
- 15.Ganapathy ME, Huang W, Wang H, Ganapathy V, Leibach FH. 1998. Valacyclovir: a substrate for the intestinal and renal peptide transporters PEPT1 and PEPT2. Biochem. Biophys. Res. Commun. 246:470–475. 10.1006/bbrc.1998.8628. [DOI] [PubMed] [Google Scholar]
- 16.Faria TN, Timoszyk JK, Stouch TR, Vig BS, Landowski CP, Amidon GL, Weaver CD, Wall DA, Smith RL. 2004. A novel high-throughput PepT1 transporter assay differentiates between substrates and antagonists. Mol. Pharm. 1:67–76. 10.1021/mp034001k. [DOI] [PubMed] [Google Scholar]
- 17.Smith DE, Clemencon B, Hediger MA. 2013. Proton-coupled oligopeptide transporter family SLC15: physiological, pharmacological and pathological implications. Mol. Aspects Med. 34:323–336. 10.1016/j.mam.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perry JR, Basrai MA, Steiner HY, Naider F, Becker JM. 1994. Isolation and characterization of a Saccharomyces cerevisiae peptide transport gene. Mol. Cell. Biol. 14:104–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hauser M, Narita V, Donhardt AM, Naider F, Becker JM. 2001. Multiplicity and regulation of genes encoding peptide transporters in Saccharomyces cerevisiae. Mol. Membr. Biol. 18:105–112. 10.1080/713816114. [DOI] [PubMed] [Google Scholar]
- 20.Barnes D, Lai W, Breslav M, Naider F, Becker JM. 1998. PTR3, a novel gene mediating amino acid-inducible regulation of peptide transport in Saccharomyces cerevisiae. Mol. Microbiol. 29:297–310. 10.1046/j.1365-2958.1998.00931.x. [DOI] [PubMed] [Google Scholar]
- 21.Forsberg H, Gilstring CF, Zargari A, Martinez P, Ljungdahl PO. 2001. The role of the yeast plasma membrane SPS nutrient sensor in the metabolic response to extracellular amino acids. Mol. Microbiol. 42:215–228. 10.1046/j.1365-2958.2001.02627.x. [DOI] [PubMed] [Google Scholar]
- 22.Forsberg H, Hammar M, Andreasson C, Moliner A, Ljungdahl PO. 2001. Suppressors of ssy1 and ptr3 null mutations define novel amino acid sensor-independent genes in Saccharomyces cerevisiae. Genetics 158:973–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Forsberg H, Ljungdahl PO. 2001. Genetic and biochemical analysis of the yeast plasma membrane Ssy1p-Ptr3p-Ssy5p sensor of extracellular amino acids. Mol. Cell. Biol. 21:814–826. 10.1128/MCB.21.3.814-826.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Byrd C, Turner GC, Varshavsky A. 1998. The N-end rule pathway controls the import of peptides through degradation of a transcriptional repressor. EMBO J. 17:269–277. 10.1093/emboj/17.1.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Turner GC, Du F, Varshavsky A. 2000. Peptides accelerate their uptake by activating a ubiquitin-dependent proteolytic pathway. Nature 405:579–583. 10.1038/35014629. [DOI] [PubMed] [Google Scholar]
- 26.Hauser M, Kauffman S, Naider F, Becker JM. 2005. Substrate preference is altered by mutations in the fifth transmembrane domain of Ptr2p, the di/tri-peptide transporter of Saccharomyces cerevisiae. Mol. Membr. Biol. 22:215–227. 10.1080/09687860500093248. [DOI] [PubMed] [Google Scholar]
- 27.Newstead S, Drew D, Cameron AD, Postis VL, Xia X, Fowler PW, Ingram JC, Carpenter EP, Sansom MS, McPherson MJ, Baldwin SA, Iwata S. 2011. Crystal structure of a prokaryotic homologue of the mammalian oligopeptide-proton symporters, PepT1 and PepT2. EMBO J. 30:417–426. 10.1038/emboj.2010.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Solcan N, Kwok J, Fowler PW, Cameron AD, Drew D, Iwata S, Newstead S. 2012. Alternating access mechanism in the POT family of oligopeptide transporters. EMBO J. 31:3411–3421. 10.1038/emboj.2012.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Doki S, Kato HE, Solcan N, Iwaki M, Koyama M, Hattori M, Iwase N, Tsukazaki T, Sugita Y, Kandori H, Newstead S, Ishitani R, Nureki O. 2013. Structural basis for dynamic mechanism of proton-coupled symport by the peptide transporter POT. Proc. Natl. Acad. Sci. U. S. A. 110:11343–11348. 10.1073/pnas.1301079110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jones JS, Prakash L. 1990. Yeast saccharomyces cerevisiae selectable markers in pUC18 polylinkers. Yeast 6:363–366. 10.1002/yea.320060502. [DOI] [PubMed] [Google Scholar]
- 31.Gietz RD, Sugino A. 1988. New yeast-escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene 74:527–534. 10.1016/0378-1119(88)90185-0. [DOI] [PubMed] [Google Scholar]
- 32.Abe F, Iida H. 2003. Pressure-induced differential regulation of the two tryptophan permeases Tat1 and Tat2 by ubiquitin ligase Rsp5 and its binding proteins, Bul1 and Bul2. Mol. Cell. Biol. 23:7566–7684. 10.1128/MCB.23.21.7566-7584.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kanda N, Abe F. 2013. Structural and functional implications of the yeast high-affinity tryptophan permease Tat2. Biochemistry 52:4296–4307. 10.1021/bi4004638. [DOI] [PubMed] [Google Scholar]
- 34.Sali A, Blundell TL. 1993. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234:779–815. 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 35.Lambert C, Leonard N, De Bolle X, Depiereux E. 2002. ESyPred3D: prediction of proteins 3D structures. Bioinformatics 18:1250–1256. 10.1093/bioinformatics/18.9.1250. [DOI] [PubMed] [Google Scholar]
- 36.DeLano W. 2002. The PyMOL molecular graphics system, version 1.5.0.4 schrödinger, LLC (http://www.pymol.org/). The PyMOL user's manual. DeLano Scientific, San Carlos, CA. [Google Scholar]
- 37.Bolger MB, Haworth IS, Yeung AK, Ann D, von Grafenstein H, Hamm-Alvarez S, Okamoto CT, Kim KJ, Basu SK, Wu S, Lee VH. 1998. Structure, function, and molecular modeling approaches to the study of the intestinal dipeptide transporter PepT1. J. Pharm. Sci. 87:1286–1291. 10.1021/js980090u. [DOI] [PubMed] [Google Scholar]
- 38.Ito K, Hikida A, Kawai S, Lan VT, Motoyama T, Kitagawa S, Yoshikawa Y, Kato R, Kawarasaki Y. 2013. Analysing the substrate multispecificity of a proton-coupled oligopeptide transporter using a dipeptide library. Nat. Commun. 4:2502. 10.1038/ncomms3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wiles AM, Cai H, Naider F, Becker JM. 2006. Nutrient regulation of oligopeptide transport in Saccharomyces cerevisiae. Microbiology 152:3133–3145. 10.1099/mic.0.29055-0. [DOI] [PubMed] [Google Scholar]
- 40.Soetens O, De Craene JO, Andre B. 2001. Ubiquitin is required for sorting to the vacuole of the yeast general amino acid permease, Gap1. J. Biol. Chem. 276:43949–43957. 10.1074/jbc.M102945200. [DOI] [PubMed] [Google Scholar]
- 41.Suzuki A, Mochizuki T, Uemura S, Hiraki T, Abe F. 2013. Pressure-induced endocytic degradation of the Saccharomyces cerevisiae low-affinity tryptophan permease Tat1 is mediated by Rsp5 ubiquitin ligase and functionally redundant PPxY motif proteins. Eukaryot. Cell 12:990–997. 10.1128/EC.00049-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Beck T, Schmidt A, Hall MN. 1999. Starvation induces vacuolar targeting and degradation of the tryptophan permease in yeast. J. Cell Biol. 146:1227–1238. 10.1083/jcb.146.6.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Omura F, Kodama Y, Ashikari T. 2001. The N-terminal domain of the yeast permease Bap2p plays a role in its degradation. Biochem. Biophys. Res. Commun. 287:1045–1050. 10.1006/bbrc.2001.5697. [DOI] [PubMed] [Google Scholar]
- 44.Lyons JA, Parker JL, Solcan N, Brinth A, Li D, Shah ST, Caffrey M, Newstead S. 2014. Structural basis for polyspecificity in the POT family of proton-coupled oligopeptide transporters. EMBO Rep. 15:886–893. 10.15252/embr.201338403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cai H, Hauser M, Naider F, Becker JM. 2007. Differential regulation and substrate preferences in two peptide transporters of Saccharomyces cerevisiae. Eukaryot. Cell 6:1805–1813. 10.1128/EC.00257-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xia Z, Turner GC, Hwang CS, Byrd C, Varshavsky A. 2008. Amino acids induce peptide uptake via accelerated degradation of CUP9, the transcriptional repressor of the PTR2 peptide transporter. J. Biol. Chem. 283:28958–28968. 10.1074/jbc.M803980200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hwang CS, Varshavsky A. 2008. Regulation of peptide import through phosphorylation of Ubr1, the ubiquitin ligase of the N-end rule pathway. Proc. Natl. Acad. Sci. U. S. A. 105:19188–19193. 10.1073/pnas.0808891105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kitamura K, Nakase M, Tohda H, Takegawa K. 2012. The ubiquitin ligase Ubr11 is essential for oligopeptide utilization in the fission yeast Schizosaccharomyces pombe. Eukaryot. Cell 11:302–310. 10.1128/EC.05253-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Island MD, Perry JR, Naider F, Becker JM. 1991. Isolation and characterization of S. cerevisiae mutants deficient in amino acid-inducible peptide transport. Curr. Genet. 20:457–463. 10.1007/BF00334772. [DOI] [PubMed] [Google Scholar]
- 50.Marzluf GA. 1997. Genetic regulation of nitrogen metabolism in the fungi. Microbiol. Mol. Biol. Rev. 61:17–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Keener JM, Babst M. 2013. Quality control and substrate-dependent downregulation of the nutrient transporter Fur4. Traffic 14:412–427. 10.1111/tra.12039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Risinger AL, Cain NE, Chen EJ, Kaiser CA. 2006. Activity-dependent reversible inactivation of the general amino acid permease. Mol. Biol. Cell 17:4411–4119. 10.1091/mbc.E06-06-0506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cain NE, Kaiser CA. 2011. Transport activity-dependent intracellular sorting of the yeast general amino acid permease. Mol. Biol. Cell 22:1919–1929. 10.1091/mbc.E10-10-0800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Simossis VA, Heringa J. 2005. PRALINE: a multiple sequence alignment toolbox that integrates homology-extended and secondary structure information. Nucleic Acids Res. 33:W289–W294. 10.1093/nar/gki390. [DOI] [PMC free article] [PubMed] [Google Scholar]