

Abstract
Many plasma membrane transporters are downregulated by ubiquitylation, endocytosis, and delivery to the lysosome in response to various stimuli. We report here that two amino acid transporters of Saccharomyces cerevisiae, the general amino acid permease (Gap1) and the arginine-specific permease (Can1), undergo ubiquitin-dependent downregulation in response to their substrates and that this downregulation is not due to intracellular accumulation of the transported amino acids but to transport catalysis itself. Following an approach based on permease structural modeling, mutagenesis, and kinetic parameter analysis, we obtained evidence that substrate-induced endocytosis requires transition of the permease to a conformational state preceding substrate release into the cell. Furthermore, this transient conformation must be stable enough, and thus sufficiently populated, for the permease to undergo efficient downregulation. Additional observations, including the constitutive downregulation of two active Gap1 mutants altered in cytosolic regions, support the model that the substrate-induced conformational transition inducing endocytosis involves remodeling of cytosolic regions of the permeases, thereby promoting their recognition by arrestin-like adaptors of the Rsp5 ubiquitin ligase. Similar mechanisms might control many other plasma membrane transporters according to the external concentrations of their substrates.
INTRODUCTION
A widespread mechanism for inhibiting the functions of specific plasma membrane receptors, transporters, and channels is selective sorting of these proteins into endocytic vesicles, followed by endosomal trafficking and delivery to the lysosome, where they are degraded. Studies initiated in yeast and extended to mammalian cells have illustrated the key role of ubiquitin (Ub) in this downregulation (1, 2). In the yeast Saccharomyces cerevisiae, ubiquitylation of cell surface proteins is mediated by the HECT family Rsp5 Ub ligase. More recent analysis of several yeast plasma membrane permeases and receptors has shown that arrestin-like adaptors named Art (for arrestin-related trafficking) are essential to recruiting Rsp5 to these proteins (3–5).
Several yeast amino acid permeases (yAAPs) have been shown to undergo ubiquitylation and endocytosis in response to diverse stimuli. For instance, the general amino acid permease Gap1 is stable and active at the plasma membrane in cells grown on a poor nitrogen source (e.g., proline). Under these conditions, Gap1 is not ubiquitylated because the Bul1 and Bul2 arrestin-like adaptors of Rsp5 mediating Gap1 ubiquitylation are phosphorylated by the Npr1 kinase, causing their binding to and inhibition by the 14-3-3 proteins (Fig. 1A). When a preferential nitrogen source, such as ammonium ions, is provided to the cells, the internal amino acid concentration increases, leading to activation of the TORC1 kinase complex. Stimulated TORC1 inhibits Npr1 by phosphorylation (6) and targets the PP2A-type Sit4 phosphatase to the Bul proteins, which are then dephosphorylated (and monoubiquitylated) and dissociate from the 14-3-3 proteins, thus leading to Rsp5-mediated ubiquitylation of Gap1 (7). This control by internal amino acids of the Bul proteins, along with the role of Art4/Rod1 in glucose-induced endocytosis of the Jen1 lactate permease (8) and that of Art1 in cycloheximide-induced endocytosis of the Can1 arginine permease (9), illustrates that ubiquitylation of permeases can be stimulated through direct regulation of their Rsp5 adaptors.
FIG 1.

The Gap1-112 mutant resists downregulation via the TORC1/Npr1 pathway. (A) Model of the regulation of Gap1 ubiquitylation by internal amino acids (aai) via the TORC1/Npr1 pathway (see the text). (B) (Inset) General topology of Gap1. The region colored blue has been enlarged to show the Gap1 residues replaced with alanines in the mutants used in this study. (C) gap1Δ (EK008) and gap1Δ npr1Δ (30794d) cells expressing Gap1-GFP (pJOD010) or Gap1-112–GFP (pMA074) were grown on Gal Pro medium. Glucose was added for 1.5 h before analysis by fluorescence microscopy. w-t, wild type (corresponding to strain EK008); DIC, differential interference contrast. (D) gap1Δ (EK008) and gap1Δ npr1(ts) (MA003) cells expressing Gap1-GFP (pJOD010) or Gap1-112–GFP (pMA074) were grown at 25°C on Gal Pro medium. The cells were transferred to 37°C for 2 h before analysis by fluorescence microscopy. (E) The Gap1-126 mutant is inactive. (Left) gap1Δ (EK008) and gap1Δ ssy1Δ (32501d) strains expressing Gap1-GFP (pJOD010), Gap1-126–GFP (pMA065), or no Gap1 protein (pFL38) (−) were grown for 4 days on solid medium containing citrulline (1 mM), proline (10 mM) plus d-histidine (0.05%), or phenylalanine (1 mM) as the sole nitrogen source. Citrulline and the toxic d-isomer of histidine enter cells only via Gap1. Phenylalanine (1 mM) enters cells via Gap1 and permeases (mainly Agp1) under the positive control of the Ssy1 sensor of amino acids. The results show that wild-type Gap1 complements the phenotypes due to the gap1Δ mutation, whereas the Gap1-126 variant does not. (Right) gap1Δ cells (EK008) were grown on Gal Pro medium, glucose was added for 2 h, and [14C]-citrulline (20 μM) was added to the medium before measurement of the incorporated radioactivity at various times. prot, protein. (F) (Right) Diagram of the experiment. Phe added to cells expressing the inactive Gap1-126 mutant is detected by the Ssy1 sensor. This leads to induction of permeases, such as Agp1, which incorporate Phe into the cells, thus causing endocytosis of Gap1-126. (Left) gap1Δ (EK008), gap1Δ ssy1Δ (32501d), and gap1Δ agp1Δ (30633c) cells expressing Gap1-126–GFP (pMA065) or Gap1-126-112–GFP (pES009) were grown on Gal Pro medium, glucose was added for 1.5 h, and Am (20 mM) or Phe (5 mM) was then added for 2 h or 3 h before analysis by fluorescence microscopy.
Several transporters, including yAAPs, undergo Ub-dependent downregulation under heat stress conditions, and in this case, permease misfolding is the event proposed to promote their recognition by the Rsp5 adaptors as part of a quality control system (10, 11). Permease misfolding has also been illustrated in the case of the Gap1 permease synthesized under conditions inhibiting sphingolipid biogenesis, and this causes its Ub-dependent downregulation (12). Inhibition by rapamycin of TORC1, as well as various stress conditions likely reducing TORC1 activity, also promotes Gap1 downregulation. In this case, the arrestin-like Aly1 and Aly2 proteins contribute, together with Bul1 and Bul2, to Gap1 ubiquitylation. However, these proteins remain phosphorylated and probably bound to the 14-3-3 proteins, suggesting that the mechanism stimulating their role as Rsp5 adaptors when TORC1 is inhibited differs from the one observed upon TORC1 stimulation by internal amino acids (13).
Another condition promoting the endocytosis of many yeast permeases is an excess of substrates. This mode of downregulation, originally illustrated in the case of the inositol permease Itr1 (14), has been described for several metal transporters (Alr1, Zrt1, Smf1 and -2, and Ctr1), the uracil permease Fur4, the phosphate permease Pho84, and the high-affinity hexose transporter Hxt6/7 (15–17). It has also been reported for Gap1 (18, 19) and several other yAAPs, e.g., Can1 (20), Lyp1 (21), Dip5 (22), Tat2 (17), and Mup1 (21). Still, it remains to be determined whether the signal inducing the endocytosis of these yAAPs is the internal accumulation of the transported amino acid (e.g., inhibiting the Npr1 kinase and thereby activating arrestin-like adaptors) or the catalytic process of amino acid transport itself. That transport catalysis might induce the Ub-dependent endocytosis of specific yAAPs is conceivable, as such a mechanism has been described for at least two fungal permeases, the uracil permease Fur4 (23) and the uric acid/xanthine permease UapA of Aspergillus nidulans (24). Recent work suggests that uracil-induced ubiquitylation of Fur4 is due to a conformational change causing disruption of an interaction between the membrane-proximal N-terminal tail and internal loops of the permease, thereby rendering N-terminal lysines more accessible to ubiquitylation by Rsp5 (25).
In this study, we show that the Gap1 and Can1 permeases undergo Ub-dependent endocytosis in response to their substrates and that this endocytosis is due to transport catalysis rather than intracellular substrate accumulation. Further investigation of this endocytosis mechanism suggests that it requires transition of the permeases to a sufficiently populated conformational state preceding substrate release into the cell and likely involving remodeling of permease regions exposed to the cytosol.
MATERIALS AND METHODS
Yeast strains and growth conditions.
All yeast strains used in this study (Table 1) derive from strain Σ1278b. The rsp5(npi1) mutant is a viable rsp5 mutant in which the expression of the RSP5 gene is severely reduced as a result of a Ty element inserted in its upstream control region (26). Cells were grown at 29°C on a minimal buffered medium, pH 6.1 (27), with galactose (Gal) (2%) or glucose (3%) as a carbon source. The nitrogen sources present in the growth media used in most experiments were ammonium (Am) in the form of (NH4)2SO4 (10 mM), proline (10 mM), or urea (10 mM) (in the case of strain MS001, which is unable to use proline). The amino acids used as sole nitrogen sources in growth tests were added at 5 mM final concentration, except for serine and valine (1 mM). Toxic compounds were added to the growth media used in specific experiments: d-histidine (0.05%), thialysine (0.1 mM [see Fig. 5B] and 5 mM [see Fig. 5C]), and canavanine (10 μg/ml).
TABLE 1.
Yeast strains used in this study
| Strain | Genotype | Reference or source |
|---|---|---|
| 23344c | ura3 | Laboratory collection |
| EK008 | gap1Δ ura3 | Laboratory collection |
| 30794b | gap1Δ npr1Δ ura3 | Laboratory collection |
| MA003 | gap1Δ npr1(ts) ura3 | 7 |
| 32501d | gap1Δ ssy1Δ ura3 | 31 |
| FB092 | gap1Δ ssy5Δ ura3 | 44 |
| 30633c | gap1Δ agp1Δ ura3 | 31 |
| 27038a | ura3 rsp5(npi1) | 26 |
| 22Δ8AA | gap1-1 put4-1 uga4-1 can1Δ lyp1Δ alp1Δ hip1Δ dip5Δ ura3 | 45 |
| MS001 | gap1-1 put4-1 uga4-1 can1Δ lyp1Δ alp1Δ hip1Δ dip5Δ ssy1Δ ura3 | 29 |
| JA493 | gap1Δ bul1Δ bul2Δ ura3 | This study |
| MA022 | gap1Δ bul1Δ ura3 | 7 |
| MA024 | gap1Δ bul2Δ ura3 | 7 |
| JA937 | gap1Δ art1Δ ura3 | This study |
| ES006 | gap1Δ can1 ART1-HA ura3 | This study |
| 35237c | gap1Δ can1 ART1-HA rsp5(npi1) ura3 | This study |
| MA032 | gap1Δ BUL2-HA ura3 | 7 |
| 21983c | gap1-1 can1-1 ura3 | 46 |
FIG 5.

Substrate-induced endocytosis of Can1 can occur without transport. (A) Wild-type cells expressing Can1 (pCJ563), Can1(E184A) (pCJ573), or Can1(E184Q) (pCJ559) fused to GFP were grown on Gal Pro medium. Glucose was added to the cultures for 0.5 h before addition of Arg (5 mM) for 2 h. (B) Mutant strain 22Δ8AA, lacking Gap1, Can1, Lyp1, and five other yAAPs and expressing the indicated permeases, was grown for 3 days on solid glucose Am medium with or without thialysine (0.1 mM). (C) Conditions as in Fig. 4F, except that thialysine (5 mM) was added instead of Arg or Lys.
Plasmids used in this study.
The plasmids used in this study are listed in Table 2. In most experiments, the GAP1 gene was expressed under the control of the GAL1 promoter (p416 GAL1-derived plasmids). Cells were grown on galactose proline (Gal Pro) medium, and glucose was added for 1 to 1.5 h before transferring cells under conditions inducing Gap1 endocytosis. Glucose addition causes repression of GAP1 expression so that the secretory pathway is devoid of Gap1 protein. The CAN1 gene was expressed under its natural promoter or the GAL1 promoter.
TABLE 2.
Plasmids used in this study
| Plasmid | Description | Reference or source |
|---|---|---|
| pFL38 | CEN-ARS (URA3) | 47 |
| p416 GAL1 | CEN-ARS-GAL1 (URA3) | 48 |
| pJOD10 | CEN-ARS GAL1-GAP1-GFP (URA3) | 49 |
| pCJ038 | CEN-ARS GAL1-GAP1(K9R-K16R)-GFP (URA3) | 50 |
| pMA074 | CEN-ARS GAL1–GAP1-112–GFP (URA3) | 30 |
| pMA065 | CEN-ARS GAL1–GAP1-126–GFP (URA3) | 30 |
| pMA108 | CEN-ARS GAL1–GAP1-126-112–GFP (URA3) | This study |
| pES009 | CEN-ARS GAL1–GAP1-126-112–GFP (URA3) | This study |
| pKG057 | CEN-ARS GAL1–GAP1-112(K9R-K16R)–GFP (URA3) | This study |
| pMS018 | CEN-ARS GAP1(G107N)-GFP | This study |
| pEL003 | CEN-ARS GAP1-GFP (URA3) | 12 |
| pMS023 | CEN-ARS GAP1(W179L)-GFP (URA3) | This study |
| pKG078 | CEN-ARS GAL1-GAP1(W179L)-GFP (URA3) | This study |
| pKG068 | CEN-ARS GAL1–GAP1-112(W179L)–GFP (URA3) | This study |
| pMA019 | CEN-ARS GAL1–GAP1-124–GFP (URA3) | 30 |
| pNG047 | CEN-ARS GAL1–GAP1-167–GFP (URA3) | 30 |
| pMA142 | CEN-ARS GAL1–GAP1-124(K9R-K16R)–GFP (URA3) | 30 |
| pMA148 | CEN-ARS GAL1–GAP1-167(K9R-K16R)–GFP (URA3) | This study |
| pKG001 | CEN-ARS CAN1 (URA3) | 29 |
| pKG036 | CEN-ARS CAN1-GFP (URA3) | 29 |
| pKG013 | CEN-ARS CAN1(T180R) (URA3) | This study |
| pKG045 | CEN-ARS CAN1(T180R)-GFP (URA3) | This study |
| pKG046 | CEN-ARS CAN1(T456S)-GFP (URA3) | 29 |
| pKG026 | CEN-ARS CAN1(S176N-T456S) (URA3) | 29 |
| pKG066 | CEN-ARS CAN1(S176N-T456S)-GFP (URA3) | 29 |
| pKG035 | CEN-ARS CAN1(E184Q)-GFP (URA3) | 29 |
| pES003 | CEN-ARS CAN1(E184A)-GFP (URA3) | 29 |
| pES002 | CEN-ARS CAN1(E184D) (URA3) | This study |
| pES006 | CEN-ARS CAN1(E184D)-GFP (URA3) | This study |
| pCJ563 | CEN-ARS GAL1-CAN1-GFP (URA3) | This study |
| pCJ573 | CEN-ARS GAL1-CAN1(E184A)-GFP (URA3) | This study |
| pCJ559 | CEN-ARS GAL1-CAN1(E184Q)-GFP (URA3) | This study |
| pKG062 | CEN-ARS LYP1 (URA3) | 29 |
Permease activity assays.
Gap1 and Can1 transport activities, and their apparent Michaelis constant (Km) and maximal velocity (Vmax) values, were determined by measuring the initial uptake rate of 14C-labeled amino acids (purchased from Perkin-Elmer, Boston, MA) as previously described (28, 29).
Fluorescence microscopy.
The subcellular location of Gap1-green fluorescent protein (GFP) and Can1-GFP proteins was determined in cells growing exponentially in liquid medium. In each figure, we typically show only a few cells that are representative of the largest cell subpopulation observed. However, some cell-to-cell variation was also visible, especially in the case of Can1-GFP. This variation was quantified in representative experiments and is discussed in detail in the supplemental material. Labeling of the vacuolar membrane with FM4-64 fluorescent dye was performed as described previously (27). Cells were laid on a thin layer of 1% agarose and viewed at room temperature with a fluorescence microscope (Eclipse E600; Nikon) equipped with a 100× differential interference contrast, numerical aperture (NA) 1.40 Plan-Apochromat objective (Nikon) and appropriate fluorescence light filter sets. Images were captured with a digital camera (DXM1200; Nikon) and ACT-1 acquisition software (Nikon) and processed with Photoshop CS (Adobe Systems).
Protein extracts and Western blotting.
For Western blot analysis, crude cell extracts were prepared as previously described (26). Proteins were transferred to a nitrocellulose membrane (Schleicher and Schuell; catalog number NBA085B) and probed with mouse anti-GFP (Roche; catalog number 11 814 460 001), antihemagglutinin (anti-HA) (12CA5; Roche), anti-Pma1 (29), or anti-yeast 3-phosphoglycerate kinase (anti-PGK) (Invitrogen). Primary antibodies were detected with horseradish peroxidase-conjugated anti-mouse or anti-rabbit immunoglobulin G secondary antibody (GE Healthcare), followed by enhanced chemiluminescence (Roche; catalog number 12 015 196 001).
Construction and validation of Gap1 3D models and docking calculations.
The three-dimensional (3D) structure of Gap1 was modeled following the same protocol that was used to model Can1 (29). The full procedure and data are presented in the supplemental material, together with the methods of docking calculations.
RESULTS
The Gap1-112 mutant is resistant to downregulation stimulated by internal amino acids via the TORC1/Npr1 pathway.
We previously performed a systematic mutational analysis of the intracellular regions of the Gap1 permease, i.e., of its N- and C-terminal tails and of five intracellular loops connecting transmembrane (TM) domains. In each of these Gap1 mutants, three or four successive residues were replaced with alanines (30). Four of them (Gap1-109 to -112), altered in a 16-amino-acid span of the N-terminal tail (Fig. 1B), were found to be resistant to the downregulation triggered when Am is provided to cells growing on proline as the sole nitrogen source (Pro medium). Further analysis of one of these mutants, Gap1-112, revealed that it is not ubiquitylated in response to Am addition (30), suggesting that Gap1 mutants altered in this N-terminal region are insensitive to ubiquitylation and endocytosis via the TORC1/Npr1 pathway (Fig. 1A). To further assess this interpretation, we expressed Gap1-112 in an npr1Δ mutant (Fig. 1C). When the wild-type Gap1 was expressed in this mutant growing on Pro medium, it was targeted to the vacuole, as expected and as confirmed by colabeling with the FM4-64 fluorescent dye. In contrast, the Gap1-112 mutant was found mostly at the cell surface in npr1Δ cells (Fig. 1C). Similarly, shifting of a thermosensitive npr1 mutant to the restrictive temperature caused downregulation of Gap1, whereas Gap1-112 remained stable at the plasma membrane (Fig. 1D). We next examined whether the Gap1-112 mutation might also protect Gap1 against the endocytosis triggered when amino acids enter the cells via other permeases. For this, we set up an experimental system in which an inactive Gap1 variant, Gap1-126, is downregulated after uptake of a single amino acid, phenylalanine (Phe), via permeases such as Agp1, which are induced by the Ssy1 sensor of external amino acids (Fig. 1F) (31). We first confirmed in growth tests and [14C]Phe uptake assays that Gap1-126, altered in an N-terminal region close to the first TM (Fig. 1B), is inactive (Fig. 1E). The Gap1-126 mutant permease was found to be present at the plasma membrane and normally downregulated by Am (Fig. 1F), showing that the TORC1/Npr1 pathway efficiently targets inactive Gap1 forms, as expected. The Gap1-126 mutant was also downregulated after Phe addition, and this downregulation was impaired in the agp1Δ mutant, as well as in the ssy1Δ or ssy5Δ mutant, unable to induce Agp1 and other Phe-transporting permeases (Fig. 1F) (data not shown). Hence, the inactive Gap1-126 was efficiently downregulated after Phe transport mainly via the Agp1 permease. The Gap-126-112 double mutant, however, was found not to be downregulated by Phe (Fig. 1F), even when expressed in wild-type cells expressing the endogenous Gap1 permease, which mediates high-rate Phe uptake (data not shown). Taken together, these data show that the Gap1-112 mutation renders Gap1 insensitive to downregulation stimulated by internal amino acids.
The Gap1-112 permease is ubiquitylated and downregulated in response to substrate transport.
As a control for the above-described experiments, we analyzed the influence of Phe on the Gap1-112 mutant. Remarkably, this active Gap1 mutant, resistant to Am-induced endocytosis, was downregulated by Phe (Fig. 2A). This endocytosis, less efficient than that of wild-type Gap1 (see below), was mostly visible in about 75% of the cells, with a significant cell-to-cell variation with respect to the intracellular distribution of the fluorescence and the intensity of residual signal present at the cell surface (see the supplemental material for details). It also occurred in the ssy1Δ and ssy5Δ mutants, where Phe uptake via Agp1 and other Phe-transporting permeases is defective (Fig. 2B). The fact that this Phe-induced downregulation was not observed in the inactive Gap1-126-112 mutant shows that Gap1-112 is downregulated upon catalyzing Phe uptake. Furthermore, as the Gap1-112 mutation renders Gap1 resistant to endocytosis induced by internal amino acids, including Phe, we deduce that Phe transport-induced endocytosis of Gap1-112 is not stimulated by Phe accumulation in the cells but by the process of Phe transport itself. We repeated the same experiments with other amino acids (e.g., isoleucine and leucine), and the results were similar to those observed with Phe (data not shown). From these data, we conclude that Gap1 undergoes downregulation in response to substrate transport.
FIG 2.

The Gap1-112 mutant is ubiquitylated and downregulated in response to substrate transport. (A) Fluorescence microscopy analysis of gap1Δ cells (EK008) expressing Gap1-GFP (pJOD010) or Gap1-112–GFP (pMA074) under the same conditions as for Fig. 1F. (B) gap1Δ ssy1Δ (32501d) and gap1Δ ssy5Δ (FB092) cells expressing Gap1-GFP (pJOD010) or Gap1-112–GFP (pMA074) were grown on Gal Pro medium. Glucose was added for 1.5 h, and Phe (5 mM) was then added for 1.5 h before analysis by fluorescence microscopy. (C) (Top) gap1Δ cells (EK008) expressing Gap1-112 (pMA074), Gap1-112-K9R-K16R (pKG057), or Gap1-126-112 (pMA108) fused to GFP were grown on Gal Pro medium and treated with Phe (5 mM) for 15 min. Crude cell extracts were prepared and immunoblotted with anti-GFP antibodies. (Middle) Conditions as for panel A, with gap1Δ cells (EK008) expressing Gap1 (pJOD010) or Gap1-112 (pMA074). (Bottom) Conditions as for panel A, except that cells were examined at several time intervals. (D) Fluorescence microscopy analysis of gap1Δ cells (EK008) expressing Gap1-112–GFP (pMA074) or Gap1-112-K9R-K16R–GFP (pKG057). Conditions were as in panel B, except that the cells were examined 3 h after Phe addition. (E) Diagram illustrating Gap1 ubiquitylation and endocytosis by internal and external amino acids. Internal amino acids (AAi) act via the TORC1/Npr1 pathway and derive from Am uptake or from direct uptake of external amino acids (AAe) via other permeases and via Gap1 itself. AAe transport by Gap1 itself also promotes ubiquitylation and endocytosis of the permease.
Downregulation of Gap1-112 upon Phe transport coincided with ubiquitylation of the permease, as shown by the Phe-induced appearance of upper bands above the immunodetected Gap1-112 signal, which were not detected if both Ub acceptor lysines of the permease, K9 and K16, were replaced with arginines (Fig. 2C). Furthermore, this Gap1-112-K9R-K16R mutant was not downregulated by Phe (Fig. 2D). The Gap1-126-112 mutant also failed to be ubiquitylated, even after a long incubation with Phe, confirming that ubiquitylation of Gap1-112 is stimulated upon Phe transport (Fig. 2C).
Taken together, these data indicate that Gap1-112 is ubiquitylated and downregulated upon transport of its amino acid substrates and that this is not due to internal accumulation of the transported amino acid. This is reminiscent of what happens with the uric acid and xanthine permease (UapA) of A. nidulans, which undergoes ubiquitylation and endocytosis in response to Am but also to transport of its substrates (24). Substrate transport-induced endocytosis has also been demonstrated in the case of Fur4, the yeast uracil permease (23, 25). The situation of Gap1 is more complex, since amino acids provided to the cells simultaneously stimulate two downregulation pathways, the TORC1/Npr1 pathway responding to internal amino acids and the pathway elicited by Gap1-mediated transport (Fig. 2E). This might explain why Phe-induced ubiquitylation and downregulation are more pronounced for wild-type Gap1 (sensitive to both pathways) than for Gap1-112 (sensitive only to the substrate transport-induced pathway) and Gap1-126 (sensitive only to the TORC1/Npr1 pathway) (Fig. 2A and C and data not shown).
A single substitution in the substrate-binding site abolishes the activity and substrate-induced endocytosis of Gap1.
We sought to determine whether a Gap1 mutant altered in its substrate-binding site might be resistant to substrate transport-induced downregulation. As the Gap1 structure is unknown, we performed structural modeling on the basis of the X-ray structure of the arginine/agmatine (AdiC) antiporter of Escherichia coli (32, 33). Like Gap1 and most other yAAPs, this bacterial amino acid permease is a member of the APC superfamily of transporters. We have recently shown that the AdiC structure in the outward-facing (OF) occluded arginine-bound state (34) provides a good template for building structural models of Can1, the yeast arginine-specific permease, and for studying its substrate-binding site (29). We thus used a similar approach to build a structural model of the Gap1 permease (also transporting arginine) (Fig. 3A; see the supplemental material).
FIG 3.

A single substitution in the substrate-binding site abolishes the activity and substrate-induced endocytosis of Gap1. (A) Side (left) and top (right) views of the best energy-scored 3D model of Gap1 in the OF occluded state. The protein is depicted as a ribbon diagram, with the five TMs shaping the substrate-binding site highlighted in color (TM1, yellow; TM3, orange; TM6, purple; TM8, blue; and TM10, green). TM11 and 12 are not part of the 5 plus 5 fold. The phenylalanine substrate is shown as spheres colored according to the following scheme: cyan, carbon; red, oxygen; blue, nitrogen. (B) Close view of the binding pocket bound to phenylalanine. The substrate Phe and Gap1 residues within 4 Å of the substrate are shown, respectively, as ball-and-stick and stick models. The residues are colored as in panel A. (C) gap1Δ (EK008) and gap1Δ ssy1Δ (32501d) cells expressing Gap1-GFP (pEL003) or Gap1(G107N)-GFP (pMS018) were grown on glucose Pro medium; then, Phe (5 mM) or Am (20 mM) was added for 2 h before analysis by fluorescence microscopy. (D) Strain MS001, lacking eight amino acid permease genes and the SSY1 amino acid sensor gene, was transformed with a plasmid containing the wild-type GAP1 gene (pEL003), the GAP1(G107N) gene (pMS018), or no GAP1 gene (pFL38) (−). The strains were grown for 4 days on solid medium containing the indicated nitrogenous compounds as sole nitrogen sources. (E) Time course of accumulation of [14C]arginine (top) or [14C]phenylalanine (bottom) (initial concentration, 10 μM) measured in cells of strain MS001 grown on glucose-urea medium and transformed with a plasmid containing the wild-type GAP1 gene (pEL003), the GAP1(G107N) mutant gene (pMS018), or no GAP1 gene (pFL38).
The substrate-binding pocket at the center of the Gap1 structure is shaped mainly by residues of TM1, -3, -6, -8, and -10 (Fig. 3A). We next performed induced-fit docking of Arg and Phe and predicted the residues of Gap1 interacting with them (Table 3). As for Phe, the backbone of the amino acid donates several hydrogen bonds to residues in TM1 and TM6, whereas its side chain is sandwiched between the aromatic residues F294 (TM6) and W179 (TM3) via hydrophobic and π-π interactions, respectively (Fig. 3B and Table 3). The OF occluded structure of Arg-bound AdiC also features such an aromatic sandwich configuration, formed by W202 and W293 (34).
TABLE 3.
Hydrogen bonds and hydrophobic, cation-π, and π-π interactions formed between arginine or phenylalanine and residues of the substrate-binding site of either AdiC or Gap1 in the outward-open-occluded conformationa
| Amino acid substrate location | AdiCb, Arg substrate |
Gap1 3D model |
||||
|---|---|---|---|---|---|---|
| Arg substrate |
Phe substrate |
|||||
| Protein residue backbone | Protein residue side chain | Protein residue backbone | Protein residue side chain | Protein residue backbone | Protein residue side chain | |
| Backbone | ||||||
| NH3+ | HBc Ile23 (TM1) | HB Phe294 (TM6) | HB Phe294 (TM6) | |||
| HB Trp202 (TM6) | HB Ser295 (TM6) | HB Ser295 (TM6) | ||||
| HB Ile205 (TM6) | HB Ala297 (TM6) | HB Ala297 (TM6) | ||||
| COO− | HB Gly27 (TM1) | HB Ser26 (TM1) | HB Gly106 (TM1) | |||
| HB Gly107 (TM1) | HB Gly107 (TM1) | |||||
| Side chain | ||||||
| CH2 | PHOBd Trp202 (TM6) | PHOB Phe294 (TM6) | PHOB Phe294 (TM6) | |||
| Guanidinium | HB Ala96 (TM3) | HB Asn101 (TM3) | HB Gln178 (TM3) | |||
| HB Cys97 (TM3) | HB Ser357 (TM10) | HB Glu301 (TM8) | ||||
| Cation-π Trp293 (TM8) | Cation-π Trp179 (TM3) | |||||
| Aromatic ring | π-π Trp179 (TM3) | |||||
See the text.
Protein Data Bank (PDB) ID, 3L1L.
HB, hydrogen bond.
PHOB, hydrophobic.
The G107 residue is part of the glycine-rich region of TM1 shown in the Gap1 3D model to establish interactions with the backbone of amino acid substrates (Table 3). We replaced this glycine with an asparagine and found that the resulting Gap1(G107N) mutant is normally targeted to the plasma membrane (Fig. 3C) but largely inactive, as judged by the results of amino acid uptake assays and of growth tests on 16 amino acids, each supplied as the sole nitrogen source (Fig. 3D and E). Gap1(G107N) displayed normal downregulation by Am (Fig. 3C), showing that it can be efficiently ubiquitylated and sorted into the endocytic pathway. However, it displayed no downregulation by Phe in ssy1Δ mutant cells, where Phe does not enter via other permeases (Fig. 3C). These results show that the G107N substitution, which directly and severely affects the substrate-binding pocket of Gap1 without altering its overall structure to any observable extent (e.g., primary sorting to the plasma membrane and TORC1-mediated ubiquitylation and subsequent internalization are undisturbed), impairs substrate-induced endocytosis of the permease.
Substrate-induced endocytosis of the Can1 permease.
We next determined whether substrate transport elicits downregulation of other yAAPs. Previous work has shown that the lysine (Lyp1), methionine (Mup1), arginine (Can1), tryptophan (Tat2), and glutamate/aspartate (Dip5) permeases are downregulated when their substrates are provided to cells at high concentration (17, 20–22). It remains to be determined, however, whether this endocytosis is caused by the process of amino acid transport catalyzed by these permeases, by internal accumulation of the transported amino acid, or by a combination of these effects. We addressed this question in the case of Can1, the arginine-specific permease. In cells growing on Pro medium, Can1 was present at the plasma membrane and active (Fig. 4A and data not shown). After addition of Am, causing an increase in internal amino acids and TORC1 stimulation (7), Can1 remained stable at the plasma membrane (Fig. 4A). Furthermore, Can1-GFP was normally present at the plasma membrane of npr1Δ cells (Fig. 4B). It therefore seems that, unlike Gap1, Can1 does not undergo endocytosis upon stimulation of the TORC1/Npr1 pathway under the tested conditions. In contrast, and in keeping with previous observations (20), Can1-GFP was internalized after addition of Arg, since the fluorescence initially present at the cell surface was targeted to the vacuole (Fig. 4A). This redistribution of Can1-GFP from the plasma membrane to the vacuole (as exemplified by cells shown in Fig. 4A) was most visible in about half of the cell population. We also noticed that Arg caused a marked increase in the size of the cell vacuoles. In some cells, this enlargement was so pronounced that it was difficult to clearly distinguish the vacuole from the surrounding cytoplasm (more details on this cell-to-cell variation are available in the supplemental material). The Arg-induced downregulation of Can1-GFP is Ub dependent, since it was found to be defective in the rsp5(npi1) mutant (Fig. 4C), where Rsp5 Ub ligase-promoted ubiquitylation of permeases is defective (2).
FIG 4.

Substrate-induced endocytosis of the Can1 permease. (A) Wild-type cells (23344c) expressing Can1-GFP (pKG036) were grown on glucose Pro medium. Am (20 mM) or Arg (5 mM) was added for 3 h before analysis by fluorescence microscopy. (B) Wild-type (23344c) and gap1Δ npr1Δ (30794d) cells expressing Can1-GFP (pKG036) were grown on glucose Pro medium and examined by fluorescence microscopy. (C) rsp5(npi1) cells (27038a) expressing Can1-GFP (pKG036) were grown on glucose Pro medium. Arg (5 mM) was added to the medium, and the cells were examined after 3 h by fluorescence microscopy. (D) (Top) Two close views of the binding pocket of wild-type Can1 and Can1(T180R) bound to arginine (purple). F295 and W177 are shown as they sandwich the arginine side chain. The residue at position 180 is colored green. (Bottom) Wild-type cells (23344c) expressing Can1-GFP (pKG036) or Can1(T180R)-GFP (pKG013) were grown on glucose Pro medium before addition of Arg (5 mM) for 3 h. (E) (Top) Wild-type (23344c) and gap1-1 can1-1 (21983c) cells expressing the indicated Can1 variants or no Can1 were grown for 2 days on solid medium containing Arg or Am as the sole nitrogen source and on Am medium supplemented with canavanine (10 μg/ml). (Bottom) Time course of [14C]arginine accumulation (initial concentration, 10 μM) measured in 22Δ8AA mutant cells (lacking Gap1, Can1, Lyp1, and five other yAAPs) expressing Can1 (pKG036), Can1(T180R) (pKG013), or no Can1 (pFL38) and grown on glucose Am medium. (F) Wild-type cells (23344c) expressing Can1-GFP (pKG036) or Can1(S176N-T456S)-GFP (pKG066) were grown on glucose Pro medium before addition of Arg or Lys (5 mM) for 3 h.
In the 3D model of Can1 permease we recently built (29), Thr180 of TM3 is one of the residues interacting with the side chain of the arginine substrate, and its replacement with an Arg residue is predicted to markedly reduce the volume of the substrate-binding site (Fig. 4D). Accordingly, Can1(T180R) proved inactive, as judged by the results of growth tests and [14C]arginine uptake assays (Fig. 4E). This inactive Can1(T180R) mutant protein was found to be normally targeted to the plasma membrane but not downregulated by Arg (Fig. 4D). In this experiment, cells expressing the Can1(T180R)-GFP construct from a plasmid-borne gene also expressed the endogenous Can1 and Gap1 permeases. This means that Arg entered the cells at a high rate. This, however, did not induce endocytosis of Can1(T180R), indicating that internal Arg does not elicit Can1 endocytosis. The same was true for other internal amino acids, since Can1(T180R) also resisted endocytosis induced by the addition of Casamino Acids to wild-type cells, conditions under which Can1 was efficiently downregulated (see the supplemental material). It is thus very likely that Arg-induced downregulation of Can1 is due to a substrate transport-induced pathway similar to that described above for Gap1.
We have recently reported that the T456S-S176N double substitution in the substrate-binding pocket is sufficient to convert Can1 to a high-affinity lysine permease unable to transport Arg (29). We thus examined whether this Can1(T456S-S176N) mutant might be downregulated by Lys (Fig. 4F). The wild-type Can1 permease was found not to be internalized in the presence of Lys. In contrast, the Can1(T456S-S176N) mutant proved to be efficiently downregulated by Lys and to have lost the ability to undergo Arg-induced endocytosis. Note that in these experiments, the cells expressed the endogenous Gap1 and Lyp1 permeases. This means that they incorporated Lys at a high rate and hence indicates that internal Lys does not promote Can1 endocytosis. Taken together, these results indicate that Can1 undergoes downregulation in response to substrate transport rather than internal substrate accumulation.
Substrate-induced endocytosis of Can1 can occur without transport.
The fact that Can1 activity is dependent on the plasma membrane H+ gradient (35) suggests that, as proposed for other yAAPs, amino acid transport by Can1 is coupled to proton symport. Our previous work identified E184 of TM3 as a residue of the Can1 substrate-binding pocket, potentially involved in H+ coupling. This residue is highly conserved among yAAPs, and its protonation strongly influences the orientation of the Arg side chain in docking calculations. Furthermore, replacement of E184 with alanine results in total loss of Can1 activity (Fig. 4E) (29). The results presented in Fig. 5A show that this inactive Can1(E184A) mutant fails to be downregulated by Arg. We next examined a Can1 mutant in which an E184Q substitution mimics the irreversibly protonated form of E184. Growth tests and uptake assays had previously shown that this E184Q substitution abolishes Can1 activity (29) (Fig. 4E), but we found the inactive Can1(E184Q) mutant to be efficiently targeted to the vacuole after Arg addition (Fig. 5A). This result shows that Arg translocation across the membrane is not essential to Arg-induced endocytosis of Can1. It suggests, rather, that binding of Arg to the Can1 substrate-binding pocket triggers a conformational change preceding Arg release into the cytosol and promoting endocytosis of the permease. Furthermore, the observation that Can1(E184Q) undergoes normal endocytosis but is inactive supports the view that this structural change precedes deprotonation of E184 and that this deprotonation, likely coupled to protonation of another Can1 residue, is essential to Arg release into the cell.
The fact that an amino acid can induce Can1 endocytosis without being transported was further illustrated by analyzing the influence of thialysine, a highly toxic lysine analog. This analog was found to poison cells expressing Lyp1, the lysine-specific permease (Fig. 5B), but it failed to inhibit the growth of cells expressing Can1(S176N-T456S), the Can1 mutant converted to a lysine-specific permease, indicating that Can1(S176N-T456S) transports lysine but not thialysine. Remarkably, thialysine nevertheless induced endocytosis of Can1(S176N-T456S) but not that of wild-type Can1 (Fig. 5C). Hence, thialysine appears to switch Can1(S176N-T456S) into the conformation inducing its endocytosis without being efficiently transported into the cell.
The Gap1(W179L) and Can1(E184D) mutants displaying reduced Vmax values of transport are largely resistant to substrate-mediated downregulation.
Previous work has focused on the substrate-induced conformational transitions of the bacterial AdiC transporter. Upon Arg binding to AdiC in the OF open conformation, the indole group of W293 establishes a cation-π interaction with the side chain of the substrate (34), and proper orientation of the two groups is crucial to initiating the transition to the next conformation, namely, the occluded state, in which Arg is sandwiched between the indole groups of W293 and W202. The role of W293 in the transition of AdiC to the OF occluded state is illustrated by the observation that the W293Y substitution mostly affects the Arg translocation rate (∼85% reduction of Vmax) and only slightly reduces its apparent binding affinity (36). In the 3D model of Gap1, the position equivalent to W293 of AdiC is also occupied by a tryptophan, W179. However, as observed for Can1 (29), this residue is provided by TM3 instead of TM8 (Table 3). We replaced W179 with different residues and found that the Gap1(W179L) variant displayed a high apparent affinity (Km value, ∼2 μM) but a strongly reduced Vmax of Phe transport (7.8 ± 0.5 versus 82 ± 7.0 nmol · min−1 · mg protein−1 of wild-type Gap1) (Fig. 6A). This strong reduction of the Vmax cannot be explained by a mislocalization or reduced abundance of Gap1(W179L) (Fig. 6B) and thus indicates that the W179L substitution reduces the Phe translocation rate. We introduced this substitution into the Gap1-112 mutant, shown to undergo Phe-induced endocytosis specifically via the substrate-induced pathway. Interestingly, the W179L mutation was found to confer strong protection against Phe-induced endocytosis (Fig. 6C). This shows that a reduction in the ability of the permease to translocate Phe may impair Phe-induced endocytosis, most likely because the conformation eliciting endocytosis is not sufficiently populated. Furthermore, the fact that the corresponding tryptophan in AdiC plays an important role in the transition to the occluded state (36) raises the possibility that shifting of Gap1 to this conformation is a crucial step of its substrate-induced endocytosis.
FIG 6.

The Gap1(W179L) and Can1(E184D) mutants display reduced Vmax values of transport and are largely resistant to substrate-mediated downregulation. (A) Concentration-dependent uptake rates of [14C]Phe in the MS001 strain grown on glucose urea medium and expressing Gap1-GFP (●) (pEL003) or Gap1(W179L)-GFP (▲) (pMS023). The error bars indicate standard deviations. (B) (Top) gap1Δ cells (EK008) expressing Gap1-GFP (pEL003) or Gap1(W179L)-GFP (pMS023) under their natural promoter were grown on glucose Pro medium before analysis by fluorescence microscopy. (Bottom) gap1Δ ssy1Δ cells (32501d) expressing Gap1-GFP (pJOD010) or Gap1(W179L)-GFP (pKG078) under the GAL promoter were grown on Gal Pro medium before addition of glucose for 1.5 h. Crude cell extracts were prepared and immunoblotted with anti-GFP antibodies. The indicated values correspond to relative quantification of the signal intensities. (C) gap1Δ cells (EK008) expressing Gap1-112 (pMA074) or Gap1-112-W179L (pKG068) fused to GFP were grown on Gal Pro medium before addition of glucose for 1.5 h. Phe (5 mM) was then added for 3 h. (D) Concentration-dependent uptake rates of [14C]arginine in 22Δ8AA cells grown on glucose Am medium and expressing Can1(E184D)-GFP (pES003). (E) Wild-type cells (23344c) expressing the GFP-fused Can1(E184D) variant (pES002) were grown on glucose Pro medium before addition of Arg (5 mM) for 3 h.
The view that a reduction of the amino acid translocation rate can impair substrate-elicited endocytosis is further substantiated in the case of Can1. We replaced E184 of Can1 with an aspartate, which should be less readily protonated given its higher pKa value in solution. The Can1(E184D) mutant was found to display a high apparent affinity (Km value, 1 ± 0.3 μM) but a reduced Vmax (2.8 ± 0.2 versus 10.5 ± 0.4 nmol · min−1 · mg protein−1 for wild-type Can1) for Arg transport (Fig. 6D) and proved much less sensitive to Arg-induced downregulation (Fig. 6E).
The Can1(T456S) mutant displaying higher Vmax values for arginine and lysine transport fails to be downregulated in response to these amino acids.
The considerable resistance of Gap1(W179L) and Can1(E184D) to substrate-induced endocytosis suggests that the conformation eliciting endocytosis is only scarcely populated in these low-Vmax mutant forms. In contrast, data obtained with Can1(E184Q) indicate that this inactive variant is able to shift to the endocytosis-signaling state but fails to release Arg into the cell. Interestingly, we found Arg-induced endocytosis to be reproducibly more efficient in this Can1(E184Q) variant than in wild-type Can1 (data not shown). We reasoned that as Can1(E184Q) fails to translocate Arg across the membrane, its endocytosis-eliciting state might be more densely populated than in wild-type Can1, thus accelerating its global downregulation. Conversely, a mutant permease featuring a poorly populated endocytosis-inducing conformation, because transition to the next conformation is somehow facilitated, should display a higher Vmax for transport and be less sensitive or insensitive to substrate-elicited endocytosis. We have recently reported that the Can1(T456S) mutant displays an ∼2-fold-higher Vmax for Arg transport that is not due to higher expression of the permease (29). Structural-modeling analysis has revealed, instead, that the T456S substitution introduces a void between TM3 (exposing S176) and TM10 (exposing T456) that might facilitate structural transitions of the permease during transport (Fig. 7A) (29). Remarkably, this hyperactive Can1(T456S) mutant proved largely resistant to Arg-induced endocytosis (Fig. 7B). Furthermore, Can1(T456S) cannot distinguish Arg from Lys and thus also transports Lys with a high apparent affinity (Km value, 13 ± 1 μM), similar to that of Can1(S176N-T456S), the Can1 mutant converted to a lysine-specific permease (Km value, 13.2 ± 1.8 μM). A difference between these two lysine-transporting Can1 variants, however, is that Can1(T456S) displays a significantly higher Vmax for transport than Can1(T456S-S176N) (13.1 versus 3.6 nmol · min−1 · mg protein−1) that is not due to a higher expression level (29). We thus compared the abilities of the two Can1 mutants to be downregulated by Lys. As shown above, Can1(T456S-S176N) was efficiently endocytosed in the presence of Lys, whereas Can1(T456S) remained largely stable at the plasma membrane after Lys addition (Fig. 7C). These results show that Can1 can catalyze substrate transport without undergoing endocytosis. As this particular phenotype was observed for a Can1 mutant displaying a higher Vmax for transport and predicted to more readily undergo a structural transition, we propose that Can1(T456S) fails to populate the endocytosis-eliciting state densely enough to undergo efficient downregulation.
FIG 7.
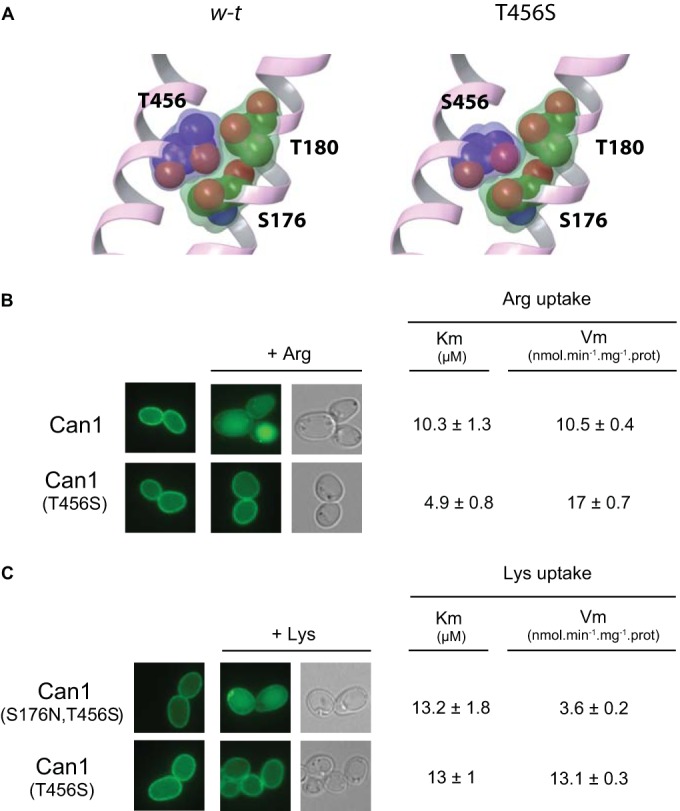
The Can1(T456S) mutant displaying higher Vmax values of arginine and lysine transport is not downregulated by these amino acids. (A) Close view of three residues (456 from TM10 and 176 and 180 from TM3, depicted as balls) of the binding pocket of wild-type Can1 (left) and of Can1(T456S) (right). Replacement of T456 with serine introduces a void between the two TMs that could facilitate the conformational transition required for transport (Table 2). (B) Wild-type cells (23334c) expressing the GFP-fused Can1 (pKG036) or Can1(T456S) (pKG046) were grown on glucose Am medium before addition of Arg (5 mM) for 3 h and subsequent analysis by fluorescence microscopy. Vm, Vmax. (C) Same as panel B, except that cells expressing the GFP-fused Can1(T456S) (pKG046) or Can1(S176N-T456S) (pKG026) were examined after addition of Lys (5 mM) to the medium.
Arrestin-like adaptors of the Rsp5 ubiquitin ligase promote substrate-induced downregulation of Gap1 and Can1.
Ubiquitylation of the Gap1 and Can1 permeases is mediated by the Rsp5 Ub ligase (21, 37). It is now established that Rsp5 requires arrestin-like adaptors to target plasma membrane permeases (2, 3). For instance, ubiquitylation of Gap1 via the TORC1/Npr1 pathway requires at least one of the redundant Bul1 and Bul2 adaptors. To determine whether the Bul proteins are also involved in substrate transport-induced endocytosis of Gap1, we expressed Gap1-112 (sensitive only to this endocytosis pathway) in the bul1Δ bul2Δ mutant and added Phe to the cells. We found Gap1-112 not to be ubiquitylated and to remain at the plasma membrane (Fig. 8A), indicating that the Bul adaptors also play an essential role in substrate-induced endocytosis. Furthermore, Phe-induced downregulation of Gap1-112 was visible in the bul1Δ and bul2Δ single mutants, indicating that Bul1 and Bul2 are largely redundant. We also examined the role of arrestin-like adaptors in Arg-induced Can1 endocytosis. It had been reported previously that cycloheximide-induced endocytosis of Can1 requires the Art1 protein (21). We thus tested Arg-induced endocytosis of Can1 in the art1Δ mutant and found Can1 not to be downregulated (Fig. 8B). Furthermore, Lys-induced endocytosis of the Can1(T456S-S176N) mutant, which transports Lys instead of Arg, was also defective in the art1Δ mutant (Fig. 8B).
FIG 8.

Arrestin-like adaptors of Rsp5 promote substrate-induced downregulation of Gap1 and Can1. (A) (Top) gap1Δ (EK008) and gap1Δ bul1Δ bul2Δ (JA493) cells expressing Gap1-112–GFP (pMA074) were grown on Gal Pro medium before addition of glucose for 0.5 h. Phe (5 mM) was then added for 2 h. (Bottom) The same cells and growth conditions, except that Phe was added for 0.5 h. Crude cell extracts were then prepared and immunoblotted with anti-GFP antibodies. (B) Wild-type (23344c) and gap1Δ art1Δ (JA937) cells expressing GFP-fused Can1 (pKG036) or Can1(S176N-T456S) (pKG066) were grown on glucose Am medium (hence, Gap1 synthesis is repressed in the wild type). Arg or Lys was then added for 3 h before analysis by fluorescence microscopy. (C) (Top) gap1Δ can1 (ES006) and gap1Δ can1 rsp5(npi1) (35237c) cells expressing Art1-HA under the natural promoter of the ART1 gene were grown on glucose Pro medium. Crude cell extracts were then prepared and immunoblotted with anti-HA antibodies. (Bottom) gap1Δ can1 ART1-HA cells (ES006) transformed with a CAN1 plasmid (pKG036) were grown on glucose Pro medium. Arg (5 mM) or Am (20 mM) was added for 20 min, and crude cell extracts were prepared and immunoblotted with anti-HA antibodies. (D) (Top) gap1Δ cells (EK008) expressing Gap1-112–GFP (pMA074) were grown on Gal Pro medium before addition of glucose for 0.5 h (left), Am (20 mM) for 2 h (middle), and Phe (5 mM) for 2 h (right) to half of the culture. (Bottom) Cells expressing Bul2-HA under the natural promoter of the BUL2 gene (MA032) and Gap1-112–GFP (pMA074) were grown on Gal Pro medium and treated as described above for the indicated time intervals. Crude cell extracts were prepared after the indicated times and immunoblotted with anti-HA antibodies.
Recent studies have shown that permease ubiquitylation can be controlled through direct regulation of these adaptors by phosphorylation and/or ubiquitylation and that these modifications lead to detectable changes in migration through a gel (7–9, 21). We thus tested whether substrate transport via Gap1 and Can1 might coincide with detectable changes in the electrophoretic mobility of the Bul and Art1 adaptors, respectively. In keeping with a previous study (21), the Art1-HA signals corresponded to nonubiquitylated and ubiquitylated forms, as judged by the reduced intensity of the upper bands in the rsp5(npi1) mutant (Fig. 8C). Interestingly, the electrophoretic mobility of these Art1-HA proteins was not altered after Arg addition to the cells (Fig. 8C). Analyzing the influence of Phe transport on the Bul proteins was more complex, since providing Phe to cells stimulates Gap1 endocytosis via both the TORC1/Npr1 and the substrate-induced pathways. We thus adopted a protocol taking advantage of the resistance of Gap1-112 to downregulation stimulated by internal amino acids (Fig. 8D). As expected, after addition of Am to cells expressing Gap1-112, the permease remained stable at the plasma membrane, but Bul2-HA displayed the band migration changes typical of its dephosphorylation (conversion of the initial signals to a faster-migrating band) and partial monoubiquitylation (appearance of a slowly migrating band) (Fig. 8D) (7). We then added Phe to the medium. This resulted, as expected, in progressive endocytosis of Gap1-112, followed by its targeting to the vacuole (Fig. 8D). However, this substrate-induced endocytosis of Gap1-112 did not coincide with any novel change in the Bul2-HA migration profile. The only detected difference was a possibly increased stability of the upper form corresponding to monoubiquitylated Bul2-HA (Fig. 8D).
In conclusion, conditions stimulating substrate-induced endocytosis of Gap1 or Can1 did not coincide with any detectable change in the posttranslational modification state of the corresponding adaptor(s) (Bul or Art1, respectively). This contrasts with the influence of internal amino acids on the Bul proteins. Although these proteins might undergo subtle modifications not visible in our experiments or be activated by some other trans-acting mechanism, a simpler model accounts for the above-described data: that the predicted conformational changes undergone by the permeases upon substrate binding are sufficient for adaptor recruitment and Rsp5-mediated ubiquitylation.
Two Gap1 mutants undergo constitutive endocytosis via a mechanism involving the Bul arrestin-like adaptors.
If substrate-induced endocytosis of Gap1 and Can1 is elicited by a shift to a particular conformation recognized by arrestin-like adaptors of the Rsp5 Ub ligase, certain mutant forms of these permeases might adopt a structure mimicking this conformation. Such mutants should undergo Bul-dependent ubiquitylation and downregulation even if their substrates are not present in the medium. In regard to this hypothesis, two other Gap1 mutants obtained by systematic mutagenesis of cytosolic regions, Gap1-124 and Gap1-167, are particularly interesting, since they reach the plasma membrane properly but undergo constitutive endocytosis and targeting to the vacuole, whereas the K9R-K16R double substitution stabilizes them at the cell surface (30) (Fig. 9A). These Gap1 mutants, mutated in the N-terminal tail and second internal loop, respectively (Fig. 1B), might thus undergo constitutive endocytosis via the substrate-induced pathway. Alternatively, they might misfold and be recognized by a plasma membrane-associated quality control system promoting their downregulation. The latter scenario seems less likely for three reasons. First, analysis of both proteins in the end3Δ mutant defective for endocytosis (30) and of the corresponding variants with K9R-K16R substitutions (Fig. 9A) shows that they travel normally to and accumulate at the cell surface, i.e., without any apparent stacking in the endoplasmic reticulum or direct sorting from the Golgi apparatus to the vacuole, phenotypes frequently observed when permeases are misfolded. Second, although some of the stresses recently reported to cause misfolding and endocytosis of multiple yeast permeases (10) also cause Gap1 downregulation, the latter still occurred in the bul1Δ bul2Δ mutant, as it also involves the Aly1 and Aly2 arrestin-like proteins (13); in contrast, the Gap1-124 and Gap1-167 permease variants were found to stabilize at the plasma membrane in the bul1Δ bul2Δ mutant (Fig. 9A), as observed for Gap1-112 after Phe transport, thus indicating that the Bul proteins promote the constitutive endocytosis of the two mutant permeases. Third, Gap1-124 and Gap1-167 stabilized at the cell surface were found to catalyze amino acid uptake, although they showed lower activity than wild-type Gap1 (Fig. 9B), mainly as a result of a reduced Vmax of transport not caused by mislocalization (Fig. 9A) or by lower expression of the mutant permeases (Fig. 9C). The amino acid regions altered in these two Gap1 mutants thus also seem important for maximal Gap1 transport activity. The properties of Gap1-124 and Gap1-167 thus suggest a model in which the N terminus and second internal loop of Gap1 contribute to the structural transitions normally associated with transport catalysis, and these structural changes could also favor recognition of the permeases by the Bul adaptors, thus leading to permease ubiquitylation and endocytosis.
FIG 9.

Two Gap1 mutants undergo constitutive Bul-dependent endocytosis. (A) gap1Δ (EK008) and gap1Δ bul1Δ bul2Δ (JA479) cells expressing the GFP-fused Gap1 (pJOD010), Gap1-124 (pMA019), Gap1-167 (pNG047), Gap1-124-(K9R-K16R) (pMA142), or Gap1-167-(K9R-K16R) (pMA148) were grown on Gal Pro medium, glucose was added for 1.5 h, and the cells were examined by fluorescence microscopy. (B) Concentration-dependent rates of [14C]Phe uptake in gap1Δ ssy1Δ cells (32501d) expressing Gap1(K9R-K16R)–GFP (pCJ038) (●), Gap1-124-(K9R-K16R)–GFP (pMA142) (○), or Gap1-167-(K9R-K16R)–GFP (pMA148) (▽). (C) The reduced Vmax values for Gap1-124 and Gap1-167 stabilized at the plasma membrane are not due to reduced amounts of the permeases. gap1Δ ssy1Δ cells (32501d) expressing GFP-fused Gap1(K9R-K16R), Gap1-124-(K9R-K16R), or Gap1-167-(K9R-K16R) (plasmids as in panel B) and gap1Δ bul1Δ bul2Δ (JA493) expressing GFP-fused Gap1, Gap1-124, or Gap1-167 (plasmids as in panel A) were grown on Gal Pro medium, and glucose was added for 2 h. Crude cell extracts were prepared and immunoblotted with anti-GFP antibodies.
DISCUSSION
The main conclusion of this study is that two yAAPs, Gap1 and Can1, undergo Ub-dependent endocytosis in response to external substrates. The simplest model accounting for the data is that this endocytosis is elicited by a conformational change induced by binding of the amino acid substrates to the substrate-binding pocket (Fig. 10). Accordingly, the Can1(S176N-T456S) variant converted to a lysine-specific permease is downregulated by lysine instead of arginine, and substitutions in the substrate-binding pocket of Gap1 and Can1 impair the substrate-induced endocytosis of these permeases. Substrate-induced endocytosis of Gap1 involves the redundant Bul1 and Bul2 proteins, and that of Can1 requires Art1. These Rsp5 adaptors, however, did not undergo any apparent modification upon stimulation of substrate-mediated endocytosis. This apparent lack of modification was also observed when the Bul proteins promote constitutive endocytosis of the Gap1-124 and Gap1-167 mutant proteins (see the supplemental material). The mechanisms promoting substrate-induced endocytosis of Gap1 and Can1, therefore, differ from that which stimulates Gap1 endocytosis in response to internal amino acids, which involves TORC1-dependent dephosphorylation of the Bul adaptors coupled to their partial monoubiquitylation (Fig. 1A) (7). Similarly, cycloheximide-induced Can1 endocytosis involves changes in the phosphorylation and ubiquitylation states of Art1 (9, 21). Our data, rather, support a model where conformational changes undergone by Gap1 and Can1 upon substrate binding would favor a particular structural state recognized by the corresponding arrestin-type adaptor(s) (Bul or Art1, respectively), which in turn would promote Rsp5-dependent ubiquitylation of the permeases. The N-terminal region of Gap1, altered by the -112 substitutions, is essential to downregulation via the TORC1/Npr1 pathway but not for substrate-induced endocytosis. Hence, this N-terminal region displays the properties expected of a permanently accessible binding site for the dephosphorylated Bul proteins. In response to external substrates, the Bul adaptors likely act via one or several other cytosolic regions of the permease, accessible only after the conformational shift. Although the above-described model is compatible with the experimental data, more complex scenarios remain possible. For instance, the conformational transition of permeases induced by substrate binding could stimulate a signaling pathway that activates the Bul and Art1 adaptors. The Gap1 permease is reported to activate protein kinase A (PKA), a property not shared by Can1 (38, 39), and this kinase might thus catalyze subtle phosphorylation of the Bul proteins not visible in our experiments. Alternatively, PKA could modulate yet another protein modulating the function of the Bul proteins. The last two models, however, seem less likely, because the Bul adaptors target other permeases (17), which would thus potentially be downregulated along with Gap1 in response to Gap1-mediated transport.
FIG 10.

Hypothetical model of substrate-induced ubiquitylation and endocytosis of yeast amino acid permeases. (Top) Upon amino acid binding (a and b), the permease shifts to the outward-open-occluded (c) and then the inward-open-occluded (d) state. An inward open conformation preceding substrate release into the cytosol is prone to ubiquitylation (e and f). Substitutions in Can1 or Gap1 impeding shifts to different states (arrows) of the transport cycle are indicated. In the case of Can1(T456S), the state favoring ubiquitylation is not stable enough to promote efficient endocytosis (d to g direct shift). In the cases of Can1(E184Q) in the presence of Arg and Can1(S176N-T456S) and in the presence of thialysine, the permeases shift to the state favoring ubiquitylation without releasing the substrate into the cell. The conformational change favoring ubiquitylation involves the N-terminal tail, possibly interacting with an internal loop of the permease. The permease in the state labeled e or f can shift to the states leading to substrate release (g and a). (Bottom) Substitutions in cytosol-facing regions (e.g., Gap1-124 and -167) can mimic the conformation prone to ubiquitylation.
The finding that the Can1(E184Q) variant is inactive but remains competent for substrate-induced endocytosis and the observation that thialysine promotes endocytosis of Can1(T456S-S176N) without being transported indicate that the conformation inducing endocytosis precedes substrate release into the cell. However, the exact nature of this endocytosis-inducing state remains unknown. The yAAPs belong to the “5+5” structural superfamily of transporters including well-studied proteins, such as the bacterial LeuT and AdiC proteins and the dopamine transporter. Proteins of the 5+5 superfamily have been crystallized in different conformations, including the outward-open, outward-occluded, and inward-open states (40). As transition of AdiC from the outward-open to the outward-occluded state does not seem to significantly reorganize its cytosolic regions, we propose that the endocytosis-inducing conformation of yAAPs more likely corresponds to an inward state, for instance, the inward-occluded state preceding substrate release into the cytosol (Fig. 10). A shift to this conformation would thus alter the structure of cytosolic regions of yAAPs, thereby promoting their interaction with the arrestin-like adaptors. In particular, the N-terminal tail and second intracellular loop could contribute to these structural changes, as substitutions in these regions (as in the Gap1-124 and -167 mutant proteins) result in constitutive endocytosis of Gap1 via mechanisms resembling that induced by substrate binding. Interestingly, structural analysis of the dopamine transporter and LeuT has shown that membrane-proximal residues of the N-terminal tail interact with intracellular loops when the proteins are in the outward-open conformation and that these interactions are disrupted upon shifting to the inward-open conformation. In particular, TM1 undergoes a large movement that could induce a significant conformational change in the directly connected N-terminal tail (41, 42). Furthermore, a recent study of uracil permease (Fur4) supports a model in which the N-terminal tail interacts with internal loops. This interaction is also proposed to be disrupted as a result of uracil transport, thereby exposing distal lysines in the N-terminal tail to ubiquitylation (25). Hence, structural remodeling of the N-terminal tail interacting with intracellular loops could be the key conformational feature inducing substrate-elicited ubiquitylation of yAAPs, and other transporters as well. In the cases of Gap1 and Can1, this interaction could more particularly involve the membrane-proximal residues of the N-terminal tail, as this region is remarkably well conserved among yAAPs.
According to our proposed model, the permease conformation eliciting endocytosis is transient but must be stable enough to be sufficiently populated and to allow efficient recognition of the substrate-bound permeases by the arrestin-like adaptors. At the scale of individual permeases, this means that the transporter must reside long enough in this conformation to increase its probability of establishing contacts with these adaptors. In this context, the Can1(T456S) variant is of particular interest, as it displays a higher intrinsic Vmax transport value but is largely resistant to substrate-induced endocytosis. We propose that for this Can1 variant, the probability of occupancy of the endocytosis-eliciting conformation is reduced because transition to the next state, leading to substrate release, is facilitated, for instance, because the energy barrier to this substrate-releasing conformation is lower. This scenario is supported by our observation that T456S introduces a void between TM3 and TM10, which might facilitate a structural transition (29). The selection during evolution of a Thr residue at position 456 in Can1, while reducing the transport velocity of the permease (an effect that could be readily compensated for by increasing its expression level), might thus confer the advantage of stabilizing a transient conformation of the transport cycle recognized by cytosolic factors promoting its downregulation (Fig. 10).
The properties of Can1(E184Q), the inactive Can1 mutant that still responds to external Arg, are reminiscent of those of the Ssy1 sensor of external amino acids. This protein is similar to yAAPs but lacks transport activity. It can nevertheless detect external amino acids and initiate a signaling response (43). Remarkably, the glutamate residue corresponding to E184 in Can1 and conserved in most yAAPs corresponds to a glutamine in Ssy1. It is therefore tempting to propose that Ssy1 might originate from a yAAP having lost its activity as a result of an E-to-Q substitution at this position. Like Can1(E184Q), this mutant would have kept the ability to undergo a conformational change in response to amino acid binding, a property that was exploited during evolution to convert this inactive yAAP to a sensor able to initiate a cellular response to external amino acids.
Our results also raise the question of why yAAPs undergo endocytosis in response to their external substrates. The high-affinity Gap1 permease is best adapted for high-affinity uptake of many different amino acids under poor nitrogen supply conditions, and its downregulation induced by excess amino acids coincides with Ssy1-dependent induction of other, lower-affinity yAAPs. Downregulation of Gap1 by external (and internal) amino acids is thus part of a mechanism allowing cells to adjust their yAAP repertoires according to the external concentration of amino acids. Downregulation of Gap1 when amino acids are abundant could also avoid detrimental hyperactivation of PKA in cells growing on a poor nitrogen source, as Gap1 is reported to activate this kinase (38, 39). Regarding Can1, we have observed that its downregulation by Arg is not as complete/efficient as that of Gap1. Partial endocytosis of Can1 in response to Arg might thus adjust the amount of this permease at the plasma membrane so as to avoid a metabolic imbalance due to excess Arg uptake. For similar reasons, many other yAAPs, and also other transporters in yeast and more complex organisms, might undergo a similar substrate-induced endocytosis.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Catherine Jauniaux for her contribution to isolating strains and plasmids. We also thank members of the laboratory for fruitful discussions and Lydia Spedale for her technical assistance.
M.P. is senior research associate and E.-M.K. is a postdoctoral researcher of the FNRS. This work was supported by an FRSM grant (3.4.592.08.F) and an ARC grant (AUWB 2010-15-2) from the Fédération Wallonie-Bruxelles.
Footnotes
Published ahead of print 29 September 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00699-14.
REFERENCES
- 1.Mukhopadhyay D, Riezman H. 2007. Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science 315:201–205. 10.1126/science.1127085. [DOI] [PubMed] [Google Scholar]
- 2.Lauwers E, Erpapazoglou Z, Haguenauer-Tsapis R, André B. 2010. The ubiquitin code of yeast permease trafficking. Trends Cell Biol. 20:196–204. 10.1016/j.tcb.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 3.Becuwe M, Herrador A, Haguenauer-Tsapis R, Vincent O, Léon S. 2012. Ubiquitin-mediated regulation of endocytosis by proteins of the arrestin family. Biochem. Res. Int. 2012:242764. 10.1155/2012/242764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.MacGurn JA, Hsu P-C, Emr SD. 2012. Ubiquitin and membrane protein turnover: from cradle to grave. Annu. Rev. Biochem. 81:231–259. 10.1146/annurev-biochem-060210-093619. [DOI] [PubMed] [Google Scholar]
- 5.Alvaro CG, O'Donnell AF, Prosser DC, Augustine AA, Goldman A, Brodsky JL, Cyert MS, Wendland B, Thorner J. 2014. Specific α-arrestins negatively regulate Saccharomyces cerevisiae pheromone response by down-modulating the G-protein coupled receptor Ste2. Mol. Cell. Biol. 34:2660–2681. 10.1128/MCB.00230-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schmidt A, Beck T, Koller A, Kunz J, Hall MN. 1998. The TOR nutrient signalling pathway phosphorylates NPR1 and inhibits turnover of the tryptophan permease. EMBO J. 17:6924–6931. 10.1093/emboj/17.23.6924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Merhi A, André B. 2012. Internal amino acids promote Gap1 permease ubiquitylation via TORC1/Npr1/14-3-3-dependent control of the Bul arrestin-like adaptors. Mol. Cell. Biol. 32:4510–4522. 10.1128/MCB.00463-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Becuwe M, Vieira N, Lara D, Gomes-Rezende J, Soares-Cunha C, Casal M, Haguenauer-Tsapis R, Vincent O, Paiva S, Léon S. 2012. A molecular switch on an arrestin-like protein relays glucose signaling to transporter endocytosis. J. Cell Biol. 196:247–259. 10.1083/jcb.201109113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.MacGurn JA, Hsu P-C, Smolka MB, Emr SD. 2011. TORC1 regulates endocytosis via Npr1-mediated phosphoinhibition of a ubiquitin ligase adaptor. Cell 147:1104–1117. 10.1016/j.cell.2011.09.054. [DOI] [PubMed] [Google Scholar]
- 10.Zhao Y, MacGurn JA, Liu M, Emr S. 2013. The ART-Rsp5 ubiquitin ligase network comprises a plasma membrane quality control system that protects yeast cells from proteotoxic stress. Elife 2:e00459. 10.7554/eLife.00459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Babst M. 2014. Quality control: quality control at the plasma membrane: one mechanism does not fit all. J. Cell Biol. 205:11–20. 10.1083/jcb.201310113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lauwers E, Grossmann G, André B. 2007. Evidence for coupled biogenesis of yeast Gap1 permease and sphingolipids: essential role in transport activity and normal control by ubiquitination. Mol. Biol. Cell 18:3068–3080. 10.1091/mbc.E07-03-0196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crapeau M, Merhi A, André B. 2014. Stress conditions promote yeast Gap1 permease ubiquitylation and down-regulation via the arrestin-like Bul and Aly proteins. J. Biol. Chem. 289:22103–22116. 10.1074/jbc.M114.582320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lai K, Bolognese CP, Swift S, McGraw P. 1995. Regulation of inositol transport in Saccharomyces cerevisiae involves inositol-induced changes in permease stability and endocytic degradation in the vacuole. J. Biol. Chem. 270:2525–2534. 10.1074/jbc.270.6.2525. [DOI] [PubMed] [Google Scholar]
- 15.Haguenauer-Tsapis R, André B. 2004. Membrane trafficking of yeast transporters: mechanisms and physiological control of downregulation, p 273–323 In Topics in current genetics, vol 9 Springer, Berlin, Germany. [Google Scholar]
- 16.Krampe S, Stamm O, Hollenberg CP, Boles E. 1998. Catabolite inactivation of the high-affinity hexose transporters Hxt6 and Hxt7 of Saccharomyces cerevisiae occurs in the vacuole after internalization by endocytosis. FEBS Lett. 441:343–347. 10.1016/S0014-5793(98)01583-X. [DOI] [PubMed] [Google Scholar]
- 17.Nikko E, Pelham HRB. 2009. Arrestin-mediated endocytosis of yeast plasma membrane transporters. Traffic 10:1856–1867. 10.1111/j.1600-0854.2009.00990.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stanbrough M, Magasanik B. 1995. Transcriptional and posttranslational regulation of the general amino acid permease of Saccharomyces cerevisiae. J. Bacteriol. 177:94–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rubio-Texeira M, Van Zeebroeck G, Thevelein JM. 2012. Peptides induce persistent signaling from endosomes by a nutrient transceptor. Nat. Chem. Biol. 8:400–408. 10.1038/nchembio.910. [DOI] [PubMed] [Google Scholar]
- 20.Opekarová M, Caspari T, Pinson B, Bréthes D, Tanner W. 1998. Post-translational fate of CAN1 permease of Saccharomyces cerevisiae. Yeast 14:215–224. . [DOI] [PubMed] [Google Scholar]
- 21.Lin CH, MacGurn JA, Chu T, Stefan CJ, Emr SD. 2008. Arrestin-related ubiquitin-ligase adaptors regulate endocytosis and protein turnover at the cell surface. Cell 135:714–725. 10.1016/j.cell.2008.09.025. [DOI] [PubMed] [Google Scholar]
- 22.Hatakeyama R, Kamiya M, Takahara T, Maeda T. 2010. Endocytosis of the aspartic acid/glutamic acid transporter Dip5 is triggered by substrate-dependent recruitment of the Rsp5 ubiquitin ligase via the arrestin-like protein Aly2. Mol. Cell. Biol. 30:5598–5607. 10.1128/MCB.00464-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Séron K, Blondel MO, Haguenauer-Tsapis R, Volland C. 1999. Uracil-induced down-regulation of the yeast uracil permease. J. Bacteriol. 181:1793–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gournas C, Amillis S, Vlanti A, Diallinas G. 2010. Transport-dependent endocytosis and turnover of a uric acid-xanthine permease. Mol. Microbiol. 75:246–260. 10.1111/j.1365-2958.2009.06997.x. [DOI] [PubMed] [Google Scholar]
- 25.Keener JM, Babst M. 2013. Quality control and substrate-dependent downregulation of the nutrient transporter Fur4. Traffic 14:412–427. 10.1111/tra.12039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hein C, Springael JY, Volland C, Haguenauer-Tsapis R, André B. 1995. NPl1, an essential yeast gene involved in induced degradation of Gap1 and Fur4 permeases, encodes the Rsp5 ubiquitin-protein ligase. Mol. Microbiol. 18:77–87. 10.1111/j.1365-2958.1995.mmi_18010077.x. [DOI] [PubMed] [Google Scholar]
- 27.Jacobs P, Jauniaux JC, Grenson M. 1980. A cis-dominant regulatory mutation linked to the argB-argC gene cluster in Saccharomyces cerevisiae. J. Mol. Biol. 139:691–704. 10.1016/0022-2836(80)90055-8. [DOI] [PubMed] [Google Scholar]
- 28.Grenson M, Mousset M, Wiame JM, Béchet J. 1966. Multiplicity of the amino acid permeases in Saccharomyces cerevisiae. I. Evidence for a specific arginine-transporting system. Biochim. Biophys. Acta 127:325–338. [DOI] [PubMed] [Google Scholar]
- 29.Ghaddar K, Krammer E-M, Mihajlovic N, Brohée S, André B, Prévost M. 2014. Converting the yeast arginine Can1 permease to a lysine permease. J. Biol. Chem. 289:7232–7246. 10.1074/jbc.M113.525915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Merhi A, Gérard N, Lauwers E, Prévost M, André B. 2011. Systematic mutational analysis of the intracellular regions of yeast Gap1 permease. PLoS One 6:e18457. 10.1371/journal.pone.0018457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Iraqui I, Vissers S, Bernard F, De Craene JO, Boles E, Urrestarazu A, André B. 1999. Amino acid signaling in Saccharomyces cerevisiae: a permease-like sensor of external amino acids and F-Box protein Grr1p are required for transcriptional induction of the AGP1 gene, which encodes a broad-specificity amino acid permease. Mol. Cell. Biol. 19:989–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gao X, Lu F, Zhou L, Dang S, Sun L, Li X, Wang J, Shi Y. 2009. Structure and mechanism of an amino acid antiporter. Science 324:1565–1568. 10.1126/science.1173654. [DOI] [PubMed] [Google Scholar]
- 33.Fang Y, Jayaram H, Shane T, Kolmakova-Partensky L, Wu F, Williams C, Xiong Y, Miller C. 2009. Structure of a prokaryotic virtual proton pump at 3.2 A resolution. Nature 460:1040–1043. 10.1038/nature08201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao X, Zhou L, Jiao X, Lu F, Yan C, Zeng X, Wang J, Shi Y. 2010. Mechanism of substrate recognition and transport by an amino acid antiporter. Nature 463:828–832. 10.1038/nature08741. [DOI] [PubMed] [Google Scholar]
- 35.Opekarová M, Caspari T, Tanner W. 1993. Unidirectional arginine transport in reconstituted plasma-membrane vesicles from yeast overexpressing CAN1. Eur. J. Biochem. 211:683–688. 10.1111/j.1432-1033.1993.tb17596.x. [DOI] [PubMed] [Google Scholar]
- 36.Kowalczyk L, Ratera M, Paladino A, Bartoccioni P, Errasti-Murugarren E, Valencia E, Portella G, Bial S, Zorzano A, Fita I, Orozco M, Carpena X, Vázquez-Ibar JL, Palacín M. 2011. Molecular basis of substrate-induced permeation by an amino acid antiporter. Proc. Natl. Acad. Sci. U. S. A. 108:3935–3940. 10.1073/pnas.1018081108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Springael JY, André B. 1998. Nitrogen-regulated ubiquitination of the Gap1 permease of Saccharomyces cerevisiae. Mol. Biol. Cell 9:1253–1263. 10.1091/mbc.9.6.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Donaton MCV, Holsbeeks I, Lagatie O, Van Zeebroeck G, Crauwels M, Winderickx J, Thevelein JM. 2003. The Gap1 general amino acid permease acts as an amino acid sensor for activation of protein kinase A targets in the yeast Saccharomyces cerevisiae. Mol. Microbiol. 50:911–929. 10.1046/j.1365-2958.2003.03732.x. [DOI] [PubMed] [Google Scholar]
- 39.Kriel J, Haesendonckx S, Rubio-Texeira M, Van Zeebroeck G, Thevelein JM. 2011. From transporter to transceptor: signaling from transporters provokes re-evaluation of complex trafficking and regulatory controls: endocytic internalization and intracellular trafficking of nutrient transceptors may, at least in part, be governed by their signaling function. Bioessays 33:870–879. 10.1002/bies.201100100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jeschke G. 2013. A comparative study of structures and structural transitions of secondary transporters with the LeuT fold. Eur. Biophys. J. 42:181–197. 10.1007/s00249-012-0802-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kniazeff J, Shi L, Loland CJ, Javitch JA, Weinstein H, Gether U. 2008. An intracellular interaction network regulates conformational transitions in the dopamine transporter. J. Biol. Chem. 283:17691–17701. 10.1074/jbc.M800475200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Krishnamurthy H, Gouaux E. 2012. X-ray structures of LeuT in substrate-free outward-open and apo inward-open states. Nature 481:469–474. 10.1038/nature10737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ljungdahl PO. 2009. Amino-acid-induced signalling via the SPS-sensing pathway in yeast. Biochem. Soc. Trans. 37:242–247. 10.1042/BST0370242. [DOI] [PubMed] [Google Scholar]
- 44.Bernard F, André B. 2001. Genetic analysis of the signalling pathway activated by external amino acids in Saccharomyces cerevisiae. Mol. Microbiol. 41:489–502. 10.1046/j.1365-2958.2001.02538.x. [DOI] [PubMed] [Google Scholar]
- 45.Fischer W-N, Loo DDF, Koch W, Ludewig U, Boorer KJ, Tegeder M, Rentsch D, Wright EM, Frommer WB. 2002. Low and high affinity amino acid H+-cotransporters for cellular import of neutral and charged amino acids. Plant J. 29:717–731. 10.1046/j.1365-313X.2002.01248.x. [DOI] [PubMed] [Google Scholar]
- 46.Hein C, André B. 1997. A C-terminal di-leucine motif and nearby sequences are required for NH4(+)-induced inactivation and degradation of the general amino acid permease, Gap1p, of Saccharomyces cerevisiae. Mol. Microbiol. 24:607–616. 10.1046/j.1365-2958.1997.3771735.x. [DOI] [PubMed] [Google Scholar]
- 47.Bonneaud N, Ozier-Kalogeropoulos O, Li GY, Labouesse M, Minvielle-Sebastia L, Lacroute F. 1991. A family of low and high copy replicative, integrative and single-stranded S. cerevisiae/E. coli shuttle vectors. Yeast 7:609–615. 10.1002/yea.320070609. [DOI] [PubMed] [Google Scholar]
- 48.Mumberg D, Müller R, Funk M. 1994. Regulatable promoters of Saccharomyces cerevisiae: comparison of transcriptional activity and their use for heterologous expression. Nucleic Acids Res. 22:5767–5768. 10.1093/nar/22.25.5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nikko E, Marini AM, André B. 2003. Permease recycling and ubiquitination status reveal a particular role for Bro1 in the multivesicular body pathway. J. Biol. Chem. 278:50732–50743. 10.1074/jbc.M306953200. [DOI] [PubMed] [Google Scholar]
- 50.Lauwers E, André B. 2006. Association of yeast transporters with detergent-resistant membranes correlates with their cell-surface location. Traffic 7:1045–1059. 10.1111/j.1600-0854.2006.00445.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.