

Abstract
Brown fat generates heat through uncoupled respiration, protecting against hypothermia and obesity. Adult humans have brown fat, but the amounts and activities are substantially decreased in obesity, by unknown mechanisms. Here we show that elevated microRNA 34a (miR-34a) in obesity inhibits fat browning in part by suppressing the browning activators fibroblast growth factor 21 (FGF21) and SIRT1. Lentivirus-mediated downregulation of miR-34a in mice with diet-induced obesity reduced adiposity, improved serum profiles, increased the mitochondrial DNA copy number, and increased oxidative function in adipose tissue in both BALB/c and C57BL/6 mice. Remarkably, downregulation of miR-34a increased coexpression of the beige fat-specific marker CD137 and the browning marker UCP1 in all types of white fat, including visceral fat, and promoted additional browning in brown fat. Mechanistically, downregulation of miR-34a increased expression of the FGF21 receptor components, FGFR1 and βKL, and also that of SIRT1, resulting in FGF21/SIRT1-dependent deacetylation of PGC-1α and induction of the browning genes Ucp1, Pgc-1α, and Prdm16. Importantly, anti-miR-34a-mediated beneficial effects, including decreased adiposity, are likely from multiple tissues, since downregulation of miR-34a also improves hepatic FGF21 signaling and lipid oxidation. This study identifies miR-34a as an inhibitor of beige and brown fat formation, providing a potential target for treating obesity-related diseases.
INTRODUCTION
The global epidemic of obesity has greatly increased research interest in adipose biology, particularly that of energy-dissipating brown fat (1). While white adipose tissue (WAT) stores excess chemical energy, leading to weight gain, brown adipose tissue (BAT) expends energy as heat through uncoupled respiration via the action of mitochondrial uncoupled protein 1 (UCP1), protecting against hypothermia and obesity (2–5). Recent studies discovered a second type of brown-like fat, “beige fat,” which is present in WAT and is genetically distinct from classical BAT (6). Interestingly, recent human studies using 2-fluorodeoxyglucose coupled with positron-emission tomography (PET) scanning showed that adult humans have brown fat and that the activities and amounts of brown fat are inversely related to the body mass index (BMI) and substantially decreased in obesity (3, 7). Increasing energy expenditure by promoting production of brown fat in BAT and also beige fat in WAT, particularly in obese individuals, would be an appealing option for weight reduction and for treating obesity-related diseases.
Brown and beige fat depots develop in response to various activators, including cold exposure, hormones, exercise, and transcriptional regulators, such as PRDM16, PPARγ, SIRT1, and PGC-1α (8–11). Fibroblast growth factor 21 (FGF21) has been shown to beneficially affect metabolism and energy balance by enhancing fatty acid β-oxidation during prolonged fasting and also by promoting fat browning in WAT, as well as in BAT, in response to cold exposure in mice (12–16). Importantly, a recent human study showed that cold exposure increased circulating levels of the brown fat activators FGF21 and irisin and that treatment with either of these endocrine regulators upregulated browning genes and promoted thermogenesis (17).
Emerging evidence indicates that microRNAs (miRs) also function as important regulators that inhibit brown remodeling of adipocytes. Muscle-enriched miR-133a directly downregulated expression of the key transcriptional activator of brown fat differentiation, PRDM16, and cold exposure decreased miR-133a levels and promoted brown fat cell differentiation (18, 19). Brown adipocyte-enriched miR-155 was also shown to inhibit brown fat cell differentiation by directly targeting the browning transcription factor C/EBPβ (20). A recent miR expression profiling analysis revealed that miR-34a levels are elevated in adipocytes from obese individuals (21), but the functional role of elevated miR-34a in obesity remains unknown.
In this study, we show that elevated miR-34a in obesity acts as an inhibitor of beige and brown fat formation. Lentivirus-mediated downregulation of miR-34a in mice with diet-induced obesity increased markers of beige fat in all types of WATs and promoted additional browning in BAT. In mechanistic studies, we showed that downregulation of miR-34a increased expression of the FGF21 receptor complex (FGFR1-βKL) and of SIRT1, contributing to the activation of the browning transcriptional program via FGF21/SIRT1-dependent deacetylation of PGC-1α. Importantly, in addition to fat browning in adipose tissue, anti-miR-34a-mediated beneficial effects, including decreased adiposity, are likely from multiple tissues, since downregulation of miR-34a improves hepatic FGF21 signaling and expression of fat oxidation genes.
MATERIALS AND METHODS
Reagents and materials.
Antisense miR-34a, anti-scrambled RNA, a TaqMan miRNA assay kit, and small interfering RNAs (siRNAs) for βKL (s96291 and s96292), FGFR1 (s66023 and s66025), and SIRT1 (s96764 and s174220) were purchased from Applied Biosystems. Antibodies to βKL (AF2619) were purchased from R&D Systems. Antibodies to FGFR1 (sc-121), SIRT1 (sc-15404), PGC-1α (sc-13067), RNA polymerase II (sc-9001), tubulin (sc-8085), and actin (sc-1616) were purchased from Santa Cruz Biotech, and antibodies for phosphorylated extracellular signal-regulated kinase (p-ERK) (9101), t-ERK (4695), phosphorylated AMP kinase (p-AMPK) (2531), t-AMPK (2532), and pan-acetyl-Lys (9441) were purchased from Cell Signaling Technology. Antibodies for CD137 (ab64836) and UCP1 (ab10983 and MAB6158) were purchased from Abcam and R&D Systems. A NAD/NADH quantitation kit was purchased from Biovision. Antibodies for βKL (LS-B3568) and FGFR1 (nb600-1287), used in immunohistochemistry (IHC) studies, were purchased from LS Biology and Novus Biological, respectively. Recombinant FGF21 was expressed in Escherichia coli and purified as previously described (12).
Animal experiments.
To produce mice with diet-induced obesity, 6- to 8-week-old male BALB/c mice (10 mice/group) were fed a high-fat diet (HFD) (60% fat; Research Diets, Inc.) for 16 to 24 weeks. Male C57BL/6 mice (6 mice/group) that had been fed a normal diet (ND) or HFD for 14 weeks were purchased from the Jackson Laboratory. In previous studies, we downregulated elevated miR-34a in mice by using antisense oligonucleotides with 2′-O-methoxyethyl (MOE)-modified ribose sugars (22, 23). Because of the high cost of these oligonucleotides for in vivo studies and the potential stress due to multiple injections (3 to 5 times), we utilized a lentivirus expressing antisense miR-34a in the current study. Lentiviral vectors encoding antisense miR-34a (anti-miR-34) or scrambled controls (anti-sc) were purchased from System Biosciences Inc. Lentivirus packaging plasmids pMD2.G and psPAX2 (Addgene) were used to make Lenti-E, containing anti-scrambled miRs, and Lenti-miR-34ai. Mice were injected with lentivirus via the tail vein at doses of 1 × 109 to 2 × 109 PFU in 100 μl phosphate-buffered saline (PBS) and were sacrificed 14 to 21 days after the injection, and adipose tissue, liver, and serum samples were collected for analysis. For in vivo FGF21 signaling studies, BALB/c mice (3 mice/group) were injected intraperitoneally (i.p.) with FGF21 (0.05 or 0.1 mg/kg of body weight), and 30 min later, adipose tissues from 3 mice were pooled for preparation of adipose extracts. All animal use protocols were approved by the Institutional Animal Care and Use and Institutional Biosafety Committees at the University of Illinois at Urbana-Champaign.
IHC studies.
Primary antibodies to UCP1 and CD137 were visualized using a biotin-labeled secondary antibody and a peroxidase-coupled streptavidin kit (Abcam), and the samples were imaged with a NanoZoomer scanner (Hamamatsu). For fluorescence IHC, secondary antibodies labeled with either Alexa Fluor 647 or 488 were used. The nucleus was stained with DAPI (4′,6-diamidino-2-phenylindole; Invitrogen). The samples were imaged by confocal microscopy (Zeiss LSM 700).
Mitochondrial DNA copy number.
Total DNA was isolated using a DNA extraction kit (Omega Bio-Tek). A serial dilution standard curve was prepared, and DNA was quantified by quantitative PCR (qPCR). The mitochondrial DNA copy number was calculated from the ratio of the DNA of the COX II gene, a mitochondrial gene, to that of the cyclophilin A gene, a single-copy nuclear gene.
CS activity assay.
Mitochondrial citrate synthase (CS) activity was measured by use of a kit (Sigma-Aldrich) according to the manufacturer's instructions. A total of 1 to 2 μg of protein from subcutaneous inguinal WAT (scWAT), gonadal WAT (gWAT), or BAT was used for the assay.
NAD+ and NADH measurements.
NAD+ and NADH levels were measured by their enzymatic reactions according to the manufacturer's instructions (Biovision, Inc.), as previously described (23).
Differentiation of 3T3-L1 cells.
3T3-L1 cells were cultured in M1 medium (Dulbecco's modified Eagle's medium [DMEM], 10% fetal bovine serum [FBS], 4.5 mg/ml glucose, 4 mM glutamine, and 1 mM sodium pyruvate). Two days after cells reached confluence, adipocyte differentiation was induced by supplementation with M2 medium (M1 medium containing 0.5 mM 3-isobutyl-1-methylxanthine, 1 μM dexamethasone, and 1.5 μg/ml insulin). Two days later, the medium was replaced with M3 medium (M1 medium containing 1.5 μg/ml insulin). M3 medium was replaced every 2 days. Differentiation of 3T3-L1 fat cells was confirmed by Oil Red O staining.
Measurement of oxygen consumption rate.
Differentiated 3T3 cells were transfected with anti-miR-34a oligonucleotide, and 48 h later, palmitic acid (2 mM) was added. Cells were further treated at 30-min intervals with oligomycin (14 μM), the pharmacological uncoupler FCCP (10 μM), or the complex III and I inhibitor antimycin A (4 μM [each]). The oxygen consumption rate was measured with an XF analyzer (Seahorse Bioscience, Inc.).
Construction of Fgfr1 3′UTR-luc plasmids and luciferase assay.
A SpeI/HindIII fragment containing the 3′ untranslated region (3′UTR) of FGFR1 was amplified and inserted into the pMIR plasmid. Mutations were made by site-directed mutagenesis and were confirmed by DNA sequencing. Luciferase assays were performed as previously described (23–25).
qRT-PCR.
MicroRNAs were isolated using a miRNA isolation kit (Ambion). Levels of miR-34a were determined by quantitative reverse transcription-PCR (qRT-PCR) and normalized to snoRNA202. For mRNA levels, total RNA was isolated with TRIzol, and levels were determined by qRT-PCR normalized to 36B4. Primer sequences are available in Table S1 in the supplemental material.
PGC-1α acetylation and ChIP assays.
WAT tissues were pooled from 3 mice, and extracts were prepared and immediately used for immunoprecipitation (IP) and immunoblotting (IB) acetylation assays as previously described (23–25). The IP buffer contained 1 μM trichostatin A (TSA) and 10 mM N-acetylmuramic acid (NAM) to inhibit deacetylation. Combined anti-miR-34a and siRNA experiments were performed in 3T3-L1 adipocytes to determine the effects of FGFR1, BKL, SIRT1, and miR-34a on FGF21 signaling, AMPK activation, PGC-1α deacetylation, and the mitochondrial DNA copy number. The effects on occupancy of PGC-1α and RNA polymerase II at browning genes were determined by chromatin IP (ChIP) as previously described (23–25).
RESULTS
Downregulation of miR-34a reduces adiposity in mice with diet-induced obesity.
To investigate functional consequences of elevated adipocyte miR-34a in obesity (21), we downregulated miR-34a by lentivirus-mediated expression of antisense miR-34a in obese BALB/c mice fed a high-fat diet (HFD). Expression of anti-miR-34a in obese mice reduced adipocyte miR-34a levels in WAT and BAT to levels near those in lean mice (Fig. 1A) and significantly reduced body weight (Fig. 1B), while food intake was not changed (Fig. 1C). Consistent with weight reduction, the amounts of abdominal fat (Fig. 1D) and different types of visceral epididymal WATs (eWATs), such as perirenal WAT (prWAT) and gonadal WAT (gWAT), as well as subcutaneous inguinal WAT (scWAT), were substantially reduced (Fig. 1E and F). The weights of adipose tissue and adipocyte sizes, particularly in eWAT, were markedly reduced in mice expressing anti-miR-34a (Fig. 1E to H). Notably, all types of WATs, including visceral gWAT and subcutaneous scWAT, and also BAT, had a brownish color in obese mice expressing anti-miR-34a (Fig. 1F). Consistent with decreased adiposity, circulating levels of triglycerides (TG) and free fatty acids were substantially decreased, serum insulin and fasting glucose levels were decreased, and serum FGF21 levels were modestly decreased in anti-miR-34a-expressing mice (Fig. 1I). These results indicate that in vivo silencing of miR-34a in mice with diet-induced obesity reduced adiposity and possibly promoted fat browning in both WAT and BAT.
FIG 1.

Downregulation of miR-34a reduced adiposity and improved serum profiles in mice with diet-induced obesity. Male BALB/c mice that had been fed a normal diet (ND) or a high-fat diet (HFD) for 20 weeks were infected with either empty lentivirus (L-E) or a virus expressing anti-miR-34a (L-34ai), as indicated (10 mice/group). (A) miR-34a levels in WAT and BAT. (B and C) Body weights (B) and food intake changes (C). Statistical significance between HFD mice injected with L-E or L-34ai was determined by the Student t test. Data are means and standard errors of the means (SEM) (n = 10). *, P < 0.05; **, P < 0.01; NS, statistically not significant. (D) Images of abdominal regions. (E and F) Weights (E) and images (F) of different types of WATs and BAT. (G) Oil Red O and hematoxylin and eosin (H&E) staining of eWAT and BAT. (H) The sizes of adipocytes were measured and quantified from the images of WAT stained with H&E. (I) Serum levels of the indicated lipid metabolites. For statistical analysis, means and SEM were determined (n = 5 for ND [L-E], 7 for HFD [L-E], and 8 for HFD [L-34ai]). *, P < 0.05; **, P < 0.01; NS, statistically not significant.
Downregulation of miR-34a in obese mice increases beige fat markers in WAT and increases browning in BAT.
We examined in more detail the potential effects of anti-miR-34a on fat browning. Remarkably, dramatic increases were observed in brown fat-like depots, detected by UCP1 expression, in visceral eWATs, such as prWAT and gWAT, and in subcutaneous scWAT, as well as in BAT (Fig. 2A). Furthermore, in all types of WATs, multiocular fat droplets, characteristic of brown fat cells, were evident in obese mice expressing anti-miR-34a (Fig. 2A). Consistent with increased mitochondrial UCP1 protein levels in WAT and BAT, the mitochondrial DNA copy number was also increased (Fig. 2B), and the activity of citrate synthase (CS), a marker of mitochondrial oxidative function, was also highly elevated in both WAT and BAT in anti-miR-34a-expressing mice (Fig. 2C). In addition, the mRNA levels of the browning genes Ucp1, Pgc-1α, and Prdm16 were increased in each type of WAT (prWAT, gWAT, and scWAT), as well as in BAT, by downregulation of miR-34a (Fig. 2D to F). Notably, while expression of the key browning gene activator PRDM16 was increased in WAT (Fig. 2F) and BAT (Fig. 2D), expression of TLE3, a white fat-selective cofactor antagonizing PRDM16 action (26), was decreased in eWAT (Fig. 2F) from mice expressing anti-miR-34a. We also examined expression of lipid-handling genes in adipose tissues. The expression of most genes involved in lipolysis, fatty acid β-oxidation, and lipid uptake was increased, whereas that of all tested lipogenic genes and the inflammatory genes Tnfα and IL-6 was decreased, in eWAT from mice expressing anti-miR-34a (Fig. 2G). These findings indicate that silencing of miR-34a in mice with diet-induced obesity promoted beige-like browning in all types of WATs, both visceral and subcutaneous fat, and increased browning in BAT.
FIG 2.

Downregulation of miR-34a increased mitochondrial function and expression of UCP1 in all types of WATs and BAT. BALB/c mice were treated as described in the legend to Fig. 1. (A) Immunostaining of UCP1 by IHC. (B) Mitochondrial DNA copy numbers. mDNA and MtDNA, mitochondrial DNA; nDNA, nuclear DNA. (C) Mitochondrial citrate synthase activities (n = 3). *, P < 0.05; **, P < 0.01. (D to G) mRNA levels of genes involved in thermogenesis and lipid metabolism as determined by qRT-PCR. Data are means and SEM (n = 5). *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Anti-miR-34a-mediated beneficial effects are also observed in obese C57BL/6 mice.
C57BL/6 mice are more prone to diet-induced obesity and are used frequently in obesity and type 2 diabetes studies (27, 28). To determine whether the anti-miR-34a-mediated beneficial effects in BALB/c mice (Fig. 1 and 2) also occur in C57BL/6 mice, we carried out in vivo studies with these mice. Tail vein injection of a lentivirus expressing anti-miR-34a decreased miR-34a levels in WAT and BAT in obese C57BL/6 mice, toward those in lean mice (Fig. 3A), and decreased body weight, without changes in food intake (Fig. 3B and C). The amounts of adipose tissues were substantially reduced (not shown), and abdominal fat levels were also decreased in C57BL/6 mice expressing anti-miR-34a (Fig. 3D). Consistent with the decreased adiposity, serum TG and fasting glucose levels (Fig. 3E) were reduced, and glucose tolerance (Fig. 3F) was also significantly improved in these mice. Intestinal TG levels were also reduced (Fig. 3E). Serum aspartate transaminase (AST) and alanine aminotransferase (ALT) levels were only slightly elevated in mice injected with lentivirus compared to those in uninfected mice (Fig. 3G), indicating that viral infection did not cause significant liver toxicity. Downregulation of miR-34a also increased body temperature, mitochondrial DNA copy number, and CS activity (Fig. 3H to J), and expression of the browning-related genes Ucp1, Pgc-1a, and Prdm16 was increased in WAT and BAT (Fig. 3K). Consistent with these effects, downregulation of miR-34a increased the oxygen consumption rate in differentiated 3T3-L1 fat cells (Fig. 3L). These results indicate that anti-miR-34a-mediated beneficial effects, including weight loss and enhanced mitochondrial functions, are observed in both C57BL/6 and BALB/c mice.
FIG 3.

Anti-miR-34a-mediated antiadipogenic and thermogenic effects in obese C57BL/6 mice. Male C57BL/6 mice (6 mice/group) that had been fed an ND or HFD for 14 weeks were injected via the tail vein with a lentivirus expressing anti-miR-34a, and 28 days later, tissues and serum were collected. (A) miR-34a levels in WAT and BAT. (B and C) Body weights (B) and food intake changes (C). (D) Images of abdominal regions. (E) Serum glucose and serum and intestinal TG levels. (F) Glucose tolerance test (GTT) results. Serum glucose levels after i.p. injection of glucose are shown. (G) Serum AST and ALT levels. (H) Rectal body temperature was measured 1 day before sacrifice. (I) Mitochondrial DNA copy numbers. (J) Citrate synthase activities in WAT and BAT. (K) Expression of browning-related genes in WAT and BAT as measured by qRT-PCR. Statistical significance was determined by the Student t test. Data are means and SEM (n = 6). *, P < 0.05; **, P < 0.01; NS, statistically not significant. (L) Effects of downregulation of miR-34a on oxygen consumption rate (OCR) in differentiated 3T3-L1 fat cells.
Downregulation of miR-34a in obesity promotes beige fat production in all types of WATs.
Recent evidence indicates that there are two distinct types of brown fat cells: an myf-5-positive, UCP1-positive classical brown fat cell and an myf-5-negative, UCP1-positive beige fat cell that is present in WAT (5, 29, 30). Importantly, gene expression patterns in beige fat cells are distinct from those in either classical BAT or WAT (6). To determine whether the browning observed in WAT was due to beige fat derived from myf-5-negative precursor cells, we examined the coexpression of beige fat-specific CD137 (6) and the general fat browning marker UCP1. While CD137 was not detected in BAT, as expected (6), expression of both CD137 and UCP1 was increased in all types of WATs in mice expressing anti-miR-34a (Fig. 4A to D). These results indicate that in vivo silencing of elevated miR-34a in obese mice promoted CD137/UCP1-positive beige fat cell differentiation in all types of WATs.
FIG 4.

Downregulation of miR-34a promoted beige-like fat production in all types of WATs in mice with diet-induced obesity. IHC staining results show the beige cell-specific marker CD137 (red), the browning marker UCP1 (green), and merged images for gWAT, prWAT, and scWAT (A to C) and for BAT (D) from BALB/c mice fed an ND or HFD and injected with control lentivirus (L-E) or a lentivirus expressing anti-miR-34a (L-34ai).
miR-34a inhibits cold temperature-induced fat browning in 3T3-L1 adipocytes.
Recent evidence indicates that miRs function as important regulators of brown fat remodeling in response to cold exposure (18, 19). Since downregulation of miR-34a in obesity increased markers for beige and brown fats, we examined the effects of cold exposure on miR-34a levels and further examined the potential role of miR-34a in expression of the fat browning marker UCP1 in differentiated 3T3-L1 adipocytes (Fig. 5A). Exposure of 3T3-L1 fat cells to cold temperature substantially decreased miR-34a levels and dramatically increased the mitochondrial DNA copy number (Fig. 5B and C). The mRNA levels of the browning-related genes Ucp1, Pgc-1a, and Prdm16 and the protein levels of UCP1 detected by IHC were also markedly increased upon cold exposure (Fig. 5D and E). Exogenous expression of miR-34a markedly reversed all these cold temperature-induced effects (Fig. 5B to E). These results suggest that miR-34a may function as a general inhibitor of fat browning not only under the obesity conditions presented above (Fig. 1 to 4) but also upon cold exposure.
FIG 5.
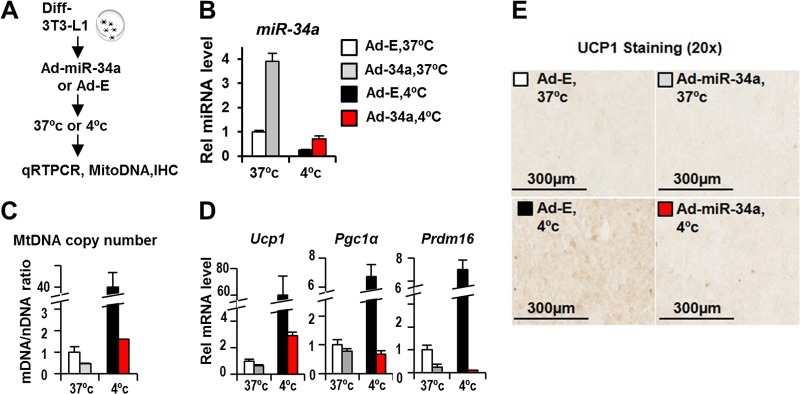
miR-34a inhibits the cold temperature-induced brown fat marker UCP1 in 3T3-L1 fat cells. (A) Experimental outline for differentiated 3T3-L1 adipocytes. (B to E) Effects of cold exposure and adenovirus-mediated expression of miR-34a on miR-34a levels (B), mitochondrial DNA copy number (C), mRNA levels of browning-related genes (D), and expression of the brown fat marker UCP1, detected by IHC (E).
Adipocyte miR-34a and FGFR1-βKL levels are inversely correlated.
We next investigated the possible mechanisms of anti-miR-34a-mediated fat browning in obese mice. Recent studies have shown that FGF21 promotes brown and beige fat formation in adaptive thermogenesis (14, 17). Surprisingly, a conserved miR-34a seed sequence was detected within the 3′UTR of the FGF21 receptor (FGFR1) (Fig. 6A). Furthermore, in previous studies, we showed that miR-34a attenuated hepatic FGF19 signaling by directly targeting βKL (22). βKL is the membrane coreceptor for FGF19 in the liver but also for FGF21 in the liver and in adipose tissue (31). We therefore hypothesized that miR-34a inhibits adiposity and fat browning at least in part by targeting the adipocyte FGF21 receptor complex (FGFR1-βKL). Indeed, in WAT of obese mice, there was an inverse correlation between expression of miR-34a and FGFR1-βKL in WAT (Fig. 6B), and the inverse correlation was also detected in 3T3-L1 fat cells (Fig. 6C and D), supporting this hypothesis.
FIG 6.

miR-34a downregulates the adipocyte FGF21 receptor complex, FGFR1-βKL, by directly targeting the 3′UTRs of the genes. (A) Potential miR-34a target sequences in FGFR1 3′UTR sequences of different species. GC, AU, and GU wobble base pairing is indicated. (B) Levels of miR-34a and of FGFR1 and βKL mRNAs in WAT of mice fed an ND or HFD were detected by qRT-PCR. Data are means and SEM (n = 6). **, P < 0.01. (C and D) Differentiated 3T3-L1 cells were infected with Ad-empty or Ad-miR-34a or transfected with increasing amounts of scrambled RNA (anti-sc) or anti-miR-34a. The miR-34a levels were measured by qRT-PCR, and protein levels of FGFR1 and βKL were detected. Data are means and SEM (n = 6). **, P < 0.01. (E to G) Cells were transfected with a luciferase reporter plasmid containing either the WT or mutated 3′UTR of Fgfr1 and with anti-miR-34a or pCDH-miR-34a, as indicated. Luciferase activities were normalized to β-galactosidase activities. Data are means and SEM (n = 6). *, P < 0.05; **, P < 0.01; NS, statistically not significant.
miR-34a attenuates FGF21 signaling by directly targeting FGFR1.
To determine whether FGFR1 is a direct target of miR-34a, the miR-34a binding sequence in the FGFR1 3′UTR or a mutated sequence was inserted into a luciferase reporter (Fig. 6E). Downregulation of miR-34a increased reporter activity for the wild-type (WT) sequence but not the mutated sequence, and conversely, overexpression of miR-34a inhibited the reporter activity only for the WT sequence (Fig. 6F and G). These results indicate that miR-34a directly downregulates FGFR1 and that the inhibition is likely mediated by binding of miR-34a to the 3′UTR of the FGFR1 mRNA.
Downregulation of miR-34a in dietary obesity increases FGF21 signaling.
We next examined the effects of miR-34a on FGF21 signaling by performing gain- and loss-of-function experiments in differentiated 3T3-L1 fat cells. FGF21-mediated phosphorylation of ERK is a measure of FGF21 responsiveness (31, 32). FGF21 treatment increased levels of activated p-ERK, but these effects were not detected in cells overexpressing miR-34a, and conversely, downregulation of miR-34a increased p-ERK levels (Fig. 7A and B), indicating that miR-34a attenuates adipocyte FGF21 signaling.
FIG 7.

In vivo downregulation of miR-34a in obesity improved adipocyte FGF21 signaling and SIRT1 function, which resulted in increased deacetylation of PGC-1α. (A and B) FGF21 signaling studies in 3T3-L1 cells. Effects of overexpression (A) or downregulation (B) of miR-34a on p-ERK levels are shown. Data are means and SEM (n = 3). (C to H) FGF21 signaling and PGC-1α acetylation studies performed in vivo. (C) Experimental outline for in vivo FGF21 signaling. (D) Expression of FGFR1 and βKL and of early target genes of FGF21 signaling. (E and F) WAT tissues from 3 mice were pooled, and adipose extracts were prepared. Protein levels of FGFR1-βKL and p-ERK in WAT were measured. (G) SIRT1 protein in WAT from mice fed ND or HFD, measured by IB. (H) NAD+ and NADH levels in WAT (n = 6). **, P <0.01; NS, statistically not significant. (I) Acetylated PGC-1α levels were measured by the IP/IB method.
We further examined whether miR-34a acts similarly in eWAT in mice in vivo (Fig. 7C). Expression of anti-miR-34a in mice with diet-induced obesity increased mRNA and protein levels of the FGF21 receptor complex (FGFR1-βKL) (Fig. 7D and E) and also increased mRNA levels of the early FGF21 target genes and indicators of FGF21 responsiveness Erg-1 and c-Fos (32) (Fig. 7D). In vivo FGF21 treatment increased p-ERK levels in adipose tissue in lean mice, but these responses were blunted in obese mice (Fig. 7F). Importantly, lentivirus-mediated expression of anti-miR-34a increased p-ERK levels in WAT in obese mice in response to FGF21 treatment (Fig. 7F). These results indicate that adipocyte FGF21 signaling is impaired in obesity and that downregulation of miR-34a improves FGF21 sensitivity in adipose tissue.
In vivo silencing of miR-34a increases adipocyte SIRT1 levels and deacetylation of PGC-1α.
The cold-inducible transcriptional coactivator PGC-1α has a crucial role in mitochondrial functions (33), browning of WAT in adaptive thermogenesis upon cold exposure, and induction of a beige fat-promoting mitokine, irisin, during exercise (10, 14). Transcriptional activity of PGC-1α is profoundly increased by NAD+-dependent, SIRT1-dependent deacetylation (34). A recent study showed that FGF21 signaling enhances SIRT1 deacetylase activity by increasing cellular NAD+ levels, which results in increased PGC-1α activity as a consequence of SIRT1-dependent deacetylation (35). Since SIRT1 is a direct target of miR-34a in the liver (23, 25, 36), we asked whether downregulation of miR-34a increases NAD+ levels and SIRT1 expression in WAT. Indeed, NAD+ levels and protein levels of adipocyte SIRT1 in WAT, which are reduced in obese mice, were significantly increased in mice expressing anti-miR-34a (Fig. 7G and H), and acetylated PGC-1α levels (Fig. 7I) were substantially decreased by anti-miR-34a expression in both control and FGF21-treated mice. These findings suggest that downregulation of SIRT1 and the FGF21 receptor complex by miR-34a increases PGC-1α acetylation, which would result in decreased transcriptional activity of PGC-1α.
Effects of downregulation of miR-34a on hepatic FGF21 signaling and metabolic gene expression.
Since lentiviruses target multiple tissues, we examined miR-34a levels in metabolic tissues in mice with diet-induced obesity that were injected with a lentivirus expressing anti-miR-34a. The miR-34a levels were highly elevated in liver and adipose tissues and markedly in the pancreas but not in muscle, and injection of the lentivirus expressing anti-miR-34a led to a substantial decrease in miR-34 levels in liver and adipose tissues (Fig. 8A). Since the overall phenotype with regard to decreased body weight results from integrated actions from multiple metabolic tissues, we examined the effects of anti-miR-34a on expression of liver metabolic genes. Increased expression of hepatic genes involved in fatty acid oxidation (Cpt1, Mcad, Eci, CytC, and Pparα) and decreased expression of lipogenic genes (Fas and Srebp-1c) and gluconeogenic genes (G6Pase and Pepck) were detected in mice expressing anti-miR-34a (Fig. 8B and C). In addition to improving adipocyte FGF21 signaling (Fig. 7F), overexpression of miR-34a compromised FGF21 signaling in hepatocytes (Fig. 8D), and conversely, downregulation of miR-34a improved it (Fig. 8E). These results suggest that multiple tissues, including liver and adipose tissues, contribute to the anti-miR-34a-mediated overall beneficial metabolic outcomes, including reduced adiposity.
FIG 8.
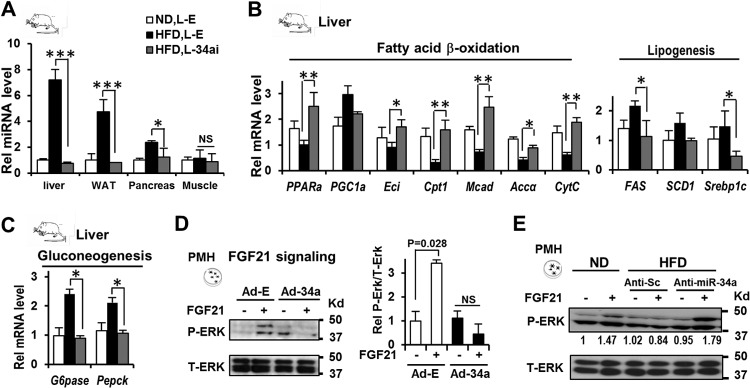
Effects of downregulation of miR-34a on hepatic FGF21 signaling and metabolic gene expression. BALB/c mice (5 to 8 mice/group) that had been fed a normal diet (ND) or a high-fat diet (HFD) for 20 weeks were infected with either empty lentivirus (L-E) or a virus expressing anti-miR-34a (L-34ai), and 21 days later, livers were collected. (A) Effects on miR-34a levels in metabolic tissues in mice with diet-induced obesity. (B and C) Effects of downregulation of miR-34a on expression of hepatic metabolic genes. Statistical significance was determined by the Student t test. Data are means and SEM (n = 5 to 8). *, P < 0.05; **, P < 0.01; ***, P < 0.001; NS, statistically not significant. (D and E) Effects of overexpression or downregulation of miR-34a on FGF21 signaling in isolated primary mouse hepatocytes. FGF21 signaling was measured by detecting p-ERK in hepatocytes.
Both FGF21 signaling and SIRT1 are critical for PGC-1α deacetylation and the browning gene program induced by anti-miR-34a.
To determine the relative importance of FGF21 signaling and SIRT1 for deacetylation of PGC-1α and the browning gene program, we performed combined antisense miR-34a and siRNA experiments in 3T3-L1 adipocytes (Fig. 9A). FGF21 treatment increased p-ERK levels, but these increases were abolished when either FGFR1 or βKL was downregulated by siRNA (Fig. 9B and C). Moreover, treatment with FGF21 increased activated p-AMPK levels, and downregulation of FGFR1, βKL, or SIRT1 reversed the increase (Fig. 9D), suggesting a potential role of AMPK in linking FGF21 signaling and increased PGC-1α activity in anti-miR-34a-treated cells. Notably, anti-miR-34a decreased levels of acetylated PGC-1α, and the decrease was reversed by SIRT1 siRNA, as expected (Fig. 9E). Surprisingly, treatment with siRNA for FGFR1 or βKL also resulted in increased acetylation of PGC-1α (Fig. 9E), suggesting that deacetylation of PGC-1α is dependent on both FGF21 and SIRT1 functions. Relevant to these findings, a recent study showed that FGF21 enhances mitochondrial oxidative function by activating the AMPK/SIRT1/PGC-1α pathway (35).
FIG 9.

Anti-miR-34a increased PGC-1α browning gene activity via FGF21/SIRT1-dependent deacetylation. (A) Experimental outline for 3T3-L1 fat cells. (B) Decreased protein levels by siRNA treatment. (C to H) Effects of downregulation of FGFR1, βKL, or SIRT1 by siRNA on p-ERK levels (C), p-AMPK levels (D), acetylated PGC-1α levels (E), PGC-1α occupancy at browning genes (F), mRNA levels of browning genes (G), and mitochondrial DNA copy number (H). Data are means and SEM (n = 3). *, P < 0.05; **, P < 0.01; ***, P < 0.001. (I) Immunostaining of UCP1 protein. (J) (Left) In mice with diet-induced obesity, elevated miR-34a attenuates FGF21 signaling and the browning gene program at least in part by directly targeting both components of the FGF21 receptor complex (FGFR1 and βKL) and the SIRT1 deacetylase. (Right) Downregulation of miR-34a in obese mice improves FGF21 signaling by increasing expression of FGFR1 and βKL and also increases expression and activity of SIRT1, which contributes to deacetylation of PGC-1α and induction of PGC-1α target browning-related genes.
Consistent with increased acetylation and decreased transcriptional activity of PGC-1α (34), downregulation of FGFR1-βKL or SIRT1 substantially reduced the occupancy of PGC-1α and RNA polymerase II at the browning-related genes Ucp1, Prdm16, Pparγ, and C/ebpβ (Fig. 9F) and decreased expression of these browning genes (Fig. 9G), the mitochondrial DNA copy number (Fig. 9H), and UCP1 protein levels (Fig. 9I). These results, together with findings from the in vivo studies described above, indicate that downregulation of elevated miR-34a in obesity promotes beige and brown fat formation and that both improved adipocyte FGF21 signaling and SIRT1 functions likely contribute to the induction of browning genes by increasing activity of PGC-1α via protein deacetylation.
DISCUSSION
This study identifies elevated miR-34a in obesity as an inhibitor of fat browning and a potential target for weight loss and for treatment of obesity-related metabolic diseases. Downregulation of miR-34a in mice with diet-induced obesity increased expression of both components of the adipocyte FGF21 receptor complex, FGFR1 and βKL, and also that of SIRT1 deacetylase, which resulted in FGF21/SIRT1-dependent deacetylation of PGC-1α and induction of PGC-1α target browning-related genes (Fig. 9J). These molecular events contributed to increased CD137-positive beige fat formation in all types of WATs, including even, abdominal visceral fat, and promoted additional browning in BAT.
FGF21 has received substantial attention recently because of its lipid-lowering and insulin-sensitizing effects (12–16, 35). FGF21 shows beneficial impacts on energy homeostasis by promoting mitochondrial oxidative metabolism and brown and beige fat production (12–16, 35). Notably, circulating FGF21 levels are highly elevated in obese individuals with metabolic syndrome (37), suggesting that FGF21 signaling might be impaired in obesity (32), but FGF21 resistance in obesity has been controversial in mouse studies (16, 32, 38). Our data, however, indicate that FGF21 signaling in adipose tissue and liver hepatocytes is impaired in mice with diet-induced obesity under our experimental conditions, in agreement with such resistance in obesity (32). We further provide a mechanistic basis for FGF21 resistance by showing that elevated miR-34a contributes to attenuated FGF21 signaling in obese mice by downregulation of expression of the adipocyte FGF21 receptor complex components FGFR1 and βKL, in part through direct binding to the 3′UTRs of their transcripts. Elevated tumor necrosis factor alpha (TNF-α) in obesity inhibits expression of βKL and impairs FGF21 action in adipocytes (39), so it is also possible that mechanisms in addition to elevated miR-34a downregulate βKL levels in obesity.
In addition to promoting beige and brown fat formation in mice with diet-induced obesity, downregulation of miR-34a led to weight loss and improved serum lipid and glucose profiles and increased glucose tolerance. These beneficial metabolic effects are likely due to integrated actions from multiple tissues. In addition to restoring FGF21 signaling in adipose tissue, we also observed that downregulation of miR-34a improved FGF21 signaling in hepatocytes and increased expression in vivo of hepatic genes involved in fatty acid oxidation. Anti-miR-34a-mediated beneficial effects on lipid metabolism and fat browning in the present study are consistent with our previous studies (22, 23) showing that downregulation of miR-34a ameliorates the liver steatosis that is observed in obese mice which have aberrantly elevated hepatic miR-34a levels and, consequently, impaired FGF19 signaling in the liver. In addition to thermogenesis in adipose tissue, anti-miR-34a-mediated effects, including improved FGF21 signaling and stimulation of lipid oxidation in hepatocytes, would contribute to the overall beneficial impacts, including decreased adiposity.
Although the present study focused on the effect of miR-34a downregulation in obese animals on fat browning, we also observed that cold exposure of differentiated 3T3-L1 fat cells resulted in decreased miR-34a levels and dramatic increases in mitochondrial DNA copy numbers, expression of the browning genes Ucp1, Pgc-1α, and Prdm16, and browning differentiation and, furthermore, that overexpression of miR-34a completely reversed these cold temperature-induced browning effects. Consistent with these results, a recent miR expression profiling study showed that miR-34a was among the miRs that were downregulated upon cold temperature exposure (18). Our findings from cold exposure studies of 3T3-L1 fat cells, together with in vivo anti-miR-34a studies on mice with diet-induced obesity and the effects of anti-miR-34a on increased thermogenic gene expression, mitochondrial oxidative activity, and body temperature, suggest that miR-34a may function as a general inhibitor of brown fat differentiation in adaptive thermogenesis as well as under obese conditions.
PGC-1α was recently shown to be critical for FGF21-mediated fat browning upon cold exposure, and FGF21 was shown to increase PGC-1α protein levels without affecting protein stability (14), but how the transcriptional activity of PGC-1α is upregulated during brown adipocyte differentiation is still unclear. Our mechanistic studies suggest that a posttranslational modification of PGC-1α (deacetylation) is important for linking FGF21 signaling and induction of PGC-1α target browning genes. PGC-1α acetylation levels were increased by downregulation of SIRT1, as expected (34, 40), but, surprisingly, also by downregulation of FGFR1-βKL, suggesting that both FGF21 signaling and SIRT1 function are important for deacetylation of PGC-1α and its browning gene activity. The precise mechanism by which FGF21 signaling promotes SIRT1-dependent deacetylation of PGC-1α requires further investigation, but our findings suggest that increased AMPK activity may play a role, which is consistent with recent studies showing that FGF21 enhances mitochondrial oxidation through the AMPK/SIRT1/PGC-1α pathway (35) and that AMPK increases energy expenditure by upregulating SIRT1 activity (41). Future studies will be required to understand how the FGF21 and SIRT1 signaling pathways are integrated to regulate the acetylation status of PGC-1α and the browning gene program. Notably, each of these components, FGFR1, βKL, and SIRT1, is a direct target of miR-34a, suggesting that miR-34a, which is aberrantly elevated in obesity, is a key upstream regulator of genes in the cellular network that controls energy metabolism.
Increasing energy expenditure by promoting heat-generating brown fat production in BAT would be an appealing option for weight loss and for treatment of obesity-related metabolic disorders. However, there are limited amounts of BAT in adult humans, and more importantly, BAT is substantially reduced and WAT is increased in obese individuals (7, 42, 43), so it is not clear whether targeting BAT would have a significant impact on overall energy balance and weight reduction in obese patients. Moreover, it is well known that abdominal visceral adiposity strongly correlates with metabolic syndrome and insulin resistance (44). In this regard, one of the exciting and therapeutically relevant findings from the present study is that in vivo silencing of miR-34a in mice with diet-induced obesity not only increased brown fat depots in BAT but also substantially increased beige-like fat depots in all types of WATs, including visceral fat. To our knowledge, this is the first report showing that fat browning and thermogenesis are promoted in mice with diet-induced obesity by downregulation of an elevated microRNA without exposure of the animals to cold temperature. The effects of anti-miR-34a on increased expression of the brown fat marker UCP1 in all types of adipose tissues, together with its beneficial metabolic effects on insulin sensitivity and also its amelioration of liver steatosis (22, 23), suggest that targeting a single microRNA in obesity, miR-34a, may provide an effective therapeutic option for combating obesity-related metabolic disorders.
Supplementary Material
ACKNOWLEDGMENTS
We thank Kai Ge at the NIH for technical guidance for 3T3-L1 fat cell experiments. We also thank Mayandi Shivaguru and Donna Epps, from the core facility center of the Institute of Genomic Biology, and Karen Doty, from the College of Veterinary Medicine Histology Laboratory, both at the University of Illinois at Urbana-Champaign (UIUC), for their help with the histology studies. We also thank Lucas Li, from the metabolomics center at the UIUC, for measurement of serum fatty acids.
This study was supported by grants from the National Institutes of Health (DK62777 and DK95842) and the American Diabetes Association (Basic Science Award) to J.K.K.
T.F., S.S., S.C., and J.K.K. designed research; T.F., S.S., S.C., and Z.H. performed experiments; K.S.-P. and H.E.X. provided purified FGF21 for in vivo experiments; T.F., S.S., S.C., B.K., and J.K.K. analyzed data; and T.F., B.K., and J.K.K. wrote the paper.
We declare that we have no conflicts of interest.
Footnotes
Published ahead of print 2 September 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00596-14.
REFERENCES
- 1.Rosen ED, Spiegelman BM. 2006. Adipocytes as regulators of energy balance and glucose homeostasis. Nature 444:847–853. 10.1038/nature05483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cannon B, Nedergaard J. 2004. Brown adipose tissue: function and physiological significance. Physiol. Rev. 84:277–359. 10.1152/physrev.00015.2003. [DOI] [PubMed] [Google Scholar]
- 3.Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB, Kuo FC, Palmer EL, Tseng YH, Doria A, Kolodny GM, Kahn CR. 2009. Identification and importance of brown adipose tissue in adult humans. N. Engl. J. Med. 360:1509–1517. 10.1056/NEJMoa0810780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kajimura S, Seale P, Spiegelman BM. 2010. Transcriptional control of brown fat development. Cell Metab. 11:257–262. 10.1016/j.cmet.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harms M, Seale P. 2013. Brown and beige fat: development, function and therapeutic potential. Nat. Med. 19:1252–1263. 10.1038/nm.3361. [DOI] [PubMed] [Google Scholar]
- 6.Wu J, Bostrom P, Sparks LM, Ye L, Choi JH, Giang AH, Khandekar M, Virtanen KA, Nuutila P, Schaart G, Huang K, Tu H, van Marken Lichtenbelt WD, Hoeks J, Enerback S, Schrauwen P, Spiegelman BM. 2012. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell 150:366–376. 10.1016/j.cell.2012.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vijgen GH, Bouvy ND, Teule GJ, Brans B, Schrauwen P, van Marken Lichtenbelt WD. 2011. Brown adipose tissue in morbidly obese subjects. PLoS One 6:e17247. 10.1371/journal.pone.0017247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seale P, Conroe HM, Estall J, Kajimura S, Frontini A, Ishibashi J, Cohen P, Cinti S, Spiegelman BM. 2011. Prdm16 determines the thermogenic program of subcutaneous white adipose tissue in mice. J. Clin. Invest. 121:96–105. 10.1172/JCI44271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ohno H, Shinoda K, Spiegelman BM, Kajimura S. 2012. PPARgamma agonists induce a white-to-brown fat conversion through stabilization of PRDM16 protein. Cell Metab. 15:395–404. 10.1016/j.cmet.2012.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bostrom P, Wu J, Jedrychowski MP, Korde A, Ye L, Lo JC, Rasbach KA, Bostrom EA, Choi JH, Long JZ, Kajimura S, Zingaretti MC, Vind BF, Tu H, Cinti S, Hojlund K, Gygi SP, Spiegelman BM. 2012. A PGC1-alpha-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 481:463–468. 10.1038/nature10777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qiang L, Wang L, Kon N, Zhao W, Lee S, Zhang Y, Rosenbaum M, Zhao Y, Gu W, Farmer SR, Accili D. 2012. Brown remodeling of white adipose tissue by SirT1-dependent deacetylation of Ppargamma. Cell 150:620–632. 10.1016/j.cell.2012.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Inagaki T, Dutchak P, Zhao G, Ding X, Gautron L, Parameswara V, Li Y, Goetz R, Mohammadi M, Esser V, Elmquist JK, Gerard RD, Burgess SC, Hammer RE, Mangelsdorf DJ, Kliewer SA. 2007. Endocrine regulation of the fasting response by PPARalpha-mediated induction of fibroblast growth factor 21. Cell Metab. 5:415–425. 10.1016/j.cmet.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 13.Badman MK, Pissios P, Kennedy AR, Koukos G, Flier JS, Maratos-Flier E. 2007. Hepatic fibroblast growth factor 21 is regulated by PPARalpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 5:426–437. 10.1016/j.cmet.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 14.Fisher FM, Kleiner S, Douris N, Fox EC, Mepani RJ, Verdeguer F, Wu J, Kharitonenkov A, Flier JS, Maratos-Flier E, Spiegelman BM. 2012. FGF21 regulates PGC-1alpha and browning of white adipose tissues in adaptive thermogenesis. Genes Dev. 26:271–281. 10.1101/gad.177857.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dutchak PA, Katafuchi T, Bookout AL, Choi JH, Yu RT, Mangelsdorf DJ, Kliewer SA. 2012. Fibroblast growth factor-21 regulates PPARgamma activity and the antidiabetic actions of thiazolidinediones. Cell 148:556–567. 10.1016/j.cell.2011.11.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Canto C, Auwerx J. 2012. Cell biology. FGF21 takes a fat bite. Science 336:675–676. 10.1126/science.1222646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee P, Linderman JD, Smith S, Brychta RJ, Wang J, Idelson C, Perron RM, Werner CD, Phan GQ, Kammula US, Kebebew E, Pacak K, Chen KY, Celi FS. 2014. Irisin and FGF21 are cold-induced endocrine activators of brown fat function in humans. Cell Metab. 19:302–309. 10.1016/j.cmet.2013.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trajkovski M, Ahmed K, Esau CC, Stoffel M. 2012. MyomiR-133 regulates brown fat differentiation through Prdm16. Nat. Cell Biol. 14:1330–1335. 10.1038/ncb2612. [DOI] [PubMed] [Google Scholar]
- 19.Yin H, Pasut A, Soleimani VD, Bentzinger CF, Antoun G, Thorn S, Seale P, Fernando P, van Ijcken W, Grosveld F, Dekemp RA, Boushel R, Harper ME, Rudnicki MA. 2013. MicroRNA-133 controls brown adipose determination in skeletal muscle satellite cells by targeting Prdm16. Cell Metab. 17:210–224. 10.1016/j.cmet.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen Y, Siegel F, Kipschull S, Haas B, Frohlich H, Meister G, Pfeifer A. 2013. miR-155 regulates differentiation of brown and beige adipocytes via a bistable circuit. Nat. Commun. 4:1769. 10.1038/ncomms2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ortega FJ, Moreno-Navarrete JM, Pardo G, Sabater M, Hummel M, Ferrer A, Rodriguez-Hermosa JI, Ruiz B, Ricart W, Peral B, Fernandez-Real JM. 2010. MiRNA expression profile of human subcutaneous adipose and during adipocyte differentiation. PLoS One 5:e9022. 10.1371/journal.pone.0009022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fu T, Choi SE, Kim DH, Seok S, Suino-Powell KM, Xu HE, Kemper JK. 2012. Aberrantly elevated microRNA-34a in obesity attenuates hepatic responses to FGF19 by targeting a membrane coreceptor beta-Klotho. Proc. Natl. Acad. Sci. U. S. A. 109:16137–16142. 10.1073/pnas.1205951109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Choi SE, Fu T, Seok S, Kim DH, Yu E, Lee KW, Kang Y, Li X, Kemper B, Kemper JK. 2013. Elevated microRNA-34a in obesity reduces NAD levels and SIRT1 activity by directly targeting NAMPT. Aging Cell 12:1062–1072. 10.1111/acel.12135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kemper JK, Xiao Z, Ponugoti B, Miao J, Fang S, Kanamaluru D, Tsang S, Wu S, Chiang CM, Veenstra TD. 2009. FXR acetylation is normally dynamically regulated by p300 and SIRT1 but constitutively elevated in metabolic disease states. Cell Metab. 10:392–404. 10.1016/j.cmet.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee J, Padhye A, Sharma A, Song G, Miao J, Mo YY, Wang L, Kemper JK. 2010. A pathway involving farnesoid X receptor and small heterodimer partner positively regulates hepatic sirtuin 1 levels via microRNA-34a inhibition. J. Biol. Chem. 285:12604–12611. 10.1074/jbc.M109.094524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Villanueva CJ, Vergnes L, Wang J, Drew BG, Hong C, Tu Y, Hu Y, Peng X, Xu F, Saez E, Wroblewski K, Hevener AL, Reue K, Fong LG, Young SG, Tontonoz P. 2013. Adipose subtype-selective recruitment of TLE3 or Prdm16 by PPARgamma specifies lipid storage versus thermogenic gene programs. Cell Metab. 17:423–435. 10.1016/j.cmet.2013.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Surwit RS, Kuhn CM, Cochrane C, McCubbin JA, Feinglos MN. 1988. Diet-induced type II diabetes in C57BL/6J mice. Diabetes 37:1163–1167. [DOI] [PubMed] [Google Scholar]
- 28.Montgomery MK, Hallahan NL, Brown SH, Liu M, Mitchell TW, Cooney GJ, Turner N. 2013. Mouse strain-dependent variation in obesity and glucose homeostasis in response to high-fat feeding. Diabetologia 56:1129–1139. 10.1007/s00125-013-2846-8. [DOI] [PubMed] [Google Scholar]
- 29.Wu J, Cohen P, Spiegelman BM. 2013. Adaptive thermogenesis in adipocytes: is beige the new brown? Genes Dev. 27:234–250. 10.1101/gad.211649.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kajimura S, Seale P, Tomaru T, Erdjument-Bromage H, Cooper MP, Ruas JL, Chin S, Tempst P, Lazar MA, Spiegelman BM. 2008. Regulation of the brown and white fat gene programs through a PRDM16/CtBP transcriptional complex. Genes Dev. 22:1397–1409. 10.1101/gad.1666108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kurosu H, Choi M, Ogawa Y, Dickson AS, Goetz R, Eliseenkova AV, Mohammadi M, Rosenblatt KP, Kliewer SA, Kuro-o M. 2007. Tissue-specific expression of betaKlotho and fibroblast growth factor (FGF) receptor isoforms determines metabolic activity of FGF19 and FGF21. J. Biol. Chem. 282:26687–26695. 10.1074/jbc.M704165200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fisher FM, Chui PC, Antonellis PJ, Bina HA, Kharitonenkov A, Flier JS, Maratos-Flier E. 2010. Obesity is a fibroblast growth factor 21 (FGF21)-resistant state. Diabetes 59:2781–2789. 10.2337/db10-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. 1998. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 92:829–839. 10.1016/S0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- 34.Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. 2005. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 434:113–118. 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 35.Chau MD, Gao J, Yang Q, Wu Z, Gromada J. 2010. Fibroblast growth factor 21 regulates energy metabolism by activating the AMPK-SIRT1-PGC-1alpha pathway. Proc. Natl. Acad. Sci. U. S. A. 107:12553–12558. 10.1073/pnas.1006962107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Choi SE, Kemper JK. 2013. Regulation of SIRT1 by microRNAs. Mol. Cells 36:385–392. 10.1007/s10059-013-0297-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mraz M, Bartlova M, Lacinova Z, Michalsky D, Kasalicky M, Haluzikova D, Matoulek M, Dostalova I, Humenanska V, Haluzik M. 2009. Serum concentrations and tissue expression of a novel endocrine regulator fibroblast growth factor-21 in patients with type 2 diabetes and obesity. Clin. Endocrinol. (Oxford) 71:369–375. 10.1111/j.1365-2265.2008.03502.x. [DOI] [PubMed] [Google Scholar]
- 38.Hale C, Chen MM, Stanislaus S, Chinookoswong N, Hager T, Wang M, Veniant MM, Xu J. 2012. Lack of overt FGF21 resistance in two mouse models of obesity and insulin resistance. Endocrinology 153:69–80. 10.1210/en.2010-1262. [DOI] [PubMed] [Google Scholar]
- 39.Diaz-Delfin J, Hondares E, Iglesias R, Giralt M, Caelles C, Villarroya F. 2012. TNF-alpha represses beta-Klotho expression and impairs FGF21 action in adipose cells: involvement of JNK1 in the FGF21 pathway. Endocrinology 153:4238–4245. 10.1210/en.2012-1193. [DOI] [PubMed] [Google Scholar]
- 40.Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P, Geny B, Laakso M, Puigserver P, Auwerx J. 2006. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 127:1109–1122. 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 41.Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. 2009. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458:1056–1060. 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Frontini A, Cinti S. 2010. Distribution and development of brown adipocytes in the murine and human adipose organ. Cell Metab. 11:253–256. 10.1016/j.cmet.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 43.Lidell ME, Betz MJ, Dahlqvist Leinhard O, Heglind M, Elander L, Slawik M, Mussack T, Nilsson D, Romu T, Nuutila P, Virtanen KA, Beuschlein F, Persson A, Borga M, Enerback S. 2013. Evidence for two types of brown adipose tissue in humans. Nat. Med. 19:631–634. 10.1038/nm.3017. [DOI] [PubMed] [Google Scholar]
- 44.Despres JP, Lemieux I, Bergeron J, Pibarot P, Mathieu P, Larose E, Rodes-Cabau J, Bertrand OF, Poirier P. 2008. Abdominal obesity and the metabolic syndrome: contribution to global cardiometabolic risk. Arterioscler. Thromb. Vasc. Biol. 28:1039–1049. 10.1161/ATVBAHA.107.159228. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.