

Abstract
14-3-3ζ promotes cell survival via dynamic interactions with a vast network of binding partners, many of which are involved in stress regulation. We show here that hypoxia (low glucose and oxygen) triggers a rearrangement of the 14-3-3ζ interactome to favor an interaction with the core autophagy regulator Atg9A. Our data suggest that the localization of mammalian Atg9A to autophagosomes requires phosphorylation on the C terminus of Atg9A at S761, which creates a 14-3-3ζ docking site. Under basal conditions, this phosphorylation is maintained at a low level and is dependent on both ULK1 and AMPK. However, upon induction of hypoxic stress, activated AMPK bypasses the requirement for ULK1 and mediates S761 phosphorylation directly, resulting in an increase in 14-3-3ζ interactions, recruitment of Atg9A to LC3-positive autophagosomes, and enhanced autophagosome production. These data suggest a novel mechanism whereby the level of autophagy induction can be modulated by AMPK/ULK1-mediated phosphorylation of mammalian Atg9A.
INTRODUCTION
The 14-3-3 protein family includes seven human isoforms (β, ε, γ, η, σ, τ, and ζ) that participate in the regulation of diverse processes, including metabolism, cell cycle control, protein trafficking, cell motility, and apoptosis. 14-3-3 proteins function by binding and modulating the activity of other proteins. The biological effect of 14-3-3 binding on the target protein varies from activation, suppression, and sequestration to scaffold-like activity, any of which depends on the protein in question and the cellular context. Nearly all 14-3-3 interactions are mediated by one or two serine/threonine phosphorylations on the interacting protein. Given this dependence on phosphorylation, 14-3-3 binding is inherently dynamic. Changes in the cellular environment (i.e., nutrient availability, adhesion, and cell-cell interactions) alter kinase signaling pathways, which in turn alter 14-3-3 interactions. This dynamic aspect of 14-3-3 biology, together with the fact that 14-3-3 proteins regulate core biological pathways (e.g., metabolism, cell proliferation, and motility), suggests a potential role for 14-3-3 proteins in promoting cellular adaptation to different environmental conditions.
The various biological functions of 14-3-3 proteins are largely dispersed between the different isoforms, and a growing body of data suggests that the ζ isoform plays a central role in modulating cell survival pathways (reviewed in reference 1). 14-3-3ζ (gene symbol YWHAZ) protects cells from numerous stresses, including chemotherapy-induced death, anoikis, and growth factor deprivation (2–6). Furthermore, 14-3-3ζ expression is upregulated in many cancers, and high-level expression has been shown to correlate with poor clinical outcomes in breast cancer (4, 5, 7, 8). Moreover, overexpression of 14-3-3ζ promotes breast tumor invasiveness in both Her2-positive and -negative breast cancer settings (3, 5). For these reasons, 14-3-3ζ has emerged as a potential therapeutic target in cancer (9). 14-3-3ζ, like other 14-3-3 isoforms, interacts with a large network of proteins, some of which are directly involved in survival/apoptosis signaling. These include RAF kinases, Bax, Bad, and caspase-2. Other cancer-associated pathways that depend on 14-3-3ζ for activity include mTOR, Akt, and glucose receptor trafficking.
14-3-3ζ promotes survival signaling in cells exposed to a wide range of conditions, including hypoxia (6, 10–12), in which both oxygen and glucose are limited. Because nutrient deprivation has widespread effects on kinase signaling pathways, we posited that such conditions likely alter the 14-3-3ζ interactome. Thus, using a proteomics approach, we found that treatment of cells with 1% O2 and 2 mM glucose (here referred to as hypoxia) triggered a rearrangement in the 14-3-3ζ interactome. Many of the well-characterized 14-3-3-interacting proteins, including AS160, Bad, and PFK2, showed decreased interactions in hypoxia, while only a small subset of proteins showed hypoxia-induced interactions with 14-3-3ζ. Given the role of macroautophagy (referred to here as autophagy) in promoting cell survival in response to nutrient deprivation (13–15), we became interested in a novel hypoxia-triggered interaction between 14-3-3ζ and Atg9A, a core regulator of autophagy.
Autophagy is an adaptive catabolic process of self-digestion in which cellular components, including organelles and cytoplasmic materials, are assembled in membrane-enclosed structures called autophagosomes and carried to the lysosome for recycling. The maturation and expansion of autophagosomes are orchestrated by a complex network of proteins, many of which carry the Atg designation and are activated in response to nutrient stress. Atg9A is a multimembrane-spanning protein required for induction of autophagy, and previously reported data suggest that it drives autophagic flux by delivering membrane to growing autophagosomes (16, 17).
We found that the interaction between 14-3-3ζ and Atg9A is mediated by phosphorylation at Ser761 within a conserved 14-3-3ζ binding site in the cytoplasmic C-terminal domain of Atg9A. Under nutrient-replete normoxic conditions, this phosphorylation is mediated by Unc51-like kinase 1 (ULK1) and AMP-activated protein kinase (AMPK), which maintain a low level of 14-3-3ζ–Atg9A interactions, but under hypoxic conditions, the full activation of AMPK results in a progressive increase in Ser761 phosphorylation and 14-3-3ζ binding. S761 phosphorylation is also triggered by other autophagy-inducing stresses, including chemotherapy treatment. Importantly, an S761A mutant of Atg9A, which is unable to bind 14-3-3ζ, is defective in autophagy and fails to localize to LC3-positive (LC3+) autophagosomes in nutrient-deprived cells. Taken together, these data highlight the dynamic nature of 14-3-3ζ interactions and demonstrate an adaptive role for 14-3-3ζ in promoting autophagy via regulation of Atg9A in hypoxia. Moreover, these results reveal a novel mechanism linking nutrient stress and AMPK activation to the direct modulation of core autophagy machinery via phosphorylation of Atg9A. We propose that continued characterization and comparison of the 14-3-3 interaction network in response to diverse stimuli will reveal biologically important phosphorylation events and novel pathways through which 14-3-3 proteins modify the cellular response to a wide range of environmental conditions.
MATERIALS AND METHODS
Cell culture and transfection.
HEK-293 cells and U2OS cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) at 37°C in a 5% CO2 incubator. HeLa cells stably expressing green fluorescent autophagy reporter protein GFP-LC3, kindly provided by Beth Levine at the University of Texas Southwestern Medical Center (18), were cultured in DMEM supplemented with 10% FBS at 37°C in a 5% CO2 incubator. For all hypoxia treatments, cells were treated in glucose-free DMEM supplemented with 2 mM glucose and 10% dialyzed FBS (Invitrogen) at 37°C in a 1% O2 incubator. HEK-293 cells, U2OS cells, and HeLa cells were transiently transfected with red fluorescent protein (RFP)-Atg9A, hemagglutinin (HA)-Atg9A, HA–14-3-3ζ, and/or AMPK alpha2 K45R (Addgene plasmid number 15992) plasmids by using FuGENE6 (Promega), TurboFect (Thermo Scientific), or polyethylenimine (PEI) (Polyscience, Inc.) according to the manufacturer's protocols.
Antibodies and chemicals.
The following antibodies were used. Anti-HA (F-7), anti-FLAG (D-8), anti-14-3-3ζ (C-16), anti-actin R (I-19), and antikinesin (H-50) were purchased from Santa Cruz Biotechnology. Antibodies for phospho-acetyl coenzyme A (acetyl-CoA) carboxylase (catalog number 3661), phospho-Thr172 of AMPK α1 (catalog number 2535), LC3B (catalog number 2775S), ULK1 (catalog number 8054S), the phosphoserine 14-3-3ζ motif (“pMotif”) (9601S), Atg9A (catalog number 9730S), and As160 (catalog number 2447S) were purchased from Cell Signaling. Antibodies for AMPK α1 and AMPK α2 were custom made by Affinity BioReagents. Anti-ULK2 (catalog number ab56736) was purchased from Abcam. The following inhibitors were used. Complete protease inhibitors were purchased from Roche and used according to the manufacturer's procedures. SB203580 (catalog number 13067), Dynasore (catalog number 14062), wortmannin (catalog number 10010591), and bafilomycin A1 (catalog number 11038) were purchased from Cayman Chemicals. Compound C (catalog number 171260) and UCN-01 (catalog number 539644) were purchased from Calbiochem. Site-directed mutagenesis kits (catalog number 200521-5) were purchased from Agilent Technologies. The ULK1 enzyme system (catalog number U3521) was purchased from Promega. For in vitro AMPK assays, in vitro kinase assay buffer, recombinant α2/β2/γ2 AMPK (rAMPK) heterotrimers, and liver kinase B1 (LKB1)-STRAD-MO25 (catalog number 14-596) were purchased from Upstate-Millipore. Cell Light Golgi-GFP Bac Mam 2.0 (catalog number C10592) was purchased from Life Technologies. A Lambda phosphatase enzyme kit (catalog number P0753S) was purchased from New England BioLabs, Inc.
Plasmids.
14-3-3ζ-HA was cloned into pCDNA3.1. Atg9A-HA and Atg9A-RFP plasmids were kindly provided by Sharon Tooze, London Research Institute, United Kingdom. The dominant negative pAMPK alpha2 K45R construct was obtained from Addgene (catalog number 15992).
RNA interference.
ULK1 small interfering RNA (siRNA) (catalog number M-005049-00) and PRKAA1 siRNA (catalog number M-005027-02) were purchased from Thermo Scientific (Dharmacon siGENOME SMARTpool). ULK2 siRNA (catalog number 956) and PRKAA2 siRNA (catalog number 103599) were purchased from Ambion by Life Technologies, Inc. siRNA was transfected by using Lipofectamine RNAiMAX (Life Technologies) at a 66 nM or 100 nM final concentration, according to the manufacturer's instructions.
Immunoprecipitation and immunoblotting.
To prepare whole-cell extracts, cells were washed twice and harvested with ice-cold phosphate-buffered saline (PBS). Cell pellets were resuspended in either coimmunoprecipitation (co-IP) buffer (10 mM HEPES [pH 7.5], 150 mM KCl, 0.1% NP-40) or Atg9A lysis buffer (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.3% [wt/vol] Triton X-100, and 5 mM EDTA) supplemented with protease and phosphatase inhibitors and incubated for 15 min on ice or at 4°C with gentle rotation. Lysates were syringed through a 25-gauge needle 10 times and centrifuged at 21,000 rpm for 10 min at 4°C. For coimmunoprecipitation of 14-3-3ζ with Atg9A-HA or Atg9A with 14-3-3ζ–HA, lysates of cells transfected with Atg9A-HA or 14-3-3ζ–HA were incubated with anti-HA–agarose beads for 1 h at 4°C with gentle rotation. The beads were then washed once with lysis buffer and three times with cold PBS. The coimmunoprecipitated proteins were eluted with modified Laemmli sample buffer by boiling at 100°C for 5 min. The proteins were analyzed/quantified, followed by immunoblotting using infrared fluorescent secondary antibodies and a Li-Cor Odyssey imaging system.
In vitro kinase assay.
rAMPK heterotrimers were prepared as described previously (19). Activation of 4 μg rAMPK was accomplished by incubation for 60 min at 30°C with 0.1 μg recombinant LKB1-STRAD-MO25 (catalog number 14-596; Upstate-Millipore) in 25 μl of kinase reaction buffer (40 mM HEPES, 0.2 mM AMP, 80 mM NaCl, 8% glycerol, 0.8 mM EDTA, 0.8 mM dithiothreitol [DTT], 5 mM MgCl2, 0.2 mM ATP [pH 7.0]). The immunoprecipitated Atg9A-HA on anti-HA–agarose beads was incubated with Lambda phosphatase (400 U) for 10 min at 30°C. The beads were then washed three times with ice-cold PBS and incubated with the in vitro kinase reaction buffer (40 mM HEPES, 0.2 mM AMP, 80 mM NaCl, 8% glycerol, 0.8 mM EDTA, 0.8 mM DTT, 5 mM MgCl2, 0.2 mM ATP [pH 7.0]) containing LKB1, activated AMPK with LKB1, or ULK1 for 30 min at 30°C. The proteins were eluted with modified Laemmli sample buffer by boiling at 100°C for 5 min and analyzed/quantified, followed by immunoblotting using infrared fluorescent secondary antibodies and the Li-Cor Odyssey imaging system.
Inhibitor assays.
The following inhibitors and concentrations were used in cell assays: LY294002 at 50 μM; UCN01 at 1 μM, 500 nM, and 300 nM; compound C at 20 μM; SB203580 at 2 μM; and U0126 at 20 μM.
Immunostaining and confocal microscopy.
Cells were seeded into 6-well plates on collagenized glass coverslips and allowed to grow overnight. Following transfection of Atg9A-RFP and/or infection with Cell Light Golgi-GFP Bac Mam 2.0 (catalog number C10592), cells were washed three times with PBS. Cells were then fixed in 4% paraformaldehyde in PBS for 15 min with light agitation at room temperature. Following fixation, cells were washed three times with PBS and then permeabilized and blocked for 30 min with 1% whole goat serum in 0.1% Tween in PBS (0.1% PBS-T). Following permeabilization and blocking, 4′,6-diamidino-2-phenylindole (DAPI) (Molecular Probes) was added for 30 min in 0.1% PBS-T with light agitation at room temperature. Following secondary antibody incubation, cells were washed three times for 10 min with PBS and mounted onto slides with ProLong Gold Antifade reagent (Molecular Probes). Micrographs were taken with an Olympus FluoView FV1000 confocal laser scanning microscope mounted on an Olympus IX81 inverted microscope with an oil-immersed 60× objective. Sequential scans were taken with consistent filter settings for DAPI, GFP, and RFP.
Gel filtration assay.
Cells transfected with HA–14-3-3ζ were treated with normoxia or hypoxia for 4 h before lysis by Dounce homogenization in hypotonic lysis buffer (20 mM HEPES [pH 7.5], 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, and 5 μg/ml each aprotinin and leupeptin). Lysates were centrifuged at 4°C for 15 min at 13,200 rpm, and the protein concentration of the supernatant was determined by using the Bio-Rad protein assay reagent. Five hundred microliters of 8 mg/ml of lysates was loaded onto a Superdex 200 column at a flow rate of 0.3 ml/min to collect fractions.
Mass spectrometry to identify 14-3-3ζ-interacting partners.
HA-tagged 14-3-3ζ (or mock control) coimmunoprecipitates were digested in solution after normalization to equivalent total protein concentrations by using a mini-Bradford assay (Bio-Rad, Inc.). Briefly, samples were supplemented with 0.25% Rapigest SF (Waters Corp.) in 50 mM ammonium bicarbonate (AmBic) and then reduced at 80°C with 20 mM dithiothreitol, followed by alkylation at room temperature with 40 mM iodoacetamide. Finally, digestion with trypsin (sequencing grade, modified; Promega) was performed overnight at 37°C (50:1 substrate/enzyme ratio). Digestion was quenched and Rapigest was degraded by using acidification to 1% (vol/vol) trifluoroacetic acid (TFA) for 2 h at 60°C. Finally, samples were dried and resuspended at 1 mg/ml in 200 mM ammonium formate (pH 10) for analysis by two-dimensional liquid chromatography-tandem mass spectrometry (LC/LC-MS/MS).
Multidimensional LC-MS/MS.
Three microliters of peptide sample digests was analyzed by using a nanoAcquity UPLC system with 2D Technology coupled to a Synapt G2 HDMS mass spectrometer (Waters Corp., Milford, MA), using a method that generated five fractions at pH 10 with approximately equal loading in each fraction, which was similar to methods described previously (20–22). The sample was first trapped at 2 μl/min at a 97/3 (vol/vol) water/methyl cyanide (MeCN) ratio in 20 mM ammonium formate (pH 10) on a 5-μm XBridge BEH130 C18 300-μm by 50-mm column. Two-minute steps at 2 μl/min to the following percentages of MeCN were used to generate the 5 fractions at pH 10, with the composition returning to 97/3/0.1 during the analytical second dimension: 10.8%, 14.0%, 16.7%, 20.4%, and 50.0%. At each fraction step, the flowthrough from the first dimension was diluted online 10-fold with a 99.8/0.1/0.1 (vol/vol/vol) ratio of water/MeCN/formic acid and trapped on a 5-μm Symmetry C18 300-μm by 180-mm trapping column. Separations for each fraction were then performed on a 1.7-μm Acquity BEH130 C18 75-μm by 150-mm column (Waters), using a 37-min gradient of 7 to 35% acetonitrile with 0.1% formic acid at a flow rate of 0.5 μl/min and a 35°C column temperature. We conducted one five-fraction data-independent analysis using ion mobility high-definition mass spectrometry (HDMSE) analysis and one five-fraction traditional data-dependent analysis (DDA). The HDMSE runs used a 0.6-s cycle time, alternating between low collision energy (6-V) and a high-collision-energy ramp in the transfer region (27 to 50 V). The DDA mode utilized a 0.6-s MS scan followed by MS/MS acquisition on the top three ions with charges of >1. MS/MS scans for each ion used an isolation window of approximately 2.3 Da, 0.6-s scans with a maximum of 3 s per precursor, and dynamic exclusion for 120 s within 1.2 Da of the selected precursor m/z.
LC-MS data processing and quantitation.
The data were searched against the UNIPROT database with Homo sapiens taxonomy and reviewed status (http://www.uniprot.org/). The database was appended with several common contaminant or internal standard proteins (ALBU_BOVIN, ADH1_YEAST, ENO1_YEAST, PYGM_RABIT, and CASA1_BOVIN) and a 1× reverse database for peptide false-discovery-rate determination. MS/MS spectra generated by DDA were compiled into .mgf format by using Mascot Distiller v2.2 and submitted to the Mascot v2.2 (Matrix Sciences, Inc.) search engine. For HDMSE and MSE data, ProteinLynx Global Server 2.5 (Waters Corp.) was used to generate searchable files, which were then submitted to the IdentityE search engine (Waters Corp., Milford, MA). The automated merge function within PLGS 2.5 was used to combine spectral information from each of the five fractions prior to searches with IdentityE. The precursor ion mass tolerances were 10 ppm and 5 ppm for Mascot and PLGS searches, respectively, and the product ion tolerances were 0.04 Da for Mascot and 13 ppm for PLGS. For both search engines, the enzyme specificity was set to tryptic, and a maximum of 2 missed cleavages were allowed. Carbamidomethyl cysteine was included as a fixed modification, and variable modifications included oxidized methionine, deamidated asparagine and glutamine, acetylated lysine, and phosphorylated serine and threonine. PLGS searches were performed by requiring at least 3 products per precursor, 7 product ions per protein, and 1 peptide per protein. Qualitative data were curated in Scaffold v4 (Proteome Software, Inc.). Relative quantitation between hypoxia and normoxia conditions was performed based on first analyzer (MS1) extracted-ion chromatograms by using the Skyline v2.5.1 software environment (23). The raw data associated with this analysis are available on the Panorama server (http://goo.gl/aO68ge).
Detection of hypoxia-induced phosphorylation of Atg9A by LC-MS/MS.
HA-Atg9A immunoprecipitates were prepared for mass spectrometry by using a modified version of filter-aided sample preparation (24). Briefly, HA-tagged Atg9A was overexpressed and immunoprecipitated from HEK-293 cells. Next, it was reduced with dithiothreitol (DL; Sigma-Aldrich) and alkylated with iodoacetamide (BioUltra; Sigma-Aldrich). It was then digested on the filter in 50 mM ammonium bicarbonate (ReagentPlus; Sigma-Aldrich) for 5 to 6 h with ∼0.1 μg trypsin (sequencing grade; Promega). Samples were acidified with formic acid (Optima; Fisher Scientific) to ∼1% (vol/vol) prior to mass spectral analysis.
Mass spectrometry was performed by using an LTQ Orbitrap XL mass spectrometer with the following parameters. Peptides were eluted over a 90-min H2O–acetonitrile–0.1% formic acid gradient flowing at 325 μl/min by using the Eksigent NanoLC Ultra instrument with a Waters Peptide Separation Technology C18 column. MS1 scans were analyzed at a resolution setting of 60,000 (at 400 m/z) with the Orbitrap instrument. The top 10 most abundant precursor ions were subject to multistage activation in the LTQ mass spectrometer with neutral loss masses corresponding to a phosphate loss (32.66, 48.99, and 97.97 Da) (25). Dynamic exclusion was used with a 180-s exclusion duration.
Data analysis was performed with Mascot, PhosphoRS 3.0, and Percolator by using the Proteome Discoverer version 1.4 software package (26–28). Precursor mass tolerances were set to 10 ppm, and fragment mass tolerances were set to 0.8 Da. Carbomethylation (C), oxidation (M), and phosphorylation (STY) were set as dynamic modifications. We allowed up to 2 missed trypsin cleavages and searched against the UNI_Human database. A strict false discovery rate was set by using a Percolator q-value threshold of 0.01. Additionally, PhosphoRS 3.0 phosphorylation site localization software assigned S761 a posttranslational modification (PTM) score of 76.6, whereas other potential phosphorylation sites on the same peptide had a PTM score of <12.
RESULTS
Hypoxia triggers a dynamic rearrangement of the 14-3-3ζ interactome.
In addition to its role in promoting chemoresistance (29), 14-3-3ζ promotes cell survival under a wide range of conditions, including those that mimic tumor hypoxia. Cellular adaptation and survival under hypoxic conditions are known to generate chemoresistant and invasive tumors (reviewed in reference 30), yet the regulatory mechanisms underlying cell survival under such conditions are poorly understood. Given that glucose/oxygen deprivation induces vast alterations in kinase pathways, including the activation (e.g., protein kinase C [PKC] and AMPK) and inhibition (e.g., Akt and Erk) of kinases known to mediate 14-3-3ζ interactions, we posited that these changes would alter the 14-3-3ζ interactome, potentially revealing novel phosphorylation-triggered interactions involved in cell survival signaling.
To begin to broadly characterize how 14-3-3ζ interactions change in hypoxia, we performed gel filtration using extracts derived from HEK-293T cells treated with hypoxia (1% O2, 2 mM glucose) or left under normoxic conditions (20 mM glucose, atmospheric oxygen). In normoxia, 14-3-3ζ spanned a wide range of molecular masses, from ∼670 kDa to ∼30 kDa (the approximate size of the 14-3-3ζ monomer), consistent with its binding to numerous protein complexes (31, 32). In contrast, hypoxic conditions induced a marked shift of 14-3-3ζ toward lower-molecular-mass fractions (Fig. 1A), indicating a potential rearrangement or loss of 14-3-3ζ interactions. To identify and characterize specific hypoxia-triggered changes in 14-3-3ζ-interacting partners, we used a coimmunoprecipitation (co-IP)-based proteomics approach. Under normoxic conditions, we identified 133 14-3-3ζ-interacting partners (see Fig. S1 in the supplemental material), many of which have been reported previously, including Bad, PFK2, AS160, IRS4, kinesin, AKAP13, and RAF kinases (reviewed in reference 33). As expected given the gel filtration results, peptide counts suggested that the vast majority of these interactions decreased in hypoxia, and quantitative analysis of a subset of 14-3-3ζ-interacting proteins confirmed the trend toward decreased interactions (Fig. 1B). Figure 1C shows Western blot validation of selected interacting proteins. Based on peptide counts, only a small subset of interacting proteins showed increased peptide counts in hypoxia, one of which was Atg9A (as confirmed by quantitative analysis in Fig. 1B; see Fig. S1 in the supplemental material for a comprehensive list of interactions). In addition, database search results for interacting proteins can be uploaded as a Scaffold 3 (.sf3) file via the following link: https://discovery.genome.duke.edu/express/resources/3211/3211_HDMSE_080212.sf3.
FIG 1.

Hypoxia triggers a rearrangement of the 14-3-3ζ interactome. (A) HA–14-3-3ζ was overexpressed in HEK-293 cells, and cells were treated with normoxia or hypoxia for 4 h. Cell lysates were subjected to gel filtration, and the resulting fractions were immunoblotted for HA. (B) Relative quantitation of the change in 14-3-3ζ interactions for the indicated binding partners in normoxia and hypoxia. MS1 ion chromatograms for the indicated interactors were quantified by using Skyline software, as described in Materials and Methods. (C) HA–14-3-3ζ was overexpressed in HEK-293 cells and treated with normoxia or hypoxia for 6 h, followed by co-IP and immunoblotting for interacting partners. The co-IPs in the bottom panels were performed as described above except with HEK-293 cells cotransfected with Bad and HA–14-3-3ζ constructs.
The interaction between 14-3-3ζ and Atg9A is increased in hypoxia.
Of the novel 14-3-3ζ-interacting partners identified in hypoxia, our attention was drawn to Atg9A due to the role of autophagy in promoting cell survival during nutrient stress (13–15). Furthermore, the regulation autophagy modulates tumorigenesis and promotes chemoresistance (18, 34), phenotypes that are also ascribed to 14-3-3ζ. To validate the proteomics result, tagged 14-3-3ζ or Atg9A was expressed in HEK-293 cells and then subject to co-IP and immunoblotting for their endogenous binding partner. As shown in Fig. 2A and B, these experiments confirmed that the interaction between 14-3-3ζ and Atg9A is increased in hypoxia.
FIG 2.
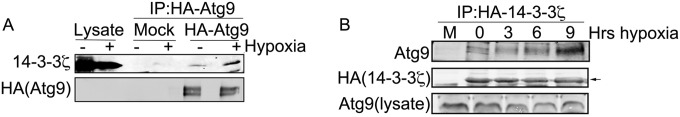
The interaction between 14-3-3ζ and Atg9A is increased in hypoxia. (A) HA-Atg9A was overexpressed in HEK-293 cells, and cells were treated with normoxia or hypoxia for 12 h. HA-Atg9A was immunoprecipitated and immunoblotted for endogenous 14-3-3ζ. (B) HA–14-3-3ζ was overexpressed in HEK-293 cells, and cells were treated with normoxia or hypoxia for the indicated time periods (M, mock). HA–14-3-3ζ was immunoprecipitated and immunoblotted for endogenous Atg9A.
Hypoxia-induced phosphorylation of Atg9A at S761 mediates 14-3-3ζ binding.
Since 14-3-3ζ interactions are typically phosphorylation dependent, we examined phosphorylation of Atg9A over a time course of hypoxia treatment with an antibody that recognizes phosphoserines and phosphothreonines within 14-3-3ζ binding consensus sequences (RXXpS/TXP). Indeed, phosphorylation of Atg9A increased over time in hypoxia, correlating with a progressive increase in 14-3-3ζ binding (Fig. 3A). Analysis of Atg9A sequences revealed a highly conserved 14-3-3ζ binding consensus site at S761 (human Atg9A numbering) (Fig. 3B). Furthermore, bioinformatics analysis of 60 14-3-3 interaction motifs showed that the sequence surrounding S761 conforms to the most prominent amino acids at the critical −3, −2, and +2 amino acid positions within the 14-3-3 binding consensus sequence (Fig. 3C).
FIG 3.

Atg9A bears a conserved canonical 14-3-3ζ binding phosphomotif. (A) HA-Atg9A was overexpressed in HEK-293 cells, and cells were treated with hypoxia for the indicated times. Immunoprecipitated HA-Atg9A was immunoblotted with an anti-14-3-3ζ phosphomotif antibody (pMotif) and for endogenous 14-3-3ζ. (B) Sequence alignment of Atg9A sequences from different species. Highlighted letters indicate the conserved canonical 14-3-3ζ binding motif. (C) Consensus logo for a canonical 14-3-3ζ binding motif generated by aligning 60 verified 14-3-3 binding motifs using WebLogo (http://weblogo.berkeley.edu/).
LC-MS/MS analysis of immunoprecipitated HA-Atg9A from hypoxic cells showed increased phosphorylation at S761 compared to that of Atg9A under normoxic conditions (Fig. 4A), and mutation of S761 to alanine abrogated the hypoxia-triggered interaction between Atg9A and 14-3-3ζ (Fig. 4B). Furthermore, the S761A mutation completely abolished Atg9A phosphorylation in HEK-293 cells and the osteosarcoma cell line U2OS (Fig. 4C and D). In addition, we detected an increase in the phosphosignal on endogenous Atg9A in lysates from cells treated with hypoxia (Fig. 4E). These data suggest that the hypoxia-triggered phosphorylation of Atg9A at S761 is necessary for 14-3-3ζ binding. Moreover, we observed increased S761 phosphorylation in response to other stresses, such as paclitaxel treatment and Earle's balanced salt solution (EBSS) starvation (Fig. 4F), suggesting that phosphorylation of S761 is not limited to hypoxia but rather may be a general response to autophagy-inducing conditions.
FIG 4.

Hypoxia-induced phosphorylation of Atg9A at S761 mediates 14-3-3ζ binding. (A) HA-Atg9A was overexpressed in HEK-293 cells, and cells were treated with normoxia or hypoxia for 12 h. HA-Atg9A was immunoprecipitated and analyzed by LC-MS/MS. The signal intensities of the S761 phosphopeptide were measured by using software described in Materials and Methods. (B) HA-tagged WT Atg9A, an S761A mutant, and an S656A/S761A double mutant (DM) were overexpressed in HEK-293 cells, and cells were treated with normoxia or hypoxia for 12 h. Proteins were subjected to co-IP and immunoblotting for endogenous 14-3-3ζ. (C) HA-tagged WT Atg9A and the S761A mutant were treated as described above for panel B and then immunoblotted with an anti-14-3-3ζ phosphomotif antibody. (D) HA-tagged WT Atg9A and the Atg9A S761A mutant were overexpressed in U2OS cells, and cells were then treated and analyzed as described above for panel C. (E) HEK-293 cells treated with or without hypoxia for 12 h were lysed and immunoblotted with the anti-14-3-3ζ phosphomotif antibody to detect phosphorylation of endogenous Atg9A. (F) HA-tagged WT Atg9A and the S761A mutant were overexpressed in HEK-293 cells and subjected to the indicated treatments for 12 h. Immunoprecipitated proteins were immunoblotted with the anti-14-3-3ζ phosphomotif antibody.
AMPK and ULK1 are differentially required for S761 phosphorylation under basal and stressed conditions.
To identify the candidate kinase(s) responsible for phosphorylation of S761, we started with a panel of inhibitors against kinases activated by metabolic stresses and involved in autophagy regulation. AMPK and the class III member of the phosphatidylinositide-dependent kinase (PI3K III) family are established regulators of autophagy, and AMPK phosphorylations, in particular, are known to mediate 14-3-3 interactions (35, 36). Furthermore, S761 lies within a canonical AMPK phosphorylation motif (LXRXXS/T), with mouse and human Atg9A bearing a leucine and an isoleucine in the −5 position, respectively. As shown in Fig. 5A, the kinase inhibitor UCN01, at concentrations that potently inhibit AMPK (37, 38), abolished hypoxia-induced phosphorylation at S761. In addition, the AMPK inhibitor compound C also suppressed phosphorylation at S761, while LY294002, a PI3K inhibitor, had no effect (Fig. 5A, overlaid image showing phosphorylation in green and loading in red).
FIG 5.

AMPK and ULK1 are differentially required for S761 phosphorylation under basal and stressed conditions. (A) HA-Atg9A was overexpressed in HEK-293 cells and treated with hypoxia for 12 h with the indicated inhibitors (UCN01 [0.3 μM], UCN01 [1 μM], compound C [20 μM], and LY294002 [50 μM]). Immunoprecipitated Atg9A from each treatment was immunoblotted with the phosphorylated 14-3-3 motif (pMotif) antibody. (B) HEK-293 cells expressing HA-Atg9A and transfected with the indicated siRNAs were treated with normoxia or hypoxia for 12 h. HA-Atg9A was immunoprecipitated and immunoblotted for phosphorylated S761. (C) HEK-293 cells expressing HA-Atg9A and transfected with the indicated siRNAs were treated with normoxia. HA-Atg9A was immunoprecipitated and immunoblotted for phosphorylated S761 and the indicated controls. (D) HEK-293 cells expressing HA-Atg9A with or without the AMPK alpha2 K45R mutant (a dominant negative AMPK construct) were treated with hypoxia for 12 h. Immunoprecipitated HA-Atg9A was immunoblotted for endogenous 14-3-3ζ. (E) Cells treated as described above for panel C were subjected to co-IP with HA-agarose resin followed by immunoblotting for phosphorylated S761. (F) HEK-293 cells expressing HA-Atg9A and transfected with siRNAs against the α1 and α2 subunits of AMPK were treated with normoxia or hypoxia for 12 h. HA-Atg9A was immunoprecipitated and immunoblotted for phosphorylated S761. Phosphorylated acetyl-CoA carboxylase (pACC), an AMPK substrate, and AMPK subunits were assessed by Western blotting in lysates to validate AMPK depletion. (G) Resin-bound HA-Atg9A purified from HEK-293 cells was treated with λ phosphatase for 10 min, thoroughly washed, and then incubated with the indicated recombinant enzymes for 30 min. After further washing, resin-bound HA-Atg9A was resolved by SDS-PAGE and immunoblotted with phospho-S761 antibody.
Based on these results, we suspected that AMPK could be the S761-targeted kinase. Previous studies have shown that ULK1 is upstream of Atg9A function (39, 40), and a recent report showed that yeast Atg9A is phosphorylated by ULK1 (41). Of note, however, yeast and mammalian Atg9A show very little sequence homology, and none of the known phosphorylations on yeast Atg9A align, even remotely, with S761. Nevertheless, to test the possibility that ULK1 mediates S761 phosphorylation, we measured phospho-S761 in cells treated with control or ULK1 siRNA. Strikingly, ULK1 depletion consistently resulted in a loss of the basal phospho-S761 signal in normoxic cells but failed to reduce S761 phosphorylation in hypoxic cells (Fig. 5B). Given that ULK2 can substitute for ULK1 in certain cell types, we also measured the phosphorylation of S761 in cells treated with ULK1 siRNA alone or ULK1 and ULK2 siRNA together (Fig. 5C). However, we did not observe any increased phosphorylation with combined siRNA treatment. In contrast to ULK1, expression of a dominant negative AMPK construct (AMPK-DN) abrogated both the hypoxia-induced Atg9A–14-3-3ζ interaction (Fig. 5D) and S761 phosphorylation under normoxic and hypoxic conditions (Fig. 5E). Similarly, depletion of the α1 and α2 AMPK subunits abrogated basal and hypoxia-induced phosphorylation at S761 (Fig. 5F). In vitro assays with purified Atg9A and recombinant AMPK and ULK1 showed that both kinases are capable of phosphorylating S761 (Fig. 5G). Active liver kinase B1 (LKB1), an upstream regulator of AMPK, was used as a control and failed to phosphorylate S761 (Fig. 5G). Our data suggest a model in which phosphorylation of S761 is dependent on ULK1 and AMPK, potentially in complex with each other (42), under basal conditions but is coopted by activated AMPK under conditions of nutrient stress.
Phosphorylation of S761 is required for autophagy induction and Atg9A localization to LC3+ autophagosomes in hypoxia.
To begin to assess the biological relevance of S761 phosphorylation and 14-3-3ζ binding to Atg9A, we measured LC3 lipidation (LC3-II) levels as a marker of autophagy in cells expressing wild-type (WT) Atg9A or the 14-3-3ζ-binding-defective Atg9A S761A mutant with or without bafilomycin A1, an inhibitor of autophagosome-lysosome fusion. While WT Atg9A expression resulted in an increase in the LC3-II/LC3-I ratio in hypoxia, expression of the S761A mutant inhibited conversion of LC3-I to LC3-II. These data led us to compare WT Atg9A and S761A mutant functions more specifically by assessing their subcellular distribution. Atg9A normally associates with the trans-Golgi network (TGN) and early endosomes (EE) and then redistributes to LC3-positive autophagosomal structures and the cell periphery upon induction of autophagy to promote the growth of autophagosomes (16, 40, 43). Consistent with previously reported data (40, 44), hypoxia induced a shift in WT Atg9A from a partially perinuclear/diffuse pattern to a more punctate localization in U2OS cells. However, the Atg9A S761A mutant remained predominately perinuclear/diffuse regardless of the treatment (Fig. 6B), suggesting a fundamental defect in the function of the phosphomutant. Indeed, we failed to observe any notable Atg9A S761A punctate structures in the hypoxia-treated cells (Fig. 6B).
FIG 6.

Phosphorylation of S761 is required for autophagy induction and Atg9A localization to LC3-positive autophagosomes in hypoxia. (A) HEK-293 cells expressing WT Atg9A or the S761A mutant or that were mock transfected were treated with hypoxia for 12 h with or without 100 nM bafilomycin A1. Cells were lysed and immunoblotted for endogenous LC3-I and LC3-II. The intensity of LC3 bands was quantified by Li-Cor infrared imaging in order to determine the LC3-II/LC3-I ratio. The combined results from five separate experiments are shown in the bar graph with means ± standard errors. (B) U2OS cells infected with Golgi-GFP Bac Mam (Life Technologies, Inc.) were transfected with the RFP-tagged Atg9A WT or S761A mutant and treated with hypoxia or normoxia for 12 h. After fixing, cells were analyzed by confocal microscopy. (C) HeLa cells stably expressing GFP-LC3 were transfected with the RFP-tagged Atg9A WT or S761A mutant and treated with hypoxia or normoxia for 12 h. After fixing, cells were analyzed by using confocal microscopy. The panel below the images shows phosphorylation at S761 in hypoxia-treated (12 h) HeLa cells. (D) LC3 punctate structures were counted in RFP-tagged Atg9A WT- or S761A mutant-expressing cells. Means ± standard errors are shown. The statistical significance of the differences between the WT and the S761A mutant was calculated by using two-tailed unpaired Student's t test with equal variance (n = 86 [WT normoxia], n = 76 [S761A mutant normoxia], n = 84 [WT hypoxia], and n = 87 [S761A mutant hypoxia]). *, P value of 1.43E−22. (E) Percentage of GFP-LC3 punctate structures colocalizing with RFP-Atg9A (WT or S761A mutant) determined by manually counting punctate structures in multiple fields (the number of GFP-LC3 and RFP-Atg9A puncta divided by the total number of GFP-LC3 puncta). Means ± standard errors are shown. The statistical significance of the difference between the WT and the S761A mutant was calculated by using two-tailed unpaired Student's t test with equal variance (n = 86 [WT normoxia], n = 76 [S761A mutant normoxia], n = 84 [WT hypoxia], and n = 87 [S761A mutant hypoxia]). *, P value of 3.87E−13.
To more accurately assess whether the failure of the S761A mutant to form punctate structures reflects an overall defect in its autophagic activity, we analyzed Atg9A localization in HeLa cells stably transfected with GFP-LC3 (18). Under normal conditions, LC3 is diffusely expressed in the cytoplasm and becomes punctate only as it incorporates into autophagosomal structures during autophagy. Thus, GFP-LC3 puncta are a marker for mature autophagosomes (45). Consistent with our data from U2OS cells, the S761A mutant and WT Atg9A showed a striking difference in localization, with the S761A mutant adopting a more perinuclear/diffuse pattern in hypoxia. Furthermore, while hypoxia treatment produced an increase in the number LC3-positive autophagosomes in WT Atg9A-expressing cells, the autophagosome numbers were reduced to background levels in hypoxic cells expressing the S761A mutant (Fig. 6D). A more precise and specific measure of Atg9A function is its ability to redistribute to autophagosomes during autophagy. Importantly, the percentage of LC3-positive autophagosomes that colocalized with WT Atg9A increased by nearly 3-fold in hypoxia (Fig. 6E), which is consistent with data from previous studies (40, 43). In contrast, the S761A mutant showed a lack of response to hypoxia, with the percentage of LC3- and Atg9A S761A mutant-copositive structures hovering below the baseline levels for WT Atg9A in normoxia (Fig. 6E). Together, these data suggest that phosphorylation of Atg9A at S761 is required for the association of Atg9A with autophagosomal structures and that, in the absence of phosphorylation and 14-3-3ζ binding, Atg9A fails to properly mobilize in response to autophagic signals.
DISCUSSION
In this study, we set out to identify stress-induced 14-3-3ζ-interacting proteins. Given the dynamic and generally prosurvival nature of 14-3-3ζ binding, we reasoned that this approach would select for biologically important phosphorylation events that help cells adapt to and survive under stress conditions. The data presented here reveal a previously uncharacterized mode of autophagy regulation that links ULK1 and AMPK directly to the core autophagy protein Atg9A. Our data suggest that Atg9A localization to autophagosomes requires phosphorylation on the C-terminal portion of Atg9A at S761, which creates a 14-3-3ζ docking site. Under basal conditions, this phosphorylation is maintained at a low level by ULK1 and AMPK. However, upon induction of hypoxic stress (low glucose and oxygen), activated AMPK bypasses the requirement for ULK1 and mediates S761 phosphorylation, resulting in increased 14-3-3ζ interactions and recruitment of Atg9A to LC3-positive autophagosomes (see the model in Fig. 7). To our knowledge, this is the first reported regulatory mechanism that involves phosphorylation of mammalian Atg9A. Moreover, this study expands our understanding of the role of AMPK in the regulation of core autophagic machinery.
FIG 7.
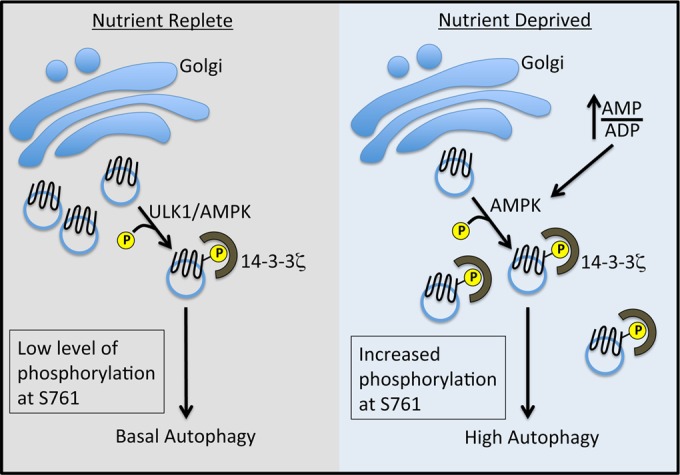
Model of ULK1- and AMPK-mediated regulation of Atg9A via phosphorylation at S761. Under nutrient-replete conditions, a low level of phosphorylation is dependent on ULK1 and AMPK, suggesting a relationship between these kinases under basal conditions (42). We favor the hypothesis that AMPK is directly responsible for Atg9A phosphorylation, with ULK1 playing an ancillary role (see Discussion). We posit that this low level of phosphorylation maintains a basal level of autophagy. Under nutrient deprivation, an increased AMP-to-ADP ratio results in the full activation of AMPK and increased phosphorylation at S761 independent of ULK1, leading to increased autophagy, localization of Atg9A to autophagosomes, and autophagosome biogenesis.
In recent years, our understanding of the role of AMPK in autophagy has rapidly expanded. AMPK was initially linked to autophagy indirectly through inhibition of the mTOR complex (36, 46), and early work on autophagy placed the yeast ortholog of AMPK, Snf1, upstream of Atg1 (the ortholog of ULK1) (47). Recent reports have expanded the picture by showing that AMPK associates with and phosphorylates ULK1 (35, 39, 48, 49). However, the precise nature of the AMPK-ULK1 link is not completely clear, with conflicting reports about whether AMPK-mediated phosphorylation of ULK1 stimulates or suppresses autophagy.
An intriguing observation from our study is that ULK1 and AMPK are required for basal (nutrient-replete) phosphorylation of Atg9A at S761. It should be noted that Kim et al. found previously that ULK1 activity is inhibited by mTOR under nutrient-replete conditions (48), which seems to conflict with our observation that ULK1 knockdown decreases S761 phosphorylation under basal conditions. However, while data from the in vitro ULK1 kinase assay (Fig. 5G) suggest that ULK1 is capable of phosphorylating Atg9A, it is possible that ULK1 is simply part of a complex with AMPK and Atg9A (potentially bridging the interaction with Atg9A) under basal conditions. Indeed, our data do not rule out the possibility the ULK1 plays a ancillary role, independent of its kinase activity, to AMPK in S761 phosphorylation. Consistent with this idea, Shang et al. found that ULK1 and AMPK form a complex that exists only under nutrient-replete conditions and is lost upon starvation (42). Furthermore, the sequence surrounding S761 forms a canonical AMPK motif but does not exactly conform to known ULK1 phosphorylation motifs, which often possess a hydrophobic residue at the −3 position (as opposed to the positively charged arginine near S761) (41). In addition, Mack et al. found that the ULK1 protein is required for Atg9A cycling under nutrient-replete conditions but not under starved conditions, an observation that might be explained by ULK1-mediated control of basal S761 phosphorylation (39). Future work will determine whether ULK1 is acting in parallel or in cooperation with AMPK to mediate S761 phosphorylation.
A recent study by Papinski and colleagues found that Atg1 (ULK1) directly phosphorylates yeast Atg9A at multiple serines within its cytoplasmic C-terminal region (41). Those authors also observed that nonphosphorylatable mutants of Atg9A were unable to localize to autophagosomes. The similarity between their observations and our data suggests that the ULK1-Atg9A link is conserved. However, there is very little sequence conservation between yeast and mammalian Atg9A, and we found no sequence in yeast Atg9A with even remote similarity to the motif surrounding S761. Thus, it is possible that the role of phosphorylation within the C-terminal region of Atg9A is functionally conserved between yeast and mammals but that regulation of mammalian Atg9A phosphorylation has been coopted by AMPK during nutrient stress.
This study also sheds light on the dynamic role of 14-3-3ζ in promoting the adaptation of cells to stress and highlights the potential of 14-3-3 interactomics to identify novel phosphorylation-based regulatory mechanisms involved in the stress response. Based on our observations, in addition to work by other groups (31, 32, 50), it is clear that the 14-3-3ζ interactome is highly context dependent. For example, in contrast to 14-3-3ζ-mediated Atg9 activation in hypoxia, 14-3-3ζ was reported to interact with the class III PI3K human vacuolar sorting protein 34 (hVps34) to suppress autophagy under glucose-fed conditions (51). Conversely, amino acid withdrawal induces an interaction between 14-3-3 and Raptor (which we also detected in our hypoxic proteomics experiments), a component of the mTOR complex, which leads to Raptor sequestration, mTOR inhibition, and activation of autophagy (36). Other studies have highlighted a role for 14-3-3 in the regulation of glucose receptor trafficking in response to insulin (31, 52, 53). Thus, 14-3-3ζ, acting dynamically at multiple levels, promotes a prosurvival cellular program appropriate for the changing physiological conditions of the cell.
In summary, our data show that hypoxic conditions trigger a rearrangement of the 14-3-3ζ interactome. These experiments led to the discovery of a novel mechanism of autophagy regulation via phosphorylation of mammalian Atg9A. While our data indicate that S761 phosphorylation is required for the Atg9A-mediated growth of autophagosomes and Atg9A colocalization with LC3-positive autophagosomal structures, several important questions remain. At which step in the Atg9A trafficking pathway does S761 phosphorylation occur? Does S761 phosphorylation/14-3-3ζ binding orchestrate an interaction between Atg9A and proteins known to mediate Atg9A trafficking or the release of Atg9A from a membrane (e.g., endoplasmic reticulum, Golgi, or endosomal) tether? Moreover, is there an opposing mechanism to dampen AMPK-induced S761 phosphorylation, such as phosphatase activation, to avoid excessive autophagy? Answers to these questions will surely add more pieces to the complex puzzle of autophagy regulation in mammals.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by university start-up funds and an Alexander and Margeret Stewart Trust fellowship to J.L.A.
We thank members of the Andersen laboratory for critical analysis of data and reading of the manuscript. We also thank Sharon Tooze, Jonathan J. Lee, and Daniel Simmons for technical help and key reagents.
Footnotes
Published ahead of print 29 September 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00740-14.
REFERENCES
- 1.Yang X, Cao W, Zhang L, Zhang W, Zhang X, Lin H. 2012. Targeting 14-3-3zeta in cancer therapy. Cancer Gene Ther. 19:153–159. 10.1038/cgt.2011.85. [DOI] [PubMed] [Google Scholar]
- 2.Li Z, Zhao J, Du Y, Park HR, Sun SY, Bernal-Mizrachi L, Aitken A, Khuri FR, Fu H. 2008. Down-regulation of 14-3-3zeta suppresses anchorage-independent growth of lung cancer cells through anoikis activation. Proc. Natl. Acad. Sci. U. S. A. 105:162–167. 10.1073/pnas.0710905105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lu J, Guo H, Treekitkarnmongkol W, Li P, Zhang J, Shi B, Ling C, Zhou X, Chen T, Chiao PJ, Feng X, Seewaldt VL, Muller WJ, Sahin A, Hung MC, Yu D. 2009. 14-3-3zeta cooperates with ErbB2 to promote ductal carcinoma in situ progression to invasive breast cancer by inducing epithelial-mesenchymal transition. Cancer Cell 16:195–207. 10.1016/j.ccr.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maxwell SA, Li Z, Jaye D, Ballard S, Ferrell J, Fu H. 2009. 14-3-3zeta mediates resistance of diffuse large B cell lymphoma to an anthracycline-based chemotherapeutic regimen. J. Biol. Chem. 284:22379–22389. 10.1074/jbc.M109.022418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neal CL, Yao J, Yang W, Zhou X, Nguyen NT, Lu J, Danes CG, Guo H, Lan KH, Ensor J, Hittelman W, Hung MC, Yu D. 2009. 14-3-3zeta overexpression defines high risk for breast cancer recurrence and promotes cancer cell survival. Cancer Res. 69:3425–3432. 10.1158/0008-5472.CAN-08-2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nutt LK, Buchakjian MR, Gan E, Darbandi R, Yoon SY, Wu JQ, Miyamoto YJ, Gibbons JA, Andersen JL, Freel CD, Tang W, He C, Kurokawa M, Wang Y, Margolis SS, Fissore RA, Kornbluth S. 2009. Metabolic control of oocyte apoptosis mediated by 14-3-3zeta-regulated dephosphorylation of caspase-2. Dev. Cell 16:856–866. 10.1016/j.devcel.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li Y, Zou L, Li Q, Haibe-Kains B, Tian R, Li Y, Desmedt C, Sotiriou C, Szallasi Z, Iglehart JD, Richardson AL, Wang ZC. 2010. Amplification of LAPTM4B and YWHAZ contributes to chemotherapy resistance and recurrence of breast cancer. Nat. Med. 16:214–218. 10.1038/nm.2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nishimura Y, Komatsu S, Ichikawa D, Nagata H, Hirajima S, Takeshita H, Kawaguchi T, Arita T, Konishi H, Kashimoto K, Shiozaki A, Fujiwara H, Okamoto K, Tsuda H, Otsuji E. 2013. Overexpression of YWHAZ relates to tumor cell proliferation and malignant outcome of gastric carcinoma. Br. J. Cancer 108:1324–1331. 10.1038/bjc.2013.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matta A, Siu KW, Ralhan R. 2012. 14-3-3 zeta as novel molecular target for cancer therapy. Expert Opin. Ther. Targets 16:515–523. [DOI] [PubMed] [Google Scholar]
- 10.Datta SR, Katsov A, Hu L, Petros A, Fesik SW, Yaffe MB, Greenberg ME. 2000. 14-3-3 proteins and survival kinases cooperate to inactivate BAD by BH3 domain phosphorylation. Mol. Cell 6:41–51. 10.1016/S1097-2765(05)00012-2. [DOI] [PubMed] [Google Scholar]
- 11.DeYoung MP, Horak P, Sofer A, Sgroi D, Ellisen LW. 2008. Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev. 22:239–251. 10.1101/gad.1617608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lynn EG, McLeod CJ, Gordon JP, Bao J, Sack MN. 2008. SIRT2 is a negative regulator of anoxia-reoxygenation tolerance via regulation of 14-3-3 zeta and BAD in H9c2 cells. FEBS Lett. 582:2857–2862. 10.1016/j.febslet.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu YL, DeLay M, Jahangiri A, Molinaro AM, Rose SD, Carbonell WS, Aghi MK. 2012. Hypoxia-induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma. Cancer Res. 72:1773–1783. 10.1158/0008-5472.CAN-11-3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Samokhvalov V, Scott BA, Crowder CM. 2008. Autophagy protects against hypoxic injury in C. elegans. Autophagy 4:1034–1041. 10.4161/auto.6994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wilkinson S, O'Prey J, Fricker M, Ryan KM. 2009. Hypoxia-selective macroautophagy and cell survival signaled by autocrine PDGFR activity. Genes Dev. 23:1283–1288. 10.1101/gad.521709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Puri C, Renna M, Bento CF, Moreau K, Rubinsztein DC. 2013. Diverse autophagosome membrane sources coalesce in recycling endosomes. Cell 154:1285–1299. 10.1016/j.cell.2013.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reggiori F, Tooze SA. 2012. Autophagy regulation through Atg9 traffic. J. Cell Biol. 198:151–153. 10.1083/jcb.201206119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wei Y, Zou Z, Becker N, Anderson M, Sumpter R, Xiao G, Kinch L, Koduru P, Christudass CS, Veltri RW, Grishin NV, Peyton M, Minna J, Bhagat G, Levine B. 2013. EGFR-mediated Beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell 154:1269–1284. 10.1016/j.cell.2013.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taylor EB, Ellingson WJ, Lamb JD, Chesser DG, Compton CL, Winder WW. 2006. Evidence against regulation of AMP-activated protein kinase and LKB1/STRAD/MO25 activity by creatine phosphate. Am. J. Physiol. Endocrinol. Metab. 290:E661–E669. 10.1152/ajpendo.00313.2005. [DOI] [PubMed] [Google Scholar]
- 20.Fan Y, Thompson JW, Dubois LG, Moseley MA, Wernegreen JJ. 2013. Proteomic analysis of an unculturable bacterial endosymbiont (Blochmannia) reveals high abundance of chaperonins and biosynthetic enzymes. J. Proteome Res. 12:704–718. 10.1021/pr3007842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoos MD, Richardson BM, Foster MW, Everhart A, Thompson JW, Moseley MA, Colton CA. 2013. Longitudinal study of differential protein expression in an Alzheimer's mouse model lacking inducible nitric oxide synthase. J. Proteome Res. 12:4462–4477. 10.1021/pr4005103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saka HA, Thompson JW, Chen YS, Kumar Y, Dubois LG, Moseley MA, Valdivia RH. 2011. Quantitative proteomics reveals metabolic and pathogenic properties of Chlamydia trachomatis developmental forms. Mol. Microbiol. 82:1185–1203. 10.1111/j.1365-2958.2011.07877.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, MacCoss MJ. 2010. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26:966–968. 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wisniewski JR, Zougman A, Nagaraj N, Mann M. 2009. Universal sample preparation method for proteome analysis. Nat. Methods 6:359–362. 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- 25.Schroeder MJ, Shabanowitz J, Schwartz JC, Hunt DF, Coon JJ. 2004. A neutral loss activation method for improved phosphopeptide sequence analysis by quadrupole ion trap mass spectrometry. Anal. Chem. 76:3590–3598. 10.1021/ac0497104. [DOI] [PubMed] [Google Scholar]
- 26.Kall L, Canterbury JD, Weston J, Noble WS, MacCoss MJ. 2007. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods 4:923–925. 10.1038/nmeth1113. [DOI] [PubMed] [Google Scholar]
- 27.Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. 1999. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20:3551–3567. 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 28.Taus T, Kocher T, Pichler P, Paschke C, Schmidt A, Henrich C, Mechtler K. 2011. Universal and confident phosphorylation site localization using phosphoRS. J. Proteome Res. 10:5354–5362. 10.1021/pr200611n. [DOI] [PubMed] [Google Scholar]
- 29.Neal CL, Yu D. 2010. 14-3-3zeta as a prognostic marker and therapeutic target for cancer. Expert Opin. Ther. Targets 14:1343–1354. 10.1517/14728222.2010.531011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilson WR, Hay MP. 2011. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 11:393–410. 10.1038/nrc3064. [DOI] [PubMed] [Google Scholar]
- 31.Dubois F, Vandermoere F, Gernez A, Murphy J, Toth R, Chen S, Geraghty KM, Morrice NA, MacKintosh C. 2009. Differential 14-3-3 affinity capture reveals new downstream targets of phosphatidylinositol 3-kinase signaling. Mol. Cell. Proteomics 8:2487–2499. 10.1074/mcp.M800544-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pozuelo Rubio M, Geraghty KM, Wong BH, Wood NT, Campbell DG, Morrice N, Mackintosh C. 2004. 14-3-3-affinity purification of over 200 human phosphoproteins reveals new links to regulation of cellular metabolism, proliferation and trafficking. Biochem. J. 379:395–408. 10.1042/BJ20031797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gardino AK, Yaffe MB. 2011. 14-3-3 proteins as signaling integration points for cell cycle control and apoptosis. Semin. Cell Dev. Biol. 22:688–695. 10.1016/j.semcdb.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Song J, Qu Z, Guo X, Zhao Q, Zhao X, Gao L, Sun K, Shen F, Wu M, Wei L. 2009. Hypoxia-induced autophagy contributes to the chemoresistance of hepatocellular carcinoma cells. Autophagy 5:1131–1144. 10.4161/auto.5.8.9996. [DOI] [PubMed] [Google Scholar]
- 35.Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, Asara JM, Fitzpatrick J, Dillin A, Viollet B, Kundu M, Hansen M, Shaw RJ. 2011. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331:456–461. 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. 2008. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 30:214–226. 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Davies SP, Reddy H, Caivano M, Cohen P. 2000. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 351:95–105. 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Komander D, Kular GS, Bain J, Elliott M, Alessi DR, Van Aalten DM. 2003. Structural basis for UCN-01 (7-hydroxystaurosporine) specificity and PDK1 (3-phosphoinositide-dependent protein kinase-1) inhibition. Biochem. J. 375:255–262. 10.1042/BJ20031119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mack HI, Zheng B, Asara JM, Thomas SM. 2012. AMPK-dependent phosphorylation of ULK1 regulates ATG9 localization. Autophagy 8:1197–1214. 10.4161/auto.20586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Young AR, Chan EY, Hu XW, Kochl R, Crawshaw SG, High S, Hailey DW, Lippincott-Schwartz J, Tooze SA. 2006. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J. Cell Sci. 119:3888–3900. 10.1242/jcs.03172. [DOI] [PubMed] [Google Scholar]
- 41.Papinski D, Schuschnig M, Reiter W, Wilhelm L, Barnes CA, Maiolica A, Hansmann I, Pfaffenwimmer T, Kijanska M, Stoffel I, Lee SS, Brezovich A, Lou JH, Turk BE, Aebersold R, Ammerer G, Peter M, Kraft C. 2014. Early steps in autophagy depend on direct phosphorylation of Atg9 by the Atg1 kinase. Mol. Cell 53:471–483. 10.1016/j.molcel.2013.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shang L, Chen S, Du F, Li S, Zhao L, Wang X. 2011. Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc. Natl. Acad. Sci. U. S. A. 108:4788–4793. 10.1073/pnas.1100844108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Orsi A, Razi M, Dooley HC, Robinson D, Weston AE, Collinson LM, Tooze SA. 2012. Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol. Biol. Cell 23:1860–1873. 10.1091/mbc.E11-09-0746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Webber JL, Tooze SA. 2010. Coordinated regulation of autophagy by p38alpha MAPK through mAtg9 and p38IP. EMBO J. 29:27–40. 10.1038/emboj.2009.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mizushima N, Yoshimori T, Levine B. 2010. Methods in mammalian autophagy research. Cell 140:313–326. 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Inoki K, Zhu T, Guan KL. 2003. TSC2 mediates cellular energy response to control cell growth and survival. Cell 115:577–590. 10.1016/S0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 47.Wang Z, Wilson WA, Fujino MA, Roach PJ. 2001. Antagonistic controls of autophagy and glycogen accumulation by Snf1p, the yeast homolog of AMP-activated protein kinase, and the cyclin-dependent kinase Pho85p. Mol. Cell. Biol. 21:5742–5752. 10.1128/MCB.21.17.5742-5752.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim J, Kundu M, Viollet B, Guan KL. 2011. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13:132–141. 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee JW, Park S, Takahashi Y, Wang HG. 2010. The association of AMPK with ULK1 regulates autophagy. PLoS One 5:e15394. 10.1371/journal.pone.0015394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pozuelo-Rubio M. 2010. Proteomic and biochemical analysis of 14-3-3-binding proteins during C2-ceramide-induced apoptosis. FEBS J. 277:3321–3342. 10.1111/j.1742-4658.2010.07730.x. [DOI] [PubMed] [Google Scholar]
- 51.Pozuelo-Rubio M. 2011. Regulation of autophagic activity by 14-3-3zeta proteins associated with class III phosphatidylinositol-3-kinase. Cell Death Differ. 18:479–492. 10.1038/cdd.2010.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen S, Synowsky S, Tinti M, MacKintosh C. 2011. The capture of phosphoproteins by 14-3-3 proteins mediates actions of insulin. Trends Endocrinol. Metab. 22:429–436. 10.1016/j.tem.2011.07.005. [DOI] [PubMed] [Google Scholar]
- 53.Neukamm SS, Toth R, Morrice N, Campbell DG, Mackintosh C, Lehmann R, Haering HU, Schleicher ED, Weigert C. 2012. Identification of the amino acids 300-600 of IRS-2 as 14-3-3 binding region with the importance of IGF-1/insulin-regulated phosphorylation of Ser-573. PLoS One 7:e43296. 10.1371/journal.pone.0043296. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.