

Abstract
Hemangioendotheliomas are categorized as intermediate-grade vascular tumors that are commonly localized in the lungs and livers. The regulation of this tumor cell's proliferative and apoptotic mechanisms is ill defined. We recently documented an important role for Hippo pathway signaling via endothelial cell adhesion molecules in brain microvascular endothelial cell proliferation and apoptosis. We found that endothelial cells lacking cell adhesion molecules escaped from contact inhibition and exhibited abnormal proliferation and apoptosis. Here we report on the roles of adherens junction molecule modulation of survivin and the Hippo pathway in the proliferation and apoptosis of a murine hemangioendothelioma (EOMA) cell. We demonstrated reduced adherens junction molecule (CD31 and VE-cadherin) expression, increased survivin and Ajuba expression, and a reduction in Hippo pathway signaling resulting in increased proliferation and decreased activation of effector caspase 3 in postconfluent EOMA cell cultures. Furthermore, we confirmed that YM155, an antisurvivin drug that interferes with Sp1-survivin promoter interactions, and survivin small interference RNA (siRNA) transfection elicited induction of VE-cadherin, decreased Ajuba expression, increased Hippo pathway and caspase activation and apoptosis, and decreased cell proliferation. These findings support the importance of the Hippo pathway in hemangioendothelioma cell proliferation and survival and YM155 as a potential therapeutic agent in this category of vascular tumors.
INTRODUCTION
Tumors derived from endothelial cells span a broad range of lesions, from benign hemangiomas, including capillary and cavernous hemangiomas, lymphangiomas, and vascular ectasias, and intermediate-grade lesions, including Kaposi sarcomas and hemangioendotheliomas, to malignant lesions, including angiosarcomas and hemangiopericytomas (1, 2). Whether benign, intermediate, or malignant, these lesions share a feature of robust proliferation at some stage during their development, although their regressive behaviors vary widely. Hemangioendotheliomas are defined as proliferations arising from endothelial cells exhibiting behavior intermediate between highly malignant angiosarcomas and benign hemangiomas (3). Hoak et al. isolated hemangioma tissue from the 129/J mouse strain that in many ways mimicked the presentation of Kasabach-Merrit syndrome and developed an animal model of this condition (4). Following this, tumor tissue was able to be passaged from mouse to mouse, and endothelial cells derived from the tumor tissue gave rise to hemangioendotheliomas when reinjected into mice and were able to be serially passaged up to 40 times (5). Obeso et al. later isolated and characterized endothelial cells from this tumor and established a cell line (EOMA) (6).
In this report, we examined the proliferation, apoptosis, morphological, cell adhesion, and Hippo pathway parameters, comparing wild-type brain microvascular endothelial cells (7, 8) with EOMA cells, both of which were derived from murine sources. We documented significant differences in proliferation, apoptosis, contact inhibition, adhesion molecule and matrix metalloprotease (MMP) expression, and Hippo pathway component expression and localization (nuclear Yes-associated protein [YAP], cytoplasmic phospho-YAP [P-YAP], and Ajuba) and their modulation by treatment with the survivin inhibitor YM155 and survivin small interference RNAs (siRNAs). The importance of the Hippo pathway and its components and its potential as a therapeutic target are discussed.
MATERIALS AND METHODS
Cell culture.
Murine hemangioendothelioma (EOMA) cells were obtained from Robert Auerbach (University of Wisconsin, Madison, WI) (6, 9). Brain endothelial cells (BEC) were isolated from cerebral microvessels of C57BL/6 wild-type BEC (WT-BEC) and CD44 knockout BEC (CD44KO-BEC) mice [B6.129(Cg)-Cd44tmlHbg/J] (The Jackson Laboratory, Bar Harbor, ME), as described previously (7). CD31 knockout endothelial cells were isolated from brain (CD31KO-BEC) as described previously (10). EOMA and endothelial cells were cultured on 1.5% gelatin (catalog no. G8-500; Thermo Fisher Scientific Inc., Waltham, MA)-coated plates in endothelial cell medium (Dulbecco's modified Eagle's medium [DMEM] with high glucose [Life Technologies, Grand Island, NY] containing 10% fetal bovine serum [FBS], 2 mM l-glutamine, 0.1 mM nonessential amino acids, 1 mM sodium pyruvate, 10 mM HEPES [pH 7.4], 10−5 M β-mercaptoethanol, 100 U/ml penicillin, and 100 μg/ml streptomycin [Life Technologies]) in 8% CO2 at 37°C (8). Cells were used between passages 21 and 23 and cultured under normoxic (20% O2) conditions.
Cell proliferation analysis.
We demonstrated growth curves of WT-BEC and EOMA cells. Each cell line was plated at 3,000 cells per well in 96-well plates (n = 4 each). At 24, 48, 72, 96, 120, 144, and 168 h after plating, the wells were washed with phosphate-buffered saline (PBS) (pH 7.4). After freeze-thawing, wells were treated with 200 μl of dye/cell lysis buffer by using a CyQUANT cell proliferation assay kit (Life Technologies). After incubation at room temperature for 5 min, sample fluorescence was measured by using the Wallac 1420 fluorescence microplate reader (PerkinElmer Inc., Turku, Finland) with filters for 485 nm (8). For proliferation rate analysis, initial and secondary proliferation rates were determined as follows: initial proliferation rates = (average cell number at 72 h − average cell number at 48 h)/24 and secondary proliferation rates = (average cell number at 168 h − average cell number at 72 h)/96. Cells were used at passage 22.
Antibodies.
Antibodies against the mouse CD31 ectodomain (affinity-purified SL-4) were raised in rabbit and purified as described previously (11). Rabbit polyclonal antibodies against mouse caspase 3 (catalog no. 9662), caspase 7 (catalog no. 9492), caspase 10 (catalog no. 9752), cleaved caspase 3 (Asp175) (catalog no. 9661), P120-catenin (catenin δ-1) (catalog no. 4989), and Ajuba (catalog no. 4897); rabbit monoclonal antibodies against cleaved caspase 8 (Asp387) (clone D5B2) (catalog no. 8592), survivin (clone 71G4B7) (catalog no. 2808), P-YAP (Ser127) (clone D9W2I) (catalog no. 13008), and YAP/tafazzin (TAZ) (clone D24E4) (catalog no. 8418); and a mouse monoclonal antibody against PCNA (proliferating cell nuclear antigen) (clone PC10) (catalog no. 2586) were purchased from Cell Signaling Technology Inc. (Danvers, MA). A rat monoclonal antibody against VE-cadherin (clone 11D4.1) (catalog no. 550548) and mouse monoclonal antibodies against α-catenin (clone 5) (catalog no. 610193) and γ-catenin (clone 15/γ-catenin) (catalog no. 610254) were purchased from BD Biosciences (San Jose, CA). Rabbit polyclonal antibodies against β-catenin (N terminus) (catalog no. 1061) were purchased from ECM Biosciences (Versailles, KY). Mouse monoclonal antibodies against mouse β-actin (clone AC-15) (catalog no. ab6276) and MMP2 (clone 6E3F8) (catalog no. ab86607) and rabbit polyclonal antibodies against VE-cadherin (catalog no. ab33168), Ki-67 (catalog no. ab15580), and MMP9 (catalog no. ab38898) were purchased from Abcam (Cambridge, MA). Rabbit polyclonal antibodies against histone H1 (FL-219) (catalog no. sc-10806) and P53 (FL-393) (catalog no. sc-6243) and secondary antibodies raised in donkey against rabbit IgG (catalog no. sc-2313) and mouse IgG (catalog no. sc-2318), which were conjugated to horseradish peroxidase (HRP), were purchased from Santa Cruz Biotechnology Inc. (Dallas, TX). Every caspase antibody was confirmed to detect full-length caspase as well as the cleaved (active) form (8).
Preparation of cell lysates.
Confluent 60-mm dishes of cells were rinsed twice with cold PBS (pH 7.4) containing 1 mM sodium orthovanadate (Na3VO4) and then lysed with radioimmunoprecipitation assay (RIPA) buffer (EMD Millipore, Billerica, MA) supplemented with complete EDTA-free protease inhibitor (catalog no. 04693132001; Roche Diagnostics, Indianapolis, IN), phosphatase inhibitor cocktail sets I and II (catalog no. 524624 and 524625; EMD Millipore), and 1 mM phenylmethylsulfonyl fluoride (PMSF) (8). Cell lysate samples were placed on ice for 30 min, vortexed thoroughly, and then centrifuged at 14,000 rpm for 15 min to remove insoluble materials. After the total protein concentration was determined by using a Pierce BCA protein assay kit (Thermo Fisher Scientific Inc., Rockford, IL), an aliquot of 12 μg of protein samples was resuspended in SDS sample buffer and boiled for 5 min.
Nuclear and cytoplasmic fractionations.
Confluent 60-mm dishes of cells were harvested with 0.05% trypsin–EDTA (Life Technologies) and centrifuged. Cells were washed twice with cold PBS and pelleted by centrifugation. Nuclear and cytoplasmic fractionation was performed by using NE-PER nuclear and cytoplasmic extraction reagents (catalog no. 78833; Thermo Fisher Scientific Inc.) according to the manufacturer's instructions. The protease and phosphatase inhibitors mentioned above were added to cytoplasmic extraction reagent I (CER-I) and nuclear extraction reagent (NER) (8). Protein concentrations of nuclear and cytoplasmic fractions were determined by using a BCA protein assay kit, and an aliquot of 12 μg of protein was resuspended in SDS sample buffer and boiled for 5 min.
Western blotting.
Samples suspended in sample buffer were subjected to 10 to 15% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) under reducing conditions, and the gels were transferred onto polyvinylidene difluoride (PVDF) membranes (EMD Millipore). The membranes were incubated with 1% bovine serum albumin (BSA) in 50 mM Tris-buffered saline (pH 7.4) containing 0.1% Tween 20 (TTBS) for 1 h at room temperature to block nonspecific protein binding, followed by incubation of the membranes overnight in TTBS containing primary antibody diluted 1:200 (anti-P53 and -histone H1), 1:250 (anti-α-catenin), 1:1,000 (anti-caspases 3, 7, 8, and 10; anti-VE-cadherin [catalog no. ab33168]; anti-β-catenin; anti-P120-catenin; antisurvivin; anti-MMP2; anti-MMP9; anti-P-YAP; anti-YAP/TAZ; and anti-Ajuba), 1:2,000 (anti-PCNA), 1:2,500 (anti-CD31), 1:5,000 (anti-β-actin), and 1:6,000 (anti-γ-catenin). After washing with TTBS, the membranes were reacted with HRP-conjugated secondary antibodies diluted 1:10,000 in TTBS for 1 h at room temperature. Target protein bands were detected by using Western Lightning enhanced chemiluminescence substrate (PerkinElmer) according to the manufacturer's instructions. Quantitation was performed on scanned densitometric images by using Quantity One software (Bio-Rad Laboratories, Hercules, CA) in triplicate experiments.
YM155 treatment.
Confluent 60-mm dishes of WT-BEC, EOMA cells, CD44KO-BEC, and CD31KO-BEC were treated with YM155 {4,9-dihydro-1-(2-methoxyethyl)2-methyl-4,9-dioxo-3-(2-pyrazinylmethyl)-1H-naphth[2,3-d]imidazolium, bromide} (12) (CAS registry no. 781661-94-7) (item no. 11490; Cayman Chemical, Ann Arbor, MI) dissolved in dimethyl sulfoxide (DMSO) at different final concentrations (0, 10, 50, 100, 200, and 500 nM) for 24 h. After incubation with YM155, cells were lysed, and survivin, caspase 8, caspase 3, caspase 7, P53, PCNA, CD31, VE-cadherin, Ajuba, cytoplasmic P-YAP, and nuclear YAP concentrations were determined by Western blotting as mentioned above. Furthermore, treated cells grown on 12-well plates were fixed, and survivin, cleaved caspase 3, Ki-67, P-YAP, YAP, CD31, VE-cadherin, and Ajuba expression/localization modes were determined by immunofluorescence. To examine the effect of YM155 on contact inhibition, 2.0 × 105 cells (WT-BEC and EOMA cells) were plated onto 6-well plates. After 6 h, floating cells and medium were removed, and fresh medium with YM155 (0 nM, 50 nM, and 500 nM) was added. This time point is defined as 0 h in Fig. 4M. Every 24 h (0, 24, 48, 72, 96, and 120 h), medium with YM155 was replaced by fresh medium, and phase-contrast microscopic photographs were taken by using Hoffman interference reflection microscopy (13) and Photoshop CS2 software (Adobe, San Jose, CA) on a Windows 7 computer.
FIG 4.

WT-BEC survival and the Hippo pathway are unaffected by YM155 treatment. (A to K) Western blot analyses reveal that survivin (A), cytoplasmic P-YAP (B), nuclear YAP (C), CD31 (D), VE-cadherin (E), cleaved caspase 8 (CC8) (F), cleaved caspase 3 (CC3) (G), MMP2 (I), MMP9 (J), and PCNA (K) levels in WT-BEC were not affected in the presence of YM155 (0, 50, and 500 nM). In contrast, in EOMA cells, MMP9 was not affected in the presence of YM155 (J), and survivin (A) and MMP2 (I) expression levels were reduced in the presence of 500 nM YM155, while CD31 (D), VE-cadherin (E), cleaved caspase 3 (G), and cytoplasmic P-YAP (B) expression levels were increased and nuclear YAP (C) and PCNA (K) expression levels were decreased. Data represent the mean ratios of survivin (A), P-YAP (B), CD31 (D), VE-cadherin (E), MMP2 (I), MMP9 (J), and PCNA (K) to β-actin; YAP (C) to histone H1; and cleaved caspase 8 (F) and caspase 3 (G) to full-length caspase from triplicate samples for each cell type (WT-BEC and EOMA cells) with different concentrations of YM155 (0, 50, and 500 nM) ± SD (*, P < 0.05; **, P < 0.01; ***, P < 0.001). In panel H, numbers of apoptotic EOMA cells were significantly increased in the presence of YM155. Data are means ± SD from triplicate experiments (**, P < 0.01). (L) Phase-contrast and merged DAPI and survivin, DAPI and P-YAP, DAPI and YAP, DAPI and CD31, DAPI and VE-cadherin, and DAPI and cleaved caspase 3 immunofluorescence micrographs of WT-BEC treated with 0 nM and 500 nM YM155 confirming our Western blot analyses. Bar = 100 μm. (M) Morphological analysis of WT-BEC and EOMA cells with YM155 treatment (0, 50, and 500 nM) plated at low cell density (0 h) (top), just at confluence (48 h) (middle), and until overconfluent (120 h) (bottom), using Hoffman interference reflection microscopy. EOMA cells were contact inhibited in the presence of 500 nM YM155 and modestly inhibited at 50 nM YM155. WT-BEC morphology was not affected by YM155 treatment. Bar = 100 μm.
Immunofluorescence.
Confluent cultures (high cell density) of WT-BEC and EOMA cells, grown on 12-well plates, were fixed with 4% paraformaldehyde in 50 mM HEPES buffer (pH 7.3) for 20 min and permeabilized for 20 min with 0.2% Triton X-100 in PBS (pH 7.4) at room temperature. For Ajuba immunostainings, cells were fixed with 100% methanol for 20 min and permeabilized for 20 min with 0%, 0.2%, or 0.5% Triton X-100 in PBS. To prevent nonspecific protein binding, cells were incubated with 5% BSA in PBS containing 0.05% Triton X-100 (T-PBS) for 1 h at room temperature (8). The cells were then incubated with the primary antibodies (VE-cadherin [clone 11D4.1] and α-catenin [clone 5] diluted 1:50 in T-PBS; β-catenin diluted 1:150; CD31 [SL-4] and YAP [clone D24E4] diluted 1:200; MMP2 and MMP9 diluted 1:300; cleaved caspase 3 and survivin [clone 71G4B7] diluted 1:400; cleaved caspase 8, Ki-67, and Ajuba diluted 1:500; γ-catenin [clone 15/γ-catenin] diluted 1:1,000; and P-YAP [clone D9W2I] diluted 1:2,000) overnight at 4°C and incubated further with secondary antibodies [Alexa Fluor 488-conjugated goat anti-rabbit IgG(H+L), anti-mouse IgG(H+L), or anti-rat IgG(H+L) (Life Technologies)] diluted 1:100 and Alexa Fluor 594 phalloidin (catalog no. A12381; Life Technologies) diluted 1:40 in T-PBS for 1 h at room temperature. Finally, cells were counterstained with DAPI (4′,6-diamidino-2-phenylindole) (catalog no. D9564; Sigma-Aldrich, St. Louis, MO) diluted 1:5,000 in T-PBS for 10 min at room temperature and coverslipped with Dako fluorescence mounting medium (catalog no. S3023; Dako, Carpentaria, CA). Digital fluorescence images were captured and phase-contrast microscopic photographs were taken on an Olympus IX71 inverted microscope equipped with a MicroFire camera and PictureFrame 1.0 software for Macintosh (Optronics) with Photoshop CS2 software (Adobe) on a Windows 7 computer.
Cell apoptosis assay.
Apoptotic cells in the confluent monolayer of WT-BEC and EOMA cells treated with or without 500 nM YM155 on 12-well plates were stained and detected by using annexin V conjugates for apoptosis detection and Alexa Fluor 488 (catalog no. A13201; Life Technologies) and 7-aminoactinomycin D (7-AAD) (catalog no. 30060; Biotium Inc., Hayward, CA). Unfixed cells on 12-well plates were washed 3 times with washing buffer (50 mM HEPES [pH 7.4], 150 mM NaCl) and 2 times with binding buffer (10 mM HEPES [pH 7.4], 150 mM NaCl, 2.5 mM CaCl2). After washing, cells were treated with annexin V (1:10) and 7-AAD (1:1,000) diluted in binding buffer and incubated for 1 h at room temperature in the dark. Stained cells were washed 2 times with binding buffer and 3 times with washing buffer. Finally, cells were counterstained with DAPI (Sigma-Aldrich) and coverslipped with Dako fluorescence mounting medium. Annexin V-positive/7-AAD-negative apoptotic cells were counted on the digital fluorescence images by using ImageJ 1.48v software (National Institutes of Health) on a Windows 7 computer. Cells were used at passage 21.
RNA interference.
Small interference RNA-mediated survivin knockdown experiments were performed by using a Stealth RNA interference (RNAi)-siRNA duplex oligoribonucleotide system (Life Technologies). The following three mouse siRNAs targeting survivin (GenBank accession no. NM_001012273.1) sense sequences were designed by using BLOCK-iT RNAi Designer (Life Technologies): 5′-GCGCGAUUUGAAUCCUGCGUUUGAG-3′ (siSurvivin1), 5′-CCUGUCACGUGAAGUUGAUUGGGAA-3′ (siSurvivin2), and 5′-GCUCAUCUGUUACCCUCGAACUGUU-3′ (siSurvivin3). The Stealth RNAi siRNA negative control (siNC), Med GC (catalog no. 12935-300; Life Technologies), and sterile Milli-Q water (Mock) were used as negative controls. Stealth siRNA transfection was performed with Lipofectamine RNAiMAX reagents (Life Technologies) by a reverse transfection method, according to the manufacturer's instructions. Efficiencies of RNAi-mediated survivin knockdown were evaluated by quantitative real-time PCR (qRT-PCR), Western blotting, and immunofluorescence staining.
qRT-PCR.
Cells were grown to confluence and lysed with TRIzol reagent (Life Technologies), and mRNA was extracted according to the manufacturer's instructions (7). cDNA was synthesized by using an iScript cDNA synthesis kit (Bio-Rad) according to the manufacturer's instructions (7). qRT-PCR was carried out on the iCycler iQ real-time PCR detection system (catalog no. 170-8740; Bio-Rad) by using iQ SYBR green Supermix, and each sample was run in triplicate (7). Threshold cycle (CT) values were normalized against β-actin values (14), and data were analyzed by using delta-CT (7) and relative expression methods. The following primer sets for mouse Ajuba (GenBank accession no. NM_010590.5), survivin (accession no. NM_001012273.1), and β-actin (accession no. NM_007393.1) were designed by using NCBI Primer-BLAST (http://www.ncbi.nlm.nih.gov/tools/primer-blast/) and synthesized by Integrated DNA Technologies Inc. (Coralville, IA): forward primer 5′-GGGCTGTGAGGACATTGTGA-3′ and reverse primer 5′-GCAACCTTCCTCGTCACTCA-3′ for Ajuba, forward primer 5′-ATCGCCACCTTCAAGAACTG-3′ and reverse primer 5′-CAGGGGAGTGCTTTCTATGC-3′ for survivin, and forward primer 5′-AAGAGCTATGAGCTGCCTGA-3′ and reverse primer 5′-TACGGATGTCAACGTCACAC-3′ for β-actin.
Cell adhesion assay.
To understand WT-BEC and EOMA cell adhesiveness compared to YM155 treatment or survivin knockdown, WT-BEC, EOMA cells, cells treated with 500 nM YM155, and Stealth siRNA-transfected cells (EOMA-siNC and EOMA-siSurvivin1) were plated at 2 × 105 cells in 200 μl of endothelial cell medium per well on 96-well microplates (n = 8 each) coated with 1.5% gelatin and cultured in CO2 incubators. At 0, 30, 60, 90, 120, 240, and 480 min after plating, nonadherent cells were washed with cold PBS (pH 7.4). After freeze-thawing, wells were treated with 200 μl of dye/cell lysis buffer by using a CyQUANT cell proliferation assay kit (Life Technologies). After incubation at room temperature for 5 min, the sample fluorescence was measured by using the Wallac 1420 fluorescence microplate reader (PerkinElmer Inc.) with filters for 485 nm. Cells were used at passage 22.
Migration and invasion assays.
Cell migration and invasion were demonstrated by using Falcon cell culture inserts in a 24-well companion tissue culture plate system with an 8-μm-pore-size polyethylene terephthalate membrane (catalog no. 353097; BD Biosciences). For invasion assays, 24-well BioCoat Matrigel Invasion Chamber systems (catalog no. 354480; BD Biosciences) were used after rehydration. For migration and invasion assays, cells were seeded in 500 μl of endothelial cell medium without FBS (2 × 104 cells/ml) in the insert chamber, and the lower chamber was filled with 750 μl of endothelial cell medium, according to the manufacturer's instructions. After 24 h of culture in CO2 incubators, the nonmigrating and noninvading cells were carefully removed from the upper surface of the membrane with cotton swabs, and the bottom membrane was fixed with 5% paraformaldehyde (PFA) in 50 mM HEPES (pH 7.4) for 20 min and stained with Mayer's hematoxylin (Lillie's modification) (catalog no. S3309; Dako). After coverslipping with mounting medium (Dako), numbers of migrating and invading cells were counted under a microscope in 1 mm2 of three different fields of filter membranes from triplicate experiments (total of 9 different fields). Cells were used at passages 22 and 23.
Statistical analysis.
All data are means ± standard deviations (SD) from a series of experiments. Statistical analysis was performed by using unpaired Student's t test using GraphPad Prism version 6.03 for Windows (GraphPad Software Inc., La Jolla, CA). P values of <0.05 were considered significant (8). Significant P values are mentioned in the figure legends.
RESULTS
EOMA cells are not contact inhibited and exhibit decreased apoptosis.
EOMA cells were not contact inhibited and were found to exhibit an overriding morphology, forming tube-like and cyst-like structures above the monolayers after confluence (Fig. 1A, bottom, areas outlined by white dashed lines). In contrast, WT-BEC were contact inhibited, exhibiting well-formed confluent monolayers (Fig. 1A, left). Growth curve and cell proliferation rate analyses indicated that EOMA cells exhibited a higher secondary proliferation rate, which is consistent with our evidence for a loss of contact inhibition (Fig. 1B and C). In a high-cell-density culture, EOMA cells exhibited higher survivin (Fig. 1D and 2M), Ki-67 (Fig. 2M), and PCNA (Fig. 1E) expression levels than did WT-BEC, resulting in higher cell proliferative potentials in EOMA cells. Regarding caspase-mediated apoptosis, EOMA cells exhibited lower initiator caspase 8 (Fig. 1F) and effector caspase 3 (Fig. 1G) activation than did WT-BEC. Levels of activation of initiator caspases 9, 10, and 12 and effector caspases 6 and 7 were not appreciably different between WT-BEC and EOMA cells (data not shown). Expression levels of full-length caspase were also not appreciably different between WT-BEC and EOMA cells (data not shown). Apoptosis was also evaluated by using annexin V/7-AAD staining. This analysis indicated that EOMA cells exhibited a significantly lower percentage of apoptotic cells than did WT-BEC (Fig. 1H).
FIG 1.
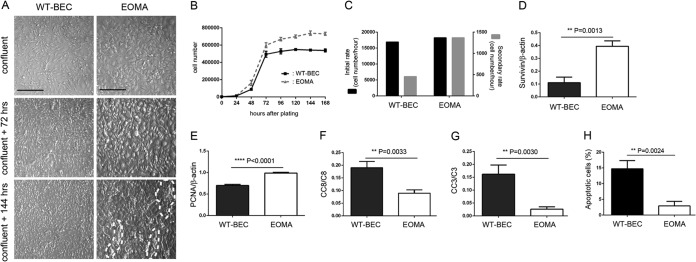
EOMA cells exhibit a loss of contact inhibition and reduced apoptosis compared to wild-type brain microvascular endothelial cells (WT-BEC). (A) Morphological analysis of WT-BEC and EOMA cells plated at high cell density (top, confluent; middle, 72 h postconfluence; bottom,144 h postconfluence) using Hoffman interference reflection microscopy. EOMA cells (right) were not contact inhibited, exhibiting overgrowth and cyst and tube formation (denoted by white dashed lines), in contrast to WT-BEC (left), which were contact inhibited. Bar = 200 μm. (B) Growth curves of WT-BEC and EOMA cells for 168 h. All data are means ± SD from triplicate experiments. (C) Analyses of initial and secondary proliferation rates of WT-BEC and EOMA cells. EOMA cells exhibited a higher secondary proliferation rate (72 to 168 h) than did WT-BEC. (D and E) Western blot expression analyses of survivin (D) and PCNA (E) between WT-BEC and EOMA cells. EOMA cells exhibit increased expression levels of survivin and proliferating nuclear antigen. Data represent the mean survivin (D)- or PCNA (E)-versus-β-actin ratios from triplicate assays for each cell type ± SD (**, P < 0.01; ****, P < 0.0001). (F and G) Western blot analyses revealing robust decreased initiator caspase 8 (CC8) (F) and effector caspase 3 (CC3) (G) activation in EOMA cells compared with WT-BEC. (H) Apoptosis was evaluated by using annexin V/7-AAD staining. Analyses indicated that EOMA cells exhibited a significantly lower percentage of apoptotic cells than did WT-BEC. All data represent the mean ratios of cleaved caspase to full-length caspase from triplicate assays for each cell type ± SD (**, P < 0.01).
FIG 2.

EOMA cells exhibit decreased levels of adherens junction-related proteins, increased MMP expression levels, and reciprocal levels of cytoplasmic and nuclear YAP compared to wild-type cells. (A to L) Western blot analyses revealed that the adherens junction-related molecules CD31 (A), VE-cadherin (B), α-catenin (C), β-catenin (D), γ-catenin (E), and P120-catenin (F) were attenuated and that MMP2 (G) and MMP9 (H) expression levels were increased in EOMA cells compared to WT-BEC. (I and J) In EOMA cells, cytoplasmic P-YAP levels (I) were downregulated, and nuclear YAP levels (J) were upregulated compared to levels in WT-BEC. Data represent the mean ratios of CD31 (A), VE-cadherin (B), α-catenin (C), β-catenin (D), γ-catenin (E), P120-catenin (F), MMP2 (G), MMP9 (H), and P-YAP (I) to β-actin and the mean ratios of YAP (J) and TAZ (L) to histone H1 from triplicate samples for each cell type ± SD (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001). (K) Representative Western blots of YAP and TAZ cytoplasmic and nuclear fractions of WT-BEC and EOMA cells illustrating the differences in YAP cytoplasmic and nuclear fractions in WT-BEC and EOMA cells, while no appreciable changes are noted for TAZ cytoplasmic and nuclear fractions in samples of the two cell lines. (L) Western blot analysis of TAZ levels in the nuclear fractions of WT-BEC and EOMA cell lysates illustrating no differences between samples of the two cell lysates. (M) Phase-contrast and merged DAPI and F-actin, DAPI and CD31, DAPI and VE-cadherin, DAPI and α-catenin, DAPI and β-catenin, DAPI and γ-catenin, DAPI and MMP2, DAPI and Ki-67, DAPI and survivin, DAPI and cleaved caspase 8 (CC8), DAPI and cleaved caspase 3 (CC3), DAPI and P-YAP, and DAPI and YAP immunofluorescence micrographs of WT-BEC and EOMA cells confirming our Western blot analyses. Insets in CD31 and VE-cadherin panels illustrate differences in labeling intensities and localization patterns between WT-BEC and EOMA cells. Bar = 50 μm.
EOMA cells exhibit decreased expression levels of adherens junction-related proteins and diminished Hippo pathway activity at high cell density.
In a high-cell-density culture, EOMA cells expressed decreased levels of CD31 (Fig. 2A and M), VE-cadherin (Fig. 2B and M), α-catenin (Fig. 2C and M), β-catenin (Fig. 2D and M), γ-catenin (Fig. 2E and M), and P120-catenin (Fig. 2F), which are major components of adherens junctions. Interestingly, this diminished adherens junction formation in EOMA cells correlated with increased expression levels of MMP2 (Fig. 2G and M) and MMP9 (Fig. 2H). Additionally, reciprocal expression/localization of cytoplasmic P-YAP (Fig. 2I and M) and nuclear YAP (Fig. 2J and M) compared to that in WT-BEC was noted and is consistent with Hippo pathway activation being diminished in EOMA cells at a high cell density. No appreciable changes in ZO-1 and CD44 expression levels between WT-BEC and EOMA cells were noted (data not shown). Interestingly, while YAP cytoplasmic and nuclear localizations were reciprocal when WT-BEC were compared with EOMA cells, TAZ cytoplasmic and nuclear localizations were unchanged (Fig. 2K and L).
The survivin inhibitor YM155 modulates Hippo pathway activation and regulates EOMA cell survival.
At high cell density in culture, EOMA cells were treated with YM155 (a known survivin inhibitor) for 24 h. YM155 suppressed survivin (Fig. 3A and L), PCNA (Fig. 3F), and Ki-67 (Fig. 3L) expression, elicited no changes in cleaved caspase 8 or 7 activation (Fig. 3B and D), and upregulated effector caspase 3 activation (Fig. 3C and L) and P53 expression (Fig. 3E), consistent with a decreased proliferation rate and an increased apoptotic rate. Interestingly, YM155 induced CD31 (Fig. 3G and L) and VE-cadherin (Fig. 3H and L) expression and adherens junction formation (as noted by the appearance of membrane staining of VE-cadherin at the junctions between the cells [Fig. 3L, inset]). Additionally, CD31 staining was noted to be more intense as well, forming more distinct membrane staining between cells albeit in a less uniform and organized pattern than for VE-cadherin (Fig. 3L, inset). Furthermore, increased Hippo pathway activation was noted, evidenced by increased cytoplasmic P-YAP localization (Fig. 3I and L) and decreased nuclear YAP translocation (Fig. 3J and L). Similar to our observations shown in Fig. 2K and L, YM155 treatment had no effect on the TAZ nuclear fraction (Fig. 3K). Treatment with YM155 was also noted to abrogate EOMA cell overgrowth and the formation of tube-like and cyst-like structures and to promote monolayer contact inhibition (Fig. 4M).
FIG 3.

YM155 regulates EOMA cell survival via modulation of the Hippo pathway. (A to I) Western blot analyses reveal that YM155 treatment downregulated survivin (A) and PCNA (F) expression and nuclear YAP localization (J) and upregulated cleaved caspase 3 (CC3) (C), P53 (E), CD31 (G), and VE-cadherin (H) expression and cytoplasmic P-YAP localization (I) in EOMA cells. (K) The TAZ nuclear fraction was not affected by YM155 treatment. Data represent the mean ratios of survivin (A), P53 (E), PCNA (F), CD31 (G), VE-cadherin (H), and P-YAP (I) to β-actin; cleaved caspase 8 (CC8) (B), caspase 3 (CC3) (C), and caspase 7 (CC7) (D) to full-length caspase; and YAP (J) and TAZ (K) to histone H1 from triplicate samples for each YM155 concentration (0, 10, 50, 100, 200, and 500 nM) ± SD (*, P < 0.05; **, P < 0.01). (L) Merged DAPI and survivin, DAPI and cleaved caspase 3 (CC3), DAPI and Ki-67, DAPI and P-YAP, DAPI and YAP, DAPI and CD31, and DAPI and VE-cadherin immunofluorescence micrographs of EOMA cells treated with 0 nM and 500 nM YM155 confirming our Western blot analyses. Insets in the CD31 and VE-cadherin panels illustrate relative expression levels and localizations. Bar = 50 μm.
YM155 does not affect WT-BEC survival and Hippo pathway modulation.
In order to ascertain if and how YM155 treatment affects WT-BEC cultures, we performed a dose-response study using cells that we used in our previous studies (7, 8). In a high-cell-density WT-BEC culture, survivin (Fig. 4A and L), CD31 (Fig. 4D and L), VE-cadherin (Fig. 4E and L), MMP2 (Fig. 4I), and MMP9 (Fig. 4J) expression; cytoplasmic P-YAP localization (Fig. 4B and L); nuclear YAP translocation (Fig. 4C and L); and caspase 3 activation (Fig. 4G and L) were not affected by YM155 treatment compared to EOMA cells, in which overgrowth and tube- and cyst-like structures were also abrogated and monolayer contact inhibition was observed in the presence of YM155 (Fig. 4M, bottom right). In WT-BEC, as well as in EOMA cells, caspase 8 activation was not affected by YM155 treatment (Fig. 4F).
EOMA cells exhibit increased expression levels of Ajuba mRNA and protein compared to WT-BEC, which are inhibited by YM155 treatment in a dose-specific fashion.
Compared to WT-BEC, whose Ajuba expression levels were negligible as evidenced by qRT-PCR, Western blotting, and immunofluorescence, EOMA cells exhibited significantly increased expression levels of Ajuba by qRT-PCR and Western blotting (Fig. 5A and B). Immunofluorescence analysis revealed that the expression of Ajuba was in a cytoplasmic localization (Fig. 5C). EOMA cell cytoplasmic Ajuba expression was significantly reduced following YM155 treatment in a dose-specific manner, as illustrated by Western blotting (Fig. 5D) and immunofluorescence (Fig. 5E).
FIG 5.

EOMA cells exhibit increased expression of Ajuba mRNA and protein compared to WT-BEC, which is inhibited by YM155 treatment in a dose-specific fashion. (A) qRT-PCR analyses reveal a significant increase in Ajuba mRNA levels in EOMA cells compared to those in WT-BEC. (B) Western blotting reveals that Ajuba protein levels in EOMA cells were also significantly increased compared to those in WT-BEC. (C) Immunofluorescence microscopy reveals increased cytoplasmic Ajuba levels in permeabilized EOMA cells compared with WT-BEC. Insets in the 0.2% and 0.5% Triton X-100 treatment panels illustrate relative expression levels and localizations. (D) Western blotting illustrates that YM155 treatment of EOMA cells reduced Ajuba expression in a dose-specific fashion. (E) Immunofluorescence microscopy reveals decreased cytoplasmic Ajuba in permeabilized EOMA cells that were treated with YM155, confirming our Western blot analyses. Insets in panels C and E illustrate relative expression levels and localizations. Bar = 50 μm. Data represent the mean Ajuba ΔCT (A) or Ajuba-versus-β-actin ratios (B and D) from triplicate assays ± SD (*, P < 0.05; **, P < 0.01).
Survivin siRNA modulates Hippo pathway activation and regulates EOMA cell survival similarly to the inhibitor YM155.
In high-cell-density cultures, EOMA cells, EOMA-Mock cells (mock-transfected cells), EOMA-siNC cells (control siRNA-transfected cells), EOMA-siSurvivin1-3 cells (cells transfected with three different survivin siRNAs), EOMA-YM155 cells (500 nM) (EOMA cells treated with YM155), and WT-BEC were cultured for 72 h and analyzed. The relative survivin gene expression level was significantly reduced in EOMA cells transfected with all three siRNAs tested and in EOMA cells treated with YM155 to similar levels as those found in WT-BEC (Fig. 6A). Similarly, the relative survivin protein expression level was also significantly reduced in EOMA cells transfected with all three siRNAs tested and EOMA cells treated with YM155, approaching the levels found in WT-BEC (Fig. 6B). Figure 6C illustrates reduced survivin expression in EOMA nuclei for all three survivin siRNAs tested. Western blot analyses of CD31 and VE-cadherin revealed significant increases in levels of both proteins in survivin siRNA-transfected EOMA cells compared to control siRNA-transfected EOMA cells (Fig. 6D, E, and I). Additionally, the cytoplasmic fraction of P-YAP was significantly increased in survivin siRNA-transfected EOMA cells compared to control siRNA-transfected EOMA cells, while the YAP nuclear fraction was decreased (Fig. 6F, G, and I). Similar to data accrued by using YM155 (Fig. 4G and K), Ki-67 staining was significantly reduced, and cleaved caspase 3 accumulation was increased in survivin siRNA-transfected EOMA cells compared to control siRNA-transfected EOMA cells (Fig. 6I).
FIG 6.

Survivin siRNA and YM155 modulate EOMA cell migration and invasion. (A) qRT-PCR analyses reveal significant decreases in survivin mRNA levels in EOMA cells transfected with survivin siRNA constructs, reducing them to levels comparable to those noted following YM155 treatment and those observed in WT-BEC. Mock- and control construct (siNC)-transfected cultures expressed survivin mRNA levels equal to those of untreated EOMA cells. All data are means ± SD from triplicate experiments (**, P < 0.01). (B) Western blotting reveals that survivin protein levels in EOMA cells were also significantly decreased in EOMA cells transfected with survivin siRNA constructs, reducing them to levels comparable to those noted following YM155 treatment and those observed in WT-BEC. Again, mock- and siNC-transfected cultures expressed survivin mRNA levels equal to those of untreated EOMA cells. All data are means ± SD from triplicate experiments (**, P < 0.01). (C) Immunofluorescence microscopy reveals decreased survivin levels in EOMA cells transfected with survivin siRNA constructs compared with WT-BEC. Bar = 100 μm. (D to H) Compared to control construct-transfected cells, Western blotting illustrated that survivin siRNA constructs exhibited increased CD31 (D) and VE-cadherin (E) expression levels as well as increased cytoplasmic P-YAP (F) and decreased nuclear YAP (G) levels but no change in nuclear TAZ expression levels (H). All data are means ± SD from triplicate experiments (*, P < 0.05). (I) Compared to control construct-transfected cells, immunofluorescence microscopy revealed that EOMA cells transfected with survivin siRNA constructs exhibited increased CD31 and VE-cadherin expression levels, decreased Ki-67 expression levels, increased cleaved caspase 3 (CC3) expression levels, increased P-YAP expression levels, and decreased YAP expression levels, confirming our Western blot analyses. Bar = 100 μm. (J and K) Adhesion studies on a gelatin substrate reveal no appreciable changes in adhesion among untreated WT-BEC and EOMA cells or cells treated with YM155 or transfected with either the control construct or survivin siRNA. All data are means ± SD from triplicate experiments. (L) Migration through 8-μm-pore-size uncoated Transwells of WT-BEC, EOMA cells, and EOMA cells treated with YM155 or transfected with survivin siRNA reveals that WT-BEC, EOMA cells, and control construct-transfected EOMA cells exhibited similar migrations, while EOMA cells treated with YM155 or transfected with survivin siRNA exhibited significantly reduced migrations. All data are means ± SD from triplicate experiments (**, P < 0.01; ****, P < 0.0001). (M) Representative hematoxylin-stained filters illustrating the numbers of differently treated cells migrating through the pores of naked membranes. Bar = 100 μm. (N) Invasion through 8-μm-pore-size Matrigel-coated Transwells of WT-BEC, EOMA cells, and EOMA cells treated with YM155 or transfected with survivin siRNA reveals that EOMA cells and control construct-transfected EOMA cells exhibited high relative migrations, while EOMA cells treated with YM155 or transfected with survivin siRNA exhibited significantly lower invasion, comparable to that noted for WT-BEC. All data are means ± SD from triplicate experiments (*, P < 0.05; **, P < 0.01; ***, P < 0.0001). (O) Representative hematoxylin-stained filters illustrating the numbers of differently treated cells invading the pores of Matrigel-coated membranes. Bar = 100 μm.
Survivin siRNA and YM155 modulate EOMA cell migration and invasion.
Prior to assessing migration and invasion in EOMA cells and WT-BEC, we assessed their adhesion properties on Transwell inserts and found no appreciable differences among untreated and YM155-treated WT-BEC and EOMA cells (Fig. 6J) and between survivin siRNA-transfected EOMA cells and control siRNA-transfected EOMA cells (Fig. 6K). Survivin siRNA-transfected EOMA cells and EOMA cells treated with YM155 exhibited significantly reduced migration through uncoated 8-μm Transwells compared to WT-BEC, EOMA cells, and EOMA cells transfected with control siRNA. Interestingly, EOMA cells treated with YM155 exhibited a greater reduction in migration through uncoated 8-μm Transwells than did the survivin siRNA-transfected EOMA cells (Fig. 6L and M). Similarly, survivin siRNA-transfected EOMA cells and EOMA cells treated with YM155 also exhibited significantly reduced invasion through Matrigel-coated 8-μm Transwells compared to EOMA cells and EOMA cells transfected with control siRNA, attaining reduced levels of invasion in the range of the levels in WT-BEC cells. As noted above for the migration assay, EOMA cells treated with YM155 exhibited a greater reduction in invasion through Matrigel-coated 8-μm Transwells than did survivin siRNA-transfected EOMA cells (Fig. 6N and O).
Deficiencies of the adhesion molecules CD44 and CD31 on BEC mimic, in part, the phenotypes observed for EOMA cells and are rescued, in part, upon treatment with YM155.
While no other murine hemangioendothelioma cell lines are available, we utilized CD44KO- and CD31KO-BEC, which we have characterized in several previous studies (7, 8, 10, 15). We have found that these two BEC lines exhibit characteristics similar to those exhibited by EOMA cells (7, 8), specifically the loss of contact inhibition and an “overriding” morphology (Fig. 7A and I); increased survivin expression, downregulated by YM155 (Fig. 7B and J); decreased VE-cadherin expression, modestly upregulated by YM155 (Fig. 7D and L); and decreased activation of caspase 3, modestly upregulated by YM155 (Fig. 7F and N), all in a dose-responsive fashion. Interestingly, we noted no changes in P-YAP and YAP cytoplasmic and nuclear localizations (Fig. 7G, H, O, and P) and no changes in proliferation (data not shown) following YM155 treatments.
FIG 7.

Deficiencies of the adhesion molecules CD44 and CD31 on BEC mimic, in part, the phenotypes observed in EOMA cells and are rescued, in part, upon treatment with YM155. We utilized CD44KO- and CD31KO-BEC, which we have previously characterized (7, 8, 10), to further assess the roles of VE-cadherin and CD31 in the modulation of Hippo pathway activation. (A) Hoffman interference reflection microscopy of a representative postconfluent culture of CD44KO-BEC illustrating a loss of contact inhibition and overriding aggregates of cells. Bar = 200 μm. (B to H) Western blot analyses of expression levels of survivin (B), CD31 (C), VE-cadherin (D), cleaved caspase 8 (CC8) (E), cleaved caspase 3 (CC3) (F), cytoplasmic P-YAP (G), and nuclear YAP (H) untreated and treated with a dose range of YM155. The survivin expression level was noted to be decreased (B), while the VE-cadherin (D) and cleaved caspase 3 (F) levels were found to be modestly increased following YM155 treatment. All data are means ± SD from triplicate experiments. (I) Hoffman interference reflection microscopy of a representative postconfluent culture of CD31KO-BEC illustrating a loss of contact inhibition and overriding aggregates of cells. Bar = 200 μm. (J to P) Western blot analyses of expression levels of survivin (J), CD31 (K), VE-cadherin (L), cleaved caspase 8 (M), cleaved caspase 3 (N), cytoplasmic P-YAP (O), and nuclear YAP (P) in untreated cells and in cells treated with a dose range of YM155. The survivin expression level was noted to be decreased (J), while the VE-cadherin (L) and cleaved caspase 3 (N) levels were found to be modestly increased following YM155 treatment. All data are means ± SD from triplicate experiments (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001).
DISCUSSION
Vascular tumors are comprised of a number of moieties spanning a range of benign entities, including hemangiomas; intermediate entities exhibiting locally aggressive behavior, such as Kaposiform hemangioendotheliomas and, rarely, metastasizing entities, including papillary intralymphatic angioendotheliomas and Kaposi sarcomas; and malignant entities, including epithelioid hemangioendotheliomas and angiosarcomas (2). A number of immunohistochemistry and immunofluorescence investigations have shed light on the cells of origin of these tumors and have influenced potential therapeutic targets (16–19). Specifically, the expression levels of selected adhesion molecules, including VE-cadherin and PECAM-1 (CD31), have been implicated in the development of vascular tumors and may be correlated with their metastatic and malignant potentials (20, 21). Recently, the importance of specific molecules associated with the regulation of apoptosis and proliferation, including survivin, Bcl-2, and Akt, in the regulation of proliferation and apoptosis of hemangiomas has been indicated (22–24).
In light of the known interactions between survivin and VE-cadherin (8, 25), survivin's interactions with p53 (26, 27), survivin's role as a regulator of smooth muscle apoptosis (28) and as a target for anticancer therapy (29), and survivin's interactions with vascular endothelial growth factor (VEGF) in endothelial cells (30), survivin is a potential therapeutic target for the control of hemangioma proliferation and apoptosis. Consistent with this possibility are reports that demonstrate Sp1 and Sp3 transactivation of the survivin promoter (27, 31). Furthermore, the small-molecule inhibitor YM155 (12), which interferes with Sp1-DNA interactions on the survivin promoter (32–34), has been used in a phase I clinical trial for advanced solid tumors (35) and was reported to elicit only modest untoward side effects (36).
Additionally, a specific inhibitor of β-adrenergic receptor activation, propranolol, has been found to affect infantile hemangioma growth and apoptosis via activation of caspases 3, 8, and 9 and cytochrome c; upregulation of p53; and increasing the Bax/BCL-xL ratio (37–39).
Interestingly, the very recent finding of a YAP1-TFE3 fusion in an epithelioid hemangioendothelioma (40) raised the possibility of the YAP protein, a crucial component of the Hippo pathway, playing a role in the proliferative and apoptotic dysregulation of vascular tumors (19). Furthermore, the LIM protein Ajuba (41, 42), a known negative regulator of the Hippo pathway, via inhibition of LATS (large tumor suppressor kinase 1) kinase (43–45), that interacts with catenin family members at cell-cell contacts (adherens junctions) and with cadherin family members (46–48), has been found to promote the epithelial-to-mesenchymal transition in colorectal cancers (49), and its lentivirus transduction into malignant mesothelioma suppresses their proliferation via modulation of the Hippo pathway (50). In addition, findings demonstrating increased expression levels of YAP and survivin and decreased LATS activity in gastric and hepatocellular carcinomas (51, 52) are consistent with Ajuba expression and localization possibly being an important regulator of the Hippo pathway in endothelia. These studies raise the possibility that modulation of the Hippo pathway has potential as a therapeutic target for hemangiomas and may provide insights into mechanisms involving the interactions of adhesion molecules and the Hippo pathway in the modulation of hemangioendothelioma proliferation and apoptosis.
Thus, using endothelial cells derived from a murine hemangioendothelioma (4–6), we tested the hypothesis that these cells exhibit dysregulated Hippo pathway activation due to reductions in levels of selected cell adhesion molecules (CD31 and VE-cadherin) (7, 8), eliciting the induction of survivin (8) and Ajuba, inactivating the Hippo pathway by inhibiting LATS kinase, and inhibiting the proteasomal degradation of YAP (53), resulting in the inhibition of caspase activation and increased proliferation (8). Furthermore, we postulate that the inhibition of survivin will rescue the EOMA phenotype, blunting its high proliferation rate and increasing its apoptotic rate.
In this report, we demonstrated that EOMA cells are larger than WT-BEC, are not contact inhibited, and exhibit a higher secondary proliferation rate and a lower level of caspase activation. Compared to WT-BEC, EOMA cells were found to express low levels of CD31 and VE-cadherin and increased levels of survivin, consistent with decreased caspase activation and increased MMP expression. Furthermore, EOMA cells exhibited increased levels of cytoplasmic Ajuba, decreased levels of cytoplasmic P-YAP, and increased levels of nuclear YAP, consistent with a reduction in Hippo pathway activation (illustrated in Fig. 8A and B). Interestingly, while the survivin inhibitor YM155 did not appreciably affect WT-BEC, it affected EOMA cells by decreasing the expression levels of survivin and Ajuba, increasing the expression levels of CD31 and VE-cadherin, increasing cytoplasmic P-YAP levels, and decreasing nuclear YAP levels, consistent with an induction of Hippo pathway activation. Furthermore, it was found to increase the expression of caspase 3 while decreasing MMP2 expression and proliferation, illustrating the interactive, dynamic roles of adhesion molecules (CD31 and VE-cadherin), survivin, and Ajuba in affecting the Hippo pathway in modulating proliferation and apoptosis in EOMA cells (Fig. 8B and C). To confirm that the effects of YM155 were due to modulation of survivin expression, we utilized survivin siRNA constructs to knock down survivin expression in EOMA cells. Survivin siRNA-transfected EOMA cells exhibited a phenotype virtually identical to that of the YM155-treated cultures, confirming the mechanism of YM155.
FIG 8.
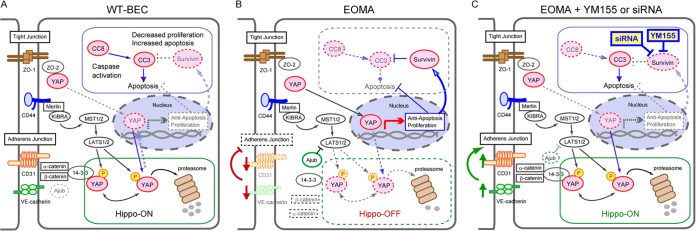
Working model of the involvement of adhesion molecule-mediated Hippo pathway modulation in the regulation of cell proliferation and survival in WT-BEC and EOMA cells as well as the role of YM155 in the modulation of the Hippo pathway in EOMA cells. (A) WT-BEC exhibit a contact-inhibited phenotype, with the cells expressing optimal levels of CD31 and VE-cadherin, low levels of survivin, nondetectable levels of Ajuba (dashed lines), moderate levels of active caspases, and reduced nuclear YAP and increased cytoplasmic P-YAP levels, consistent with an active Hippo pathway. (B) Compared to WT-BEC, EOMA cells exhibit decreased contact inhibition and junctional molecule expression, resulting in downregulation of the Hippo pathway with increases in survivin, Ajuba (thick lines), and MMP expression levels and increased YAP nuclear translocation, suppressing caspase-mediated apoptosis and increasing proliferation. (C) EOMA cells treated with YM155 or transfected with survivin siRNA exhibit increased contact inhibition and junctional molecule expression resulting from increased CD31 and VE-cadherin expression levels and decreased survivin, Ajuba (dashed lines), and nuclear YAP expression levels, resulting in upregulation of the Hippo pathway, which in turn results in increased cytoplasmic P-YAP and decreased MMP2 expression levels and caspase-mediated apoptosis. (WT-BEC are not affected by YM155 treatment.)
Our data are consistent a loss of YAP regulation by cell adhesion molecule expression and junctional assembly (54, 55). A perturbation of cell adhesion molecule-driven signaling (altered cell-cell contacts) induces survivin expression (8, 25) and the activity of the Hippo pathway (altering YAP subcellular localization), which in turn affect cell proliferation and apoptosis. Specifically, the reduction in levels of selected cell adhesion molecules (VE-cadherin and CD31) disrupts adherens and tight junction integrity, which, in addition to increasing survivin expression, results in decreased YAP cytoplasmic binding and increased expression of Ajuba, leading to inhibition of LATS and resulting in increased YAP nuclear localization and induction of proproliferative and antiapoptotic gene expression.
To support our findings implicating the cell adhesion molecules VE-cadherin and CD31 as being important components modulating Hippo pathway activation, we utilized CD44 and CD31 knockout BEC lines that we have previously characterized (7, 8, 10). Both of these BEC lines exhibit either reduced expression of VE-cadherin and CD31 (CD44KO-BEC) or a complete loss of CD31 and reduced VE-cadherin expression (CD31KO-BEC) (7, 8). Reduction or loss of these cell adhesion molecules resulted in cultures exhibiting a loss of contact inhibition, evidenced by overriding cells, decreased caspase 3 activation, and increased survivin expression. Treatment with YM155 resulted in increased VE-cadherin expression, increased caspase 3 activation, and decreased survivin expression, supporting the important role of these cell adhesion molecules in endothelial cell behaviors.
In our studies, cell matrix perturbations did not appear to play a significant role in EOMA cell proliferative and apoptotic regulation, as WT-BEC, EOMA cells, EOMA cells treated with YM155, and survivin siRNA-transfected EOMA cells all exhibited identical adhesion profiles on gelatin but did exhibit significant changes in secondary proliferation rates under high-density-culture conditions. Behaviors on other substrates have not been examined, and differences cannot be ruled out presently.
Although YAP and TAZ have essentially the same affinity for TEAD (TEA domain family member 1), differences have been found in the ways in which they interact with TEAD (56). Recent evidence suggests that YAP and TAZ do not compensate for each other (57), as evidenced by the findings that YAP and TAZ knockout mice show different phenotypes and that the phenotypes of YAP and TAZ knockdowns cannot be compensated for by the other gene (58). Our studies are consistent with these findings. Specifically, although YAP cytoplasmic and nuclear fractions differ significantly in WT-BEC and EOMA cells, TAZ subcellular localizations are not changed. The mechanism(s) for this differential regulation is as yet unknown but is in agreement with those recent reports.
Regarding our finding of a greater decrease in MMP2 expression following YM155 treatment of EOMA cells than that noted following transfection with survivin siRNA, it is known that, like survivin, the MMP2 and MMP14 promoters also lack a typical TATA or CCAAT box, suggesting that Sp1 may be involved in regulating MMP2 and, possibly, MMP14 promoter activity (59, 60), thus being consistent with YM155 also directly affecting MMP2 expression exclusive of its effects on survivin. Indeed, a recent study found that when Sp1 expression was reduced, MMP2 expression was reduced in human umbilical vein endothelial cells (59), consistent with the possibility that YM155 inhibition of at least these two proteins occurs via the same putative mechanism (33, 34).
In aggregate, these findings support the importance of adhesion molecules (VE-cadherin and CD31), survivin, and Ajuba (46, 48, 53, 61) in modulating the Hippo pathway, which regulates, in part, proliferation and survival in hemangioendotheliomas. The potential of YM155 as a therapeutic agent (Fig. 8), while an interesting possibility, is tempered by the lack of availability of additional murine hemangioendothelioma cell lines to evaluate.
ACKNOWLEDGMENTS
This work was supported in part by grant PO1-NS00344738 and an unrestricted gift from Joseph and Lucille Madri to J.A.M., as well as a grant-in-aid from a Japan Society for the Promotion of Science (JSPS) research fellowship for young scientists (no. 23-1154), a JSPS postdoctoral fellowship for research abroad (no. 513), and a Uehara Memorial Foundation postdoctoral fellowship for study abroad (no. 201230024) to M.T.
Footnotes
Published ahead of print 29 September 2014
REFERENCES
- 1.Mitchell RN, Schoen FJ. 2010. Blood vessels—tumors, p 520 In Kumar V, Abbas AK, Fausto N, Aster JC. (ed), Robbins & Cotran pathologic basis of disease, 8th ed. Saunders, Philadelphia, PA. [Google Scholar]
- 2.Fletcher CDM, Unni KK, Mertens F. (ed). 2002. World Health Organization classification of tumours: pathology and genetics of tumours of soft tissue and bone, 3rd ed, p 155–177 IARC Press, Lyon, France. [Google Scholar]
- 3.Weiss SW, Enzinger FM. 1986. Spindle cell hemangioendothelioma. A low-grade angiosarcoma resembling a cavernous hemangioma and Kaposi's sarcoma. Am. J. Surg. Pathol. 10:521–530. [PubMed] [Google Scholar]
- 4.Hoak JC, Warner ED, Cheng HF, Fry GL, Hankenson RR. 1971. Hemangioma with thrombocytopenia and microangiopathic anemia (Kasabach-Merritt syndrome): an animal model. J. Lab. Clin. Med. 77:941–950. [PubMed] [Google Scholar]
- 5.Fry GL, Czervionke RL, Hoak JC, Smith JB, Haycraft DL. 1980. Platelet adherence to cultured vascular cells: influence of prostacyclin (PGI2). Blood 55:271–275. [PubMed] [Google Scholar]
- 6.Obeso J, Weber J, Auerbach R. 1990. A hemangioendothelioma-derived cell line: its use as a model for the study of endothelial cell biology. Lab. Invest. 63:259–269. [PubMed] [Google Scholar]
- 7.Flynn KM, Michaud M, Canosa S, Madri JA. 2013. CD44 regulates vascular endothelial barrier integrity via a PECAM-1 dependent mechanism. Angiogenesis 16:689–705. 10.1007/s10456-013-9346-9. [DOI] [PubMed] [Google Scholar]
- 8.Tsuneki M, Madri JA. 2014. CD44 regulation of endothelial cell proliferation and apoptosis via modulation of CD31 and VE-cadherin expression. J. Biol. Chem. 289:5357–5370. 10.1074/jbc.M113.529313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ilan N, Tucker A, Madri JA. 2003. Vascular endothelial growth factor expression, beta-catenin tyrosine phosphorylation, and endothelial proliferative behavior: a pathway for transformation? Lab. Invest. 83:1105–1115. 10.1097/01.LAB.0000083531.84403.8B. [DOI] [PubMed] [Google Scholar]
- 10.Graesser D, Solowiej A, Bruckner M, Osterweil E, Juedes A, Davis S, Ruddle NH, Engelhardt B, Madri JA. 2002. Altered vascular permeability and early onset of experimental autoimmune encephalomyelitis in PECAM-1-deficient mice. J. Clin. Invest. 109:383–392. 10.1172/JCI13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pinter E, Barreuther M, Lu T, Imhof BA, Madri JA. 1997. Platelet-endothelial cell adhesion molecule-1 (PECAM-1/CD31) tyrosine phosphorylation state changes during vasculogenesis in the murine conceptus. Am. J. Pathol. 150:1523–1530. [PMC free article] [PubMed] [Google Scholar]
- 12.Minematsu T, Iwai M, Umehara K, Usui T, Kamimura H. 2010. Characterization of human organic cation transporter 1 (OCT1/SLC22A1)- and OCT2 (SLC22A2)-mediated transport of 1-(2-methoxyethyl)-2-methyl-4,9-dioxo-3-(pyrazin-2-ylmethyl)-4,9-dihydro-1H-naphtho[2,3-d]imidazolium bromide (YM155 monobromide), a novel small molecule survivin suppressant. Drug Metab. Dispos. 38:1–4. 10.1124/dmd.109.028142. [DOI] [PubMed] [Google Scholar]
- 13.Ilan N, Mahooti S, Rimm DL, Madri JA. 1999. PECAM-1 (CD31) functions as a reservoir for and a modulator of tyrosine-phosphorylated beta-catenin. J. Cell Sci. 112(Part 18):3005–3014. [DOI] [PubMed] [Google Scholar]
- 14.Flynn KM, Michaud M, Madri JA. 2013. CD44 deficiency contributes to enhanced experimental autoimmune encephalomyelitis: a role in immune cells and vascular cells of the blood-brain barrier. Am. J. Pathol. 182:1322–1336. 10.1016/j.ajpath.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Biswas P, Canosa S, Schoenfeld D, Schoenfeld J, Li P, Cheas LC, Zhang J, Cordova A, Sumpio B, Madri JA. 2006. PECAM-1 affects GSK-3beta-mediated beta-catenin phosphorylation and degradation. Am. J. Pathol. 169:314–324. 10.2353/ajpath.2006.051112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Requena L, Kutzner H. 2013. Hemangioendothelioma. Semin. Diagn. Pathol. 30:29–44. 10.1053/j.semdp.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 17.Aboutalebi A, Jessup CJ, North PE, Mihm MC., Jr 2012. Histopathology of vascular anomalies. Facial Plast. Surg. 28:545–553. 10.1055/s-0032-1329929. [DOI] [PubMed] [Google Scholar]
- 18.Liu L, Kakiuchi-Kiyota S, Arnold LL, Johansson SL, Wert D, Cohen SM. 2013. Pathogenesis of human hemangiosarcomas and hemangiomas. Hum. Pathol. 44:2302–2311. 10.1016/j.humpath.2013.05.012. [DOI] [PubMed] [Google Scholar]
- 19.Antonescu C. 2014. Malignant vascular tumors—an update. Modern Pathol. 27(Suppl 1):S30–S38. 10.1038/modpathol.2013.176. [DOI] [PubMed] [Google Scholar]
- 20.Martin-Padura I, De Castellarnau C, Uccini S, Pilozzi E, Natali PG, Nicotra MR, Ughi F, Azzolini C, Dejana E, Ruco L. 1995. Expression of VE (vascular endothelial)-cadherin and other endothelial-specific markers in haemangiomas. J. Pathol. 175:51–57. 10.1002/path.1711750109. [DOI] [PubMed] [Google Scholar]
- 21.Rothermel TA, Engelhardt B, Sheibani N. 2005. Polyoma virus middle-T-transformed PECAM-1 deficient mouse brain endothelial cells proliferate rapidly in culture and form hemangiomas in mice. J. Cell. Physiol. 202:230–239. 10.1002/jcp.20114. [DOI] [PubMed] [Google Scholar]
- 22.Murakami M, Sakai H, Kodama A, Mori T, Maruo K, Yanai T, Masegi T. 2008. Expression of the anti-apoptotic factors Bcl-2 and survivin in canine vascular tumours. J. Comp. Pathol. 139:1–7. 10.1016/j.jcpa.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 23.Mabeta P, Pepper MS. 2012. Inhibition of hemangioma development in a syngeneic mouse model correlates with bcl-2 suppression and the inhibition of Akt kinase activity. Angiogenesis 15:131–139. 10.1007/s10456-011-9248-7. [DOI] [PubMed] [Google Scholar]
- 24.Amin RM, Hiroshima K, Miyagi Y, Kokubo T, Hoshi K, Fujisawa T, Nakatani Y. 2008. Role of the PI3K/Akt, mTOR, and STK11/LKB1 pathways in the tumorigenesis of sclerosing hemangioma of the lung. Pathol. Int. 58:38–44. 10.1111/j.1440-1827.2007.02186.x. [DOI] [PubMed] [Google Scholar]
- 25.Iurlaro M, Demontis F, Corada M, Zanetta L, Drake C, Gariboldi M, Peiro S, Cano A, Navarro P, Cattelino A, Tognin S, Marchisio PC, Dejana E. 2004. VE-cadherin expression and clustering maintain low levels of survivin in endothelial cells. Am. J. Pathol. 165:181–189. 10.1016/S0002-9440(10)63287-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nabilsi NH, Broaddus RR, Loose DS. 2009. DNA methylation inhibits p53-mediated survivin repression. Oncogene 28:2046–2050. 10.1038/onc.2009.62. [DOI] [PubMed] [Google Scholar]
- 27.Boidot R, Vegran F, Lizard-Nacol S. 2014. Transcriptional regulation of the survivin gene. Mol. Biol. Rep. 41:233–240. 10.1007/s11033-013-2856-0. [DOI] [PubMed] [Google Scholar]
- 28.Blanc-Brude OP, Yu J, Simosa H, Conte MS, Sessa WC, Altieri DC. 2002. Inhibitor of apoptosis protein survivin regulates vascular injury. Nat. Med. 8:987–994. 10.1038/nm750. [DOI] [PubMed] [Google Scholar]
- 29.Ryan BM, O'Donovan N, Duffy MJ. 2009. Survivin: a new target for anti-cancer therapy. Cancer Treat. Rev. 35:553–562. 10.1016/j.ctrv.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 30.Mesri M, Morales-Ruiz M, Ackermann EJ, Bennett CF, Pober JS, Sessa WC, Altieri DC. 2001. Suppression of vascular endothelial growth factor-mediated endothelial cell protection by survivin targeting. Am. J. Pathol. 158:1757–1765. 10.1016/S0002-9440(10)64131-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu R, Zhang P, Huang J, Ge S, Lu J, Qian G. 2007. Sp1 and Sp3 regulate basal transcription of the survivin gene. Biochem. Biophys. Res. Commun. 356:286–292. 10.1016/j.bbrc.2007.02.140. [DOI] [PubMed] [Google Scholar]
- 32.Cheng Q, Ling X, Haller A, Nakahara T, Yamanaka K, Kita A, Koutoku H, Takeuchi M, Brattain MG, Li F. 2012. Suppression of survivin promoter activity by YM155 involves disruption of Sp1-DNA interaction in the survivin core promoter. Int. J. Biochem. Mol. Biol. 3:179–197. [PMC free article] [PubMed] [Google Scholar]
- 33.Cheung CH, Huang CC, Tsai FY, Lee JY, Cheng SM, Chang YC, Huang YC, Chen SH, Chang JY. 2013. Survivin—biology and potential as a therapeutic target in oncology. Onco Targets Ther. 6:1453–1462. 10.2147/OTT.S33374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Coumar MS, Tsai FY, Kanwar JR, Sarvagalla S, Cheung CH. 2013. Treat cancers by targeting survivin: just a dream or future reality? Cancer Treat. Rev. 39:802–811. 10.1016/j.ctrv.2013.02.002. [DOI] [PubMed] [Google Scholar]
- 35.Satoh T, Okamoto I, Miyazaki M, Morinaga R, Tsuya A, Hasegawa Y, Terashima M, Ueda S, Fukuoka M, Ariyoshi Y, Saito T, Masuda N, Watanabe H, Taguchi T, Kakihara T, Aoyama Y, Hashimoto Y, Nakagawa K. 2009. Phase I study of YM155, a novel survivin suppressant, in patients with advanced solid tumors. Clin. Cancer Res. 15:3872–3880. 10.1158/1078-0432.CCR-08-1946. [DOI] [PubMed] [Google Scholar]
- 36.Tao YF, Lu J, Du XJ, Sun LC, Zhao X, Peng L, Cao L, Xiao PF, Pang L, Wu D, Wang N, Feng X, Li YH, Ni J, Wang J, Pan J. 2012. Survivin selective inhibitor YM155 induce apoptosis in SK-NEP-1 Wilms tumor cells. BMC Cancer 12:619. 10.1186/1471-2407-12-619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ji Y, Li K, Xiao X, Zheng S, Xu T, Chen S. 2012. Effects of propranolol on the proliferation and apoptosis of hemangioma-derived endothelial cells. J. Pediatr. Surg. 47:2216–2223. 10.1016/j.jpedsurg.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 38.Tu JB, Ma RZ, Dong Q, Jiang F, Hu XY, Li QY, Pattar P, Zhang H. 2013. Induction of apoptosis in infantile hemangioma endothelial cells by propranolol. Exp. Ther. Med. 6:574–578. 10.3892/etm.2013.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kum JJ, Khan ZA. 2014. Propranolol inhibits growth of hemangioma-initiating cells but does not induce apoptosis. Pediatr. Res. 75:381–388. 10.1038/pr.2013.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Antonescu CR, Le Loarer F, Mosquera JM, Sboner A, Zhang L, Chen CL, Chen HW, Pathan N, Krausz T, Dickson BC, Weinreb I, Rubin MA, Hameed M, Fletcher CD. 2013. Novel YAP1-TFE3 fusion defines a distinct subset of epithelioid hemangioendothelioma. Genes Chromosomes Cancer 52:775–784. 10.1002/gcc.22073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goyal RK, Lin P, Kanungo J, Payne AS, Muslin AJ, Longmore GD. 1999. Ajuba, a novel LIM protein, interacts with Grb2, augments mitogen-activated protein kinase activity in fibroblasts, and promotes meiotic maturation of Xenopus oocytes in a Grb2- and Ras-dependent manner. Mol. Cell. Biol. 19:4379–4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kanungo J, Pratt SJ, Marie H, Longmore GD. 2000. Ajuba, a cytosolic LIM protein, shuttles into the nucleus and affects embryonal cell proliferation and fate decisions. Mol. Biol. Cell 11:3299–3313. 10.1091/mbc.11.10.3299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Das Thakur M, Feng Y, Jagannathan R, Seppa MJ, Skeath JB, Longmore GD. 2010. Ajuba LIM proteins are negative regulators of the Hippo signaling pathway. Curr. Biol. 20:657–662. 10.1016/j.cub.2010.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reddy BV, Irvine KD. 2013. Regulation of Hippo signaling by EGFR-MAPK signaling through Ajuba family proteins. Dev. Cell 24:459–471. 10.1016/j.devcel.2013.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun G, Irvine KD. 2013. Ajuba family proteins link JNK to Hippo signaling. Sci. Signal. 6:ra81. 10.1126/scisignal.2004324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marie H, Pratt SJ, Betson M, Epple H, Kittler JT, Meek L, Moss SJ, Troyanovsky S, Attwell D, Longmore GD, Braga VM. 2003. The LIM protein Ajuba is recruited to cadherin-dependent cell junctions through an association with alpha-catenin. J. Biol. Chem. 278:1220–1228. 10.1074/jbc.M205391200. [DOI] [PubMed] [Google Scholar]
- 47.Haraguchi K, Ohsugi M, Abe Y, Semba K, Akiyama T, Yamamoto T. 2008. Ajuba negatively regulates the Wnt signaling pathway by promoting GSK-3beta-mediated phosphorylation of beta-catenin. Oncogene 27:274–284. 10.1038/sj.onc.1210644. [DOI] [PubMed] [Google Scholar]
- 48.Nola S, Daigaku R, Smolarczyk K, Carstens M, Martin-Martin B, Longmore G, Bailly M, Braga VM. 2011. Ajuba is required for Rac activation and maintenance of E-cadherin adhesion. J. Cell Biol. 195:855–871. 10.1083/jcb.201107162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liang XH, Zhang GX, Zeng YB, Yang HF, Li WH, Liu QL, Tang YL, He WG, Huang YN, Zhang L, Yu LN, Zeng XC. 2014. The LIM protein JUB promotes epithelial-mesenchymal transition in colorectal cancer. Cancer Sci. 105:660–666. 10.1111/cas.12404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tanaka I, Osada H, Fujii M, Fukatsu A, Hida T, Horio Y, Kondo Y, Sato A, Hasegawa Y, Tsujimura T, Sekido Y. 16 December 2013. LIM-domain protein AJUBA suppresses malignant mesothelioma cell proliferation via Hippo signaling cascade. Oncogene. 10.1038/onc.2013.528. [DOI] [PubMed] [Google Scholar]
- 51.Da CL, Xin Y, Zhao J, Luo XD. 2009. Significance and relationship between Yes-associated protein and survivin expression in gastric carcinoma and precancerous lesions. World J. Gastroenterol. 15:4055–4061. 10.3748/wjg.15.4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li H, Wolfe A, Septer S, Edwards G, Zhong X, Abdulkarim AB, Ranganathan S, Apte U. 2012. Deregulation of Hippo kinase signalling in human hepatic malignancies. Liver Int. 32:38–47. 10.1111/j.1478-3231.2011.02646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Badouel C, McNeill H. 2011. SnapShot: the hippo signaling pathway. Cell 145:484–484.e1. 10.1016/j.cell.2011.04.009. [DOI] [PubMed] [Google Scholar]
- 54.Low BC, Pan CQ, Shivashankar GV, Bershadsky A, Sudol M, Sheetz M. 2014. YAP/TAZ as mechanosensors and mechanotransducers in regulating organ size and tumor growth. FEBS Lett. 588:2663–2670. 10.1016/j.febslet.2014.04.012. [DOI] [PubMed] [Google Scholar]
- 55.Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, Elvassore N, Piccolo S. 2011. Role of YAP/TAZ in mechanotransduction. Nature 474:179–183. 10.1038/nature10137. [DOI] [PubMed] [Google Scholar]
- 56.Hau JC, Erdmann D, Mesrouze Y, Furet P, Fontana P, Zimmermann C, Schmelzle T, Hofmann F, Chene P. 2013. The TEAD4-YAP/TAZ protein-protein interaction: expected similarities and unexpected differences. Chembiochem 14:1218–1225. 10.1002/cbic.201300163. [DOI] [PubMed] [Google Scholar]
- 57.Yan L, Cai Q, Xu Y. 29 July 2014. Hypoxic conditions differentially regulate TAZ and YAP in cancer cells. Arch. Biochem. Biophys. 10.1016/j.abb.2014.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhao B, Li L, Wang L, Wang CY, Yu J, Guan KL. 2012. Cell detachment activates the Hippo pathway via cytoskeleton reorganization to induce anoikis. Genes Dev. 26:54–68. 10.1101/gad.173435.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen Y, Huang Y, Huang Y, Xia X, Zhang J, Zhou Y, Tan Y, He S, Qiang F, Li A, Re OD, Li G, Zhou J. 2014. JWA suppresses tumor angiogenesis via Sp1-activated matrix metalloproteinase-2 and its prognostic significance in human gastric cancer. Carcinogenesis 35:442–451. 10.1093/carcin/bgt311. [DOI] [PubMed] [Google Scholar]
- 60.Haas TL, Stitelman D, Davis SJ, Apte SS, Madri JA. 1999. Egr-1 mediates extracellular matrix-driven transcription of membrane type 1 matrix metalloproteinase in endothelium. J. Biol. Chem. 274:22679–22685. 10.1074/jbc.274.32.22679. [DOI] [PubMed] [Google Scholar]
- 61.Oka T, Remue E, Meerschaert K, Vanloo B, Boucherie C, Gfeller D, Bader GD, Sidhu SS, Vandekerckhove J, Gettemans J, Sudol M. 2010. Functional complexes between YAP2 and ZO-2 are PDZ domain-dependent, and regulate YAP2 nuclear localization and signalling. Biochem. J. 432:461–472. 10.1042/BJ20100870. [DOI] [PubMed] [Google Scholar]