

Abstract
The yeast Saccharomyces cerevisiae has two main high-affinity inorganic phosphate (Pi) transporters, Pho84 and Pho89, that are functionally relevant at acidic/neutral pH and alkaline pH, respectively. Upon Pi starvation, PHO84 and PHO89 are induced by the activation of the PHO regulon by the binding of the Pho4 transcription factor to specific promoter sequences. We show that PHO89 and PHO84 are induced by alkalinization of the medium with different kinetics and that the network controlling Pho89 expression in response to alkaline pH differs from that of other members of the PHO regulon. In addition to Pho4, the PHO89 promoter is regulated by the transcriptional activator Crz1 through the calcium-activated phosphatase calcineurin, and it is under the control of several repressors (Mig2, Nrg1, and Nrg2) coordinately regulated by the Snf1 protein kinase and the Rim101 transcription factor. This network mimics the one regulating expression of the Na+-ATPase gene ENA1, encoding a major determinant for Na+ detoxification. Our data highlight a scenario in which the activities of Pho89 and Ena1 are functionally coordinated to sustain growth in an alkaline environment.
INTRODUCTION
Inorganic phosphate (Pi) is indispensable for the biosynthesis of key cellular components such as nucleic acids, nucleoproteins, and phospholipids and is involved in many metabolic and signaling pathways. Transport across the plasma membrane is the first step in the utilization of this essential nutrient. In the yeast Saccharomyces cerevisiae, the transport of Pi is mediated by several transporters that differ in their affinities for Pi. The low-affinity transport system (Km, ∼1 mM) is composed of the Pho87 and Pho90 H+/Pi symporters, and it is sufficient for growth at normal external Pi concentrations (1–3). High-affinity Pi uptake (Km, ∼10 μM) is mediated by the plasma membrane Pho84 and Pho89 transporters (4–6). Pho84 cotransports phosphate with H+, is mostly active at acidic pH, and is responsible for most of the high-affinity Pi uptake under normal growth conditions (7). Pho89 is a phosphate/cation symporter and works most efficiently under alkaline conditions, with an optimum pH of 9.5. Effective transport through Pho89 requires the existence of an alkali-metal cation gradient, with Na+ being preferred over K+ or Li+ (5, 8).
In the presence of sufficient Pi in the external medium, the expression levels of both the PHO84 and PHO89 transporter-encoding genes are low. The expression levels of these genes are greatly increased in response to Pi starvation, which occurs through the activation of the PHO signaling pathway (9–11). This response involves the activation of the cyclin-dependent kinase inhibitor Pho81, which leads to the inhibition of the Pho85-Pho80 cyclin-dependent kinase complex. As a result, unphosphorylated Pho4 (a basic helix-loop-helix transcription factor) accumulates in the nucleus and binds to diverse phosphate-responsive gene promoters, including PHO84 and PHO89, driving their transcriptional response. When extracellular Pi is not limiting, Pho81 is inactivated. As a consequence, the Pho85-Pho80 kinase complex becomes active and phosphorylates Pho4, resulting in its exclusion from the nucleus and thus triggering repression of the PHO-regulated genes (12–14).
In spite of the common regulatory traits mentioned above, evidence suggesting that PHO84 and PHO89 might respond differently to diverse external inputs or cellular conditions has been raised in the last few years. For instance, alkalinization of the medium results in the transcriptional activation of genes induced by phosphate starvation, including PHO89 and PHO84. However, accumulation of PHO89 mRNA occurs much faster than that of PHO84 (15, 16). It is worth noting that in some cases, such as under alkaline stress, the involvement of signaling mechanisms other than the PHO pathway has been suggested (15, 16), pointing to the possible regulation of PHO89 by the calcium-responsive calcineurin protein phosphatase pathway, which controls the activity of the Crz1/Tcn1 transcription factor (see reference 17 for a review).
Pho89 is a widely conserved protein with close homologs from bacteria to humans (18). On the basis of these precedents, we considered it necessary to explore in a systematic way the possible regulatory inputs governing the expression of the PHO89 gene. Alkaline stress was employed as a transcriptional trigger because (i) it is a condition that results in powerful induction of PHO89 expression and (ii) it has been shown to involve the modulation of a wide variety of signaling pathways in S. cerevisiae (15, 16, 19–23). Here we show that the expression patterns of Pho84 and Pho89 under high-pH stress are different. Contrary to Pho84, accumulation of Pho89 is also regulated, aside from the Pho4 contribution, by the Crz1 transcription factor in response to calcineurin activation, mainly through a calcineurin-dependent response element (CDRE) located in the PHO89 promoter at positions −273 to −267. In addition, we demonstrate that PHO89 expression is also under the control of the Mig2 and Nrg1 repressors, in a way that implicates the Rim101 and Snf1 signaling pathways, and that this complex regulatory network is identical to the one described previously for alkaline induction of the Na+-ATPase-encoding gene ENA1 (24, 25). Ena1 is a major determinant of salt tolerance, and its mutation renders cells highly sensitive to sodium and lithium cations (26–28). Interestingly, ena1 cells are also sensitive to alkaline pH even in the absence of high levels of external sodium (24, 29). Our work strongly suggests that the common signaling network that regulates Pho89 and Ena1 expression provides the molecular basis for a physiological interaction between the phosphate transporter and the sodium ATPase.
MATERIALS AND METHODS
Yeast strains and growth conditions.
S. cerevisiae cells were grown at 28°C in YPD medium (10 g/liter yeast extract, 20 g/liter peptone, and 20 g/liter dextrose) or, when carrying plasmids, in synthetic minimal medium lacking the appropriate selection requirements (30). Low-phosphate (low-Pi) medium was prepared from yeast nitrogen base (YNB)-based medium without amino acids and phosphate (catalog number CYN0803 [requested to also lack potassium salts]; Formedium Ltd., United Kingdom), which was made with 30 mM KCl and 100 μM potassium phosphate (except where otherwise stated). The equivalent high-phosphate (high-Pi) medium was made by adding 20 mM KCl and 10 mM potassium phosphate. In some experiments, YNB-based medium lacking phosphate and sodium was employed (catalog number CYN0810; Formedium Ltd.). For preparation of low-phosphate agar plates, purified agar (catalog number 1806.05; Conda) was employed.
Yeast strains used in the present study are described in Table 1. Strains RSC82, ONA1, and ASC13 were constructed by transformation of wild-type strain DBY746 with a disruption cassette amplified from the crz1::kanMX4 (3.0-kbp), pho84::kanMX4 (2.3-kbp), and pho89::kanMX4 (2.1-kbp) mutants from the systematic disruption library in the BY4741 background (31). Strain RSC38 was made by transforming strain ONA1 with a pho89::TRP1 deletion cassette obtained by amplification of the corresponding locus of strain PAM1 (7) with oligonucleotides pho89_orf_5′ and pho89_orf_3′. Strains ASC17 and ASC30 were constructed by disruption of the PHO84 and PHO89 genes with the cassettes described above in the RH16.6 (all four repeats at the ENA locus deleted) background. Strain RSC22, which carries the rim101::HIS3 deletion in the DBY746 background, was made by short-flanking replacement with a 1.3-kbp cassette amplified from plasmid pFA6a-hisMx6 (32). Strain RSC89 was constructed by transforming wild-type strain DBY746 with a reg1::kanMX4 cassette obtained by amplification from the BY4741 reg1 deletion mutant with oligonucleotides 5′-reg1_disr and 3′-reg1_disr. Strain RSC90 was prepared by disruption of SNF1 with a 3.9-kbp snf1::LEU2 cassette from BamHI- and HindIII-digested plasmid pCC107 (33) in the RSC89 background. Strain ASC15 was made by deletion of the PHO4 gene in strain RSC22 (see above) with a deletion cassette (1.6 kbp) obtained from the pho4::kanMX4 BY4741 deletion mutant. Strain ASC14 was made similarly but by transforming strain RSC10 (34) with the pho4::kanMX4 cassette.
TABLE 1.
Strains used in this work
| Strain | Genotype | Source or reference |
|---|---|---|
| DBY746 | MATα his3-1 leu2-3,112 ura3-52 trp1-289 | D. Botstein |
| MAR15 | DBY746 cnb1::kanMX4 | 15 |
| RSC82 | DBY746 crz1::kanMX4 | This work |
| MAR89 | DBY746 cnb1::TRP1 crz1::kanMX4 | 86 |
| RSC4 | DBY746 pho4::LEU2 | 15 |
| RSC10 | DBY746 snf1::LEU2 | 34 |
| RSC89 | DBY746 reg1::kanMX4 | This work |
| RSC90 | DBY746 reg1::kanMX4 snf1::LEU2 | This work |
| RSC21 | DBY746 rim101::kanMX4 | 15 |
| RSC13 | DBY746 mig1::LEU2 | 24 |
| MP010 | DBY746 mig2::TRP1 | 24 |
| MP012 | DBY746 mig1::LEU2 mig2::TRP1 | 24 |
| MP014 | DBY746 snf1::LEU2 mig1::kanMX4 | 24 |
| MP015 | DBY746 snf1::LEU2 mig2::TRP1 | 24 |
| MP016 | DBY746 snf1::LEU2 mig1::kanMX4 mig2::TRP1 | 24 |
| MAR197 | DBY746 nrg1::nat1 | This work |
| MP018 | DBY746 nrg2::TRP1 | 24 |
| MAR205 | DBY746 nrg1::nat1 nrg2::TRP1 | This work |
| MAR206 | DBY746 rim101::kanMX4 nrg1::nat1 | 24 |
| MAR195 | DBY746 rim101::kanMX4 nrg2::TRP1 | 24 |
| MAR200 | DBY746 rim101::kanMX4 nrg1::nat1 nrg2::TRP1 | 24 |
| MP020 | DBY746 snf1::LEU2 nrg1::kanMX4 | 24 |
| MP021 | DBY746 snf1::LEU2 nrg2::kanMX4 | 24 |
| MP022 | DBY746 snf1::LEU2 nrg1::kanMX4 nrg2::TRP1 | 24 |
| MAR193 | DBY746 mig2::TRP1 nrg2::kanMX4 | This work |
| MAR204 | DBY746 mig2::TRP1 nrg2::kanMX4 nrg1::nat1 | This work |
| MAR199 | DBY746 mig1::LEU2 mig2::TRP1 nrg2::kanMX4 nrg1::nat1 | 24 |
| ASC07 | DBY746 PHO89-3×HA-HIS3 | This work |
| ASC08 | DBY746 PHO89-3×HA-HIS3 cnb1::kanMX4 | This work |
| ASC09 | DBY746 PHO89-3×HA-HIS3 snf1::LEU2 | This work |
| ASC10 | DBY746 PHO89-3×HA-HIS3 pho4::LEU2 | This work |
| ASC11 | DBY746 PHO89-3×HA-HIS3 rim101::kanMX4 | This work |
| ASC13 | DBY746 pho89::kanMX4 | This work |
| ASC14 | DBY746 snf1::LEU2 pho4::kanMX4 | This work |
| ASC15 | DBY746 rim101::His3Mx pho4::kanMX4 | This work |
| ASC17 | DBY746 ena1–ena4::LEU pho84::kanMX4 | This work |
| ONA1 | DBY746 pho84::kanMX4 | This work |
| RH16.6 | DBY746 ena1–ena4::LEU2 | 29 |
| ASC28 | DBY746 ENA1-3×HA-HIS3 | This work |
| ASC30 | DBY746 ena1–ena4::LEU2 pho89::kanMX4 | This work |
| RSC38 | DBY746 pho84::kanMX4 pho89::TRP1 | This work |
| SP048 | DBY746 MIG2-GFP-HIS3 | This work |
| ASC34 | DBY746 reg1::kanMX4 MIG2-GFP-HIS3 | This work |
| BY4741 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 | 87 |
| SP002 | BY4741 MIG2-GFP-HIS3 | This work |
| SP020 | BY4741 CRZ1-3×HA-kanMX6 | This work |
| SP011 | BY4741 MIG2-3×HA-kanMX6 | This work |
| SP014 | BY4741 MIG1-3×HA-kanMX6 | This work |
| SP018 | BY4741 MIG1-GFP-kanMX6 | This work |
| ASC03 | BY4741 MIG1-3×HA-kanMX6 snf1::LEU2 | This work |
| ASC04 | BY4741 MIG2-3×HA-kanMX6 snf1::LEU2 | This work |
Strain MAR193 was made by transforming mig2 strain MP010 (24) with the nrg2::kanMX4 cassette described previously in that same report. Strains MAR197, MAR205, and MAR204 were obtained by transformation of wild-type strain DBY746, strain MP018 (nrg2), and MAR193 (mig2 nrg2), respectively, with the 2.1-kbp nrg1::nat1 cassette previously described (24). Strains ASC07, ASC08, ASC09, ASC10, and ASC11, carrying a version of PHO89 encoding a C-terminally 3× hemagglutinin (3×HA)-tagged protein, were created by transformation of strains DBY476, MAR15 (cnb1), RSC10 (snf1), RSC4 (pho4), and RSC21 (rim101), respectively, with a cassette amplified with primers PHO89-C-pFA6-rev and PHO89-C-pFA6-dir from plasmid pFA6-3HA-His3MX6 (32). Strain SP011 contains a version of MIG2 encoding a C-terminally 3×HA-tagged protein integrated into the same chromosomal locus and was constructed by transforming wild-type BY4741 cells with a cassette made by amplification of plasmid pFA6-3HA-kanMX6 (32) using oligonucleotides MIG2-C-pFA6-dir and MIG2-C-pFA6-rev. The same template was employed to amplify cassettes to generate a chromosomally encoded C-terminally 3×HA-tagged version of the MIG1 or CRZ1 protein in the BY4741 background (strains SP014 and SP020). The oligonucleotide pairs used were MIG1-C-pFA6-dir/MIG1-C-pFA6-rev (for SP014) and CRZ1-C-pFA6-dir/CRZ1-C-pFA6-rev (for SP020). Strain ASC28 was constructed by transformation of DBY746 cells with an ENA1-3×HA-His3 cassette generated by PCR with primers ENA1-C-pFA6-dir and ENA1-c-pFA6-rev and plasmid pFA6-3HA-His3MX6 (32) as the template. Because of the highly conserved sequence of the members of the ENA cluster, particular care was taken to ensure the selection of transformants carrying the tag specifically at the C terminus of the ENA1 protein. To this end, positive clones were verified with two pairs of primers: ENA1-pFA6-comp-Cter and pFA6a_3HA_rev, which amplify a 0.2-kbp region irrespective of the member of the cluster receiving the tag, and ENA1-pFA6-comp-Nter and pFA6a_3HA_rev, which yield a 3.8-kb amplification fragment specific for ENA1 integration. Strains ASC03 and ASC04 were obtained from strains SP014 and SP011, respectively, by transformation with the previously described snf1::LEU2 cassette (34). Strains expressing Mig1-green fluorescent protein (GFP) (SP018) or Mig2-GFP (SP002, SP048, and ASC34) fusions were prepared by transforming either wild-type strain BY4741, DBY746, or RSC89 (reg1) with amplification fragments obtained by using plasmid pFA6-GFP(S65T)-kanMX6 (for Mig1) or pFA6-GFP(S65T)-HIS3MX6 (for Mig2) (32) and oligonucleotide pairs MIG1-C-pFA6-dir/MIG1-C-pFA6-rev and MIG2-C-pFA6-dir/MIG2-C-pFA6-rev, respectively. Transformants were verified by yeast colony PCR as well as by fluorescence microscopy. Oligonucleotides used in this work are described in Table S2 in the supplemental material.
Plasmids and recombinant DNA techniques.
Escherichia coli DH5α cells were used as a plasmid DNA host and were grown at 37°C in LB (Luria-Bertani) broth supplemented, if necessary, with 100 μg/ml ampicillin. Recombinant DNA techniques and bacterial and yeast cell transformations were performed by using standard methods. Plasmid pPHO89-LacZ is a YEp357-based reported plasmid in which the region from positions −671 to +33 (relative to the initiating ATG codon) is translationally fused to the lacZ gene (15). Mutation of the upstream CDRE (CDRE1) in pPHO89-LacZ to generate PHO89CDRE1-LacZ was accomplished by two-step PCR mutagenesis with specific oligonucleotides PHO89_5_CDRE1 and PHO89_3_CDRE1, so the native sequence GTGGCTG was changed to GTGTATG (modified nucleotides are shown in italics). Similarly, PHO89CDRE2-LacZ contained a mutated version of the downstream CDRE (CDRE2) and required oligonucleotides pho89_5′_CDRE2_m and pho89_3′_CDRE2_m, which promoted the change from CAGCCAC to CAGTAAC. Plasmid pMM15-PHO84, which expresses the PHO84 protein from its native promoter with a 3×HA C-terminal tag, was made by PCR amplification of the relevant PHO84 locus (nucleotides [nt] −598 to +1760 from the Met initiation codon) with oligonucleotides PHO84_prom_5′ and Clon_PHO84_3′ (carrying EcoRI- and BamHI-added sites, respectively) and cloning into these same sites of the centromeric plasmid (URA3 marker). Similarly, plasmid pMM17-PHO89, expressing the PHO89 protein C-terminally fused to the 3×HA epitope, was made by amplification of the PHO89 locus (nt −671 to +1722) with oligonucleotides Pho89_prom_5′ and Pho89_3′_SacI and cloned into the EcoRI and SacI sites of plasmid pMM17 (centromeric, LEU2 marker). Plasmids pMM15 and pMM17 were generous gifts from E. Herrero (Universitat de Lleida, Spain). Plasmid pKC201, containing ENA1 sequences from nt −1385 to +35 (relative to the initiating Met) fused to lacZ, was described previously (35, 36). Plasmid pAMS366 drives the expression of the lacZ reporter gene from four copies in tandem with the CDRE present in the FKS2 promoter (37).
Preparation of yeast extracts and immunoblot analysis.
Yeast cells were subjected to high-pH, high-salt, and/or low-phosphate conditions as follows. For low-phosphate stress, 50 ml of cells was grown until the optical density at 660 nm (OD660) reached 0.6 in high-Pi medium, and cells were collected by centrifugation (3 min at 1,228 × g), rinsed with low-Pi medium, and resuspended in 50 ml of low-Pi medium. For alkaline-stress experiments, 50 ml of cultures grown on YPD or YNB-based (low- or high-Pi) medium (pH 5.8) (OD660 = 0.6) was shifted to pH 8.0 by the addition of the appropriate volume of a 1 M KOH stock solution. For salt stress, the appropriate amount of solid NaCl was added to the cultures. For Pho89, Pho84, and Ena1 detection, 5 ml of cells was collected at the indicated times by rapid vacuum filtration through 0.45-μm Metricel GN-6 filters (Pall Corp.). Cells were quickly frozen in dry ice and stored at −80°C until use. Extracts were prepared for 10% SDS-PAGE (7% gels for Ena1) as previously described (8, 38). To detect phosphorylated Snf1 and the Snf1 protein, as well as the mobility shift of Mig1 and Mig2 upon alkaline-pH stress, cultures (5 to 10 ml) were made with 5.5% trichloroacetic acid and stored on ice for 15 min, and the pellet was collected by centrifugation (2 min at 4°C at 1,200 × g), washed twice with acetone, air dried, and stored at −80°C. For Mig1 and Mig2 mobility shifts, samples were processed as previously described (39, 40), except that when breaking the cells, EDTA was omitted from the urea buffer and each cell breakage round was extended to 45 s. Extracts were centrifuged for 10 min at 4°C in a microcentrifuge, the supernatant was recovered, and the concentration of proteins was determined by the Bradford method (Sigma Chemical Co.). For Snf1 detection, samples were resuspended in 150 μl of 10 mM Tris (pH 7.5)–1 mM EDTA buffer and then processed as described previously (41). Extracts were subjected to SDS-PAGE (8 or 10% polyacrylamide gels). Samples containing tagged Mig1 or Mig2 were treated with 10 units of calf alkaline phosphatase (Roche) for 90 min at 37°C, in the presence or in the absence of 50 mM EDTA.
In all cases, proteins were transferred onto polyvinylidene difluoride (PVDF) membranes (Immobilon-P; Millipore). HA-tagged Pho89, Ena1, and Mig2 were detected by means of mouse monoclonal anti-HA antibody 12CA4 at a 1:1,000 dilution (Roche), whereas the HA.11 clone 16B12 monoclonal antibody (Covance) was employed for detection of HA-tagged Pho84 and Mig1. A 1:20,000 dilution of secondary anti-mouse IgG–horseradish peroxidase (GE Healthcare) was used. Phosphorylated Snf1 and Snf1 protein were monitored with anti-phospho-Thr172-AMPK (Cell Signaling Technology) and anti-His (GE Healthcare) antibodies, respectively. Actin was detected with the (I-19)-R rabbit polyclonal antibody (catalog number sc-1616-R; Santa Cruz Biotechnology) followed by ECL anti-rabbit IgG–horseradish peroxidase secondary antibodies (GE Healthcare). Immunoreactive proteins were visualized with the ECL Select or Prime (for Pho84-HA detection) kit (GE Healthcare).
Chromatin immunoprecipitation experiments.
For chromatin immunoprecipitation (ChIP) experiments, strain SP020, encoding a CRZ1-3×HA version, was grown on YPD to an OD600 of 0.6 to 0.8 (∼50 ml/time point). At time zero (t0), the pH of the culture was increased to 8.0 by the addition of 1 M KOH, and growth resumed. Forty milliliters of cultures was taken at the appropriate times, and formaldehyde was added to a final concentration of 1% to cross-link proteins and DNA. From this point, the procedure was performed as described previously (40), with the following modifications. (i) Chromosomal DNA was fragmented by using Bioruptor Plus UCD-300 equipment (Diagenode) provided with a cooling system (4°C) for 45 cycles (high intensity; 30 s of sonication followed by a 60-s pause) to generate fragments ≤300 bp in length. (ii) The cleared lysate was precleared by the addition of ∼20 μl of ChIP-grade protein G-Sepharose beads (catalog number 16-201; Millipore) to each sample, followed by incubation for 1 h with gentle rotation at 4°C. Samples were centrifuged at 220 × g for 1 min at 4°C, and the supernatant was transferred into screw-cap tubes. One microliter of anti-HA antibodies (catalog number ab9110; Abcam) was added to each precleared sample (except for the no-IP controls), and incubation continued overnight at 4°C. Fifty microliters to 100 μl of ChIP-grade protein G-Sepharose beads was added to each sample (including controls), and incubation continued for 1 h at 4°C until samples were centrifuged at 220 × g for 1 min at 4°C. (iii) Treatment with proteinase K was extended for 2 h at 37°C. Finally, the eluted DNA was purified as described previously (40). For PCR assays, 40 ng of immunoprecipitated DNA was used together with oligonucleotides 18Right and 23Left. The procedures for ChIP-sequencing (ChIP-Seq) using these samples will be described elsewhere (not shown). Reads (FastQ files, 150-nt paired ends) were mapped with Bowtie 2 software (42). Mapped reads (1.9 million to 3.7 million/time point) were processed with IGV Tools (v 2.3) and quantified and represented with SeqMonk software (v 0.26.0).
Determination of intracellular sodium accumulation.
Cells were grown on synthetic medium containing 30 mM HEPES (pH 5.5) in the presence of 5 mM Na+, 10 mM K+, and 10 mM Pi until the OD660 reached 0.3. Cultures were centrifuged at 1,200 × g for 5 min; washed with the same medium, in which the concentration of Pi was reduced to 0.2 mM; and resuspended in this low-Pi medium containing 15 mM K+. Cells were grown for 60 min to induce the high-affinity phosphate transport system and then centrifuged and resuspended in the same medium, which was adjusted to pH 7.8 with KOH (which increases the concentration of K+ to 18 mM). Five-milliliter aliquots were taken immediately after resuspension (t0), and subsequent samples were taken at the desired times. Samples were rapidly filtered through 25-mm-diameter, 0.45-μm Metricel GN-6 filters (Pall Corp.) previously washed with 2.5 ml of a cold 20 mM MgCl2 solution, and cells were washed with 10 ml of the MgCl2 solution. Cells were recovered with 2 ml of this solution, filtered through a fresh filter, and subjected to a final wash. Filters were immersed in 10 ml of 10 mM MgCl2–0.2 M HCl and gently shaken to resuspend the cells, and the intracellular sodium concentration was determined by atomic absorption spectrometry. Membranes used to filter medium without cells were processed in the same way and used for blank values. The number of cells in each aliquot was determined by measuring the OD660 at the moment of sampling, assuming that 1 OD unit equals 8 × 107 cells. A cellular volume of 49 pl was used for calculations, according to data described previously (43) and our own determinations for strain BY4741.
Confocal microscopy.
Log-phase yeast cultures expressing GFP fusions growing in a low-fluorescence minimal medium lacking riboflavin and folic acid (44) were stained with 1 μg/ml of 4′,6′-diamidino-2-phenylindole (DAPI) to visualize nuclei. Cell suspensions were placed onto glass-bottom dishes and allowed to settle for a couple of minutes. Alkaline stress was initiated by gentle removal of the medium by using a pipette, placing the tip on the corner of the well, and the medium was then quickly replaced by low-fluorescence medium adjusted with 1 M KOH to pH 8.0. Images of living cells were taken before and after initiation of stress at different times with a confocal laser scanning microscope (Leica TCS SP2), using a 63× oil immersion objective. Images were analyzed by using LAS AF Lite (Leica Microsystems) and Wasabi (Hamamatsu Photonics Germany GmbH) imaging software.
Other techniques.
A growth test in liquid cultures was performed as described previously (24). Growth on agar plates (dot tests) was carried out as described previously (28). lacZ reporter assays were performed essentially as described previously (15). For alkaline-pH stress, cells were collected after 90 min of shifting cells to pH 8.0. For NaCl stress, cells were exposed to 0.4 M (for 60 min) or to 0.8 M (for 90 min) NaCl.
PHO89 mRNA levels were determined by semiquantitative reverse transcription-PCR (RT-PCR) and quantitative RT-PCR (qRT-PCR) with 15 ng of total RNA and oligonucleotides RT-PHO89-up1 and RT-PHO89-do1. For semiquantitative RT-PCR, the Illustra Ready-to-Go RT-PCR bead kit (GE Healthcare) was used. Reverse transcription was performed at 42°C for 30 min, and subsequent amplification was carried out for 23 cycles (1 min at 50°C and 72°C for annealing and extension conditions, respectively). qRT-PCR was performed with a CFX96 real-time system (Bio-Rad), using the QuantiTect SYBR green RT-PCR kit (Qiagen). Actin amplification was performed similarly but with oligonucleotides RT-ACT1-up2 and RT-ACT1-do2. Changes in ENA1 mRNA levels were determined by DNA microarray experiments.
Relative Pho89 and Ena1 protein levels were calculated by the integration of immunoblot signals from YPD-grown cultures using GelAnalyzer software after background subtraction. Data for Pho89 were obtained from the integration of data from 3 experiments per time point, whereas data for Ena1 were obtained from a representative experiment.
In silico analysis of the PHO89 promoter was carried out on the nucleotide sequence of the region comprising nucleotides −800 to −1 (from the initiating ATG) of PHO89, which was previously shown to contain high-pH response elements (15) and was analyzed with the matrix-scan algorithm available at the RSAT website (45), setting a P value threshold of 0.001. Matrices for transcription factor binding sequences were obtained from the JASPAR database (46).
RESULTS
Kinetics for Pho89 and Pho84 accumulation in response to low-phosphate and high-pH stress are different.
Since high-pH stress triggers a response that mimics the situation of phosphate starvation, we considered it essential to compare the kinetics of Pho89 accumulation in cells subjected to these types of stress. To this end, a C-terminally HA-tagged version of Pho89 was engineered and introduced into its own chromosomal locus, and the resulting strain was grown in synthetic medium at standard pH (5.5) and then transferred to medium either adjusted to pH 8.0, containing low phosphate (100 μM), or with a combination of both circumstances. As shown in Fig. 1A, high pH resulted in a fast and transient accumulation of Pho89 (30 to 60 min), whereas the kinetic for Pho89 accumulation after transfer to low-phosphate medium was much slower, with detectable amounts of the transporter being found only 4 to 8 h after phosphate removal. As expected, the response to phosphate starvation was largely abolished in a pho4 mutant. Exposure of cells to high pH and phosphate scarcity simultaneously yielded a profile of Pho89 expression that was a combination of that observed for each independent stress, that is, an early and transient peak followed by a late accumulation of the transporter (Fig. 1A).
FIG 1.
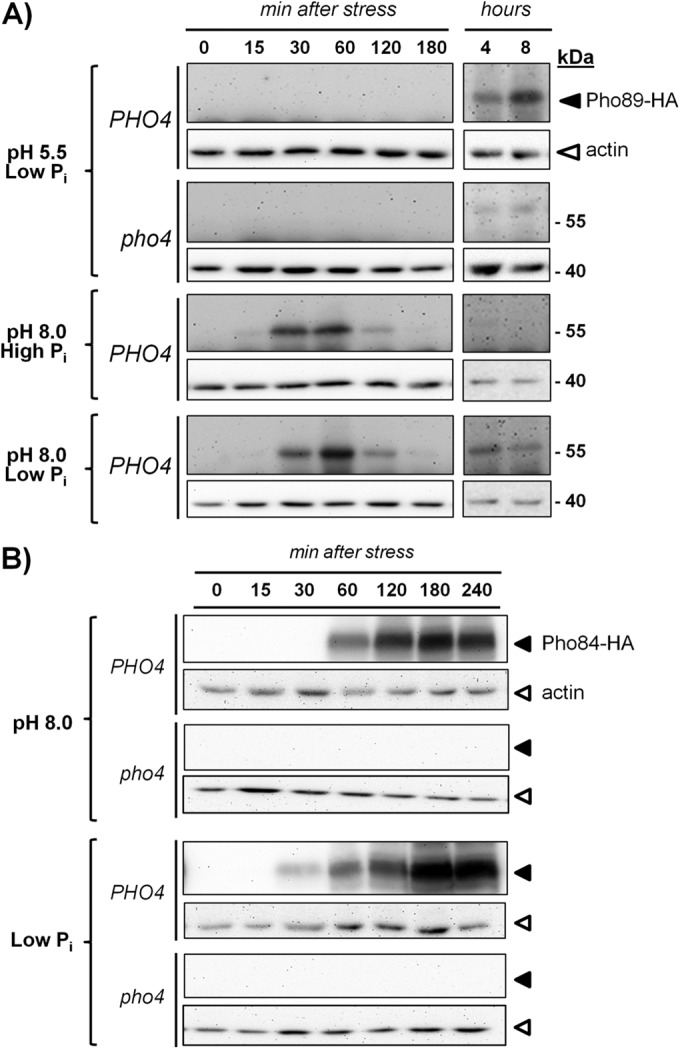
Comparative accumulations of Pho89 and Pho84 in response to phosphate starvation and high-pH stress. (A) Strains ASC07 (PHO4 PHO89-3×HA) and ASC10 (pho4::LEU2 PHO89-3×HA) were grown on synthetic high-Pi medium until the OD660 reached 0.6. Cells were collected and resuspended in synthetic medium lacking phosphate supplemented with 10 mM Pi (high Pi) or 100 μM Pi (low Pi) at pH 5.5 and/or pH 8.0 and adjusted with KOH. Growth was resumed, and samples were taken at the indicated times and processed for protein extract preparation as indicated in Materials and Methods. Equivalent amounts (30 μg of total protein) were subjected to SDS-PAGE (10% polyacrylamide gels) followed by immunoblotting using anti-HA antibodies to reveal HA-tagged Pho89 (dark triangles). Membranes were stripped and reprobed with antiactin antibodies (open triangles) for loading and transfer reference. (B) Wild-type strain DBY746 and its pho4 derivative (RSC4) were transformed with centromeric plasmid pMM15-PHO84, which expresses the C-terminally HA-tagged version of Pho84. Cells were subjected to high-pH or low-phosphate stress and collected after the indicated periods, and the amount of Pho84 was assessed by using anti-HA antibodies. Correct loading and transfer were monitored with antiactin antibodies in the stripped membranes.
In contrast, as shown in Fig. 1B, the temporal profile of Pho84 expression in response to high pH or phosphate starvation was essentially identical, peaking at ∼180 min after initiation of the stress. In this case, the response was stronger under phosphate starvation conditions, but in both circumstances, it was fully abolished in the absence of the Pho4 transcription factor. Therefore, our results point out substantial differences in the expressions of Pho89 and Pho84 in response to high-pH stress: Pho89 expression occurs faster and does not mimic the kinetics observed upon phosphate starvation.
We also investigated whether the absence of one of the transporters could influence the expression of the other under Pi starvation or high-pH-stress conditions. We observed that while the pho89 mutant displayed a Pho84 expression pattern identical to that of the wild-type strain (not shown), in pho84 cells subjected to high-pH stress, expression of Pho89 was induced slightly earlier, and a second induction peak, similar to that observed upon Pi depletion, was found (see Fig. S1 in the supplemental material). This could be due to a higher-than-normal activation of Pho4 in Pho84-deficient cells, which would fit with the previously observed derepression of PHO5 in this mutant (3).
Regulation of PHO89 expression by the calcineurin/Crz1 pathway.
Information retrieved from DNA microarray transcriptomic databases (15, 47, 48) and previous results from our laboratory (15, 16) suggested that PHO89 expression may be influenced by the calcineurin pathway. To further investigate this process, we subjected wild-type DBY746 cells and their isogenic cnb1 derivatives, lacking the regulatory subunit of the calcineurin phosphatase, to alkaline stress (pH 8.0) and prepared total RNA from cultures collected at different times. As shown in Fig. 2A, PHO89 mRNA accumulated rapidly (5 min) in response to stress, and the mRNA levels declined only after 60 min, in agreement with the kinetics of accumulation of the Pho89 protein described above. This response was largely blocked in cells lacking calcineurin, thus confirming the participation of this pathway in the control of PHO89 expression. Calcineurin activation results in dephosphorylation of the Crz1 transcription factor, its entry into the nucleus, and binding to specific sequences known as CDREs (calcineurin-dependent response elements). Therefore, a lacZ reporter gene fused to the PHO89 promoter was introduced into wild-type, cnb1, crz1, and cnb1 crz1 strains. As shown in Fig. 2B, the lack of the phosphatase, its downstream transcription factor, or both components resulted in a strong decrease, virtually identical in all cases, in the response of the promoter. As shown in Fig. 2C, when the production of the Pho89 protein was monitored in cells grown in rich medium (YPD), substantial accumulation was already observed after 15 min of exposure of the culture to high pH. This accumulation was strongly decreased in cells deficient in calcineurin activity. Therefore, our results indicate that calcineurin is involved in a major signaling pathway controlling the expression of the Pho89 transporter in response to high-pH stress. In contrast, deletion of cnb1 did not alter the expression pattern of Pho84 under low-Pi or high-pH conditions (not shown), thus confirming the absence of calcineurin-mediated regulation of the PHO84 promoter (15).
FIG 2.
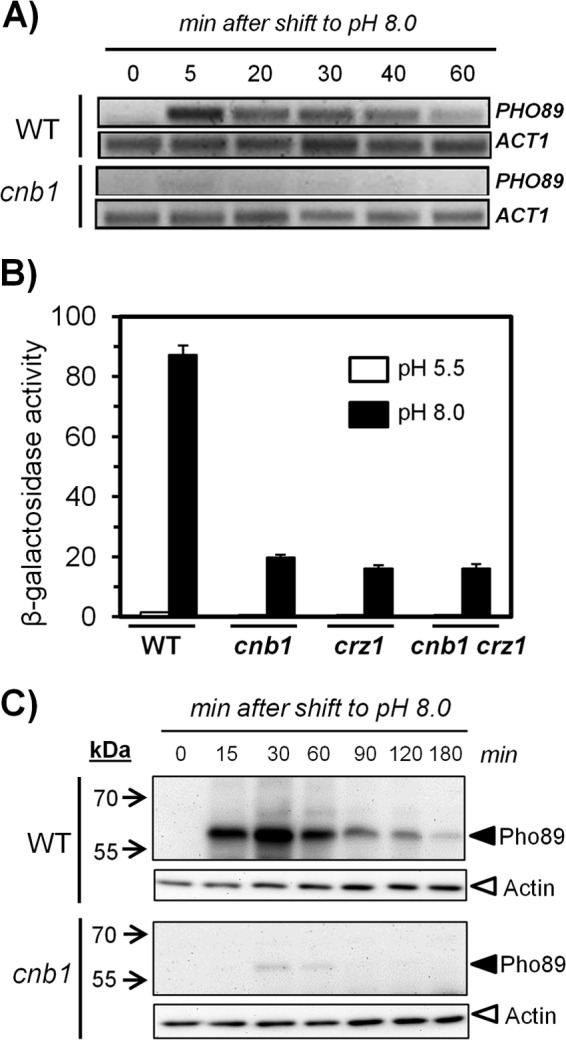
Expression of Pho89 in response to high-pH stress is controlled by the calcineurin/Crz1 pathway. (A) Wild-type (WT) DBY746 and MAR15 (cnb1) cells were collected at the indicated times after switching the medium to pH 8.0, and total RNA was prepared. Semiquantitative RT-PCR was carried out by using oligonucleotides specific for PHO89 and ACT1 (as a control), and the products were resolved in agarose gels and stained with GelRed (Biotium Inc.). (B) The indicated strains were transformed with the pPHO89-LacZ reporter, and exponentially growing cultures were exposed to pH 8.0 or maintained at pH 5.5 for 90 min. Cells were then collected, and β-galactosidase activity was measured as reported previously (15). Data are mean ± standard errors of the means from 12 determinations. (C) Cultures of strains ASC07 (PHO89-3×HA) and ASC08 (cnb1 PHO89-3×HA) were shifted to pH 8.0, and protein extracts were prepared. Samples (30 μg of protein) were resolved by SDS-PAGE and processed for immunoblotting as described in the legend to Fig. 1A.
We then searched in the PHO89 promoter for possible Crz1 binding regions as well as for sites for the Pho4 activator and other possible regulatory sites, including Nrg1/Nrg2 and Mig1/Mig2 (see Table S1 in the supplemental material). These two pairs of repressors were included in the search because they regulate the high-pH-responsive ENA1 promoter (24). As shown in Fig. 3A, two possible consensus sites each for Crz1, Pho4, and Nrg1/2 plus a single putative Mig1/2 binding site were located in the region spanning nt −520 to −250 upstream of the initiating Met codon. No Msn2/Msn4 binding sites were identified in the analyzed region, suggesting that this promoter is not under general stress response regulation.
FIG 3.

Functional characterization of two potential CDREs in the PHO89 promoter. (A) Cartoon depicting the predicted regulatory sites in the PHO89 upstream region (see the text for details). The bold discontinuous line spans the region amplified from ChIP samples shown in panel C. (B) Wild-type strain DBY746 was transformed with plasmid pPHO89-LacZ, pPHO89CDRE1-LacZ, or pPHO89CDRE2-LacZ, and cultures were subjected to pH 8.0 or maintained at pH 5.5. β-Galactosidase activity was determined as described in the legend to Fig. 2B. Data are means ± standard errors of the means from 12 determinations. (C, top) Chromatin-immunoprecipitated material from cells exposed to pH 8.0 for the indicated times was subjected to PCR amplification for the 143-nt region encompassing the CDRE2 consensus (discontinuous line in panel A). NI, sample lacking anti-HA antibodies; WCE, whole-cell extract. (Bottom) ChIP samples were subjected to massive sequencing. Mapped reads were quantified, normalized, and represented by using SeqMonk software. The thick arrow represents the PHO89 open reading frame, and the thin line represents the 0.5-kbp upstream region (note that PHO89 is on the Crick strand).
We first concentrated on the two putative CDRE binding sites, which are in fact identical sequences encoded in opposite DNA strands. The upstream sequence (CDRE1 [GTGGCTG]) was located at positions −513 to −507, whereas the second, with the sequence CAGCCAC, was found at positions −273 to −267 (CDRE2). We modified two nucleotides in each consensus sequence by site-directed mutagenesis to eliminate the conserved GCC core characteristic of the CDREs, thus changing CDRE1 to GTGTATG and converting CDRE2 to CAGTAAC (modified nucleotides are shown in italics). We then tested the ability of these modified promoters to drive transcription of a lacZ reporter. As shown in Fig. 3B, mutation of CDRE1 resulted in a relatively minor decrease in β-galactosidase activity. In contrast, modification of CDRE2 strongly interfered with the function of the promoter, since the decrease in the response was almost as strong as the effect observed upon deletion of calcineurin or Crz1 (compare Fig. 3B with 2B). This suggests that CDRE2 is a major calcineurin-dependent regulatory site for PHO89. To further confirm the participation of this regulatory element in the control of PHO89, we performed chromatin immunoprecipitation experiments using strain SP020, which carries a C-terminally HA-tagged version of Crz1. As shown in Fig. 3C (top), the transcription factor is recruited very quickly to a region of the promoter that encompasses the CDRE2 sequence, showing a peak between 2 and 5 min after exposure to alkaline pH. This very fast response is in agreement with the rapid nuclear localization, upon a shift to alkaline pH, of a version of Crz1 fused to GFP (49; our unpublished data). In an independent experiment, ChIP samples were subjected to massive sequencing to obtain a genome-wide perspective of the binding of Crz1 to alkaline-pH-responsive promoters. Among the >100 promoters recognized by Crz1 upon high-pH stress (data not shown), time-dependent recruitment of Crz1 to the PHO89 promoter was clearly detected (Fig. 3C, bottom).
Both Snf1 and Rim101 contribute to the induction of PHO89 upon alkaline stress.
Previous studies, including the characterization of the response of the ENA1 promoter, showed that both Snf1 and Rim101 pathways could be activated by high pH (22, 24, 50). To investigate the possible impact of these signaling pathways on PHO89 expression, we monitored the response of the PHO89-lacZ reporter in cells lacking Snf1 and Rim101 as well as in pho4 mutants. As shown in Fig. 4A, the absence of Snf1 decreased the response of the reporter by ∼40% compared to the wild type. This effect was similar in potency to the decrease observed upon deletion of RIM101, whereas a lack of Pho4 had a somewhat stronger impact. However, in all cases, the effect was less prominent than that observed in calcineurin-deficient cells. Deletion of SNF1 in a rim101 or a pho4 background resulted in a further reduction of promoter activity. Whereas deletion of RIM101 in the pho4 background decreased the promoter response only marginally, the lack of Rim101 in a calcineurin-deficient strain further blocked the response of the PHO89 promoter upon shifting the cells to pH 8.0. These results indicate that, in most cases, concurrent elimination of the above-mentioned components is additive, thus suggesting that they control independent signaling pathways acting on the PHO89 promoter. The loss of response in snf1 and rim101 mutants was confirmed by monitoring PHO89 mRNA levels by RT-PCR (Fig. 4B). Similarly, the amount of the Pho89 protein was determined by immunoblot analysis of snf1, rim101, and pho4 mutants. As shown in Fig. 4C, the different mutations decreased the amount of the transporter, although the pattern was not identical. For instance, mutation of RIM101 barely affected the early peak of Pho89 accumulation, although it resulted in a faster-than-normal decrease at longer times. In parallel experiments, we also observed that deletion of SNF1 or REG1 did not alter the expression of the Pho84 transporter in response to low Pi, confirming the absence of Snf1-mediated regulation in response to Pi starvation (51, 52). No major changes in the accumulation pattern of Pho84 in the snf1 mutant were observed, although a slight decrease in the overall levels of the transporter was detected in reg1 cells subjected to alkaline-pH stress (not shown).
FIG 4.

PHO89 expression under conditions of high-pH stress is regulated by Snf1 and Rim101. (A) The indicated strains, transformed with the pPHO89-LacZ reporter, were exposed to pH 8.0 or maintained at pH 5.5, and β-galactosidase activity was determined. Data are means ± standard errors of the means from 10 to 15 experiments. (B) Wild-type DBY476, RSC10 (snf1), and RSC21 (rim101) cells were collected at the indicated times after switching the medium to pH 8.0, and total RNA was prepared. Semiquantitative RT-PCR was performed as described in the legend to Fig. 2A. Signals were integrated, and bars at the bottom of each panel denote the ratios between the PHO89 and ACT1 mRNAs for each time point and strain. (C) The above-mentioned strains plus strain RSC4 (pho4) were subjected to pH 8.0 for the indicated times and processed as described in the legend to Fig. 1A, and the amount of Pho89 was revealed by immunoblotting.
Mig2 is phosphorylated in response to high-pH stress and contributes to PHO89 regulation.
We next concentrated on the role of Snf1 in PHO89 regulation. To corroborate the involvement of the kinase in the control of this promoter, we tested the PHO89 reporter in a reg1 strain, lacking the regulatory subunit of the Glc7 phosphatase, in which Snf1 cannot be dephosphorylated and, therefore, is hyperactive. The results (Fig. 5A) showed a substantial constitutive induction of the reporter in the reg1 strain, which was further increased (4-fold) upon high-pH stress. The exacerbated response of the reg1 strain was completely abolished by deletion of SNF1, suggesting that it was caused by hyperactivation of the kinase.
FIG 5.
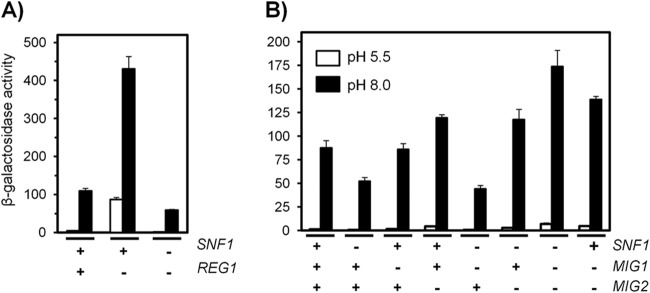
Regulation of PHO89 expression by Snf1 may be mediated by Mig2. (A) Wild-type strain DBY746 and its reg1 and reg1 snf1 derivatives were transformed with pPHO89-LacZ and subjected to high-pH stress (pH 8.0) (black bars) prior determination of β-galactosidase activity. (B) pPHO89-LacZ activity was measured in wild-type strain DBY746 and in strains containing different combinations of the snf1, mig1, and mig2 mutations. Data are means ± standard errors of the means from 9 to 12 experiments.
Snf1 can regulate Mig1 activity by phosphorylating the repressor and promoting exit from the nucleus under conditions of glucose scarcity (see reference 53 for a review). To test the possible contribution of Mig1 and its structurally and functionally related repressor Mig2, we measured the response of the pPHO89-LacZ reporter to high pH in cells lacking these repressors in the presence or absence of Snf1 (Fig. 5B). The absence of Mig2 was already detectable in nonstressed cells (1.41 ± 0.17 units for the wild type and 1.73 ± 0.18, 4.51 ± 0.23, and 4.90 ± 0.26 units for the mig1, mig2, and mig1 mig2 mutants, respectively). These results were indicative of a more prominent role for Mig2 in repression of the PHO89 promoter. As shown in Fig. 5B, deletion of MIG1 did not affect the response to alkaline stress, whereas a lack of Mig2 resulted in augmented promoter activity. Deletion of both repressors increased the response observed for the mig2 mutant only slightly. The response of the snf1 mig1 mutant was identical to that observed in snf1 cells, whereas cells lacking both snf1 and mig2 showed an induction equivalent to that displayed by the mig2 strain. These results indicate that Mig2, but not Mig1, represses the PHO89 promoter and that high-pH-induced activation of Snf1 might relieve the repression caused by Mig2. Remarkably, we observed an unexpected higher induction in the snf1 mig1 mig2 strain than in the mig1 mig2 double mutant. It should also be noted that mutation of reg1 generates a stronger derepression of the PHO89 promoter than that achieved by deletion of both the MIG1 and MIG2 repressors.
It has been reported that activation of Snf1 in response to glucose starvation results in Mig1 phosphorylation (54–57). In view of the apparent absence of a role for Mig1 in the regulation of PHO89 upon high-pH stress, we resorted to the use of a HA-tagged version of the repressor and immunoblot techniques to test whether alkalinization of the medium would result in Mig1 phosphorylation. As shown in Fig. S2A in the supplemental material, exposure to alkaline pH resulted in the rapid appearance of slower-migrating forms, indicative of hyperphosphorylated versions of the protein. This was a transient effect, since after 10 min of stress, Mig1 returned to its initial mobility. The shift was completely abolished when samples were treated prior to SDS-PAGE with alkaline phosphatase in the absence of EDTA (which blocks the phosphatase activity). The Mig1 band shift was not detected in cells lacking the Snf1 kinase, indicating a requirement for Snf1 function for alkali-induced phosphorylation of the repressor. Similarly, we investigated whether alkaline-pH stress could affect Mig1 subcellular localization by using a chromosomally integrated construct expressing a GFP-fused version of this repressor. We observed that Mig1 was mostly nuclear in nonstressed cells and that only 3 min after switching to pH 8.0, most of the fluorescence was distributed in the cytosol (see Fig. S2B in the supplemental material). This shift was transient, since after 10 min of stress, Mig1 was again concentrated in the nucleus. Therefore, alkaline-pH stress promotes transient Snf1-mediated phosphorylation and a nuclear-cytoplasmic shift of Mig1, although this does not translate into regulation of the PHO89 promoter.
We then repeated these experiments using an HA-tagged version of Mig2. As shown in Fig. 6A, the tagged protein appeared as a doublet at ∼55 kDa that very rapidly (2 min) suffered a marked shift to slower migration forms upon high-pH stress. After 10 to 15 min of stress, the pattern reversed to the original profile. Treatment of the samples with alkaline phosphatase prior to electrophoresis abolished the observed shift. When the experiment was repeated in cells lacking the Snf1 kinase, the initial mobility shift was essentially absent. For evaluation of the eventual nuclear-cytoplasmic shift of Mig2 in response to high-pH stress, we integrated a gene encoding a GFP–C-terminal fusion into the MIG2 locus of strain BY4741. However, the fluorescent signal was too low. For unequivocal localization, we constructed this fusion in the DBY746 background, which yielded a better signal. The experiments showed that the transient phosphorylation of Mig2 correlated well with the transfer of the GFP-tagged protein from the nucleus to the cytosol and the subsequent return (10 to 15 min) to its original nuclear localization (Fig. 6B). Remarkably, localization of Mig2 was essentially cytosolic in a reg1 mutant, even at pH 5.8 (Fig. 6B), and it was not altered by the shift to high pH (not shown). We then monitored the kinetics of phosphorylation of Snf1 in the wild-type strain exposed to pH 8.0. As shown in Fig. 6C, Snf1 is almost immediately phosphorylated in response to high-pH stress in a transient fashion, returning to baseline levels 15 to 20 min after the stress. Remarkably, in reg1 cells, phosphorylated Snf1 was already detected in the absence of stress. Phosphorylation was further increased by high-pH stress, and the phosphorylation levels remained high during the entire experiment. Phosphorylation of Snf1 was not induced by shifting cells to low-Pi (100 μM) medium (not shown). Altogether, our results indicate that both Mig1 and Mig2 can be rapidly and transiently phosphorylated in response to alkaline stress in an Snf1-dependent manner, although only Mig2 contributes to PHO89 regulation.
FIG 6.

High-pH stress induces transient phosphorylation and nuclear-cytoplasmic shift of Mig2. (A) A chromosomally encoded copy of Mig2 including a C-terminal 3×HA epitope tag was introduced into cells of wild-type strain BY4741 and its snf1 derivative. After cells were exposed to pH 8.0 for the indicated times, extracts were prepared and subjected to SDS-PAGE (8% polyacrylamide gels) prior to treatment with alkaline phosphatase in the absence (−) or in the presence (+) (to prevent the action of the phosphatase) of 50 mM EDTA. Immunoblot assays were performed by using anti-HA antibodies. The open triangle denotes slower (more-phosphorylated) species. (B) Strains SP048 (wild type) and ASC34 (reg1) containing chromosomally encoded C-terminal fusions of GFP with Mig2 were shifted to pH 8.0, and the localization of the repressor was monitored by fluorescence confocal microscopy (only pH 5.8 is shown for ASC34). Nuclei were stained with DAPI to illustrate the nuclear colocalization of the GFP and DAPI signals (merged). (C) Strains DBY746 (wild type) and RSC89 (reg1) were subjected to an alkaline shift for the indicated periods, and samples (10 μl) were processed for SDS-PAGE (10% gels) and immunoblotting using anti-phospho-Thr172-AMPK (phosphorylated Snf1 [P-Snf1]) or anti-His (Snf1 protein) antibodies. An extract from strain RSC10 (snf1Δ) is included as a negative control.
Combined regulation of PHO89 by Snf1 and Rim101 through Nrg1 and Nrg2.
The potential presence of Nrg1/Nrg2 consensus sites in the PHO89 promoter prompted us to investigate, with the aid of the pPHO89-LacZ reporter, the possible role of these regulatory proteins in high-pH stress. As presented in Fig. 7A, cells lacking both Nrg1 and Nrg2 showed a detectable increase in PHO89 promoter activity (5.5- ± 1.5-fold) under noninduced conditions, suggesting that these repressors could control PHO89 expression. Upon high-pH stress, the absence of Nrg1 or Nrg2 resulted in a roughly similar increase in reporter activity compared with the wild-type strain, and deletion of both genes yielded a further increase in the response, indicating that both components could have a physiological role. The response of the promoter in snf1 nrg1 and snf1 nrg2 cells was intermediate between that observed for snf1 cells and that observed for wild-type cells, whereas the activation level for the snf1 nrg1 nrg2 triple mutant was identical to that of the wild-type strain and therefore was not as high as that for the nrg1 nrg2 mutant. This could be explained by assuming that even if Snf1 can control Nrg1/2 function, in the absence of Snf1, the PHO89 promoter still suffers Mig2-mediated repression. In agreement with this notion, in an nrg1 nrg2 mig1 mig2 quadruple mutant strain, we observed a stronger response than in the nrg1 nrg2 strain (Fig. 7A).
FIG 7.
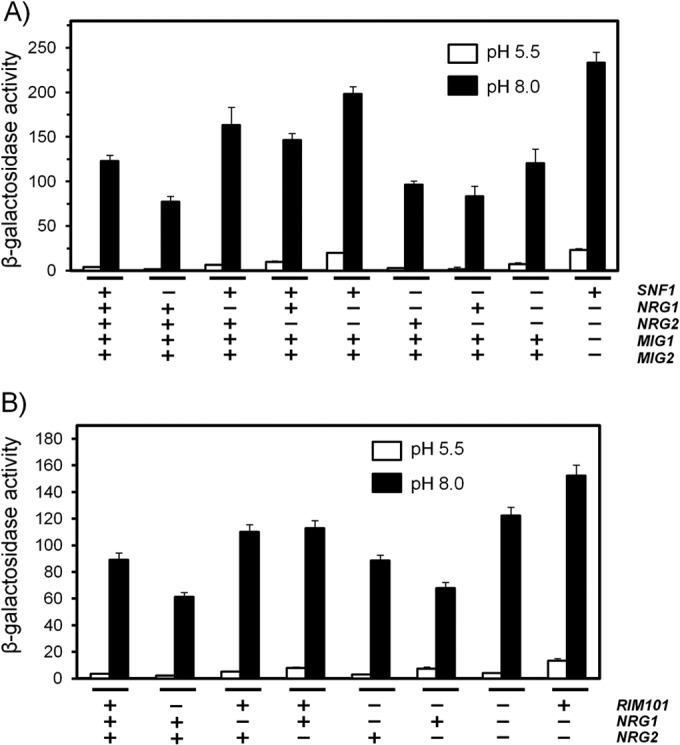
Snf1 and Rim101 control PHO89 expression through Mig2 and Nrg1. The indicated strains were transformed with plasmid pPHO89-LacZ. Exponential cultures were switched to pH 8.0, and β-galactosidase activity was determined after 90 min. Data are means ± standard errors of the means from 12 to 15 experiments (A) or 20 experiments (B).
We next investigated the mechanism by which Rim101 might regulate PHO89 induction in response to high-pH stress. Since Rim101 is known to control Nrg1 and Nrg2, we combined the rim101 mutation with mutation of NRG1 and/or NRG2 and compared the induction of the pPHO89-LacZ reporter. We observed that a lack of Nrg1 or Nrg2 causes a similarly moderate increase in the response of the reporter to high pH, which is further enhanced by removal of both repressors (Fig. 7B). However, we observed that the response in a rim101 nrg1 background was similar to that of the wild-type strain, whereas the induction level of the rim101 nrg2 mutant was only slightly higher than that of the rim101 strain. This suggests that the input on the PHO89 promoter mediated by Rim101 in response to high-pH stress would involve preferentially Nrg1 over Nrg2. We also investigated the subcellular localization of Nrg1 in response to alkalinization of the medium using an Nrg1-GFP fusion (not shown) and found that the repressor does not leave the nucleus in response to the stress. This is reminiscent of the previously reported observation that the nuclear localization of Nrg1 is not regulated by the carbon source (58).
The PHO89 promoter is high-pH but not salt responsive.
Exposure of yeast cells to high concentrations of salt triggers the activation of the calcineurin and Snf1 pathways (59–61). However, we could not find evidence in the literature for induction of PHO89 upon salt stress. To verify this point, we exposed wild-type cells carrying the pPHO89-LacZ reporter to mild (0.4 M) or strong (0.8 M) NaCl stress and compared the activities of the promoter in cells challenged by pH 8.0. As shown in Fig. S3 in the supplemental material, salt stress barely affected expression from the PHO89 promoter. In contrast, reporters containing the entire ENA1 promoter or a synthetic tandem arrangement of CDREs, which were introduced into the experiment as positive controls, were induced by both high-pH and salt stress conditions. We then examined if salt stress could affect the phosphorylation of Mig2. In Fig. S4 in the supplemental material, we show that the electrophoretic mobility of HA-tagged Mig2 was unchanged upon exposure to 0.8 M NaCl, indicating that salt stress does not trigger the phosphorylation of the repressor.
Evidence for functional coupling between Pho89 and Ena1.
The regulatory network described above, involving calcineurin, Snf1, Rim101, Mig2, and Nrg1/Nrg2, was essentially identical to that reported several years ago by our laboratory regarding the regulation of the monovalent cation ATPase gene ENA1 (24), thus suggesting the possibility of synchronic regulation. To test this, we determined the levels of mRNA of PHO89 and ENA1 and plotted the values against time after the shift to high pH (8.0). As shown in Fig. 8A, in both cases, the amounts of mRNA increased rapidly, with a peak ∼10 to 15 min after alkaline stress, and then decreased nearly in parallel. Similarly, a peak of Pho89 protein accumulation was produced 15 to 20 min after the mRNA peak, roughly at the same time that Ena1 accumulation reached its maximum. It is worth noting that whereas Pho89 levels declined quite sharply, the decrease of Ena1 accumulation was slower, allowing detection of significant levels of the transporter even 3 h after the onset of stress. These results indicated that Ena1 and Pho89 are produced at nearly the same time upon exposure to high-pH stress. Since Ena1 actively extrudes Na+ cations and Pho89 requires monovalent cations (preferably Na+) for effective phosphate cotransport, we considered the possibility that this coordinated regulation is at the basis of a functional link between both activities. To test this possibility, we compared the growth rates of strains lacking the high-affinity phosphate transporters and/or the cation ATPase gene under different pH and phosphate availability conditions. As shown in Fig. 8B, at standard (acidic) pH and in the presence of limiting amounts of phosphate (0.2 mM), the pho84, ena1 pho84, and pho84 pho89 strains could not proliferate. Under the same conditions, but at pH 7.6, the pho84 mutant grew reasonably well, likely due to the activity of Pho89, which would be induced by the lack of phosphate and the alkaline environment and, as shown here (see Fig. S1 in the supplemental material), enhanced by the absence of Pho84. As expected, further deletion of PHO89 blocked proliferation. Remarkably, virtually the same effect was achieved by deleting the ENA1 to -4 cluster, thus blocking the ability to extrude Na+ cations at alkaline pH (it should be noted that ENA1 represents the main extrusion system in the cluster and the only one induced by high pH). Therefore, the lack of ENA1 mimics the effect of mutating PHO89 in a pho84 background, thus phenocopying the situation of full deficiency of high-affinity phosphate transporters.
FIG 8.

Coregulation of Pho89 and Ena1 expression allows functional coupling between both proteins under conditions of high-pH stress. (A) Concerted coregulation of Pho89 (open circles) and Ena1 (closed circles) expression. Shown are time courses for ENA1 and PHO89 mRNA accumulation (top) or protein accumulation expressed as a percentage over the maximum signal (bottom) after shifting cells from pH 5.5 to 8.0 (see Materials and Methods for details). (B) The indicated strains at an OD660 of 0.05, plus a 10-fold dilution, were spotted onto YNB-based medium (lacking phosphate and sodium) agar plates supplemented with 5 mM NaCl and the indicated amounts of potassium phosphate and adjusted to different pHs. Growth was monitored after 3 days. (C) The indicated strains were shifted for 1 h to low-Pi (0.2 mM) medium at pH 5.5 and then shifted to pH 7.8, always in the presence of 5 mM NaCl. Samples were taken at the indicated periods and processed for determination of the intracellular Na+ content. Data are means ± standard errors of the means from 4 to 8 independent experiments. (D) Illustration of the proposed functional link between Ena1 and Pho89 under high-pH stress (see the text for explanations).
Shortly after the discovery of PHO89, it was proposed that the activity of ENA1 might serve as a Na+ motive force for phosphate transport through Pho89 (6). However, our plates shown in Fig. 8B contained 5 mM Na+, an external concentration sufficient to drive substantial phosphate influx through Pho89 (5, 8). Nevertheless, we designed an experiment to test this possibility. To this end, pho84, ena1, ena1 pho84, and ena1 pho89 cells were grown at pH 7.2 in the presence of 5 mM or 0.2 mM NaCl with either 5 mM or 0.2 mM Pi in the medium, and the 5/0.2 mM Na+ growth ratio was calculated and plotted. As shown in Fig. S5 in the supplemental material, ena1 pho84 cells grew worse at 5 mM than at 0.2 mM NaCl, and this effect was independent of the amount of Pi present in the medium, while the other mutants grew equally well under both NaCl conditions. This result raised the possibility that the growth defect of the ena1 pho84 mutant at alkaline pH is derived not from the lack of enough Na+ to sustain Pho89-mediated Pi uptake but from the intracellular accumulation of toxic Na+ cations accompanying Pi influx, which could not be eliminated in the absence of Ena1. To assess this possibility, the relevant strains were grown for 60 min in low-Pi medium (0.2 mM) in the presence of 5 mM NaCl and then shifted to pH 7.8, and samples were taken periodically and processed for determination of intracellular Na+ content. As shown in Fig. 8C, wild-type or pho84 cells did not significantly accumulate Na+ even after 8 h of growth. Mutation of ENA1 provoked a moderate, time-dependent intracellular accumulation of the cation that was distinctly augmented upon deletion of PHO84. This effect was interpreted as the result of increased Pho89 activity due to the absence of Pho84-mediated transport. This hypothesis was reinforced by the observation that an ena1 pho89 mutant accumulated very little intracellular Na+, clearly below the levels measured for the ena1 strain. Collectively, our results indicate that the accumulation of intracellular Na+ as a result of Pho89 activity can be detrimental in the absence of Ena1 and suggest that, under these conditions, the activity of the ATPase may be instrumental as a detoxification mechanism (Fig. 8D).
DISCUSSION
The capacity of yeast cells to survive and grow under changing environmental conditions is of widespread importance in the fungal pathogenicity of animals and plants and also from a biotechnological view (fungal production of antibiotics, secreted enzymes, and toxic compounds). Budding yeast has been shown to be an excellent model to study cellular strategies used to maintain appropriate intracellular ion concentrations and intracellular pH, among other important cellular parameters, when exposed to detrimental external conditions. However, little is known about the functional interactions between different ion transporters and how these possible interactions are sustained by common signaling pathways. Our work shows that Ena1 and Pho89 transporters are subjected to the same regulatory network, thus allowing coordinated expression and functional coupling upon high-pH stress.
PHO84 and PHO89 encode proteins that constitute the high-affinity phosphate transport system in budding yeast, and they are considered members of the PHO regulon, under the control of the Pho4/Pho2 transcriptional activators. We show here that under phosphate starvation, Pho84 accumulates much faster than Pho89, whereas under high-pH stress, a condition previously known to induce PHO84 and PHO89 expression (15, 16, 22), induction of PHO89 occurs earlier. Differential expression kinetics for PHO84 and PHO89 were also observed in yeast cells exposed to the cell wall-damaging agent Congo red, with the PHO89 response being faster than that of PHO84 (62), and it is known that mutation of the Ptc1 phosphatase gene or Mg2+ deprivation results in the induction of PHO89 but not of PHO84 (63, 64). In contrast, we recently observed that deprivation of potassium induces PHO84 but not PHO89 expression (65). Therefore, it seems evident that in spite of being cataloged as members of the PHO regulon, PHO84 and PHO89 exhibit distinct regulatory properties. The network regulating PHO89 expression described in our work allows explanation of these differences.
We demonstrate that in cells exposed to alkaline-pH stress, PHO89 is under the control of a complex signaling network involving (in addition to Pho4) calcineurin, Snf1, and Rim101 (see Fig. 9 for a schematic depiction of the proposed network). Previous evidence suggested that high-pH induction of PHO89, but not that of PHO84, was influenced by the presence of calcineurin. We confirm here that the Ca2+-activated phosphatase calcineurin is a major regulator of PHO89 upon alkaline-pH stress. This is explained by the previously reported observation that exposure to high pH triggers a very fast entry of calcium cations into the cell from the extracellular medium (16). Activation of calcineurin by high pH results in dephosphorylation of Crz1 and very fast entry (2 to 5 min) of the transcription factor into the nucleus (49; our unpublished results). In agreement with this kinetic, we observed recruitment of Crz1 to the PHO89 promoter in the first few minutes after shifting cells to alkaline pH. We also identify, by mutational analysis, a CDRE responsible for most of the regulatory properties of calcineurin. The evidence that PHO89 expression is potently controlled by calcineurin allows an explanation for why the basal expression level of PHO89 is higher than normal (and sensitive to the calcineurin inhibitor FK506) in cells deficient in the protein phosphatase Ppz1, since these mutants have a constitutively activated calcineurin pathway (66). Moreover, the calcineurin-mediated activation of Crz1 would be reinforced by both the inhibition of the protein kinase A (PKA) pathway (21) and the activation of the PHO pathway under high-pH stress, since protein kinase A and Pho85 are known to phosphorylate Crz1 (67, 68), thus contributing to its inactivation (Fig. 9). Interestingly, the regulation of PHO89 by calcium/calcineurin may be a common trait in fungi, since a recent report showed an important role of calcium influx in the alkaline stress response, which was also Crz1 dependent, in Candida albicans (69). This control mechanism is reminiscent of the induced expression of the sodium-dependent phosphate cotransporter Pit-1, the human homolog of Pho89, by long-term calcium treatment in smooth muscle cells (70).
FIG 9.

Schematic depiction of regulatory inputs acting on the PHO89 promoter upon high-pH stress. Elements upstream of Snf1, Rim101, Pho81, or calcineurin are not included for clarity. Discontinuous lines indicate possible physical interactions not experimentally tested. The functional Pho4 consensus site is deduced from recently reported genome-wide data (85).
Exposure of yeast cells to high-salt stress also increases intracellular calcium concentrations (71, 72) and activates calcineurin. Activation of calcineurin is responsible for part of the transcriptional response of salt-induced genes, such as the Na+-ATPase ENA1. However, our results indicate that saline stress does not induce PHO89 expression. This suggests that either the potency of the calcium signal generated by salt stress is insufficient to activate PHO89 or efficient induction of this gene also requires the concomitant derepression of its promoter, not triggered by salt stress. In this regard, it is interesting that we did not observe phosphorylation of Mig2 upon exposure to high salt (see Fig. S4 in the supplemental material), suggesting that this repressor may remain bound to the PHO89 promoter under this kind of stress.
It has been reported that Snf1, which is cytosolic in unstressed cells, becomes enriched in the nucleus when cells are subjected to alkaline pH (59). Under glucose-limiting conditions, Snf1 phosphorylates Mig1 and promotes its export from the nucleus, thus relieving transcriptional repression of a set of glucose-repressed genes. We observed that Mig1 can be rapidly and transiently phosphorylated in response to alkaline stress in an Snf1-dependent manner and that Mig1 is rapidly released from the nucleus upon shifting cells to high pH. This is in contrast to what was reported previously for high-salt stress, in which Snf1 activation is not followed by Mig1 phosphorylation (73, 74). However, our data indicate that Mig1 is barely relevant in repressing PHO89 under normal growth conditions and plays a small role in the expression change of the transporter gene upon high-pH induction. Therefore, it can be assumed that either Mig1 is not bound to the PHO89 promoter under standard growth conditions or it is unable to repress PHO89 expression. Here we present evidence that Mig2, a homolog of Mig1, is also rapidly phosphorylated in an Snf1-dependent way in response to high-pH stress. This is remarkable because for many years, it has been generally accepted that, in contrast to Mig1, Mig2 is not a substrate for the Snf1 kinase (75, 76). Recent work proposed that galactose induces fast degradation of Mig2 by a mechanism that involves Snf1-dependent Mig2 phosphorylation (77), although it is worth noting that those authors observed an almost complete depletion of Mig2 after only 20 min upon shifting of cells to galactose, whereas in our hands, the amount of Mig2 did not significantly change after 30 min of alkaline stress. In any case, our data support a role for Mig2 in repression of PHO89 expression, and it is reasonable to assume that activation of Snf1 results in a relief of such repression. In this regard, previously reported transcriptomic data based on DNA microarray studies pointed to a strong derepression of PHO89 in a mig1 mig2 mutant (78), whereas the expression of PHO89 in the mig1 mutant was similar to that of the wild type. Although those authors postulated a redundant role for mig1 and mig2 in the regulation of PHO89, their results also seem consistent with a preeminent role for Mig2 in the control of PHO89 expression. In conclusion, we postulate that Mig2, but not Mig1, seems to be relevant for mediating Snf1-controlled induction of PHO89 upon high-pH stress.
It has been shown that Snf1 interacts with the repressor Nrg1 and its closely related homolog Nrg2 (58), and both repressors have been reported to mediate Snf1-dependent responses (79, 80). NRG1 expression is downregulated by high-pH stress, and deletion of this gene results in increased ENA1 expression levels under these conditions (23, 24). In addition, Nrg1 is also under the control of Rim101 in response to salt and alkaline-pH stresses (22–24). We show here that Nrg1 is likely repressing PHO89 expression and that high-pH stress leads to derepression of the promoter in a way that involves combined control by both Snf1 and Rim101. It must be noted that in contrast to other organisms such as Aspergillus, where Rim101 directly controls the expression of alkali-inducible genes (see reference 81 for a review), in budding yeast, the effect seems to be indirect: the activation of Rim101 represses the expression of Nrg1, thus relieving the repression of Nrg1 target genes such as ENA1 (23) or PHO89 (this work). This indirect control, which would involve a 2-step mechanism, would explain why deletion of RIM101 affects the late response of PHO89 but not the early peak of accumulation of Pho89 mRNA or protein (Fig. 4B and C).
The involvement of Snf1 in the regulation of PHO89 is further reinforced by the observation that deletion of REG1 resulted in a dramatic increase of PHO89 promoter activity under both basal and high-pH stress conditions that is fully Snf1 dependent. Mutation of REG1 causes hyperactivation of Snf1 (41), and we observed constitutive and sustained phosphorylation of Snf1 and a cytosolic localization of Mig2 in the reg1 mutant (Fig. 6B and C). Remarkably, the potent derepression of PHO89 in reg1 cells was substantially greater than that achieved in the mig1 mig2 nrg1 nrg2 quadruple mutant. Therefore, the phenomenon cannot be explained only by Snf1-mediated inhibition of the repressor's function. Although we do not have a definite explanation, this observation is not totally surprising, since existing evidence indicates that Snf1 affects multiple steps in gene regulation, including transcription factor binding, RNA polymerase II activity, and cytoplasmic mRNA stability (82).
Alkaline-pH stress results in the transcriptional induction of the Ena1 Na+-ATPase, which actively extrudes Na+ cations (15, 26, 35, 61), whereas Pho89 is a Na+-driven phosphate transporter active at alkaline pH (5). Previous results from our laboratory (24) demonstrated that alkaline stress induction of ENA1 is mediated by three different pathways, involving the activation of calcineurin, Rim101 (through Nrg1), and Snf1 (through Nrg1 and Mig2). Here we provide experimental evidence that ENA1 and PHO89 are under the control of the same regulatory network (Fig. 9), thus allowing the coordinate expression of both proteins in response to high-pH stress (Fig. 8A). In addition, we demonstrate that under alkaline-pH conditions, mutation of ENA1 in a pho84 background results in the inability to grow in phosphate-depleted medium. This strongly suggests that the functions of Ena1 and Pho89 are linked. It should be noted that simply deleting ENA1 and growing cells in low-phosphate medium does not allow testing of this hypothesis, since Pho84 has a higher transport capacity than Pho89, and although it is optimally active at acidic pH, it retains a significant activity at neutral or even mild alkaline pH (8).
Shortly after the discovery of PHO89, it was proposed that Ena1 could provide a motive force for Pho89-mediated phosphate transport (6). While this notion might still hold in some circumstances, alternative scenarios can be devised. Our observation that increasing the amount of Na+ in the medium (which should diminish the role of Ena1 as a provider of driving force) did not improve growth when ENA1 was deleted in the pho84 background suggests that the main role of Ena1 would be to avoid the accumulation of Na+ cations entering through Pho89, instead of supplying Na+ cations for Pho89-mediated Pi entry. This scenario is supported by the observation that the ena1 pho84 strain accumulated high levels of Na+ but that the ena1 pho89 strain did not, pointing to the fact that the observed accumulation of Na+ is the result of the Pho89 activity. This increase in Na+ content could be detrimental to the cell for at least two reasons: (i) because of the recognized toxicity of Na+ cations and (ii) because the increase in intracellular Na+ levels would abolish or even reverse the extracellular/intracellular cation gradient that is necessary to support effective Pi entry through Pho89 (Fig. 8D). In any case, the existence of an identical high-pH-responsive signaling network controlling both genes would serve to ensure the well-timed expression of these transporters, which would be clearly advantageous for the cell to thrive in such adverse environments. Pho89-like transporters are widely spread among eukaryotes, whereas Ena1-like ATPases are found in all fungi and are extensively present in bryophyte and parasitic protozoa (83). The requirement for Ena1 to grow at alkaline pH (even in the absence of salt stress) has been proven for very diverse fungi, such as Ustilago maydis and Cryptococcus neoformans (see reference 83 and references therein), and the induction of ENA1 genes in response to a shift to high pH not only is a fungal characteristic but also is observed in bryophytes (84). Therefore, the functional and regulatory coupling between Pho89 and Ena1 transporters reported here for budding yeast might be conserved through evolution.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge the excellent technical assistance of Montserrat Robledo. We thank Maria Platara for preliminary work. We also thank Anna Barceló (Servei de Genòmica i Bioinformàtica, UAB) for support in ChIP-Seq experiments, Núria Barba (Institut de Neurociències, UAB) for helping with the microscopy experiments, Ana Salazar (Universidad de Córdoba) for support in intracellular sodium concentration determinations, and Enric Herrero (Universitat de LLeida) for plasmids pMM15 and pMM17.
This work was supported by grants BFU2011-30197-C3-01 and EUI2009-04147 (SysMo2) to J.A. (Ministry of Science and Innovation, Spain, and Fondo Europeo de Desarrollo Regional [FEDER]). J.A. is the recipient of an Ajut 2014SGR-4 award and an Institució Catalana de Recerca i Estudis Avançats (ICREA) Academia 2009 award (Generalitat de Catalunya). D.C. is the recipient of a predoctoral fellowship from the Spanish Ministry of Education.
We declare that we do not have any conflict of interest.
Footnotes
Published ahead of print 29 September 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.01089-14.
REFERENCES
- 1.Bun-ya M, Shikata K, Nakade S, Yompakdee C, Harashima S, Oshima Y. 1996. Two new genes, PHO86 and PHO87, involved in inorganic phosphate uptake in Saccharomyces cerevisiae. Curr. Genet. 29:344–351. 10.1007/s002940050055. [DOI] [PubMed] [Google Scholar]
- 2.Ghillebert R, Swinnen E, De Snijder P, Smets B, Winderickx J. 2011. Differential roles for the low-affinity phosphate transporters Pho87 and Pho90 in Saccharomyces cerevisiae. Biochem. J. 434:243–251. 10.1042/BJ20101118. [DOI] [PubMed] [Google Scholar]
- 3.Wykoff DD, O'Shea EK. 2001. Phosphate transport and sensing in Saccharomyces cerevisiae. Genetics 159:1491–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bun-ya M, Nishimura M, Harashima S, Oshima Y. 1991. The PHO84 gene of Saccharomyces cerevisiae encodes an inorganic phosphate transporter. Mol. Cell. Biol. 11:3229–3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martinez P, Persson BL. 1998. Identification, cloning and characterization of a derepressible Na+-coupled phosphate transporter in Saccharomyces cerevisiae. Mol. Gen. Genet. 258:628–638. 10.1007/s004380050776. [DOI] [PubMed] [Google Scholar]
- 6.Persson BL, Petersson J, Fristedt U, Weinander R, Berhe A, Pattison J. 1999. Phosphate permeases of Saccharomyces cerevisiae: structure, function and regulation. Biochim. Biophys. Acta 1422:255–272. 10.1016/S0304-4157(99)00010-6. [DOI] [PubMed] [Google Scholar]
- 7.Pattison-Granberg J, Persson BL. 2000. Regulation of cation-coupled high-affinity phosphate uptake in the yeast Saccharomyces cerevisiae. J. Bacteriol. 182:5017–5019. 10.1128/JB.182.17.5017-5019.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zvyagilskaya RA, Lundh F, Samyn D, Pattison-Granberg J, Mouillon JM, Popova Y, Thevelein JM, Persson BL. 2008. Characterization of the Pho89 phosphate transporter by functional hyperexpression in Saccharomyces cerevisiae. FEMS Yeast Res. 8:685–696. 10.1111/j.1567-1364.2008.00408.x. [DOI] [PubMed] [Google Scholar]
- 9.Ogawa N, Saitoh H, Miura K, Magbanua JP, Bun-ya M, Harashima S, Oshima Y. 1995. Structure and distribution of specific cis-elements for transcriptional regulation of PHO84 in Saccharomyces cerevisiae. Mol. Gen. Genet. 249:406–416. [DOI] [PubMed] [Google Scholar]
- 10.Persson BL, Lagerstedt JO, Pratt JR, Pattison-Granberg J, Lundh K, Shokrollahzadeh S, Lundh F. 2003. Regulation of phosphate acquisition in Saccharomyces cerevisiae. Curr. Genet. 43:225–244. 10.1007/s00294-003-0400-9. [DOI] [PubMed] [Google Scholar]
- 11.Auesukaree C, Homma T, Kaneko Y, Harashima S. 2003. Transcriptional regulation of phosphate-responsive genes in low-affinity phosphate-transporter-defective mutants in Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 306:843–850. 10.1016/S0006-291X(03)01068-4. [DOI] [PubMed] [Google Scholar]
- 12.Ogawa N, DeRisi J, Brown PO. 2000. New components of a system for phosphate accumulation and polyphosphate metabolism in Saccharomyces cerevisiae revealed by genomic expression analysis. Mol. Biol. Cell 11:4309–4321. 10.1091/mbc.11.12.4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaffman A, Herskowitz I, Tjian R, O'Shea EK. 1994. Phosphorylation of the transcription factor PHO4 by a cyclin-CDK complex, PHO80-PHO85. Science 263:1153–1156. 10.1126/science.8108735. [DOI] [PubMed] [Google Scholar]
- 14.Toh-e A, Tanaka K, Uesono Y, Wickner RB. 1988. PHO85, a negative regulator of the PHO system, is a homolog of the protein kinase gene, CDC28, of Saccharomyces cerevisiae. Mol. Gen. Genet. 214:162–164. 10.1007/BF00340196. [DOI] [PubMed] [Google Scholar]
- 15.Serrano R, Ruiz A, Bernal D, Chambers JR, Arino J. 2002. The transcriptional response to alkaline pH in Saccharomyces cerevisiae: evidence for calcium-mediated signalling. Mol. Microbiol. 46:1319–1333. 10.1046/j.1365-2958.2002.03246.x. [DOI] [PubMed] [Google Scholar]
- 16.Viladevall L, Serrano R, Ruiz A, Domenech G, Giraldo J, Barcelo A, Arino J. 2004. Characterization of the calcium-mediated response to alkaline stress in Saccharomyces cerevisiae. J. Biol. Chem. 279:43614–43624. 10.1074/jbc.M403606200. [DOI] [PubMed] [Google Scholar]
- 17.Cyert MS. 2003. Calcineurin signaling in Saccharomyces cerevisiae: how yeast go crazy in response to stress. Biochem. Biophys. Res. Commun. 311:1143–1150. 10.1016/S0006-291X(03)01552-3. [DOI] [PubMed] [Google Scholar]
- 18.Werner A, Kinne RK. 2001. Evolution of the Na-P(i) cotransport systems. Am. J. Physiol. Regul. Integr. Comp. Physiol. 280:R301–R312. [DOI] [PubMed] [Google Scholar]
- 19.Serrano R, Martin H, Casamayor A, Arino J. 2006. Signaling alkaline pH stress in the yeast Saccharomyces cerevisiae through the Wsc1 cell surface sensor and the Slt2 MAPK pathway. J. Biol. Chem. 281:39785–39795. 10.1074/jbc.M604497200. [DOI] [PubMed] [Google Scholar]
- 20.Casamayor A, Serrano R, Platara M, Casado C, Ruiz A, Arino J. 2012. The role of the Snf1 kinase in the adaptive response of Saccharomyces cerevisiae to alkaline pH stress. Biochem. J. 444:39–49. 10.1042/BJ20112099. [DOI] [PubMed] [Google Scholar]
- 21.Casado C, Gonzalez A, Platara M, Ruiz A, Arino J. 2011. The role of the protein kinase A pathway in the response to alkaline pH stress in yeast. Biochem. J. 438:523–533. 10.1042/BJ20110607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lamb TM, Xu W, Diamond A, Mitchell AP. 2001. Alkaline response genes of Saccharomyces cerevisiae and their relationship to the RIM101 pathway. J. Biol. Chem. 276:1850–1856. 10.1074/jbc.M008381200. [DOI] [PubMed] [Google Scholar]
- 23.Lamb TM, Mitchell AP. 2003. The transcription factor Rim101p governs ion tolerance and cell differentiation by direct repression of the regulatory genes NRG1 and SMP1 in Saccharomyces cerevisiae. Mol. Cell. Biol. 23:677–686. 10.1128/MCB.23.2.677-686.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Platara M, Ruiz A, Serrano R, Palomino A, Moreno F, Arino J. 2006. The transcriptional response of the yeast Na+-ATPase ENA1 gene to alkaline stress involves three main signaling pathways. J. Biol. Chem. 281:36632–36642. 10.1074/jbc.M606483200. [DOI] [PubMed] [Google Scholar]
- 25.Ruiz A, Arino J. 2007. Function and regulation of the Saccharomyces cerevisiae ENA sodium ATPase system. Eukaryot. Cell 6:2175–2183. 10.1128/EC.00337-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garciadeblas B, Rubio F, Quintero FJ, Banuelos MA, Haro R, Rodriguez-Navarro A. 1993. Differential expression of two genes encoding isoforms of the ATPase involved in sodium efflux in Saccharomyces cerevisiae. Mol. Gen. Genet. 236:363–368. [DOI] [PubMed] [Google Scholar]
- 27.Wieland J, Nitsche AM, Strayle J, Steiner H, Rudolph HK. 1995. The PMR2 gene cluster encodes functionally distinct isoforms of a putative Na+ pump in the yeast plasma membrane. EMBO J. 14:3870–3882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Posas F, Camps M, Arino J. 1995. The PPZ protein phosphatases are important determinants of salt tolerance in yeast cells. J. Biol. Chem. 270:13036–13041. 10.1074/jbc.270.22.13036. [DOI] [PubMed] [Google Scholar]
- 29.Haro R, Garciadeblas B, Rodriguez-Navarro A. 1991. A novel P-type ATPase from yeast involved in sodium transport. FEBS Lett. 291:189–191. 10.1016/0014-5793(91)81280-L. [DOI] [PubMed] [Google Scholar]
- 30.Adams A, Gottschling DE, Kaiser CA, Stearns T. 1997. Methods in yeast genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 31.Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, Chu AM, Connelly C, Davis K, Dietrich F, Dow SW, El Bakkoury M, Foury F, Friend SH, Gentalen E, Giaever G, Hegemann JH, Jones T, Laub M, Liao H, Davis RW. 1999. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285:901–906. 10.1126/science.285.5429.901. [DOI] [PubMed] [Google Scholar]
- 32.Longtine MS, McKenzie A, III, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. 1998. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14:953–961. [DOI] [PubMed] [Google Scholar]
- 33.Rodriguez C, Sanz P, Gancedo C. 2003. New mutations of Saccharomyces cerevisiae that partially relieve both glucose and galactose repression activate the protein kinase Snf1. FEMS Yeast Res. 3:77–84. 10.1111/j.1567-1364.2003.tb00141.x. [DOI] [PubMed] [Google Scholar]
- 34.Ruiz A, González A, García-Salcedo R, Ramos J, Arino J. 2006. Role of protein phosphatases 2C on tolerance to lithium toxicity in the yeast Saccharomyces cerevisiae. Mol. Microbiol. 62:263–277. 10.1111/j.1365-2958.2006.05370.x. [DOI] [PubMed] [Google Scholar]
- 35.Alepuz PM, Cunningham KW, Estruch F. 1997. Glucose repression affects ion homeostasis in yeast through the regulation of the stress-activated ENA1 gene. Mol. Microbiol. 26:91–98. 10.1046/j.1365-2958.1997.5531917.x. [DOI] [PubMed] [Google Scholar]
- 36.Cunningham KW, Fink GR. 1996. Calcineurin inhibits VCX1-dependent H+/Ca2+ exchange and induces Ca2+ ATPases in Saccharomyces cerevisiae. Mol. Cell. Biol. 16:2226–2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stathopoulos AM, Cyert MS. 1997. Calcineurin acts through the CRZ1/TCN1-encoded transcription factor to regulate gene expression in yeast. Genes Dev. 11:3432–3444. 10.1101/gad.11.24.3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Horak J, Wolf DH. 2001. Glucose-induced monoubiquitination of the Saccharomyces cerevisiae galactose transporter is sufficient to signal its internalization. J. Bacteriol. 183:3083–3088. 10.1128/JB.183.10.3083-3088.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Urban J, Soulard A, Huber A, Lippman S, Mukhopadhyay D, Deloche O, Wanke V, Anrather D, Ammerer G, Riezman H, Broach JR, De Virgilio C, Hall MN, Loewith R. 2007. Sch9 is a major target of TORC1 in Saccharomyces cerevisiae. Mol. Cell 26:663–674. 10.1016/j.molcel.2007.04.020. [DOI] [PubMed] [Google Scholar]
- 40.González A, Casado C, Ariño J, Casamayor A. 2013. Ptc6 is required for proper rapamycin-induced down-regulation of the genes coding for ribosomal and rRNA processing proteins in S. cerevisiae. PLoS One 8:e64470. 10.1371/journal.pone.0064470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Orlova M, Barrett L, Kuchin S. 2008. Detection of endogenous Snf1 and its activation state: application to Saccharomyces and Candida species. Yeast 25:745–754. 10.1002/yea.1628. [DOI] [PubMed] [Google Scholar]
- 42.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9:357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Navarrete C, Petrezselyova S, Barreto L, Martinez JL, Zahradka J, Arino J, Sychrova H, Ramos J. 2010. Lack of main K+ uptake systems in Saccharomyces cerevisiae cells affects yeast performance in both potassium-sufficient and potassium-limiting conditions. FEMS Yeast Res. 10:508–517. 10.1111/j.1567-1364.2010.00630.x. [DOI] [PubMed] [Google Scholar]
- 44.Verduyn C, Postma E, Scheffers WA, Van Dijken JP. 1992. Effect of benzoic acid on metabolic fluxes in yeasts: a continuous-culture study on the regulation of respiration and alcoholic fermentation. Yeast 8:501–517. 10.1002/yea.320080703. [DOI] [PubMed] [Google Scholar]
- 45.Thomas-Chollier M, Defrance M, Medina-Rivera A, Sand O, Herrmann C, Thieffry D, van Helden J. 2011. RSAT 2011: regulatory sequence analysis tools. Nucleic Acids Res. 39:W86–W91. 10.1093/nar/gkr377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Portales-Casamar E, Thongjuea S, Kwon AT, Arenillas D, Zhao X, Valen E, Yusuf D, Lenhard B, Wasserman WW, Sandelin A. 2010. JASPAR 2010: the greatly expanded open-access database of transcription factor binding profiles. Nucleic Acids Res. 38:D105–D110. 10.1093/nar/gkp950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yoshimoto H, Saltsman K, Gasch AP, Li HX, Ogawa N, Botstein D, Brown PO, Cyert MS. 2002. Genome-wide analysis of gene expression regulated by the calcineurin/Crz1p signaling pathway in Saccharomyces cerevisiae. J. Biol. Chem. 277:31079–31088. 10.1074/jbc.M202718200. [DOI] [PubMed] [Google Scholar]
- 48.Causton HC, Ren B, Koh SS, Harbison CT, Kanin E, Jennings EG, Lee TI, True HL, Lander ES, Young RA. 2001. Remodeling of yeast genome expression in response to environmental changes. Mol. Biol. Cell 12:323–337. 10.1091/mbc.12.2.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ruiz A, Serrano R, Arino J. 2008. Direct regulation of genes involved in glucose utilization by the calcium/calcineurin pathway. J. Biol. Chem. 283:13923–13933. 10.1074/jbc.M708683200. [DOI] [PubMed] [Google Scholar]
- 50.Hong SP, Leiper FC, Woods A, Carling D, Carlson M. 2003. Activation of yeast Snf1 and mammalian AMP-activated protein kinase by upstream kinases. Proc. Natl. Acad. Sci. U. S. A. 100:8839–8843. 10.1073/pnas.1533136100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Klosinska MM, Crutchfield CA, Bradley PH, Rabinowitz JD, Broach JR. 2011. Yeast cells can access distinct quiescent states. Genes Dev. 25:336–349. 10.1101/gad.2011311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huang D, Farkas I, Roach PJ. 1996. Pho85p, a cyclin-dependent protein kinase, and the Snf1p protein kinase act antagonistically to control glycogen accumulation in Saccharomyces cerevisiae. Mol. Cell. Biol. 16:4357–4365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Santangelo GM. 2006. Glucose signaling in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 70:253–282. 10.1128/MMBR.70.1.253-282.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schmidt MC, McCartney RR, Zhang X, Tillman TS, Solimeo H, Wolfl S, Almonte C, Watkins SC. 1999. Std1 and Mth1 proteins interact with the glucose sensors to control glucose-regulated gene expression in Saccharomyces cerevisiae. Mol. Cell. Biol. 19:4561–4571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Treitel MA, Kuchin S, Carlson M. 1998. Snf1 protein kinase regulates phosphorylation of the Mig1 repressor in Saccharomyces cerevisiae. Mol. Cell. Biol. 18:6273–6280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sutherland CM, Hawley SA, McCartney RR, Leech A, Stark MJ, Schmidt MC, Hardie DG. 2003. Elm1p is one of three upstream kinases for the Saccharomyces cerevisiae SNF1 complex. Curr. Biol. 13:1299–1305. 10.1016/S0960-9822(03)00459-7. [DOI] [PubMed] [Google Scholar]
- 57.Ostling J, Ronne H. 1998. Negative control of the Mig1p repressor by Snf1p-dependent phosphorylation in the absence of glucose. Eur. J. Biochem. 252:162–168. 10.1046/j.1432-1327.1998.2520162.x. [DOI] [PubMed] [Google Scholar]
- 58.Vyas VK, Kuchin S, Carlson M. 2001. Interaction of the repressors Nrg1 and Nrg2 with the Snf1 protein kinase in Saccharomyces cerevisiae. Genetics 158:563–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hong SP, Carlson M. 2007. Regulation of snf1 protein kinase in response to environmental stress. J. Biol. Chem. 282:16838–16845. 10.1074/jbc.M700146200. [DOI] [PubMed] [Google Scholar]
- 60.Nakamura T, Liu Y, Hirata D, Namba H, Harada S, Hirokawa T, Miyakawa T. 1993. Protein phosphatase type 2B (calcineurin)-mediated, FK506-sensitive regulation of intracellular ions in yeast is an important determinant for adaptation to high salt stress conditions. EMBO J. 12:4063–4071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mendoza I, Rubio F, Rodriguez-Navarro A, Pardo JM. 1994. The protein phosphatase calcineurin is essential for NaCl tolerance of Saccharomyces cerevisiae. J. Biol. Chem. 269:8792–8796. [PubMed] [Google Scholar]
- 62.Garcia R, Bermejo C, Grau C, Perez R, Rodriguez-Pena JM, Francois J, Nombela C, Arroyo J. 2004. The global transcriptional response to transient cell wall damage in Saccharomyces cerevisiae and its regulation by the cell integrity signaling pathway. J. Biol. Chem. 279:15183–15195. 10.1074/jbc.M312954200. [DOI] [PubMed] [Google Scholar]
- 63.Gonzalez A, Ruiz A, Serrano R, Arino J, Casamayor A. 2006. Transcriptional profiling of the protein phosphatase 2C family in yeast provides insights into the unique functional roles of Ptc1. J. Biol. Chem. 281:35057–35069. 10.1074/jbc.M607919200. [DOI] [PubMed] [Google Scholar]
- 64.Wiesenberger G, Steinleitner K, Malli R, Graier WF, Vormann J, Schweyen RJ, Stadler JA. 2007. Mg2+ deprivation elicits rapid Ca2+ uptake and activates Ca2+/calcineurin signaling in Saccharomyces cerevisiae. Eukaryot. Cell 6:592–599. 10.1128/EC.00382-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Barreto L, Canadell D, Valverde-Saubi D, Casamayor A, Arino J. 2012. The short-term response of yeast to potassium starvation. Environ. Microbiol. 14:3026–3042. 10.1111/j.1462-2920.2012.02887.x. [DOI] [PubMed] [Google Scholar]
- 66.Ruiz A, Yenush L, Arino J. 2003. Regulation of ENA1 Na(+)-ATPase gene expression by the Ppz1 protein phosphatase is mediated by the calcineurin pathway. Eukaryot. Cell 2:937–948. 10.1128/EC.2.5.937-948.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kafadar KA, Cyert MS. 2004. Integration of stress responses: modulation of calcineurin signaling in Saccharomyces cerevisiae by protein kinase A. Eukaryot. Cell 3:1147–1153. 10.1128/EC.3.5.1147-1153.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sopko R, Huang D, Preston N, Chua G, Papp B, Kafadar K, Snyder M, Oliver SG, Cyert M, Hughes TR, Boone C, Andrews B. 2006. Mapping pathways and phenotypes by systematic gene overexpression. Mol. Cell 21:319–330. 10.1016/j.molcel.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 69.Wang H, Liang Y, Zhang B, Zheng W, Xing L, Li M. 2011. Alkaline stress triggers an immediate calcium fluctuation in Candida albicans mediated by Rim101p and Crz1p transcription factors. FEMS Yeast Res. 11:430–439. 10.1111/j.1567-1364.2011.00730.x. [DOI] [PubMed] [Google Scholar]
- 70.Yang H, Curinga G, Giachelli CM. 2004. Elevated extracellular calcium levels induce smooth muscle cell matrix mineralization in vitro. Kidney Int. 66:2293–2299. 10.1111/j.1523-1755.2004.66015.x. [DOI] [PubMed] [Google Scholar]
- 71.Denis V, Cyert MS. 2002. Internal Ca(2+) release in yeast is triggered by hypertonic shock and mediated by a TRP channel homologue. J. Cell Biol. 156:29–34. 10.1083/jcb.200111004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Matsumoto TK, Ellsmore AJ, Cessna SG, Low PS, Pardo JM, Bressan RA, Hasegawa PM. 2002. An osmotically induced cytosolic Ca2+ transient activates calcineurin signaling to mediate ion homeostasis and salt tolerance of Saccharomyces cerevisiae. J. Biol. Chem. 277:33075–33080. 10.1074/jbc.M205037200. [DOI] [PubMed] [Google Scholar]
- 73.McCartney RR, Schmidt MC. 2001. Regulation of Snf1 kinase. Activation requires phosphorylation of threonine 210 by an upstream kinase as well as a distinct step mediated by the Snf4 subunit. J. Biol. Chem. 276:36460–36466. 10.1074/jbc.M104418200. [DOI] [PubMed] [Google Scholar]
- 74.Ye T, Elbing K, Hohmann S. 2008. The pathway by which the yeast protein kinase Snf1p controls acquisition of sodium tolerance is different from that mediating glucose regulation. Microbiology 154:2814–2826. 10.1099/mic.0.2008/020149-0. [DOI] [PubMed] [Google Scholar]
- 75.Lutfiyya LL, Iyer VR, DeRisi J, DeVit MJ, Brown PO, Johnston M. 1998. Characterization of three related glucose repressors and genes they regulate in Saccharomyces cerevisiae. Genetics 150:1377–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kaniak A, Xue Z, Macool D, Kim JH, Johnston M. 2004. Regulatory network connecting two glucose signal transduction pathways in Saccharomyces cerevisiae. Eukaryot. Cell 3:221–231. 10.1128/EC.3.1.221-231.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lim MK, Siew WL, Zhao J, Tay YC, Ang E, Lehming N. 2011. Galactose induction of the GAL1 gene requires conditional degradation of the Mig2 repressor. Biochem. J. 435:641–649. 10.1042/BJ20102034. [DOI] [PubMed] [Google Scholar]
- 78.Westholm JO, Nordberg N, Muren E, Ameur A, Komorowski J, Ronne H. 2008. Combinatorial control of gene expression by the three yeast repressors Mig1, Mig2 and Mig3. BMC Genomics 9:601. 10.1186/1471-2164-9-601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Berkey CD, Vyas VK, Carlson M. 2004. Nrg1 and Nrg2 transcriptional repressors are differently regulated in response to carbon source. Eukaryot. Cell 3:311–317. 10.1128/EC.3.2.311-317.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kuchin S, Vyas VK, Carlson M. 2002. Snf1 protein kinase and the repressors Nrg1 and Nrg2 regulate FLO11, haploid invasive growth, and diploid pseudohyphal differentiation. Mol. Cell. Biol. 22:3994–4000. 10.1128/MCB.22.12.3994-4000.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Penalva MA, Tilburn J, Bignell E, Arst HN., Jr 2008. Ambient pH gene regulation in fungi: making connections. Trends Microbiol. 16:291–300. 10.1016/j.tim.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 82.Young ET, Zhang C, Shokat KM, Parua PK, Braun KA. 2012. The AMP-activated protein kinase Snf1 regulates transcription factor binding, RNA polymerase II activity, and mRNA stability of glucose-repressed genes in Saccharomyces cerevisiae. J. Biol. Chem. 287:29021–29034. 10.1074/jbc.M112.380147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rodriguez-Navarro A, Benito B. 2010. Sodium or potassium efflux ATPase a fungal, bryophyte, and protozoal ATPase. Biochim. Biophys. Acta 1798:1841–1853. 10.1016/j.bbamem.2010.07.009. [DOI] [PubMed] [Google Scholar]
- 84.Fraile-Escanciano A, Garciadeblas B, Rodriguez-Navarro A, Benito B. 2009. Role of ENA ATPase in Na(+) efflux at high pH in bryophytes. Plant Mol. Biol. 71:599–608. 10.1007/s11103-009-9543-5. [DOI] [PubMed] [Google Scholar]
- 85.Zhou X, O'Shea EK. 2011. Integrated approaches reveal determinants of genome-wide binding and function of the transcription factor Pho4. Mol. Cell 42:826–836. 10.1016/j.molcel.2011.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gonzalez A, Casado C, Petrezselyova S, Ruiz A, Arino J. 2013. Molecular analysis of a conditional hal3 vhs3 yeast mutant links potassium homeostasis with flocculation and invasiveness. Fungal Genet. Biol. 53:1–9. 10.1016/j.fgb.2013.02.007. [DOI] [PubMed] [Google Scholar]
- 87.Brachmann CB, Davies A, Cost GJ, Caputo E, Li J, Hieter P, Boeke JD. 1998. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14:115–132. . [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.