

Abstract
Lipolysis is a delicate process involving complex signaling cascades and sequential enzymatic activations. In Caenorhabditis elegans, fasting induces various physiological changes, including a dramatic decrease in lipid contents through lipolysis. Interestingly, C. elegans lacks perilipin family genes which play a crucial role in the regulation of lipid homeostasis in other species. Here, we demonstrate that in the intestinal cells of C. elegans, a newly identified protein, lipid droplet protein 1 (C25A1.12; LID-1), modulates lipolysis by binding to adipose triglyceride lipase 1 (C05D11.7; ATGL-1) during nutritional deprivation. In fasted worms, lipid droplets were decreased in intestinal cells, whereas suppression of ATGL-1 via RNA interference (RNAi) resulted in retention of stored lipid droplets. Overexpression of ATGL-1 markedly decreased lipid droplets, whereas depletion of LID-1 via RNAi prevented the effect of overexpressed ATGL-1 on lipolysis. In adult worms, short-term fasting increased cyclic AMP (cAMP) levels, which activated protein kinase A (PKA) to stimulate lipolysis via ATGL-1 and LID-1. Moreover, ATGL-1 protein stability and LID-1 binding were augmented by PKA activation, eventually leading to increased lipolysis. These data suggest the importance of the concerted action of lipase and lipid droplet protein in the response to fasting signals via PKA to maintain lipid homeostasis.
INTRODUCTION
Fasting is one of the most prevalent environmental conditions that directly modulate whole-body energy metabolism. Caenorhabditis elegans has received a lot of attention as a genetically tractable model organism for studying the molecular mechanisms of adaptive responses to nutritional changes (1, 2). C. elegans has its own developmental programs for nutritional adaptations. For instance, at multiple stages of larval development, food deprivation halts the developmental program and promotes entry into the diapause state (3). In addition, during larval stage 2 (L2), C. elegans can enter an alternative developmental stage, called dauer, to withstand long periods of starvation (4, 5). In adult worms, food availability influences diverse aspects of physiological responses, including metabolic gene expression (6), locomotive behavior (7), pharyngeal pumping (8), germ line stem cell proliferation (9), and egg laying (10, 11). Like mammals, C. elegans stores large amounts of lipids, in intestinal cells, and expresses most key metabolic proteins, such as SREBP, AMPK, C/EBP, TOR, and nuclear hormone receptors (12–15). Therefore, it is very likely that C. elegans has complex regulatory mechanisms to coordinate systemic energy homeostasis upon nutritional changes such as fasting and feeding.
During nutritional deprivation, lipolysis plays important roles to provide an energy source (16, 17). Most neutral lipids, including triglycerides and cholesterol esters, are stored in the form of intracellular lipid droplets, highly dynamic organelles involved in cellular lipid homeostasis (18). Numerous proteins have been shown to associate with lipid droplets (19, 20). The perilipin family is one of the best-studied lipid droplet protein groups conserved from Dictyostelium to mammals (21, 22). In mammalian adipocytes, perilipin 1 (Plin1) is a major lipid droplet-binding protein that regulates lipase activity. In the basal state, Plin1 surrounds lipid droplets and blocks lipolysis. However, in response to catecholamine signals, it promotes lipolysis by inducing the translocation of hormone-sensitive lipase (HSL) into lipid droplets (23, 24). In addition, Plin1 binds to comparative gene identification-58 (CGI-58; ABHD5), a coactivator of adipose triglyceride lipase (ATGL), and releases CGI-58 to mediate lipolysis upon protein kinase A (PKA) activation (25–29). Interestingly, the genome of C. elegans lacks genes with a perilipin homology domain. Given that C. elegans is able to modulate lipid metabolism to reflect nutritional states even without perilipin (12), it is plausible to speculate that C. elegans may have unique regulatory mechanisms of lipolytic protection under basal conditions and lipolytic activation under energy-demanding conditions.
In mammals, adipocytes, as energy reservoirs, sense and integrate various endocrine signals to modulate lipolytic activity. Fatty acids released from adipocytes are subsequently transported to other target tissues and used as key substrates for energy production via fatty acid oxidation (30). On the other hand, excess accumulation of intracellular lipids in ectopic fat tissues often impairs physiological responses due to lipotoxicity (31). Thus, it is crucial to decipher how lipases are temporally and spatially regulated to access lipid droplets in response to fasting signals. Based on sequence homology, C. elegans has at least 32 genes which are annotated as potential lipases. And a subset of lipases has been reported to function in the long-term survival of dauers (32), longevity (33), induction of autophagy (34), and lysosomal lipolysis during fasting (35). Previously, we reported that endoplasmic reticulum (ER) resident proteins IRE-1 and HSP-4 are associated with induction of fil-1 and fil-2, which are required for the decrease of Nile red-positive lipid granules upon fasting (36). Recently, it has been suggested that in C. elegans, the major fat-storing organelles are distinct from lysosome-related organelles which are stained by the Nile red dye in live worms (37, 38). However, other studies have shown that Nile red staining after fixation or Oil Red O staining correlates with biochemical triglyceride content (37, 39).
In this study, we have identified key genes involved in fasting-induced lipolysis and their regulatory pathways in C. elegans. In adult worms, ATGL-1 (C05D11.7) was identified as a key lipase in the response to fasting, and a lipid droplet protein, LID-1 (lipid droplet protein 1; C25A1.12), was determined to be critical for ATGL-1 function. In worms, fasting stimulated cyclic AMP (cAMP) and PKA, which led to elevation of the levels of ATGL-1 protein for efficient lipolysis via translocation to lipid droplets with LID-1. Together, these data suggest that in C. elegans, LID-1 and ATGL-1 coordinately mediate lipolysis to dissipate stored energy sources through PKA activation during nutritional deprivation.
MATERIALS AND METHODS
Worm strains and culture.
N2 Bristol was used as the wild-type strain. The following alleles and transgenes were used: kin-2(ce179), hjIs67[atgl-1p::atgl-1::gfp], and Ex[act-5p::lid-1::gfp]. All animals were raised at 20°C in standard nematode growth medium (NGM).
Feeding RNAi.
RNA interference (RNAi) clones were obtained from the Ahringer and Vidal RNAi library. The kin-1 RNAi clone was generated by cloning the cDNA fragment of kin-1. Synchronized worms were cultured on RNAi plates until they reached the young adult stage. The efficiency of RNAi was confirmed by real-time quantitative PCR (qPCR) using appropriate PCR primers.
Fasting assays.
Synchronized L1 larvae were grown to the 1-day young-adult stage. Feeding group worms were harvested and washed with M9 buffer before proceeding to the next step. Except for the treatment of chemicals, worms were fasted in empty plates for 4 or 8 h, harvested, and washed. Then the prepared worms were resuspended with buffers appropriate for further analysis. For MG132 or forskolin (FSK) treatment, worms were preincubated in M9 buffer with Escherichia coli and 100 μM MG132 or 100 μM forskolin for 2 h. Then worms were washed and incubated for additional 4 h in M9 buffer with 100 μM MG132 or 100 μM forskolin in the presence or absence of Escherichia coli, depending feeding or fasting, respectively.
Lipid staining and quantification.
Oil Red O staining was performed as previously reported (40). Briefly, worms were harvested and resuspended in 60 μl of 1× phosphate-buffered saline (PBS; pH 7.4), 120 μl of 2× MRWB buffer {160 mM KCl, 40 mM NaCl, 14 mM Na2-EGTA, 1 mM spermidine-HCl, 0.4 mM spermine, 30 mM Na-PIPES [Na-piperazine-N,N′-bis(2-ethanesulfonic acid); pH 7.4], 0.2% β-mercaptoethanol}, and 60 μl of 4% paraformaldehyde. The worms were freeze-thawed 3 times and washed with PBS. The worms were then dehydrated in 60% isopropyl alcohol for 10 min at room temperature and stained with Oil Red O solution. All Oil Red O images from the same experiment were acquired under identical settings and exposure times for direct and fair comparisons. Relative Oil Red O intensities were quantified using ImageJ software (NIH). Ten to 15 worms from each group were randomly selected for quantification. Each image was split into red, green, and blue (RGB) channels, and the green channel was subtracted from the red channel to eliminate nonred background signals. Anterior intestinal cell areas were selected to measure Oil Red O intensities. Fixed and live Nile red staining was performed as described previously (1, 39).
Biochemical triglyceride measurement.
Triglyceride contents were measured using a triglyceride assay kit (Thermo Scientific; catalog no. TR22321). Synchronized young adult worms were resuspended in 5% Triton X-100 solution and homogenized using glass beads and a Precellys 24 homogenizer (Bertin Technologies). For complete lysis and triglyceride extraction, the homogenates were sonicated and subjected to two cycles of heating (80°C) and cooling (room temperature). Then worm extracts obtained by centrifugation were used to measure the total triglyceride amount according to the manufacturer's protocol.
cAMP measurement.
cAMP concentrations were measured using a direct cAMP enzyme-linked immunosorbent assay (ELISA) kit (Enzo Life Sciences; catalog no. 25-0114) according to the manufacturer's protocol. Harvested worms were resuspended in 0.1 M HCl to inactivate phosphodiesterase. Worms were homogenized using glass beads and the Precellys 24 homogenizer, after which the homogenates were sonicated. Then worm extracts were collected by centrifugation and used for ELISA. Results were analyzed using 4 parametric logistic curve fitting models.
In vitro kinase assay.
To perform an in vitro kinase assay, glutathione S-transferase (GST)-tagged recombinant ATGL-1 (wild type [WT]), ATGL-1 (S303A), and LID-1 proteins were produced from E. coli and purified by GST pulldown. One microgram of each GST-protein was mixed with the PKA catalytic subunit (New England BioLabs), 10× kinase buffer (500 mM Tris-HCl [pH 7.5], 1 mM EGTA, 100 mM magnesium acetate), 1 mM 32P-labeled ATP, and distilled water and incubated for 30 min at 37°C. The kinase reaction was analyzed by autoradiography after SDS-PAGE and Coomassie staining.
Oxygen consumption rate measurement.
To measure the oxygen consumption rate, synchronized worms were cultured in RNAi plates to the 1-day young-adult stage. Half the worms were harvested and cultured on empty NGM plates for 4 h to prepare fasted samples. Worms were harvested just prior to measurement and were cultured in oxygen-saturated M9 buffer. The oxygen consumption rate was monitored using a Clark-type electrode sensor, a YSI 5300A oxygen monitor (YSI Corporation). Protein content was determined using the bicinchoninic acid (BCA) method and was used to normalize the oxygen consumption rate, which is reported as relative mmol of O2/h/mg of protein.
Microscopy.
Green fluorescent protein (GFP) and Nile red images were observed using Axio Observer Z1 and a confocal LSM 700 system (Zeiss). For quantification of GFP signals, z-stack images of each worm were combined to make projections and GFP fluorescence intensities of intestinal cells were quantified using ImageJ software (NIH). Oil Red O staining was visualized using an Axioplan II microscope, and images were captured using an Axiocam HRc camera (Zeiss).
Cell culture and transfection.
For transfection, HEK293T and Cos-1 cells were grown to 70% confluence, and each expression vector was transfected using the calcium phosphate and Lipofectamine 2000 (Invitrogen). At 6 h posttransfection, the medium was replaced with Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% antibiotic-antimycotic (penicillin-streptomycin) solution. At 24 h posttransfection, FSK (Calbiochem) or an equal amount of dimethyl sulfoxide (DMSO; Amresco) in serum-free DMEM was added to the corresponding plates. After incubation for 3 h, cells were harvested.
Immunoprecipitation and Western blotting.
Cells were lysed on ice with radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride [PMSF], 1% [vol/vol] NP-40, 0.25% [wt/vol] sodium deoxycholate, and protease inhibitor cocktail) and subjected to immunoprecipitation or Western blotting. For immunoprecipitation, lysates were incubated with anti-Myc (Cell Signaling) or anti-Flag (Sigma-Aldrich) antibody for 12 h at 4°C. Immunocomplexes were collected using protein A-Sepharose beads (GE Healthcare) for 2 h at 4°C. The beads were washed 3 times with 500 μl of RIPA buffer. Proteins were eluted by boiling in SDS sample buffer for 5 min, separated by SDS-PAGE, and analyzed by Western blotting. For Western blotting, proteins separated by SDS-PAGE were transferred to polyvinylidene difluoride (PVDF) membranes (Millipore). The membranes were blocked with 5% (wt/vol) skim milk in Tris-buffered saline containing 0.1% (vol/vol) Tween 20 (TBST; 25 mM Tris-HCl [pH 8.0], 137 mM NaCl, 2.7 mM KCl, and 0.1% Tween 20) at room temperature for 30 min, followed by overnight incubation with primary antibodies at 4°C. Antibodies against Flag tag (Sigma-Aldrich), Myc tag (Cell Signaling), mouse ATGL (Cell Signaling), hemagglutinin (HA) tag (Cell Signaling), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; AbFrontier) were used. The membranes were washed 3 times with TBST and hybridized with secondary antibodies conjugated with horseradish peroxidase (Sigma-Aldrich) in 5% skim milk dissolved in TBST at room temperature for 2 h. The membranes were then washed 3 times with TBST, incubated with enhanced chemiluminescence reagents, and quantified with LuminoImager (LAS-3000) and Science Lab Image Gauge software (Fuji Photo Film). The band intensities of the lanes were quantified using ImageJ (NIH).
qRT-PCR.
Total RNA was isolated using TRIzol reagent (Invitrogen, CA) according to the manufacturer's protocol. cDNA was synthesized using Moloney murine leukemia virus (M-MuLV) reverse transcriptase with random hexamer primers (Fermentas). Quantitative RT-PCR (qRT-PCR) was performed on a CFX96 real-time system (Bio-Rad) with SYBR green (Invitrogen). Relative expression levels of all mRNAs were normalized to actin-1/3 mRNA.
Microarray analysis.
Synchronized young adult worms were divided into well-fed and 8-h-fasted samples using the fasting assay protocol described above. Total RNA was extracted using TRIzol reagent (Invitrogen) and purified using an RNeasy minikit (Qiagen). Affymetrix C. elegans genome array chip was used for all microarray experiments. DAVID analysis was used to show enrichment of specific functional categories in fasting-responsive genes (41). Pvclust was used for clustering analysis of microarray data (42).
PKA activity assay.
Synchronized young adult worms were harvested and homogenized in protein extraction buffer (20 mM Tris-HCl, 10 mM dithiothreitol [DTT], 1 mM NaF, 1 mM Na3VO4, 10 mM EDTA, 10 mM EGTA, and protease inhibitor cocktail). Worm extracts were sonicated and centrifuged to obtain soluble proteins. PKA activity was measured according to a previously reported protocol (43). Briefly, 30 μg of protein was incubated with 10× kinase assay buffer (500 mM Tris-HCl, 1 mM EGTA, 100 mM magnesium acetate), kemptide (PKA substrate peptide), and 1 mM [γ-32P]ATP at 30°C for 10 min. The reaction was terminated by spotting the reaction mixture onto P81 phosphocellulose paper. The papers were immediately immersed in 75 mM phosphoric acid. After being washed 3 times with fresh phosphoric acid, the papers were briefly rinsed with acetone and air dried. Radioactivity was measured by Cerenkov counting.
LPAAT activity assay.
Recombinant proteins used for the lysophosphatidic acid acyltransferase (LPAAT) activity assay were expressed and purified in E. coli BL21(DE3) and SM2-1(DE3). The LPAAT activity of each recombinant protein was assessed as previously reported (44), with modifications. Briefly, the reaction mixtures, containing 50 mM Tris-HCl, 10 μM [1-14C]oleoyl coenzyme A (oleoyl-CoA; 54 mCi/mmol), 1 μg of recombinant protein, and 50 μM LPA, were incubated at 30°C for 10 min. Then the reactions were terminated by lipid extraction. Lipid extraction was performed by adding 600 μl of methanol-chloroform (2:1 [vol/vol]) according to the method of Bligh and Dyer (45). After 20 min of agitation, 200 μl of chloroform and 360 μl of water were added, followed by vortexing. After phase separation, radioactivity of the organic phase containing phosphatidic acid was measured by liquid scintillation counting.
Statistical analysis.
Values are shown as means plus standard deviations (SDs). Unless otherwise mentioned, comparison of mean values was evaluated by two-way analysis of variance (ANOVA) with Bonferroni posttest. A P value of less than 0.05 was considered significant.
RESULTS
C. elegans atgl-1 is necessary for fasting-induced lipolysis.
In C. elegans, fasting significantly decreased stored lipid droplets in anterior intestinal cells, which is consistent with previous reports (data not shown) (12, 37). To identify the key lipase(s) involved in this process, we carried out reverse genetic approaches. The C. elegans genome has over 32 putative lipase genes. We selected 21 genes with available RNAi clones that target 9 class II lipase genes (lips and fil-1), 3 patatin domain-containing genes (atgl-1 and the D1054.1 and B0524.2 genes), 1 HSL homolog gene (hosl-1), 2 phospholipase A2 homolog genes (the C07E3.9 and C03H5.4 genes), and 6 lipase-related genes (lipl). After suppression of each lipase via RNAi, stored lipid droplets were assessed by Oil Red O staining in fed and fasted adult worms, and the staining intensities in the anterior intestinal region were quantified (Fig. 1A). As shown in Fig. 1B and C, suppression of atgl-1 significantly attenuated lipid droplet hydrolysis in the fasted state and substantially increased lipid droplets even in the fed state. Consistently, biochemical triglyceride measurement with total worm extracts showed similar tendencies with Oil Red O staining (Fig. 1D).
FIG 1.

ATGL-1 is a major lipase for fasting-induced lipolysis in C. elegans. (A) RNA interference (RNAi) screening of lipases involved in fasting-induced lipolysis based on Oil Red O staining. Oil Red O staining intensities of young adult worms in the RNAi groups under feeding and 8-h-fasting conditions were quantified and classified according to relative fold increase compared to the L4440 control. (B and C) Representative images and quantitation data of Oil Red O staining with or without atgl-1 RNAi in young adult worms under feeding and 8-h-fasting conditions. Marked areas were subjected to quantitation of Oil Red O staining. (D) Relative triglyceride amounts of young adult worms were measured with a biochemical triglyceride assay kit and normalized by total worm extract protein. (E) Confocal microscopic image of atgl-1(hj67) worms after fixation and Nile red staining. The inset is a magnified view. (F) mRNA levels of atgl-1 and fasting-responsive genes (fil-1, cpt-2, acs-2, and fat-7) measured by qRT-PCR and normalized to act-1/3 mRNA. (G) Confocal microscopic images of atgl-1(hj67) worms under feeding and fasting conditions at the young adult stage. Scale bars, 10 μm. White lines indicate the boundaries of worm bodies. Error bars represent standard deviations. *, P < 0.05; **, P < 0.01. N.S., not significant.
The atgl-1 gene encodes a homolog of mammalian ATGL and has been implicated in the mobilization of fat stores at the dauer stage in C. elegans (32). In accordance with previous reports (46, 47), ATGL-1 was localized at lipid droplets by showing colocalization of ATGL-1::GFP with Nile red after fixation (Fig. 1E). Compared to feeding, fasting slightly, but not significantly, changed the levels of atgl-1 mRNA, whereas the mRNA levels of other fasting-responsive genes, such as fil-1, cpt-3, acs-2, and fat-7 (6, 36), were altered in fasted worms (Fig. 1F). However, unlike atgl-1 mRNA, the levels of ATGL-1::GFP exhibited an evident increase upon fasting in transgenic worms expressing ATGL-1::GFP (Fig. 1G). These data suggest that posttranscriptional regulations of ATGL-1 would be a key step to mediate the hydrolysis of stored lipid metabolites in fasted worms.
LID-1 is a lipid droplet protein in C. elegans homologous to mammalian CGI-58.
While C. elegans conserves atgl-1 similarly to other eukaryotes, it does not have the lipid droplet binding protein perilipin. In mammals, CGI-58, which belongs to esterase/lipase/thioesterase subfamily, is a lipid droplet associated protein essential for ATGL activation. C. elegans has three genes, the C25A1.12, C37H5.2, and C37H5.3 genes, which show sequence homology with the mammalian CGI-58/ABHD5 gene (Fig. 2A). To identify an ortholog of mammalian CGI-58, we examined which C. elegans gene(s) would preserve a unique character of the CGI-58 family, a loss of active serine at the GXSXG motif in the α/β hydrolase domain (26, 48). As shown in Fig. 2B, the serine residue of the GXSXG motif is replaced by alanine only in C25A1.12 and not in C37H5.2 and C37H5.3. These results suggest that C25A1.12 (whose gene is lid-1) might be an ortholog of CGI-58, whereas C37H5.2 and C37H5.3 might be hydrolase proteins. The amino acid sequence of the lid-1 product shares approximately 40% identity with mammalian CGI-58 (Fig. 2C).
FIG 2.
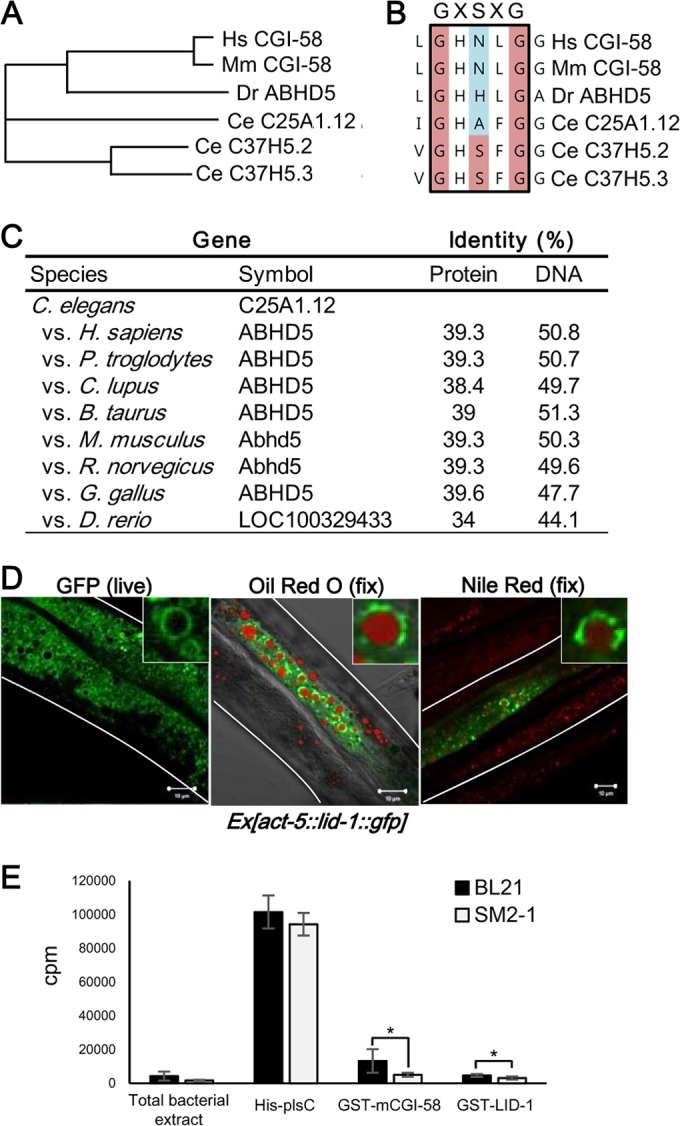
LID-1 is a homolog of CGI-58 and localizes to lipid droplets. (A) Phylogenic trees of CGI-58 gene family members on the basis of amino acid sequences from Homo sapiens (Hs), Mus musculus (Mm), Danio rerio (Dr), and Caenorhabditis elegans (Ce). Amino acid sequences encoded by all genes were subjected to multiple alignments and analyzed for phylogenetic trees obtained by the neighbor-joining method using ClustalW. (B) Alignment of the GXSXG motif in the α/β hydrolase domain of CGI-58 gene family. (C) Homology scores of LID-1 (C25A1.12) obtained from the Homologene database. (D) Confocal images showing the localization of GFP-fused LID-1 in live worms and in fixed worms costained with Oil Red O and Nile red. The stained area of the Oil Red O image is pseudocolored red. The insets are magnified views. Scale bars, 10 μm. White lines indicate the boundaries of worm bodies. (E) Lysophosphatidyl choline acyltransferase activity assay. One-microgram quantities of total bacterial protein and purified recombinant proteins were used.
To characterize the roles of LID-1 in C. elegans, we generated transgenic worms expressing LID-1::GFP in the intestinal cells. In live worms, LID-1::GFP explicitly formed ring-like structures on the surface of lipid droplets, which was confirmed by costaining with Oil Red O or Nile red after fixation (Fig. 2D). These data suggest that LID-1 is a genuine lipid droplet protein in C. elegans. However, the subcellular distribution of Nile red staining without fixation in live worms was distinct from that of LID-1::GFP, implying that LID-1-containing lipid droplets may be different from lysosome-related organelles (data not shown).
Previously, CGI-58 has been reported to possess lysophosphatidic acid acyltransferase (LPAAT) activity (49, 50). Very recently, it has been reported that LPAAT activity by recombinant CGI-58 protein would be due to the contamination of E. coli protein plsC (44). To test whether LID-1 might possess LPAAT activity, we used E. coli strain SM2-1, which lacks plsC (51), to produce recombinant proteins and performed LPAAT activity assays. As shown in Fig. 2E, both mouse CGI-58 and LID-1 did not reveal LPAAT activity. Together, these data suggest a role for LID-1 as a lipid droplet protein rather than an enzyme with LPAAT activity.
LID-1 is required for ATGL-1 function.
The finding that mammalian CGI-58 is dispersed in the cytoplasm upon lipolytic stimulation (25) led us to test whether fasting may change the levels of lid-1 mRNA or protein. In C. elegans, fasting did not alter the levels of lid-1 mRNA or overall distribution of LID-1::GFP (data not shown). Nevertheless, RNAi of lid-1 prevented the fasting-induced decrease in intestinal lipid droplets of C. elegans (Fig. 3A and B). In contrast, worms treated with RNAi of C37H5.2 or C37H5.3 showed reduced lipid droplets, as did wild-type worms (data not shown), indicating that lid-1 may have a distinct function(s) under fasting conditions. Based on these results, we hypothesized that LID-1 is a constitutive lipid droplet protein that may be required for the proper activity of ATGL-1. To elucidate the genetic interaction between the lid-1 and atgl-1 genes, we suppressed the lid-1 gene via RNAi in atgl-1-overexpressing worms. Compared to findings for wild-type worms, overexpression of atgl-1 alone decreased the intestinal lipid droplets in the basal state, whereas RNAi of lid-1 greatly inhibited the effects of atgl-1 overexpression on reduction of lipid droplets (Fig. 3C and D). Given that mammalian CGI-58 associates with ATGL (25–27), we next examined whether C. elegans LID-1 could interact with ATGL-1. As shown in Fig. 3E, LID-1 formed a protein complex with ATGL-1. Furthermore, depletion of lid-1 via RNAi attenuated the increase in ATGL-1 protein levels and translocation of ATGL-1 into lipid droplets upon fasting (Fig. 3F). These data suggest that in C. elegans, LID-1 recruits ATGL-1 to lipid droplets to stimulate lipolysis during fasting.
FIG 3.
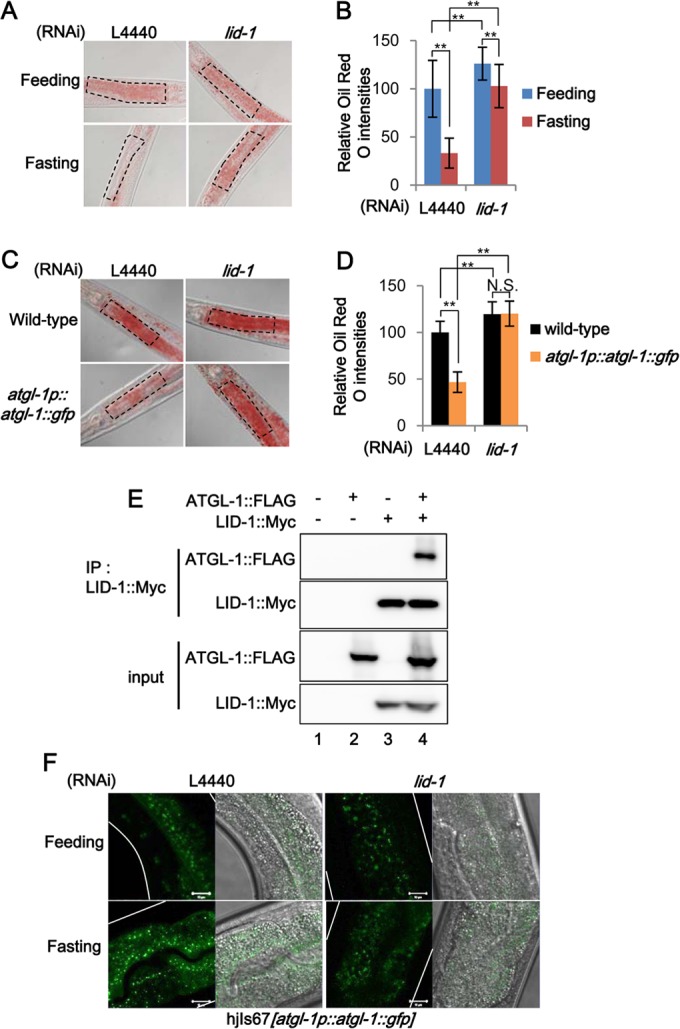
LID-1, a C. elegans homolog of CGI-58, is required for ATGL-1 activity. (A and B) Representative images and quantitation data of Oil Red O staining with or without lid-1 RNAi in young adult worms under feeding or fasting conditions. Marked areas were subjected to quantitation of Oil Red O staining. (C and D) Representative images and quantitation data of Oil Red O staining in atgl-1(hj67) worms after L4440 control and lid-1 RNAi. Marked areas were subjected to quantitation of Oil Red O staining. (E) Coimmunoprecipitation (IP) assay of HEK293T cells expressing ATGL-1-Flag and LID-1-Myc. (F) Confocal images of atgl-1(hj67) worms under feeding and fasting conditions after L4440 control or lid-1 RNAi. Scale bars, 10 μm. White lines indicate the boundaries of worm bodies. Error bars represent standard deviations. **, P < 0.01.
Fasting induces lipolysis via PKA signaling.
The observation that in C. elegans, ATGL-1 and LID-1 are involved in the hydrolysis of stored lipid droplets upon fasting prompted us to investigate which signaling pathway(s) would be involved in the response to nutritional changes. As key energy sensors, PKA and AMPK have been extensively studied (30, 52), and both of these kinases are well conserved in C. elegans. As shown in Fig. 4A and B, RNAi of kin-1, which encodes the catalytic subunit of PKA, drastically impaired fasting-induced decrease in lipid droplets. Even under feeding conditions, kin-1 RNAi substantially elevated intracellular lipid accumulation, as observed in response to atgl-1 or lid-1 suppression, implying that these genes are likely involved in the same lipolytic pathway. Because PKA is stimulated by cAMP, we measured whole-body cAMP levels. In fasted worms, cAMP levels were elevated (Fig. 4C). Thus, the C. elegans cAMP-PKA pathway appears to be well conserved to reflect energy states throughout evolution. In adult worms, AMPK catalytic subunit mutants (53), the aak-1(tm1944), aak-2(gt33), and aak-1(tm1944); aak-2(gt33) mutants, decreased lipid droplets under fasting conditions (Fig. 4D and E). Consistently, RNAi of aak-1 and aak-2 did not suppress fasting-induced lipolysis (Fig. 4F and G). Worms with mutations in AMPK exhibited a greater decrease of lipid contents upon fasting than did wild-type worms. These data suggest that in C. elegans, PKA is activated upon fasting and plays a critical role in facilitating fasting-induced lipid droplet hydrolysis, whereas AMPK signaling may attenuate fasting-induced lipolysis.
FIG 4.

PKA signaling mediates fat mobilization upon fasting in C. elegans. (A and B) Representative images and quantitation data of Oil Red O staining with or without kin-1 RNAi in young adult worms under feeding or fasting conditions. (C) Cyclic AMP (cAMP) concentrations measured by direct ELISA in total extracts of wild-type worms under feeding and 4-h-fasting conditions. (D and E) Representative images and quantitation data of Oil Red O staining in wild-type and AMPK mutant worms under feeding or fasting conditions. (F and G) Representative images and quantitation of Oil Red O intensities with control, aak-1, or aak-2 RNAi in young adult worms under feeding or fasting conditions. (H and I) Representative images and quantitation data of Oil Red O staining in the PKA hyperactive kin-2 mutant strain (ce179 strain) after RNAi of atgl-1 or lid-1. For Oil Red O staining, marked areas were subjected to quantitation. Error bars represent standard deviations. *, P < 0.05; **, P < 0.01.
To determine whether PKA could indeed modulate the activity of ATGL-1 and LID-1, we tested a kin-2 PKA regulatory subunit mutant strain. kin-2(ce179) worms contain a missense mutation (R92C) in the pseudo-substrate domain of the PKA regulatory subunit and thereby exhibit increased PKA activity (54). Compared to those in wild-type worms, the levels of accumulated lipid droplets were greatly decreased in kin-2(ce179) worms under well-fed conditions (Fig. 4H and I). A similar phenotype for kin-2(ce179) worms was confirmed by kin-2 RNAi (data not shown), indicating that PKA activity seems to be also important for the regulation of lipid metabolism under basal conditions. Intriguingly, in kin-2(ce179) worms, knockdown of atgl-1 and lid-1 restored the levels of intestinal lipid droplets to nearly those of wild-type worms (Fig. 4H and I). These data suggest that in fasted worms, PKA signaling would act as a key upstream regulator for atgl-1 and lid-1, which may be a central and conserved signaling pathway for lipolysis in most animals upon fasting.
PKA phosphorylates ATGL-1 and enhances protein stability as well as LID-1 binding.
Given that PKA acts upstream from ATGL-1 and LID-1 in fasting-induced lipolysis, we asked the question whether PKA might directly phosphorylate ATGL-1 and/or LID-1. Recently, it has been demonstrated that mammalian ATGL is phosphorylated by PKA at multiple sites (55). In vitro kinase assays revealed that PKA phosphorylated C. elegans ATGL-1 but not LID-1 (data not shown). A search for a PKA phosphorylation site(s) revealed serine 303 of ATGL-1 as one of the potential PKA phosphorylation sites (Fig. 5A). Compared with wild-type ATGL-1, mutation of serine 303 to alanine in ATGL-1 (S303A) abrogated the phosphorylation by PKA, indicating that serine 303 of ATGL-1 is one of the major phosphorylation sites for activated PKA (Fig. 5B).
FIG 5.

PKA phosphorylates and regulates ATGL-1 functions. (A) Potential PKA phosphorylation site prediction results obtained using the GPS2.1 software were sorted according to the scores indicated. (B) In vitro kinase assay with wild-type (WT) ATGL-1 and S303A ATGL-1 recombinant proteins. Purified GST-fused ATGL-1 proteins were incubated with the PKA catalytic subunit and 32P-labeled ATP before SDS-PAGE. (C) Confocal images of atgl-1(hj67) worms after L4440 control and kin-2 RNAi. (D and E) Representative images and quantitation of ATGL-1::GFP intensities of atgl-1(hj67) worms under control or forskolin (FSK; 100 μM) treatment. (F) Quantitation of Western blots of C. elegans ATGL-1 proteins in Cos-1 cells with or without forskolin (50 μM, 3 h) treatment. (G) Western blot analysis of mammalian ATGL protein levels in differentiated 3T3-L1 adipocytes treated with forskolin (20 mM) for the indicated time periods. The GAPDH protein was used as a loading control. Scale bars, 20 μm. White lines indicate the boundaries of worm bodies. Error bars represent standard deviations. **, P < 0.01.
To gain further insights into ATGL-1 phosphorylation by PKA, we examined localization of ATGL-1::GFP in kin-2-suppressed worms. Because kin-2 encodes the regulatory subunit of PKA, knockdown of kin-2 increased basal PKA activity in C. elegans (data not shown). It is of interest to note that PKA activation by kin-2 RNAi or treatment of forskolin, a PKA activator, greatly increased the levels of ATGL-1::GFP intensity, even more than those observed in fasted worms (Fig. 5C to E). Moreover, the levels of the C. elegans ATGL-1 protein were elevated by forskolin in mammalian cells, whereas the S303A mutation in ATGL-1 attenuated the effect of forskolin (Fig. 5F). Consistent with these results, the levels of mammalian ATGL protein were also increased by forskolin in differentiated adipocytes, supporting the idea that the regulation of ATGL by PKA is conserved in both mammals and C. elegans (Fig. 5G).
Because atgl-1 mRNA levels were not significantly altered upon fasting (Fig. 1E), we investigated whether the increase in ATGL-1 protein by PKA was modulated by protein stability. Forskolin treatment enhanced the half-life of ATGL-1 protein (Fig. 6A). In addition, worms were treated with MG132, a proteasome inhibitor, and the levels of ATGL-1::GFP were monitored. As shown in Fig. 6B, the inhibition of proteasomal degradation with MG132 augmented the levels of ATGL-1::GFP. Furthermore, MG132 treatment did not further elevate ATGL-1::GFP levels in forskolin-treated worms. Also, we observed that ubiquitination of ATGL-1 was decreased by PKA activation (Fig. 6C). Next, we asked whether phosphorylation of ATGL-1 by PKA may affect LID-1 binding. As shown in Fig. 6D, PKA activation with forskolin evidently potentiated the interaction between ATGL-1 and LID-1, which was drastically attenuated in ATGL-1 S303A mutant. In addition, inhibition of PKA signaling by kin-1 RNAi sufficiently blocked the increase of ATGL-1::GFP signal upon fasting (Fig. 6E). Taken together, these data suggest that ATGL-1 phosphorylation by PKA stimulates the levels of ATGL-1 protein via inhibition of proteasomal degradation and/or promoting LID-1 binding.
FIG 6.

PKA increases ATGL-1 protein stability and LID-1 binding. (A) Western blot data of C. elegans ATGL-1 proteins in Cos-1 cells treated with cycloheximide (CHX; 50 μg/ml). Forskolin (50 μM) was pretreated for 3 h. GAPDH was used as a loading control. (B) Confocal images of atgl-1(hj67) worms treated with DMSO, forskolin (100 μM), or MG132 (100 μM) under feeding or fasting conditions. (C) Ubiquitination assay of ATGL-1 in Cos-1 cells. (D) Coimmunoprecipitation assay of Cos-1 cells expressing ATGL-1-Flag and LID-1-Myc in the absence or presence of forskolin (50 μM, 3 h). (E) Confocal images of atgl-1(hj67) worms after L4440 control and kin-1 RNAi under feeding or 4-h-fasting conditions. Scale bars, 20 μm. White lines indicate the boundaries of worm bodies.
Lipid hydrolysis is required for energy production during fasting.
In general, ATP production via fatty acid oxidation demands more oxygen. To examine the roles of ATGL-1 and LID-1 in energy production during fasting, we measured oxygen consumption rate as an index of fatty acid oxidation activity. Fasted worms consumed more oxygen, presumably due to increased mitochondrial activity to oxidize lipid metabolites, as previously reported (36). Inhibition of lipolytic pathways by RNAi of atgl-1 or lid-1 alleviated the fasting-induced increase in oxygen consumption rate (Fig. 7A). These data indicate that hydrolysis of stored lipids by atgl-1 or lid-1 would eventually lead to an energy-producing process such as fatty acid oxidation.
FIG 7.
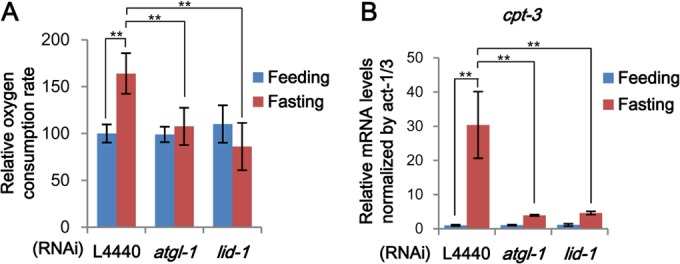
atgl-1 and lid-1 are required for energy production during fasting. (A) Oxygen consumption rates of worms in oxygen-saturated M9 buffer after feeding and fasting, normalized to the amounts of total proteins. For details, see Materials and Methods. (B) mRNA levels of cpt-3 measured by qRT-PCR and normalized to act-1/3 mRNA. Error bars represent standard deviations. **, P < 0.01.
To determine the effects of atgl-1, lid-1, and kin-1 RNAi on gene expression upon fasting, we performed microarray analyses to identify fasting-responsive gene sets. Gene set enrichment analysis using DAVID showed that various signaling pathways, including fatty acid and nutrient metabolic pathways, were significantly altered upon fasting (see the supplemental material). Clustering analysis and gene expression profiles also revealed that disruption of PKA signaling dampened fasting-induced gene expression changes, implying that PKA could mediate pleiotropic responses. In contrast, the global expression patterns of the atgl-1 and lid-1 RNAi groups were not very different from that of the control group. On the other hand, qRT-PCR analysis showed that changes in mRNA levels of genes involved in fatty acid oxidation, such as cpt-3, were blunted by RNAi of atgl-1 and lid-1 (Fig. 7B). These data suggest that the functions of both atgl-1 and lid-1 are more closely and selectively associated with lipolytic pathways and fatty acid oxidation downstream from PKA.
DISCUSSION
In fasted animals, hydrolysis of stored lipid metabolites is precisely regulated in a timely fashion to provide an energy source. Because excess lipid accumulation is often associated with metabolic dysregulation, lipase activity should be finely tuned in response to fasting signals. Our data suggest that worms would prevent unwanted lipolytic activity through proteasomal degradation of a key lipase ATGL-1, even without perilipin 1 (Plin1) protein, which plays a barrier role to inhibit lipolysis in mammals under basal conditions. Here, we demonstrated that in fasted worms, activated PKA stabilizes the ATGL-1 protein and drives ATGL-1 to localize on lipid droplets through interaction with the lipid droplet-binding protein LID-1, eventually leading to hydrolysis of stored lipid droplets in intestinal cells (Fig. 8).
FIG 8.
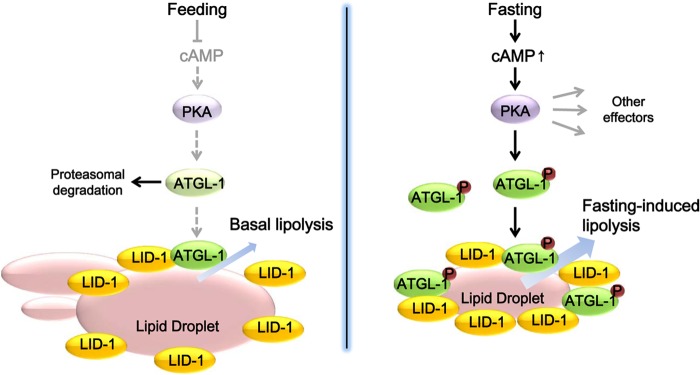
Working model. In the feeding state, ATGL-1 is actively degraded by the proteasome pathway to maintain a low rate of basal lipolysis. In the fasting state, increased cAMP levels activate PKA signaling. Then, PKA phosphorylates and stabilizes ATGL-1, which is recruited to lipid droplets via interaction with LID-1, eventually stimulating lipid hydrolysis.
Besides atgl-1, RNAi of lips-1, lips-2, lips-13, fil-1, D1054.1, and C03E3.9 also partially attenuated fasting-induced lipid hydrolysis (Fig. 1A). Thus, C. elegans may use several lipases to accommodate various nutritional changes. However, our data evidently show that knockdown of atgl-1 or lid-1 impedes fasting-induced lipolysis in adult worms. It has been well established that ATGL is essential for lipolysis in mammalian adipocytes and that ATGL-deficient mice show systemic lipid accumulation (56, 57). In addition, CGI-58, a homolog of lid-1, has been identified as a causative gene of Chanarin-Dorfman syndrome, a rare recessive genetic disease with systemic lipid accumulation and ichthyosis (26, 58). Thus, our data indicate that atgl-1 and lid-1 are functionally well conserved throughout evolution to execute and coordinate lipolytic metabolism.
Lipid droplets are dynamic cellular organelles coated with various proteins, and there are significant similarities in lipid droplet proteins between mammals and C. elegans (18, 47, 59). On the other hand, as the perilipin family of lipid droplet proteins is well conserved from Dictyostelium to humans, the lack of the perilipin domain-containing gene in C. elegans seems to place LID-1 at a unique position in the evolution of lipolytic machinery (data not shown). In this study, we proposed that LID-1 may be an ortholog of mammalian CGI-58. Nevertheless, it seems that LID-1 would possess distinct features from CGI-58 in the following aspects. First, LID-1 is constitutively localized at lipid droplets (Fig. 2D). Second, unlike mammalian CGI-58, LID-1 does not require perilipin proteins to target on lipid droplets, although LID-1 may not fulfill the role of Plin1 in C. elegans. For instance, unlike Plin1, LID-1 does not have antilipolytic function, since suppression of lid-1 slightly increased fat contents in worms (Fig. 3A and B), whereas ablation of Plin1 greatly promotes the basal lipolytic activity in mammalian adipocytes (23, 60). Given that mammalian CGI-58 requires Plin1 to localize to lipid droplets (25), it needs to be determined whether LID-1 can localize to lipid droplets by itself without other proteins. It appears that during evolution, perilipin family proteins might have emerged with the need for constituting large lipid droplets in specialized fat-storing cells with greater capability for energy source storage. Thus, it is likely that C. elegans, which lacks perilipin, may have evolved its own regulatory mechanisms using protein stability to control lipase activity upon LID-1 accessibility, which may be distinct from mammals.
Several regulatory mechanisms for lipases have evolved with different lipid droplet proteins. In other species, lipase activity can be controlled by mRNA expression (61–64), posttranslational modification (65), and translocation to lipid droplets (24, 66). Our data suggest that in C. elegans, proteasomal degradation of ATGL-1 prevents futile lipolysis during feeding, and PKA-dependent phosphorylation and stabilization of ATGL-1 protein stimulate LID-1 binding and lipase activity during fasting. Similar to the case with C. elegans ATGL-1, we observed that the mammalian ATGL protein was increased by PKA activation in adipocytes. Furthermore, it has been recently reported that mammalian ATGL protein is regulated by proteasomal degradation (67, 68), indicating the possibility of an evolutionarily conserved mechanism. Interestingly, a very recent study showed that serotonin and octopamine signaling promotes body fat loss via transcriptional upregulation of ATGL-1, suggesting an additional layer of regulatory mechanism of lipolysis (69). In regard to spatial and temporal regulation of lipase(s), it is plausible to speculate that regulation of protein stability and translocation of a certain lipase(s) would be an efficient way to activate lipolysis, ensuring an immediate response to fasting. Of course, we cannot exclude the possibility that phosphorylation of ATGL-1 affects its own hydrolase activity.
Previously, it has been reported that AMPK also phosphorylates atgl-1 at multiple sites and downregulates hydrolase activity to ration lipid reserves during long-term starvation at the dauer stage (32). Consistently, we also observed that AMPK might attenuate lipolysis in fasted adult worms, implying that PKA and AMPK may regulate atgl-1 in opposite ways with different repertoires of phosphorylation and molecular components. It appears that PKA is rapidly activated at the initial phase of fasting. Immediate activation of lipid hydrolysis by PKA upon fasting would not be sustainable for long-term starvation. In addition, we observed that cAMP levels returned to basal levels after 8 h of fasting (data not shown). On the contrary, it is likely that AMPK would restrain excessive lipolysis during fasting to prevent energy depletion. As previously suggested, it is possible that C. elegans exhibits biphasic metabolic responses in nutrient deprivations: rapid consumption of stored lipid metabolites with high metabolic activity followed by an inert preservation mode with low metabolic activity (70). Furthermore, it has been demonstrated that in mammalian cells, AMPK is activated as a consequence of lipolysis and inhibits further lipolysis, forming a negative-feedback control (71, 72). A recent study showed that hydrolase activity of mouse ATGL is enhanced via phosphorylation by PKA (55), suggesting that ATGL would be a conserved target of PKA. Further studies are required to address the cross talk between PKA and AMPK signaling in the control of lipolysis at various developmental stages of C. elegans under different nutritional conditions.
The nature of lipid droplets in C. elegans has been controversial due to heterogeneity of staining granules in intestinal cells (37). In this study, we identified that the localizations of a lipid droplet protein, LID-1, and lipid-staining dyes showed different patterns depending on the staining methods. For instance, LID-1 colocalized with Oil Red O-positive lipid droplets after fixative staining. However, LID-1 did not colocalize with the Nile red dye without fixation in live worms, whereas LID-1 colocalized with Nile red-positive granules after fixation. These results suggest that in the live state, certain biological processes in the intestinal cells of C. elegans may exclude Nile red from LID-1-positive lipid droplets. Despite these methodological discrepancies, GFP-fused LID-1, which was constitutively localized on lipid droplets, can be used for the visualization of lipid droplets in both live and fixed worms.
In conclusion, we have identified atgl-1 and lid-1 as key modulators of lipolysis in fasted adult worms. In the absence of perilipin, acting as a barrier against lipolysis, C. elegans suppresses unnecessary lipolysis by actively degrading ATGL-1 protein. Upon nutritional depletion, C. elegans activates PKA signaling to stabilize ATGL-1, which induces the association with the lipid droplet protein LID-1 to activate the lipase function of ATGL-1, resulting in energy production. For an efficient and immediate response to fasting in C. elegans, stabilized ATGL-1 forms a protein complex with LID-1 at lipid droplets, ensuring prompt lipolytic activity. Collectively, our data highlight the cooperative action of LID-1 for proper functioning of the lipase ATGL-1 to maintain energy homeostasis following nutritional changes.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by National Research Foundation of Korea (NRF) grants funded by the Korean government (MISP) (2011-0018312 and 2010-0026035).
C. elegans strains N2, KG532, and VS20 were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440). aak-1(tm1944), aak-2(gt33), and aak-1(tm1944); aak-2(gt33) strains were kindly provided by Hyeon-Sook Koo (Yonsei University). Plasmid constructs for mCGI-58 and plsC and bacterial strains used in the lysophosphatidic acid acyltransferase assay were gifted by Dawn L. Brasamele (University of New Jersey).
We have no conflict of interest to declare.
Footnotes
Published ahead of print 8 September 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00722-14.
REFERENCES
- 1.Ashrafi K, Chang FY, Watts JL, Fraser AG, Kamath RS, Ahringer J, Ruvkun G. 2003. Genome-wide RNAi analysis of Caenorhabditis elegans fat regulatory genes. Nature 421:268–272. 10.1038/nature01279. [DOI] [PubMed] [Google Scholar]
- 2.Jones KT, Ashrafi K. 2009. Caenorhabditis elegans as an emerging model for studying the basic biology of obesity. Dis. Model. Mech. 2:224–229. 10.1242/dmm.001933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johnson TE, Mitchell DH, Kline S, Kemal R, Foy J. 1984. Arresting development arrests aging in the nematode Caenorhabditis elegans. Mech. Ageing Dev. 28:23–40. 10.1016/0047-6374(84)90150-7. [DOI] [PubMed] [Google Scholar]
- 4.Cassada RC, Russell RL. 1975. The dauerlarva, a post-embryonic developmental variant of the nematode Caenorhabditis elegans. Dev. Biol. 46:326–342. 10.1016/0012-1606(75)90109-8. [DOI] [PubMed] [Google Scholar]
- 5.Golden JW, Riddle DL. 1984. The Caenorhabditis elegans dauer larva: developmental effects of pheromone, food, and temperature. Dev. Biol. 102:368–378. 10.1016/0012-1606(84)90201-X. [DOI] [PubMed] [Google Scholar]
- 6.Van Gilst MR, Hadjivassiliou H, Yamamoto KR. 2005. A Caenorhabditis elegans nutrient response system partially dependent on nuclear receptor NHR-49. Proc. Natl. Acad. Sci. U. S. A. 102:13496–13501. 10.1073/pnas.0506234102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sawin ER, Ranganathan R, Horvitz HR. 2000. C. elegans locomotory rate is modulated by the environment through a dopaminergic pathway and by experience through a serotonergic pathway. Neuron 26:619–631. 10.1016/S0896-6273(00)81199-X. [DOI] [PubMed] [Google Scholar]
- 8.Avery L, Horvitz HR. 1990. Effects of starvation and neuroactive drugs on feeding in Caenorhabditis elegans. J. Exp. Zool. 253:263–270. 10.1002/jez.1402530305. [DOI] [PubMed] [Google Scholar]
- 9.Angelo G, Van Gilst MR. 2009. Starvation protects germline stem cells and extends reproductive longevity in C. elegans. Science 326:954–958. 10.1126/science.1178343. [DOI] [PubMed] [Google Scholar]
- 10.Trent C. 1982. Genetic and behavioral studies of the egg-laying system of Caenorhabditis elegans. Ph.D. thesis Massachusetts Institute of Technology, Cambridge, MA. [Google Scholar]
- 11.Horvitz HR, Chalfie M, Trent C, Sulston JE, Evans PD. 1982. Serotonin and octopamine in the nematode Caenorhabditis elegans. Science 216:1012–1014. 10.1126/science.6805073. [DOI] [PubMed] [Google Scholar]
- 12.McKay RM, McKay JP, Avery L, Graff JM. 2003. C. elegans: a model for exploring the genetics of fat storage. Dev. Cell 4:131–142. 10.1016/S1534-5807(02)00411-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Apfeld J, O'Connor G, McDonagh T, DiStefano PS, Curtis R. 2004. The AMP-activated protein kinase AAK-2 links energy levels and insulin-like signals to lifespan in C. elegans. Genes Dev. 18:3004–3009. 10.1101/gad.1255404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang F, Vought BW, Satterlee JS, Walker AK, Jim Sun ZY, Watts JL, DeBeaumont R, Saito RM, Hyberts SG, Yang S, Macol C, Iyer L, Tjian R, van den Heuvel S, Hart AC, Wagner G, Naar AM. 2006. An ARC/Mediator subunit required for SREBP control of cholesterol and lipid homeostasis. Nature 442:700–704. 10.1038/nature04942. [DOI] [PubMed] [Google Scholar]
- 15.Jia K, Chen D, Riddle DL. 2004. The TOR pathway interacts with the insulin signaling pathway to regulate C. elegans larval development, metabolism and life span. Development 131:3897–3906. 10.1242/dev.01255. [DOI] [PubMed] [Google Scholar]
- 16.Duncan RE, Ahmadian M, Jaworski K, Sarkadi-Nagy E, Sul HS. 2007. Regulation of lipolysis in adipocytes. Annu. Rev. Nutr. 27:79–101. 10.1146/annurev.nutr.27.061406.093734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zechner R, Zimmermann R, Eichmann TO, Kohlwein SD, Haemmerle G, Lass A, Madeo F. 2012. FAT SIGNALS—lipases and lipolysis in lipid metabolism and signaling. Cell Metab. 15:279–291. 10.1016/j.cmet.2011.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martin S, Parton RG. 2006. Lipid droplets: a unified view of a dynamic organelle. Nat. Rev. Mol. Cell Biol. 7:373–378. 10.1038/nrm1912. [DOI] [PubMed] [Google Scholar]
- 19.Cermelli S, Guo Y, Gross SP, Welte MA. 2006. The lipid-droplet proteome reveals that droplets are a protein-storage depot. Curr. Biol. 16:1783–1795. 10.1016/j.cub.2006.07.062. [DOI] [PubMed] [Google Scholar]
- 20.Brasaemle DL, Dolios G, Shapiro L, Wang R. 2004. Proteomic analysis of proteins associated with lipid droplets of basal and lipolytically stimulated 3T3-L1 adipocytes. J. Biol. Chem. 279:46835–46842. 10.1074/jbc.M409340200. [DOI] [PubMed] [Google Scholar]
- 21.Miura S, Gan JW, Brzostowski J, Parisi MJ, Schultz CJ, Londos C, Oliver B, Kimmel AR. 2002. Functional conservation for lipid storage droplet association among Perilipin, ADRP, and TIP47 (PAT)-related proteins in mammals, Drosophila, and Dictyostelium. J. Biol. Chem. 277:32253–32257. 10.1074/jbc.M204410200. [DOI] [PubMed] [Google Scholar]
- 22.Brasaemle DL. 2007. Thematic review series: adipocyte biology. The perilipin family of structural lipid droplet proteins: stabilization of lipid droplets and control of lipolysis. J. Lipid Res. 48:2547–2559. 10.1194/jlr.R700014-JLR200. [DOI] [PubMed] [Google Scholar]
- 23.Tansey JT, Sztalryd C, Gruia-Gray J, Roush DL, Zee JV, Gavrilova O, Reitman ML, Deng CX, Li C, Kimmel AR, Londos C. 2001. Perilipin ablation results in a lean mouse with aberrant adipocyte lipolysis, enhanced leptin production, and resistance to diet-induced obesity. Proc. Natl. Acad. Sci. U. S. A. 98:6494–6499. 10.1073/pnas.101042998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sztalryd C, Xu G, Dorward H, Tansey JT, Contreras JA, Kimmel AR, Londos C. 2003. Perilipin A is essential for the translocation of hormone-sensitive lipase during lipolytic activation. J. Cell Biol. 161:1093–1103. 10.1083/jcb.200210169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Subramanian V, Rothenberg A, Gomez C, Cohen AW, Garcia A, Bhattacharyya S, Shapiro L, Dolios G, Wang R, Lisanti MP, Brasaemle DL. 2004. Perilipin A mediates the reversible binding of CGI-58 to lipid droplets in 3T3-L1 adipocytes. J. Biol. Chem. 279:42062–42071. 10.1074/jbc.M407462200. [DOI] [PubMed] [Google Scholar]
- 26.Lass A, Zimmermann R, Haemmerle G, Riederer M, Schoiswohl G, Schweiger M, Kienesberger P, Strauss JG, Gorkiewicz G, Zechner R. 2006. Adipose triglyceride lipase-mediated lipolysis of cellular fat stores is activated by CGI-58 and defective in Chanarin-Dorfman syndrome. Cell Metab. 3:309–319. 10.1016/j.cmet.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 27.Yamaguchi T, Omatsu N, Matsushita S, Osumi T. 2004. CGI-58 interacts with perilipin and is localized to lipid droplets. Possible involvement of CGI-58 mislocalization in Chanarin-Dorfman syndrome. J. Biol. Chem. 279:30490–30497. 10.1074/jbc.M403920200. [DOI] [PubMed] [Google Scholar]
- 28.Granneman JG, Moore HP, Krishnamoorthy R, Rathod M. 2009. Perilipin controls lipolysis by regulating the interactions of AB-hydrolase containing 5 (Abhd5) and adipose triglyceride lipase (Atgl). J. Biol. Chem. 284:34538–34544. 10.1074/jbc.M109.068478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miyoshi H, Perfield JW, II, Souza SC, Shen WJ, Zhang HH, Stancheva ZS, Kraemer FB, Obin MS, Greenberg AS. 2007. Control of adipose triglyceride lipase action by serine 517 of perilipin A globally regulates protein kinase A-stimulated lipolysis in adipocytes. J. Biol. Chem. 282:996–1002. 10.1074/jbc.M605770200. [DOI] [PubMed] [Google Scholar]
- 30.Carmen GY, Victor SM. 2006. Signalling mechanisms regulating lipolysis. Cell. Signal. 18:401–408. 10.1016/j.cellsig.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 31.Unger RH, Clark GO, Scherer PE, Orci L. 2010. Lipid homeostasis, lipotoxicity and the metabolic syndrome. Biochim. Biophys. Acta 1801:209–214. 10.1016/j.bbalip.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 32.Narbonne P, Roy R. 2009. Caenorhabditis elegans dauers need LKB1/AMPK to ration lipid reserves and ensure long-term survival. Nature 457:210–214. 10.1038/nature07536. [DOI] [PubMed] [Google Scholar]
- 33.Wang MC, O'Rourke EJ, Ruvkun G. 2008. Fat metabolism links germline stem cells and longevity in C. elegans. Science 322:957–960. 10.1126/science.1162011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O'Rourke EJ, Kuballa P, Xavier R, Ruvkun G. 2013. w-6 polyunsaturated fatty acids extend life span through the activation of autophagy. Genes Dev. 27:429–440. 10.1101/gad.205294.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O'Rourke EJ, Ruvkun G. 2013. MXL-3 and HLH-30 transcriptionally link lipolysis and autophagy to nutrient availability. Nat. Cell Biol. 15:668–676. 10.1038/ncb2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jo H, Shim J, Lee JH, Lee J, Kim JB. 2009. IRE-1 and HSP-4 contribute to energy homeostasis via fasting-induced lipases in C. elegans. Cell Metab. 9:440–448. 10.1016/j.cmet.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 37.O'Rourke EJ, Soukas AA, Carr CE, Ruvkun G. 2009. C. elegans major fats are stored in vesicles distinct from lysosome-related organelles. Cell Metab. 10:430–435. 10.1016/j.cmet.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mörck C, Olsen L, Kurth C, Persson A, Storm NJ, Svensson E, Jansson JO, Hellqvist M, Enejder A, Faergeman NJ, Pilon M. 2009. Statins inhibit protein lipidation and induce the unfolded protein response in the non-sterol producing nematode Caenorhabditis elegans. Proc. Natl. Acad. Sci. U. S. A. 106:18285–18290. 10.1073/pnas.0907117106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brooks KK, Liang B, Watts JL. 2009. The influence of bacterial diet on fat storage in C. elegans. PLoS One 4:e7545. 10.1371/journal.pone.0007545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Soukas AA, Kane EA, Carr CE, Melo JA, Ruvkun G. 2009. Rictor/TORC2 regulates fat metabolism, feeding, growth, and life span in Caenorhabditis elegans. Genes Dev. 23:496–511. 10.1101/gad.1775409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. 2003. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 4:P3. 10.1186/gb-2003-4-5-p3. [DOI] [PubMed] [Google Scholar]
- 42.Suzuki R, Shimodaira H. 2006. Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 22:1540–1542. 10.1093/bioinformatics/btl117. [DOI] [PubMed] [Google Scholar]
- 43.Hastie CJ, McLauchlan HJ, Cohen P. 2006. Assay of protein kinases using radiolabeled ATP: a protocol. Nat. Protoc. 1:968–971. 10.1038/nprot.2006.149. [DOI] [PubMed] [Google Scholar]
- 44.McMahon D, Dinh A, Kurz D, Shah D, Han GS, Carman GM, Brasaemle DL. 2014. Comparative gene identification 58/alpha/beta hydrolase domain 5 lacks lysophosphatidic acid acyltransferase activity. J. Lipid Res. 55:1750–1761. 10.1194/jlr.M051151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bligh EG, Dyer WJ. 1959. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37:911–917. 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 46.Zhang SO, Box AC, Xu N, Le Men J, Yu J, Guo F, Trimble R, Mak HY. 2010. Genetic and dietary regulation of lipid droplet expansion in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U. S. A. 107:4640–4645. 10.1073/pnas.0912308107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang P, Na H, Liu Z, Zhang S, Xue P, Chen Y, Pu J, Peng G, Huang X, Yang F, Xie Z, Xu T, Xu P, Ou G, Zhang SO, Liu P. 2012. Proteomic study and marker protein identification of Caenorhabditis elegans lipid droplets. Mol. Cell. Proteomics 11:317–328. 10.1074/mcp.M111.016345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lefèvre C, Jobard F, Caux F, Bouadjar B, Karaduman A, Heilig R, Lakhdar H, Wollenberg A, Verret JL, Weissenbach J, Ozguc M, Lathrop M, Prud'homme JF, Fischer J. 2001. Mutations in CGI-58, the gene encoding a new protein of the esterase/lipase/thioesterase subfamily, in Chanarin-Dorfman syndrome. Am. J. Hum. Genet. 69:1002–1012. 10.1086/324121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ghosh AK, Ramakrishnan G, Chandramohan C, Rajasekharan R. 2008. CGI-58, the causative gene for Chanarin-Dorfman syndrome, mediates acylation of lysophosphatidic acid. J. Biol. Chem. 283:24525–24533. 10.1074/jbc.M801783200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Montero-Moran G, Caviglia JM, McMahon D, Rothenberg A, Subramanian V, Xu Z, Lara-Gonzalez S, Storch J, Carman GM, Brasaemle DL. 2010. CGI-58/ABHD5 is a coenzyme A-dependent lysophosphatidic acid acyltransferase. J. Lipid Res. 51:709–719. 10.1194/jlr.M001917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Coleman J. 1990. Characterization of Escherichia coli cells deficient in 1-acyl-sn-glycerol-3-phosphate acyltransferase activity. J. Biol. Chem. 265:17215–17221. [PubMed] [Google Scholar]
- 52.Hardie DG, Ross FA, Hawley SA. 2012. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 13:251–262. 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee H, Cho JS, Lambacher N, Lee J, Lee SJ, Lee TH, Gartner A, Koo HS. 2008. The Caenorhabditis elegans AMP-activated protein kinase AAK-2 is phosphorylated by LKB1 and is required for resistance to oxidative stress and for normal motility and foraging behavior. J. Biol. Chem. 283:14988–14993. 10.1074/jbc.M709115200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schade MA, Reynolds NK, Dollins CM, Miller KG. 2005. Mutations that rescue the paralysis of Caenorhabditis elegans ric-8 (synembryn) mutants activate the G alpha(s) pathway and define a third major branch of the synaptic signaling network. Genetics 169:631–649. 10.1534/genetics.104.032334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pagnon J, Matzaris M, Stark R, Meex RC, Macaulay SL, Brown W, O'Brien PE, Tiganis T, Watt MJ. 2012. Identification and functional characterization of protein kinase A phosphorylation sites in the major lipolytic protein, adipose triglyceride lipase. Endocrinology 153:4278–4289. 10.1210/en.2012-1127. [DOI] [PubMed] [Google Scholar]
- 56.Zimmermann R, Strauss JG, Haemmerle G, Schoiswohl G, Birner-Gruenberger R, Riederer M, Lass A, Neuberger G, Eisenhaber F, Hermetter A, Zechner R. 2004. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science 306:1383–1386. 10.1126/science.1100747. [DOI] [PubMed] [Google Scholar]
- 57.Haemmerle G, Lass A, Zimmermann R, Gorkiewicz G, Meyer C, Rozman J, Heldmaier G, Maier R, Theussl C, Eder S, Kratky D, Wagner EF, Klingenspor M, Hoefler G, Zechner R. 2006. Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science 312:734–737. 10.1126/science.1123965. [DOI] [PubMed] [Google Scholar]
- 58.Radner FP, Streith IE, Schoiswohl G, Schweiger M, Kumari M, Eichmann TO, Rechberger G, Koefeler HC, Eder S, Schauer S, Theussl HC, Preiss-Landl K, Lass A, Zimmermann R, Hoefler G, Zechner R, Haemmerle G. 2010. Growth retardation, impaired triacylglycerol catabolism, hepatic steatosis, and lethal skin barrier defect in mice lacking comparative gene identification-58 (CGI-58). J. Biol. Chem. 285:7300–7311. 10.1074/jbc.M109.081877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xu N, Zhang SO, Cole RA, McKinney SA, Guo F, Haas JT, Bobba S, Farese RV, Jr, Mak HY. 2012. The FATP1-DGAT2 complex facilitates lipid droplet expansion at the ER-lipid droplet interface. J. Cell Biol. 198:895–911. 10.1083/jcb.201201139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Martinez-Botas J, Anderson JB, Tessier D, Lapillonne A, Chang BH, Quast MJ, Gorenstein D, Chen KH, Chan L. 2000. Absence of perilipin results in leanness and reverses obesity in Lepr(db/db) mice. Nat. Genet. 26:474–479. 10.1038/82630. [DOI] [PubMed] [Google Scholar]
- 61.Grönke S, Mildner A, Fellert S, Tennagels N, Petry S, Muller G, Jackle H, Kuhnlein RP. 2005. Brummer lipase is an evolutionary conserved fat storage regulator in Drosophila. Cell Metab. 1:323–330. 10.1016/j.cmet.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 62.Lake AC, Sun Y, Li JL, Kim JE, Johnson JW, Li D, Revett T, Shih HH, Liu W, Paulsen JE, Gimeno RE. 2005. Expression, regulation, and triglyceride hydrolase activity of Adiponutrin family members. J. Lipid Res. 46:2477–2487. 10.1194/jlr.M500290-JLR200. [DOI] [PubMed] [Google Scholar]
- 63.Kershaw EE, Hamm JK, Verhagen LA, Peroni O, Katic M, Flier JS. 2006. Adipose triglyceride lipase: function, regulation by insulin, and comparison with adiponutrin. Diabetes 55:148–157. 10.2337/diabetes.55.01.06.db05-0982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Villena JA, Roy S, Sarkadi-Nagy E, Kim KH, Sul HS. 2004. Desnutrin, an adipocyte gene encoding a novel patatin domain-containing protein, is induced by fasting and glucocorticoids: ectopic expression of desnutrin increases triglyceride hydrolysis. J. Biol. Chem. 279:47066–47075. 10.1074/jbc.M403855200. [DOI] [PubMed] [Google Scholar]
- 65.Kurat CF, Wolinski H, Petschnigg J, Kaluarachchi S, Andrews B, Natter K, Kohlwein SD. 2009. Cdk1/Cdc28-dependent activation of the major triacylglycerol lipase Tgl4 in yeast links lipolysis to cell-cycle progression. Mol. Cell 33:53–63. 10.1016/j.molcel.2008.12.019. [DOI] [PubMed] [Google Scholar]
- 66.Egan JJ, Greenberg AS, Chang MK, Wek SA, Moos MC, Jr, Londos C. 1992. Mechanism of hormone-stimulated lipolysis in adipocytes: translocation of hormone-sensitive lipase to the lipid storage droplet. Proc. Natl. Acad. Sci. U. S. A. 89:8537–8541. 10.1073/pnas.89.18.8537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dai Z, Qi W, Li C, Lu J, Mao Y, Yao Y, Li L, Zhang T, Hong H, Li S, Zhou T, Yang Z, Yang X, Gao G, Cai W. 2013. Dual regulation of adipose triglyceride lipase by pigment epithelium-derived factor: a novel mechanistic insight into progressive obesity. Mol. Cell. Endocrinol. 377:123–134. 10.1016/j.mce.2013.07.001. [DOI] [PubMed] [Google Scholar]
- 68.Olzmann JA, Richter CM, Kopito RR. 2013. Spatial regulation of UBXD8 and p97/VCP controls ATGL-mediated lipid droplet turnover. Proc. Natl. Acad. Sci. U. S. A. 110:1345–1350. 10.1073/pnas.1213738110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Noble T, Stieglitz J, Srinivasan S. 2013. An integrated serotonin and octopamine neuronal circuit directs the release of an endocrine signal to control C. elegans body fat. Cell Metab. 18:672–684. 10.1016/j.cmet.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tan KT, Luo SC, Ho WZ, Lee YH. 2011. Insulin/IGF-1 receptor signaling enhances biosynthetic activity and fat mobilization in the initial phase of starvation in adult male C. elegans. Cell Metab. 14:390–402. 10.1016/j.cmet.2011.06.019. [DOI] [PubMed] [Google Scholar]
- 71.Gauthier MS, Miyoshi H, Souza SC, Cacicedo JM, Saha AK, Greenberg AS, Ruderman NB. 2008. AMP-activated protein kinase is activated as a consequence of lipolysis in the adipocyte: potential mechanism and physiological relevance. J. Biol. Chem. 283:16514–16524. 10.1074/jbc.M708177200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Djouder N, Tuerk RD, Suter M, Salvioni P, Thali RF, Scholz R, Vaahtomeri K, Auchli Y, Rechsteiner H, Brunisholz RA, Viollet B, Makela TP, Wallimann T, Neumann D, Krek W. 2010. PKA phosphorylates and inactivates AMPKalpha to promote efficient lipolysis. EMBO J. 29:469–481. 10.1038/emboj.2009.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.