

Abstract
Recently, we demonstrated that the microRNA 424(322)/503 [miR-424(322)/503] cluster is transcriptionally controlled by transforming growth factor β (TGF-β) in the mammary epithelium. Induction of this microRNA cluster impacts mammary epithelium fate by regulating apoptosis and insulin-like growth factor 1 (IGF1) signaling. Here, we expanded our finding to demonstrate that miR-424(322)/503 is an integral component of the cell cycle arrest mediated by TGF-β. Mechanistically, we showed that after TGF-β exposure, increased levels of miR-424(322)/503 reduce the expression of the cell cycle regulator CDC25A. miR-424(322)/503-dependent posttranscriptional downregulation of CDC25A cooperates with previously described transcriptional repression of the CDC25A promoter and proteasome-mediated degradation to reduce the levels of CDC25A expression and to induce cell cycle arrest. We also provide evidence that the TGF-β/miR-424(322)/503 axis is part of the mechanism that regulates the proliferation of hormone receptor-positive (HR+) mammary epithelial cells in vivo.
INTRODUCTION
Transforming growth factor β (TGF-β) signaling has a major role in epithelial mammary gland morphogenesis (1), mainly as a potent inhibitor of proliferation (2, 3). This cytostatic effect is mediated by the ability of canonical TGF-β signaling to up-/downregulate a variety of genes involved in cell cycle control. The identities of these critical genes differ depending on the cell type (4). In mammary epithelial cells (MECs), TGF-β-dependent SMAD signaling has been shown to inhibit the G1/S transition by induction of P15INK4B (5) and repression of CDC25A and MYC (6, 7). P15INK4B binding induces an allosteric change in cyclin-dependent kinases 4 and 6 (CDK4/6) that prevents binding to D-type cyclins and inhibits the phosphorylation of retinoblastoma (Rb) family members (8). CDC25A is a dual-specificity phosphatase that exerts its cell cycle control by activating the CDK2-cyclin E and CDK2-cyclin A complexes (9).
Downregulation of CDC25A protein levels by TGF-β occurs at the transcriptional and posttranslational levels. The CDC25A promoter contains a TGF-β response element that recruits silencing E2F4-p130-histone deacetylase 1 (HDAC1) complexes to inhibit the transcriptional activity of the promoter (10). TGF-β-induced ubiquitination and proteosome-dependent degradation of the CDC25A protein are promoted by the SKP1–CULLIN–β-TrCP (SCFbeta-TrCP) complex (11). Both mechanisms cooperate to reduce CDC25A expression and sustain cell cycle arrest during TGF-β-mediated cytostasis.
Targeting of CDC25A by microRNA 424(322)/503 [miR-424(322)/503] has previously been found in endothelial (12), microglial (13), and muscle (14) cells. However, a role for the miR-424(322)/503 cluster in the regulation of CDC25A in mammary epithelial cells has not been previously described. Recently, through a systematic study that combined computational studies for target prediction with biochemical and genetic experimental validation, we described miR-424(322)/503 as a regulator of mammary epithelial involution (15). Importantly, we also demonstrated that miR-424(322)/503 is transcriptionally regulated by TGF-β (15).
Since CDC25A downregulation is a key step during TGF-β-mediated cytostasis in mammary epithelial cells and miR-424(322)/503 is transcriptionally regulated by TGF-β, we decided to investigate the hypothesis that upregulation of miR-424(322)/503 plays a role in regulating the expression of CDC25A during TGF-β-induced cell cycle arrest.
Here, we demonstrated that experimental upregulation of miR-424(322)/503 in mammary epithelial cells recruits CDC25A mRNA to RNA-induced silencing complexes (RISC), reduces the expression of CDC25A endogenous protein levels, and promotes G1 cell cycle arrest. In contrast, miR-424(322)/503 knockout (KO) cells presented with higher levels of CDC25A and a higher proliferation rate than wild-type (WT) counterparts, both in vitro and in vivo. By using reporter constructs engineered to separately evaluate the transcriptional regulation of the CDC25A promoter and miR-424(322)/503-mediated silencing of the CDC25A mRNA, we demonstrated that upon TGF-β exposure, microRNA (miRNA)-mediated silencing is necessary to achieve maximum reduction of CDC25A expression levels.
Mammary epithelial cells expressing estrogen and progesterone receptors (hormone receptor positive [HR+]) secrete growth factors that promote the growth of neighboring cells via paracrine methods. However, they are themselves protected from these stimuli by activation of TGF-β signaling (16–18). Here we found that these HR+ cells express low CDC25A and high levels of miR-424(322)/503. Importantly, miR-424−/−/miR-503−/− (KO) animals present a 2-fold increase in the number of HR+ cells that colocalize with proliferation-associated markers, suggesting that miR-424(322)/503 is part of the TGF-β signaling that restrains their proliferation. Overall, the data presented here demonstrate that the upregulation of miR-424(322)/503 is an integral part of the molecular mechanism that orchestrates the downregulation of CDC25A during the cell cycle arrest mediated by TGF-β in mammary epithelial cells.
MATERIALS AND METHODS
Cell culture.
Normal breast epithelial MCF-10A and MCF-12 cells were cultured at 37°C in 5% CO2 in Dulbecco's modified Eagle medium (DMEM)–Ham's F-12 supplemented with 1% penicillin-streptomycin, epidermal growth factor (EGF) (20 ng/ml), insulin (10 μg/ml), cholera toxin (100 ng/ml), hydrocortisone (500 ng/ml), and 5% horse serum. Wild-type (WT) and KO hTERT-immortalized mouse mammary epithelial cells (MMECs) were grown in DMEM–F-12 medium containing 1% penicillin-streptomycin, EGF (5 ng/ml), insulin (5 μg/ml), hydrocortisone (1 μg/ml), and 5% fetal bovine serum (FBS). 184B5 cells were grown in MEGM medium (Lonza/Clonetics CC-3150) supplemented with cholera toxin (1 ng/ml). Phoenix cells were grown in DMEM–10% fetal bovine serum and 1% penicillin-streptomycin at 37°C, 5% CO2. TGF-β1 was purchased from Sigma (T7039).
Viral production, infection, and in vitro Mimic/miRIDIAN hairpin inhibitor transfection conditions.
Production of miR-424(322)/503 and CDC25A-containing lentivirus was achieved by transfecting Phoenix packaging cells with linear jetPEI (101-10N; Polyplus) in combination with lentiviral plasmids[pTRIPz-424(322)/503 and pLOC-CDC25A], pCMV-dR8.91, and pMD.G helper plasmids (19) at a ratio of 2:1:1, respectively. Similarly, the same conditions were employed to produce hTERT retrovirus by combining the retroviral plasmid pBABE-hTERT with the retroviral helper plasmids pMSCV-Psi and pCMV-VSV-G. Twenty-four hours after transfection, packaging cells were cultured with regular MCF-10A growth medium for 24 h; afterwards, the medium containing the viral particles was collected.
Cells were plated at 60% confluence in a 6-well plate, and after 24 h, cells were cultured in normal medium mixed with medium containing the viral particles (1:1 ratio). Cells were reinfected 12 h later, following the same procedure. Infection medium was replaced after 12 h with fresh medium, and the cells were selected with the appropriate antibiotics.
MCF-10A cells were infected with synthetic miRIDIAN-Mimic-424 and -503 (C-300717-05 and C-300841-05, respectively; Dharmacon) at a 100 nM final concentration using the hemagglutinating virus of Japan (HVJ) envelope vector system (GN004EX; Cosmo Bio Co.) and following the manufacturer's instructions.
MCF-10A cells were infected with the synthetic miRIDIAN microRNA hairpin inhibitors anti-hsa-miR-424 and anti-hsa-miR-503 (IH-300717-07 and IH-300841-07, respectively; Dharmacon) at a 100 nM final concentration using the HVJ envelope vector system (GN004EX; Cosmo Bio Co.), following the manufacturer's instructions.
293T transfections were performed as follows. 293T cells were plated at 70% confluence in 96-well plates. Twenty-four hours later, cells were transfected with 50 ng of pMIR-REPORT constructs containing the luc-3′ untranslated region (UTR) sequences, 50 ng of a Renilla normalization control, and with 100 nM (each) individual synthetic mirVana miRNA mimics at a 100 nM final concentration using the TransIT-LT1 (2300A; Mirus Bio) and TransIT-TKO (2150; Mirus Bio) transfection reagents, following the manufacturer's instructions. After 24 h, relative luciferase units (RLU) were measured using the Dual-Glo luciferase assay system (E2949; Promega).
Western blotting.
Cells were washed with cold phosphate-buffered saline (PBS) and lysed with EZ lysis buffer (1 M Tris [pH 7], 50% glycerol, 20% SDS, 1 mM orthovanadate, 1 mM sodium fluoride, and 1 mM phenylmethylsulfonyl fluoride). Protein concentrations were determined by using the Protein Assay kit (500-0006; Bio-Rad). Equal amounts of proteins were subjected to SDS-PAGE and transferred to nitrocellulose membranes (10401197; GE Healthcare). Nonspecific binding was blocked by incubation with TBST (20 mM Tris-HCl [pH 7.4], 150 mM NaCl, and 0.1% Tween 20) plus 5% of nonfat milk. Membranes were incubated with the primary antibodies overnight at 4°C and for 1 h with secondary horseradish peroxidase (HRP)-conjugated antibodies at room temperature (NA9350V, NA931V and NA934V; Amersham). Signal was detected using the Lumi-Light Western blotting substrate (12015200001 and 12015196001; Roche).
The antibodies used in this study include: CDC25A (sc-7389; Santa Cruz), SMAD3 (9523; Cell Signaling), SMAD2 (3122; Cell Signaling), pSMAD2 (3108; Cell Signaling), pSMAD3 (9520; Cell Signaling), β-actin (A0760-40; USBiological), histone H3 (ab1791; Abcam), and phospho-histone H3 (p-histone H3) (Ser10) (H5110-14B; USBiological).
3′UTR cloning, luciferase reporter assays, and mutagenesis.
The 3′ UTR of CDC25A was cloned downstream of the luciferase reporter in the pMIR-REPORT vector (AM5795M; Life Technologies) by PCR from human genomic DNA using specific primers (CDC25A-3′UTR-Mlu-F, ACGCGTACGGAGGGGAGTAGAGAAG; CDC25A-3′UTR-HindIII-R, AAGCTTCACCTCCCACCAAATAGATA). To measure luciferase activity, Phoenix cells were plated at 70% confluence in 96-well plates. Twenty-four hours later, cells were transfected with 50 ng of pMIR-REPORT constructs containing the luc-3′-UTR sequences in combination with a Renilla normalization control and with 100 nM pLEMIR-424(322)/503 using the jPEI transfection reagent. After 24 h, the relative luciferase units (RLU) were measured using the Dual-Glo luciferase assay system (E2949; Promega).
To mutagenize the miR-424 and -503 binding sites, we used the QuikChange site-directed mutagenesis kit (200518; Agilent), following the instructions in the manual.
The following primer sequences were used for mutagenesis of miR-424/503 binding sites: CDC25AΔ1BS-UTR-F, GAAGTTACACAGAAAGGCCAAATAGCAAAG; CDC25AΔ1BS-UTR-R, CTTTGCTATTTGGCCTTTCTGTGTAACTTC; CDC25AΔ2BS-UTR-F, CTGTGGTACTGGGGCTTTAAGCCAAGAACT; CDC25AΔ2BS-UTR-R, AGTTCTTGGCTTAAAGCCCCAGTACCACAG.
Inducible hsa-miR-424/503 cluster cloning.
The human miR-424/503 cluster was cloned by PCR from human genomic DNA into a doxycycline-inducible pTRIPz recipient vector (Thermo Scientific) using restriction enzymes. Primers used were as follows: hsa-Induc-XhoI-F, CTCGAGAACTCGAGTCACCCACTACGTTGTTCCAAG; hsa-Induc-EcoRI-R, GAATTCAAGAATTCGGTGGTATTCTGATTGGGAAGG.
CDC25A promoter cloning.
The human CDC25A promoter was cloned by PCR from human genomic DNA into the pGL3-Basic recipient vector (E1751; Promega) using restriction enzymes. Primers used were as follows: hsa-CDC25Aprom-NheI-F, GCTAGCGATCCGGCCAGACCTCCACAGGTCTTC; hsa-CDC25Aprom-HindIII-R, AAGCTTCTCCCACCCGCTTGCCCAGCTC.
To mutagenize the E2F-A binding site, we used the QuikChange site-directed mutagenesis kit (Agilent 200518), following the manual instructions. The following primer sequences were used for mutagenesis of the E2F-A binding site: Prom-CDC25Amut-F, TTACTGATTGGTGGATTCCGTATAACACCAACTAGGAAAGGGGGGCGG; Prom-CDC25Amut-R, CCGCCCCCCTTTCCTAGTTGGTGTTATACGGAATCCACCAATCAGTAA.
The 3′ UTR of CDC25A was cloned downstream of the luciferase reporter in the pGL3-Basic vector by PCR from human genomic DNA using specific primers. Proper 5′-3′ orientation was analyzed by using Sanger sequencing (Sanger-seq). The following primers were used: CDC25A-3′UTR-XbaI-F, TCTAGAACGGAGGGGAGTAGAGAAG; CDC25A-3′UTR-XbaI-R, TCTAGACACCTCCCACCAAATAGATA.
To perform luciferase assays, MCF-10A cells were initially treated with TGF-β for 24 h. Afterwards, cells were transfected with linear-jPEI (101-10N; Polyplus) in the presence of TGF-β for an additional 24 h. After 24 h, relative luciferase units (RLU) were measured using the Dual-Glo luciferase assay system (E2949; Promega).
RNA extraction, reverse transcription, and real-time PCR.
To perform miR-424 and -503 expression analysis by reverse transcription-quantitative PCR (qRT-PCR), RNA was purified using the mirVana miRNA isolation kit (AM1561; Ambion) according to the instructions provided. Reverse transcription reaction and stem-loop-based quantification for human and murine microRNAs were also carried out in accordance with published approaches (20, 21). Briefly, 50 ng of RNA was retrotranscribed using the microRNA reverse transcription kit (4366596; Roche) using the specific Pul-RT primers at a 250 nM final concentration. Retrotranscription conditions include a first step at 16°C for 30 min, a second 60-repeat phase (20°C for 3 s, 42°C for 30 s, and 50°C for 2 s), and a final inactivation step at 85°C for 5 min. The real-time PCRs were performed using 2 μl of cDNA with 10 μl FastStart SYBR green master mix. The thermal cycler conditions were as follows: AmpliTaq activation, 95°C for 3 min; denaturation, 95°C for 10 s; and annealing/extension, 60°C for 30 s (repeat 40 times). Triplicate threshold cycle (CT) values were further analyzed (2−ΔΔCT) by normalizing to an endogenous reference gene (RNU6 or sno-202). Results are presented as a relative mRNA amount compared to the untreated samples.
The following miRNA primers were used: Pul-Rev (GTGTCGTGGAGTCGGC, Pul-RNU6_FW (ACACTCCACTGCGCAAGGATGACAC), Pul-RNU6-RT (CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGAAAAATAT), Pul-miR-424-FW (ACACTCCAGCTGGGCAGCAGCAATTCATGT), Pul-miR-424-RT (CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGTTCAAAAC), Pul-miR-503-FW (ACACTCCAGCTGGGTAGCAGCGGGAACAGTT), and Pul-miR-503-RT (CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGCTGCAGAA).
Experimental animals, LY2109761 treatment, mouse primary MEC purification, sorting, and immunofluorescence.
All mice used in this study were of a mixed C57BL/6 × BALB/c background, and in all experiments littermate controls were used. Mice were sacrificed using a CO2 chamber. Genotyping controls were performed with tail samples, using the Direct PCR reagent (202-Y; Viagen). Tails were incubated in 250 μl of Direct PCR reagent and proteinase K (20 mg/ml) (P8102; NEB) at 55°C for 5 h. Thereafter, samples were incubated at 85°C for 45 min, after which they were centrifuged 5 min at full speed. One microliter of sample was used to perform genotyping PCR using FastStart polymerase (04738420001; Roche). Thermal conditions were as follows: predenaturing at 94°C for 5 min, 35× (denaturing, 94°C, 30 min; annealing, 55°C, 30 min; and elongation, 72°C, 30 min), and final elongation at 72°C for 7 min. All experiments were performed under Institutional Animal Care and Use Committee (IACUC) guidelines.
The following primers were used for genotyping: Pair-1, forward, CACCAGCAGATCCTGGAAAT; reverse, CAAGTGAGGCGCTAACAACA; Pair-2, forward, AGTTTCGAAAAACGGACATA; reverse, ATTGCCTCTCATTGTACCAC.
To inhibit TGF-β signaling for the first 2 days of involution, lactating wild-type females were separated from pups. Immediately afterward, animals were subjected to LY2109761 (ApexBio) administration twice daily with 100 mg/kg of body weight by oral gavage. Mice were sacrificed as described above, and mammary glands were dissected, mechanically disrupted, and digested using a digestion solution composed of DMEM–F-12, 100 U/ml penicillin-streptomycin, 2 mg/ml collagenase A, and 100 U/ml hyaluronidase. Samples were incubated in a rotator for 4 h at 37°C. To isolate mammary epithelial cells, samples were first washed and centrifuged in PBS solution at 800 rpm for 10 min. After this first step, samples were washed and centrifuged 5 times at 1,500 rpm for 10 min. Final pellets were lysed with protein EZ lysis buffer for Western blot analysis.
Initial sample preparation for cell-sorting-based separation of MECs was performed as follows. Final pellets enriched in epithelial cell content (obtained as mentioned previously) were resuspended in red blood cell lysing buffer (R7757; Sigma) for 4 min at room temperature. Cell extracts were then resuspended in PBS and centrifuged at 1,500 rpm for 5 min. Pellets were resuspended in 3 ml of 0.25% trypsin and incubated for 3 min at 37°C. After centrifugation, cell extracts were resuspended in 2 ml of PBS containing 5 mg/ml dispase II (17105-041; Life Technologies) and 0.1 mg/ml DNase I (79254; Qiagen) and incubated at 37°C for 3 min. Finally cells were centrifuged at 1,500 rpm for 5 min and resuspended in 1 ml PBS–2% FBS. Purification of basal and luminal estrogen receptor alpha-negative (ER−) and -positive (ER+) cell populations was performed as previously described (22). In brief, nonepithelial cells were first removed by incubation of single-cell suspensions with a biotinylated antibody (Ab) cocktail containing CD31, CD45, Ter119, and BP1 using a mouse epithelial cell enrichment kit (19758; Stemcell Tech), following the manufacturer's instructions. Final labeling was achieved by incubating cell suspensions in anti-mouse Sca-1, CD49f, and EpCAM (108119, 313615, and 118205, respectively; Biolegends) on ice for a minimum of 30 min.
Immunofluorescence analysis of ER (dilution, 1:2,000) and Ki67 (dilution, 1:3,000) was achieved by staining cells with specific antibodies (ab37438 and 15580, respectively; Abcam). Briefly, after sorting, cells were resuspended in PBS–2% FBS at a final concentration of 1,500 cells/μl. Thirty microliters of cell suspension was immobilized on glass slides using a holder block (1001644; Thermo Scientific), 8-well strips (1001642; Thermo Scientific), a filter card (1001641; Thermo Scientific), a glass slide (12-550-15; Thermo Scientific), and a Cytospin 4 cytocentrifuge (A78300003; Thermo Scientific). Cells were centrifuged at 1,200 rpm for 4 min. Thereafter, slides, filter cards, and sample chambers were removed from the centrifuge and disassembled carefully. Immediately, attached cells were fixed with paraformaldehyde (4%) for 10 min and washed with PBS 3 times.
Cells were permeabilized with PBS–Triton (0.5%) for 10 min at room temperature, washed with PBS, and blocked with 10% goat serum in 2% bovine serum albumin (BSA)-PBS for 1 h. Afterwards, samples were incubated with primary antibody overnight at 4°C. After incubation, cells were washed with PBS and incubated with secondary antibody (1:1,000 Alexa Fluor 488–goat anti-rabbit antibody, A11070; Life Technologies) for 1 h at room temperature. Thereafter, cells were washed in PBS, and nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI). Finally, a cover glass was added using mounting medium for fluorescence (Vectashield, CA94010; Vector Laboratories) and sealed.
Immunohistochemistry.
Mammary glands were fixed in formalin (Fischer 175) for immunohistochemistry (IHC) analysis. Formalin-fixed paraffin-embedded samples were first heated at 100°C for 3 min on a heat block to melt the paraffin. Subsequently, samples were deparaffinized by a serial incubation with xylene for 3 min, 100% ethanol (EtOH) for 3 min, 95% EtOH for 3 min, and distilled water for 2 min. Peroxidase inactivation and antigen retrieval were achieved by incubating samples in 1% H2O2 for 15 min at room temperature and incubating slides with citric buffer (2 mM citric acid and 8 mM sodium citrate) in a steamer for 30 min. Samples were washed twice in PBS for 5 min and incubated in 10% whole goat blocking serum diluted in 2% BSA-PBS for 30 min at room temperature. Thereafter, samples were incubated in primary antibody (1:200 Ki67 [ab15580; Abcam], 1:300 anti-cytokeratin 18 antibody [ab668; Abcam], and 1:1,000 anti-cytokeratin 5 [Covance PRB-160P]) diluted in 2% BSA-PBS plus 0.01% sodium azide for 2 h at room temperature.
Samples were then washed in PBS and incubated in 1:500 biotinylated anti-rabbit IgG made in goat diluted in 2% BSA-PBS for 30 min at room temperature. Afterwards, samples were washed and exposed to peroxidase substrate (PK-6100; Vector Laboratories) for 30 min at room temperature and subsequently permeabilized with PBS-0.5% Triton. Thereafter, samples were incubated in chromogen 3,3′-diaminobenzidine (DAB) and then washed in distilled water and counterstained. Counterstaining was performed by treating samples with hematoxylin for 1 s, dipped in 1% hydrochloric acid, and finally washed in ammonia water for 1 s. Finally, dehydration was performed by incubating samples in 95% EtOH for 2 min, 100% EtOH for 2 min, and xylene for 4 to 5 min and ultimately mounted with a coverslip.
PAR-CLIP analysis.
A photoactivatable-ribonucleoside-enhanced cross-linking immunoprecipitation (PAR-CLIP) assay to measure Ago2 enrichment in miR-424(322)/503 mRNA targets was performed as described previously (23). Briefly, cells were pretreated with 50 μM 4-thiouridine (T4509; Sigma) overnight and cross-linked at 150 mJ/cm2 at 365 nm UV on ice. Cells were reconstituted in lysis buffer (2.5 mM HEPES [pH 7], 50 mM NaCl, 10% glycerol, 1% Triton X-100, proteinase inhibitor [Roche 04693159001], 0.2 mM dithiothreitol [DTT], and 1 U/μl RNAseOUT [10777-019]; Invitrogen). Mild (5 U/μl) RNase-T1 (EN0541; Fermentas) digestion was performed at 22°C for 15 min. Immunoprecipitation was performed using A beads (11719408001; Roche), G beads (11719416001; Roche), and 10 μg of anti-AGO2 antibody (H00027161-M01; Abnova) overnight at 4°C. Samples were subsequently washed twice with washing buffer 1 (50 mM Tris [pH 7.5], 150 mM NaCl, 0.1% NP-40, and 1 mM EDTA) at 4°C for 30 min, digested with RNase-T1 (20 U/μl) at 22°C for 15 min, washed once with washing buffer 2 (50 mM Tris [pH 7.5], 500 mM NaCl, and 0.1% NP-40) at 4°C for 30 min, and finally washed twice with washing buffer 3 (50 mM Tris [pH 7.5] and 500 mM NP-40) at 4°C for 30 min. Samples were centrifuged for 5 min at 5,000 rpm at 4°C, and supernatant was treated with 5 mg/ml of proteinase K (P8102; New England BioLabs) for 1 h at 50°C. Finally, extracts were lysed and RNA extracted using the mirVana miRNA isolation kit (AM1561; Ambion) according to the instructions provided. RNA was retrotranscribed as described above in “RNA extraction, reverse transcription, and real-time PCR,” and target mRNA was quantified by real-time PCR using specific primers, as follows: for CDC25A, forward, GCCATTCTAGGTAGGGTTTT; reverse, CCTAGCTTTCTGTCCGATAA; for RPL13A, forward. CCTGGAGGAGAAGAGGAAAGAGA; reverse, TTGAGGACCTCTGTGTATTTGTCAA; for GAPDH, forward, CATCTTCTTTTGCGTCGC; reverse, AAAAGCAGCCCTGGTGAC for B2M, forward, TGCTGTCTCCATGTTTGATGTATCT; reverse, TCTCTGCTCCCCACCTCTAAGT.
Flow cytometry and cell viability.
All flow cytometry experiments were performed on a BD FACSCalibur cell analyzer using the CellQuest Pro software for Mac OS X. We used the fluorescein isothiocyanate (FITC)/allophycocyanin (APC) BrdU Flow kit (559619 and 552598; BD-Pharmingen) for BrdU experiments. Luminescent detection of ATP to measure metabolically active cells and cell viability was conducted using the CellTiter-Glo luminescent cell viability assay (G7571; Promega), following the manufacturer's instructions. For BrdU experiments, MCF-10A pTRIPz-Scrambled and MCF-10A pTRIPz-miR-424(322)/503 cells were treated with doxycycline (100 ng/ml) for 5 days, trypsinized, exposed to BrdU for 4 h, and labeled using the APC BrdU Flow kit (552598; BD-Pharmingen), following the manual's instructions.
To obtain the cell cycle profile data, doxycycline-treated cells were trypsinized and fixed with 70% ethanol overnight at 4°C. Thereafter, samples were washed twice in cold PBS, centrifuged for 5 min at 5,000 rpm, and resuspended in 1 ml of PBS with RNase A (350 ng/ml) and propidium iodide (7.5 ng/ml).
RESULTS
CDC25A is targeted by miR-424(322)/503 in mammary epithelial cells.
During our previous studies, which aimed to identify microRNAs that control mammary epithelial homeostasis, we identified miR-424(322)/503 as an important regulator of epithelial involution after pregnancy. The expression of this miRNA cluster was found to be induced by TGF-β (15). Computational prediction using TargetScan identified 1,273 targets for miR-424(322) and 385 for miR-503. Since both miRNAs belong to the miR-16 family (24, 25), 90% of the targets predicted for miR-503 were part of the list predicted for miR-424 (322) (15). We found that the predicted targets that showed a reduction in the mRNA level when miR-424(322)/503 was experimentally upregulated were statistically associated with cell cycle, apoptosis, TGF-β, and canonical extracellular signal-regulated kinase (ERK)/AKT signal transduction pathways (15). Further biochemical studies validated BCL2 and insulin-like growth factor 1 receptor (IGF1R) as targets in vitro and in vivo (15).
The biological functions of miRNAs are mediated by their ability to attenuate the expression of targeted mRNAs. In mammalian cells, miRNAs function by binding to specific regions in the targeted mRNA, which are complementary to the seed sequence of the mature miRNA, mainly in the 3′ UTR (26). Computational prediction of targets based on seed sequence conservation (27) as well as nonconserved sites (26) identified two conserved target sites for miR-424(322) and miR-503 in the 3′ UTR of CDC25A (Fig. 1A). We utilized the TargetScan algorithm (http://www.targetscan.org/), which has been shown to have one of the highest specificities in preselecting putative target genes (28).
FIG 1.

Experimental upregulation of the miR-424/503 cluster targets the CDC25A 3′ UTR in mammary epithelial cells in vitro. (A) The panel shows miR-424 and miR-503 conserved and poorly conserved binding sites in the 3′ UTRs of different predicted targets (TargetScan). Predicted binding sites for the previously identified targets BCL2 and IGF1R were also included for comparison. (B) Cloning of the wild-type and mutated forms of the CDC25A 3′ UTR into a luciferase reporter vector. Subsequent 3′-UTR luciferase assays showed that both predicted miRNA binding sites are necessary for optimal targeting activity. (C) In the upper panel, Western blots show that transfection of MCF-10A cells with miR-424 and -503 mimics alone or in combination results in a decrease of endogenous CDC25A and protein levels. Previously validated miR-424(322)/503 targets BCL2 and IGF1R were included for comparison. The bottom panel shows qRT-PCR of miR-424 and -503 to monitor miRNA levels in the same cells analyzed in the upper panel. (D) Schematic representation of the pTRIPz doxycycline (Dox)-inducible system and qRT-PCR analysis measuring the induction of miR-424 and miR-503 expression after 5 days using different amounts of Dox. (E) Western blot analysis of CDC25A, BCL2, and IGF1R after exposure of MCF-10A cells to 100 nM doxycycline for 5 days. (F) PAR-CLIP analysis by qRT-PCR of CDC25A, BCL2, and IGF1R in MCF-10A-424/503miR-Dox cells demonstrates enrichment of CDC25A, BCL2, and IGF1R mRNAs upon upregulation of miR-424(322)/503. Nontargeted controls are also shown (n = 5).
In order to investigate the targeting of CDC25A by miR-424(322)/503, we first decided to assess the contribution of each of the predicted target sites. Thus, we cloned a portion of the CDC25A 3′ UTR containing the predicted wild-type binding site or mutated versions downstream of a luciferase reporter gene (Luc) (Fig. 1B) (a description of the mutated binding sites is shown in Fig. 2B). We cotransduced 293T cells with the corresponding Luc-UTR reporter (50 ng), a Renilla luciferase expression vector (Ren) lacking any UTR (50 ng) and used for normalization purposes, and microRNA mimics corresponding to miR-424(322) and miR-503 (100 nM). Twenty-four hours after transfection, we quantified the expression of the reporters. These experiments showed that although the presence of one target site is able to attenuate the expression of the corresponding mRNA, both sites are necessary to achieve the maximum inhibitory effect (Fig. 1B).
FIG 2.

Protein levels of CDC25A are linked to expression of the miR-424/503 cluster in mammary epithelial cells in vivo. (A) The panels show side by side expression levels of the CDC25A protein (Western blot) as well as mature miR-424(322) and miR-503 (qRT-PCR) in purified mammary epithelial cells during six different phases of mammary development. (B and C) Western blot analysis showing that purified primary miR-424−/−/miR-503−/− MMECs from involuting mammary glands (B) and miR-424−/−/miR-503−/− Tert-immortalized MMECs (C) present higher steady-state levels of CDC25A than wild-type counterparts. A minimum of three animals were analyzed.
Next, we asked if both microRNAs have the ability to downregulate CDC25A in mammary epithelial cells. We transfected microRNA mimics for each miRNA, individually and in combination, and we evaluated the expression levels of endogenous CDC25A. In order to use a relevant model, we performed our experiments in MCF-10A cells. MCF-10A is a nontransformed human mammary epithelial cell line that is widely used as a model to study normal mammary epithelium in vitro (6, 29, 30). Western blot studies showed that when transfected at similar levels (50 nM), both miR-424 (322) and miR-503 were competent at targeting CDC25A (Fig. 1C).
miRNAs downregulate the expression of targeted genes by recruiting mRNAs to the RNA-induced silencing complex (RISC). Thus, we performed additional validation using PAR-CLIP (23) to study whether increased cellular levels of the miRNA cluster enriched the fraction of CDC25A mRNA bound to RISC. We performed PAR-CLIP experiments with a MCF-10A variant (MCF-10A-424/503miR-Dox) that was engineered to upregulate the expression of miRNA 424/503 using a doxycycline (Dox)-inducible system (Fig. 1D). Importantly, this system allowed us to upregulate miR-424 and miR-503 to comparable levels similar to those observed in vivo in the mammary epithelium (previously, we have described that miR-424(322) and miR-503 peak at ∼2,500 and 250 molecules/cell, respectively [15]). As the first step in setting up these experiments, we quantified the number of molecules of each miRNA per MCF-10A-424/503miR-Dox cell after Dox exposure. For this, we first set up a qRT-PCR standard curve of miR-424(322) and miR-503 levels by titrating chemically synthesized mature microRNA sequences (Sigma). Next, we purified RNA from 100,000 MCF-10A-424/503miR-Dox cells that were treated with different amounts of Dox for 5 days (Fig. 1D). This RNA was used to perform qRT-PCR to measure the expression level of miR-424(322) and miR-503. Finally, those CT values were intercalated with the standard curve to estimate the number of molecules of mature microRNA/cell. These experiments revealed that the expression of miR-424(322) in MCF-10A cells is about a few hundred per cell but increases to a few thousand (about 10- to 15-fold) after Dox exposure. The mature form of miR-503 also increased proportionally, but its steady-state levels were about a log of magnitude lower than the levels of miR-424(322). We have previously described that biased expression levels between these two miRNAs also occur in vivo (15). As expected, this system efficiently targeted the expression of CDC25A (Fig. 1E). After PAR-CLIP, qRT-PCR was used to quantify the levels of captured CDC25A mRNA. Our data revealed a statistically significant enrichment for CDC25A mRNA in RISC when miR-424(322)/503 was upregulated (Dox+ versus Dox−), while no change was observed for three negative-control genes (Fig. 1F).
Finally, we compared the expression levels of CDC25A in primary mouse mammary epithelial cells (MMECs) from miR-424−/−/miR-503−/− (KO) and wild-type (WT) animals (we have previously described the generation of this KO model [15]). We showed that the expression of miR-424(322)/503 increases during mammary epithelial involution (15). Of note, Western blot studies of purified virgin and pregnant female mammary epithelial cells showed that in WT animals the expression of CDC25A is strongly downregulated during the peak of expression of miR-424(322) and miR-503 (72 h after weaning) (Fig. 2A). In contrast, KO animals presented higher levels of CDC25A than WT counterparts (Fig. 2B). As expected, Tert-immortalized KO mammary epithelial cells also presented consistently higher CDC25A protein levels than WT counterparts (Fig. 2C).
Overall, these data represent confirmatory analysis showing that miR-424(322)/503 also targets CDC25A in mammary epithelial cells due to the presence of two active binding sites in the mRNA 3′ UTR.
Upregulation of miR-424(322)/503 contributes to the reduction of CDC25A levels during TGF-β response in mammary epithelial cells.
Reduction in the expression of CDC25A in mammary epithelial cells after TGF-β exposure is a key event for proper cytostatic response (6). Downregulation of CDC25A protein levels occurs at the transcriptional (10) and posttranslational (11) levels. Previously, we demonstrated that TGF-β ligands upregulate the expression of miR-424(322)/503 (15); here, we investigated the involvement of this upregulation in decreasing the levels of CDC25A during TGF-β exposure.
In order to evaluate how promoter regulation and microRNA-mediated targeting contribute to the downregulation of CDC25A, we engineered a series of Luc reporter constructs containing wild-type and mutant versions of the endogenous CDC25A promoter (Fig. 3A) and 3′ UTR (Fig. 3B). In the mutant version of the promoter, we mutated an E2F binding motif that has been shown to be essential for transcriptional repression mediated by TGF-β (10). The mutant 3′ UTR contained a deletion of the two microRNA seed sequence binding sites that we demonstrated to be crucial for miR-424(322) and miR-503 activity (Fig. 1). These constructs (50 ng) were cotransfected in the presence or absence of TGF-β1 ligand (10 ng/ml) along with normalization vectors (50 ng) that express Renilla luciferase driven by a cytomegalovirus (CMV) promoter. Cells were pretreated with TGF-β1 ligand for 24 h before transduction of the plasmids to induce the expression of the miRNA cluster. The luciferase signal was read 24 h after plasmid transduction. These experiments revealed that full downregulation of the reporter is achieved only when both regulatory elements, the promoter and the 3′ UTR, are intact (Fig. 3C). Remarkably, the miR-424(322)/503 target sites in the 3′ UTR accounted for about 30% of the inhibitory effect.
FIG 3.

The binding sites of miR-424(322)/503 within the CDC25A 3′ UTR contribute to the reduction of the CDC25A level upon TGF-β exposure. (A) Cloning of the wild-type CDC25A promoter into the pGL3-Basic vector (WT-PROM) and mutagenesis of the repressive E2F-A binding site (MUT-PROM). (B) Addition of a wild-type and mutated CDC25A 3′ UTR tail to the WT-PROM and MUT-PROM constructs. (C) Luciferase assays show that addition of the extra wild-type 3′ UTR tail is necessary to achieve maximum CDC25A repression by TGF-β. The data represent five independent experiments. The TGF-β-mediated transcriptional repression values are calculated relative to no-TGF-β treatment for each construct.
Downregulation of CDC25A mediated by upregulation of miR-424(322)/503 induces G1 cell cycle arrest.
While the data described above showed the ability of miR-424(322)/503 to modulate the expression of CDC25A in the mammary epithelium, it was crucial to determine if this regulation was biologically relevant.
CDC25A is a key player in the progression of the cell cycle via its ability to modulate the activity of G1/S cyclin-dependent kinases (31). Reduction in CDC25A expression by several stimuli and subsequent cell cycle arrest have been described. Thus, next, we studied the role of miR-424(322)/503 cluster-mediated reduction of CDC25A expression in the cell cycle and cell proliferation. In order to perform our experiments under physiological conditions, we experimentally upregulated miR-424(322) and miR-503 to the levels found after stimulation with TGF-β. As the first step to set up these experiments, we quantified the number of molecules of each miRNA per cell before and after induction by TGF-β. Quantification of miRNAs was performed after generation of a standard curve as previously described for Fig. 1D. For this experiment, 100,000 MCF-10A cells were treated with TGF-β1 ligand (10 ng/ml) at different time points (Fig. 4A). These experiments revealed that the expression of miR-424(322) in MCF-10A increases to a few thousand molecules/cell after TGF-β exposure (Fig. 4A). As before, the mature form of miR-503 also increased proportionally after TGF-β treatment, but its steady-state levels were about a log of magnitude lower than the levels of miR-424(322). Next, we utilized our miR-424(322)/503 Dox-inducible system to artificially express mature miR-424(322) and miR-503 to the levels found after TGF-β exposure. Treatment of Dox-inducible cells with 100 ng/ml of Dox for 5 days generated a mature miRNA expression that was comparable to the levels observed after treating cells with TGF-β1 ligand for 72 h (Fig. 1D). Importantly, Western blot studies showed that this level of miRNA expression was able to reduce CDC25A protein levels by 30 to 50% (Fig. 1E).
FIG 4.

Increased miR-424/503 expression levels impair the G1/S transition and compromise cell proliferation. (A) Estimation of the numbers of molecules of miR-424 and miR-503 per cell during TGF-β exposure at several time points in MCF-10A cells as described in the main text. (B to D) MCF-10A and MCF-12A cells were lentivirally transduced with a control (pTRIPz-Scrambled) or with a Dox-inducible vector expressing miR-424(322)/503 (pTRIPz-miR-424/503). The cells were treated for 3 days with 100 ng/ml of doxycycline (+Dox) or left untreated for the same time (−Dox). Cell proliferation was then monitored for 72 extra hours using the CellTiter-Glo system. The results indicate that the doxycycline-induced miR-424/503 impairs cell proliferation due to accumulation of cells in G1 phase (C) and a reduction of BrdU-positive cells (D). The y axis in cell cycle graphics represents the percentage of cells in G1 when levels of the miRNA cluster are high (DOX+) minus the percentage of cells in G1 when levels of miRNA cluster are low (DOX−). The same is applicable for the graphics representing BrdU incorporation data. (E) Analysis by Western blotting of pT-miR-424/503-MCF-10A cells transduced with lentiviral particles containing a control vector (pLOC-Scr) or nontargetable pLOC-CDC25A devoid of the 3′ UTR. (F and G) Expression of nontargetable CDC25A in Dox-treated pT-424/503-MCF-10A cells attenuates the G1 arrest imposed by miR-424/503 (F) and the decrease in BrdU-positive cells (G). (H) Representative Western blot showing that the expression of the nontargetable CDC25A form ensures progression into mitosis in Dox-treated pT-424/503-MCF-10A cells as measured by p-histone 3.
To study the impact of this downregulation on cell cycle progression, we performed four complementary studies comparing cells with low versus high levels of mature miRNAs (−Dox versus +Dox). We performed cell proliferation (Fig. 4B), cell cycle analysis (Fig. 4C), and measurement of DNA synthesis (BrdU incorporation) (Fig. 4D) studies, and we also evaluated the presence of cells in mitosis by histone-3 phosphorylation at Ser10 (p-histone 3) (Fig. 4H). For these studies, unsynchronized and exponentially growing Dox-inducible MCF-10A control (pTRIPz-Scrambled) and miR-424(322)/503-expressing cells (pTRIPz-miR-424(322)/503) were treated with doxycycline (100 ng/ml) for 5 days before processing. For measurement of DNA synthesis, cells were exposed to BrdU for 4 h before being processed.
Our data showed that reduction of CDC25A protein levels mediated by the upregulation of miR-424(322)/503 reduces cell proliferation. This reduction was associated with the accumulation of cells in G1 and loss of replicative potential, as shown by the reduction of BrdU incorporation and p-histone 3. Similar downregulation of CDC25A led to G1 arrest and loss of proliferative potential when miR-424(322)/503 was upregulated in another nontransformed mammary epithelial cell line model (MCF-12A) (Fig. 4B to D and data not shown). Importantly, to demonstrate that this effect was mediated by the downregulation of CDC25A, we engineered our inducible model to stably express a nontargetable version of CDC25A lacking the 3′ UTR using lentiviral vectors (Fig. 4E). Despite the upregulation of the miRNA cluster after Dox addition, these cells presented cell cycle and BrdU profiles more similar to those of wild-type cells as well as an increased percentage of cells in mitosis (Fig. 4F to H).
To complement our studies, we compared cell proliferation (Fig. 5A) as well as measurement of DNA synthesis (Fig. 5B) in Tert-immortalized wild-type and miR-424−/−/miR-503−/− MMECs. As before, unsynchronized and exponentially growing cells were analyzed. For measurement of DNA synthesis, cells were exposed to BrdU for 4 h before being processed. As shown before, Tert-immortalized KO cells presented with higher levels of CDC25A (Fig. 2C). Notably, these cells also presented with lower doubling times (Fig. 5A) associated with higher DNA synthesis (Fig. 5B).
FIG 5.
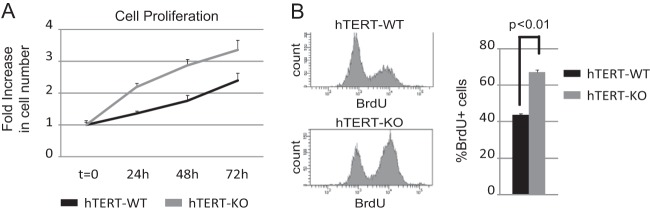
miR-424−/−/miR-503−/− mammary epithelial cells show increased proliferative potential. Tert-immortalized miR-424−/−/miR-503−/[minus] cells display increased basal proliferative potential as measured by Cell-Titer-Glo (A), accompanied by increased BrdU incorporation ratios (B).
Targeting of CDC25A by miR-424(322)/503 is necessary for the cytostatic response mediated by TGF-β in mammary epithelial cells in vitro.
Next, we investigated the role for the upregulation of miR-424(322)/503 during the cytostatic response mediated by TGF-β. Here we studied both CDC25A downregulation and G1 cell cycle arrest induced by TGF-β when the upregulation of the mature miRNAs was abrogated.
In MCF-10A cells, miRNA upregulation was prevented by transfecting miRIDIAN hairpin inhibitors (100 nM) into cells 24 h prior to TGF-β stimulation (Fig. 6A). Then, miR-424(322)/503 expression was induced by exposure to TGF-β1 ligand during 24 h. After that time, cells were collected and the levels of mature miR-424(322), miR-503, and CDC25A were evaluated. Additionally, we also performed cell cycle analysis, BrdU incorporation, and p-histone 3 studies in these cells. All these studies were started with unsynchronized and exponentially growing cells, and cells were exposed to BrdU for 4 h before being processed for measurement of DNA synthesis. As previously described (6, 10, 11), activation of TGF-β signaling induced downregulation of CDC25A levels (Fig. 6B) concomitantly with accumulation of cells in G1 as well as reduction in p-histone 3 and DNA synthesis (Fig. 6B to D). Importantly, all these effects were attenuated when the levels of mature miR-424(322) and miR-503 were reduced by hairpin inhibitors (Fig. 6B to D).
FIG 6.

MiR-424/503 inhibition attenuates TGF-β cytostatic effects. (A) MCF-10A cells were transfected with miRIDIAN Scrambled (Scr) or hairpin inhibitors (M.H.I.) for miR-424(322) and miR-503 and treated with TGF-β-1 ligand for 24 h (A, upper panel). Transfection of M.H.I. results in reduction of the levels of both miRNAs as measured by qRT-PCR analysis (A, bottom panel). (B) Analysis by Western blotting shows that M.H.I.-transfected cells are able to rescue the decrease in CDC25A protein levels after TGF-β treatment. Numeric values show densitometry quantification of the bands. Western blot analysis also shows reentry into mitosis as measured by p-histone 3. (C and D) Mir-424(322) and miR-503 knockdown cells show attenuated arrest in G1 phase (C) and a decrease in BrdU-positive cells (D) imposed by TGF-β. (E) Western blot analysis shows that both wild-type and miR-424−/−/miR-503−/− hTERT-MECs respond to TGF-β by activating p-SMAD2 and p-SMAD3 at similar levels. (F) Analysis by Western blotting shows that miR-424−/−/miR-503−/− hTERT-MECs are not able to downregulate CDC25A protein levels in response to TGF-β to the levels observed in their wild-type counterparts. Immunostaining for p-histone 3 indicates that the transition into mitosis is compromised in wild-type hTERT-MECs but not in miR-424−/−/miR-503−/− cells after TGF-β exposure. (G and H) Analysis of the percentage of cells in G1 phase (G) and BrdU incorporation ratios (H) indicates that miR-424−/−/miR-503−/− hTERT-MECs overcome growth-inhibitory effects induced by TGF-β exposure. In panels C, D, G, and H, the data are presented as the ratio between results for TGF-β-treated cells (TGFβ) and those for untreated cells (UN). All the experiments were performed in triplicate.
For a more comprehensive analysis, we also compared the response to TGF-β in Tert-immortalized WT and miR-424−/−/miR-503−/− cells. First, we studied the activation of the canonical TGF-β pathway in these cells by evaluating by Western blotting the levels of phospho-SMAD2 and -3 after addition of TGF-β1 ligand (10 ng/ml) to the medium. WT and KO cells presented comparable levels of R-SMAD activation (Fig. 6E). Next, we investigated the reduction in CDC25A as well as cell cycle analysis, BrdU incorporation, and p-histone 3 in these cells. All these studies were started with unsynchronized and exponentially growing cells. Here, 10 ng/ml of TGF-β1 was added to the medium for 24 h, and the cells were exposed to BrdU for 4 h before being processed for measurement of DNA synthesis. While the response of Tert-immortalized WT cells to TGF-β was similar to that of parental MCF-10A cells, miR-322/503-KO cells showed both higher steady-state levels and inefficient downregulation of CDC25A (Fig. 6F). Not surprisingly, miR-322/503-KO cells also displayed a reduced cytostatic response to TGF-β (Fig. 6F to H).
Inhibition of TGF-β signaling during mammary involution reduces miR-424(322)/503 expression and increases CDC25A protein expression and proliferation.
Finally, we transitioned our studies to an in vivo context. TGF-β ligands impact mammary epithelial development mainly as potent inhibitors of proliferation (2, 3). Since peak expression of miR-424(322)/503 occurs during involution, we first studied the TGF-β-miR-424(322)/503-CDC25A axis at this stage. During early and late involution phases, TGF-β3 plays an important role in regulating regression of the mammary epithelium to a prelactation stage (32, 33). We have recently described that inhibition of TGF-β signaling during involution by small-molecule inhibitors prevents miR-424(322)/503 upregulation and delays involution (15). Here, we expanded our studies to investigate the effect that blockage of TGF-β signaling has on CDC25A expression. When lactating females were treated with the TGF-β receptor type I/II (TβRI/II) dual inhibitor LY2109761 (34, 35), 100 mg/kg twice a day for 48 h after weaning, they showed an evident inhibition of involution as indicated by carmine red staining of the mammary gland and qRT-PCR levels of milk proteins (Fig. 7A and B). As expected, the upregulation of miR-424(322) and miR-503 was significantly attenuated (Fig. 7C). Importantly, Western blots of purified mammary epithelial cells also revealed that the expression of CDC25A was higher in LY2109761-treated cells (Fig. 7D). Histological analysis of mammary glands from miR-424(322)/503-KO females (10 days after weaning) revealed that positive staining for proliferation-associated markers (Ki-67 and p-histone 3 staining) was higher than that observed in the WT epithelium (Fig. 7E and F). Interestingly, follow-up studies of the mammary glands of KO animals at 60 days after weaning revealed that although these animals underwent complete involution, their mammary epithelium still presented higher levels of proliferation than that of WT counterparts (Fig. 7F).
FIG 7.

Inhibition of TGF-β signaling during mammary involution abrogates the induction of miR-424 and -503 expression. (A and B) Administration of the TβRI/II specific inhibitor LY2109761 partially inhibits mammary epithelial regression as measured by whole-mount carmine red staining (A) and quantification by qRT-PCR of the milk proteins α-lactalbumin (LALBA), whey-acidic protein (WAP), and β-casein (CSN2) in purified mammary epithelial cells (B). (C) Pharmacological inhibition of the TGF-β pathway abrogated the induction of miR-424 and -503 expression levels. The red line indicates the number of molecules observed for miR-424 and -503 during lactation. (D) Western blot showing increased protein levels of CDC25A in mammary epithelial cells treated with LY2109761. (E) Immunohistochemistry studies performed with mammary glands at 10 days of involution by using hematoxylin and eosin (H&E), Ki67, and p-histone 3 in wild-type and miR-424−/−/miR-503−/− females. These studies show a nearly complete regression in wild-type glands, while miR-424−/−/miR-503−/− females exhibit a delayed involuting phenotype coupled with an increased proliferative activity, as shown by increase in Ki67- and p-histone 3-positive cells. (F) Quantification of percentages of Ki67- and p-histone 3-positive cells in mammary glands at 10 days and 60 days postinvolution. All the experiments were performed in triplicate. A minimum of three animals were analyzed. For quantification of percentages of Ki67- and p-histone 3-positive cells, a minimum of 3,000 cells were counted per animal.
Targeting of CDC25A by miR-424(322)/503 is necessary for the cytostatic response mediated by TGF-β in luminal HR+ mammary epithelial cells in vivo.
In the normal mammary gland, luminal hormone receptor-positive (HR+) cells secrete growth factors to induce the growth of neighboring cells in a paracrine fashion. However, they are restrained from proliferating by activation of TGF-β signaling (17, 18). It has been proposed that the reduction in CDC25A expression mediated by TGF-β is responsible, at least in part, for making HR+ cells insensitive to proliferative stimuli (17). Thus, we hypothesized that activation of TGF-β signaling in luminal/HR+ mammary epithelial cells induces the expression of miR-424(322)/503, which in turn reduces the expression of CDC25A and inhibits proliferation. The lack of miR-424(322)/503 may compromise the inhibitory function of TGF-β and promote hyperproliferation of luminal/HR+ cells.
In order to investigate this model, we first purified three epithelial populations from WT and KO mammary glands: basal, luminal/HR+, and luminal/HR− cells. Then, we evaluated the level of mature miR-424(322) and miR-503, as well as the expression of CDC25A. Purification of these three populations was performed as described previously (22). In brief, after extraction of all mammary cells from female mammary glands at the estrus phase, nonepithelial cells were removed by incubation of single-cell suspensions with CD31-biotin, CD-45–biotin, Ter119-biotin, and BP-1–biotin. The remaining epithelial cells were separated by fluorescence-activated cell sorting (FACS) into three populations enriched in basal (EpCAMlo CD49fhi), luminal/HR− (EpCAMhi CD49flo Sca1lo), and luminal/HR+ (EpCAMhi CD49flo Sca1hi) subtypes (Fig. 8A). Immunofluorescence staining of estrogen receptor α in the purified cells demonstrated an enrichment in HR+ cell of >85% in the EpCAMhi CD49flo Sca1hi group and an enrichment of >90% in HR− cells in the EpCAMhi CD49flo Sca1lo group (Fig. 8B). Remarkably, luminal/HR+ cells presented the highest expression of mature miR-424(322) and miR-503 and the lowest levels of CDC25A (Fig. 8C).
FIG 8.

The miR-424/503 cluster is prominently expressed in luminal/ER+ cells, and its absence promotes hyperproliferation of the hormone-sensitive (HR+) cellular compartment. (A) Murine mammary epithelial cells were purified and fluorescently activated cell (FAC) sorted into different subpopulations using specific epithelial markers: basal (EpCAMlo CD49fhi), luminal/HR− (EpCAMhi CD49flo Sca1lo), and luminal/HR+ (EpCAMhi CD49flo Sca1hi). Relative acquisition of HR− and HR+ cells is expressed as a percentage of the total luminal subpopulation. (B) Immunofluorescence staining of the estrogen receptor α (ER) and quantification graphic showing the percentage of HR+ cells in the purified luminal subpopulations. Cells were sorted, fixed on slides, and stained as described in Materials and Methods. (C) MiR-424 and -503 expression levels were analyzed by qRT-PCR in the three mammary epithelial subpopulations. The Western blot shows the protein expression level of endogenous CDC25A in the purified luminal compartments. (D) The graphic shows the fold change in the number of Ki-67+ cells, comparing WT versus miR-424−/−/miR-503−/− virgin females at estrus for purified basal, luminal/HR+, and luminal/HR− cells. The three mammary epithelial subpopulations were purified as described in the text, and the fraction of Ki-67+ cells was determined by immunofluorescence staining. All the experiments were performed in triplicate. A minimum of three animals were analyzed. For quantification of percentages of Ki67-positive cells, a minimum of 3,000 cells were counted per animal.
Next, we investigated if the absence of miR-424(322)/503 in our knockout females had any impact on the proliferative fraction of HR+ cells. For this, we used the same FACS approach described above to purify basal, luminal/HR−, and luminal HR+ cells from WT and miR-424(322)/503-KO virgin females at estrus. Then, we performed immunofluorescence staining for Ki-67 in each of the purified cell groups. These studies revealed a statistically significant increase in the fraction of Ki67+ cells in both luminal/HR+ and luminal/HR− cells between 424(322)/503-KO animals and WT counterparts, while there were not significant differences between basal cell subpopulations (Fig. 8D).
DISCUSSION
TGF-β signaling exerts a major antiproliferative effect during epithelial mammary gland development (2, 3). Repression of CDC25A (6) by canonical TGF-β signaling has been shown to be crucial for the inhibition of the G1/S transition and the induction of cell cycle arrest. Downregulation of the protein levels of CDC25A in response to TGF-β occurs at both the transcriptional (10) and posttranslational (11) levels, but to date no evidence has been reported that demonstrates a role for miRNA-mediated silencing in this process.
We have previously reported that canonical TGF-β signaling upregulates the expression of miR-424(322)/503 due to the presence of SMAD-binding boxes (36) in the miR-424(322)/503 promoter region (15). Here we have shown that the CDC25A 3′ UTR contains two active binding sites for this miRNA cluster that mediate its downregulation. In general, miRNAs in mammals do not induce endonucleolytic cleavage and complete loss of gene expression. Instead, they block translation and increase mRNA instability to attenuate target expression levels (37, 38). This is also the case for miR-424(322)/503 and CDC25A. Using a reporter system to evaluate the transcriptional control mediated by the CDC25A promoter and miR-424(322)/503-3′ UTR sites, we found that upregulation of miR-424(322)/503 accounted for an additional 30% reduction in gene expression during TGF-β exposure. Our reporter system does not include the regulatory elements that modulate proteasome-mediated degradation of CDC25A. Consequently, the apparent ability of miR-424(322)/503 to reduce the expression of CDC25A could be biased. However, our data showed that downregulation of the CDC25A protein during TGF-β exposure was partially blocked when upregulation of miR-424 (322) and miR-503 was prevented, demonstrating the active role of these microRNAs in regulating CDC25A protein levels.
Importantly, our data also demonstrate that the reduction in CDC25A expression mediated by miR-424(322)/503 was biologically relevant. Experimental manipulation to increase the level of the miRNA cluster was able to induce cell cycle arrest dependent on CDC25A. On the other hand, cells in which the upregulation of miR-424(322)/503 was prevented/abrogated showed deficient cell cycle arrest in vitro and presented higher proliferation during TGF-β exposure in vivo.
In vivo, mammary epithelial cells expressing steroid receptors (HR+) are located in the luminal compartment, and they are rarely associated with markers of proliferation (16, 17). While these cells secrete growth factors that act on neighboring cells (HR−) to induce proliferation in a paracrine fashion, they remain insensitive to these stimuli (17). Cell biology and genetic studies have provided evidence that TGF-β activation functionally restrains HR+ cells from proliferating (18). Receptor-phosphorylated Smad2/3 are found in the nucleus in luminal/HR+ cells. Mice deficient in TGF-β1 expression presented an increase of colocalization of estrogen receptor (ERα) and proliferation markers. Furthermore, expression of constitutively active TGF-β1 was able to suppress proliferation of luminal/HR+ cells (18).
Our studies show that expression of miR-424(322)/503 is high in luminal/HR+ cells and anticorrelates with CDC25A protein levels when luminal HR+ and HR− cells are compared. Importantly, the absence of miR-424(322)/503 augments the number of luminal/HR+ cells associated with proliferation markers, while no differences were observed in the basal subpopulation (Fig. 7D). It is interesting that, although to a less extent, we also observed an increase in the number of Ki67+ luminal/HR− cells. Although the expression of miR-424(322) and miR-503 is lower in luminal/HR− cells than in luminal/HR+ cells, the increase in Ki-67+ cells could indicate that a role for miR-424(322)/503 in regulating cell proliferation also exists for this group. Alternatively, it could be mediated by an expansion of the luminal/HR+ cells, which provide paracrine proliferative stimuli. Overall, our data suggest that in vivo, miR-424(322)/503 is an integral part of the TGF-β pathway, involved in the homeostasis of luminal/HR+ cells due to its ability to modulate CDC25A expression.
A final consideration is the potential link between miR-424(322)/503 and tumorigenesis. Several members of the miR-16 family have been shown to be involved in human cancers (24, 39, 40). Furthermore, the role of CDC25 family members in the context of cancer is well known (9). Interestingly, miR-424(322)/503 is located in a chromosomal region that has been recently identified to present significant deletions in ∼8% of HR+ breast cancers (41). Thus, one unanswered question is whether loss of expression of miR-424(322)/503 can be involved in breast tumorigenesis and whether this can be linked to deregulation of CDC25A in luminal HR+ cells. Although the answer to this question is still unknown, it is interesting that both miR-424(322)/503-KO female (15) and transgenic mice with ectopic expression of CDC25A (42) develop alveolar hyperplasia, associated with increased proliferation and reduced apoptosis. Additional investigations will be required to ascertain whether these lesions progress to carcinomas or whether loss of miR-424(322)/503 expression cooperates with other genetic alteration to promote tumorigenesis. Additionally, it will be important to define the origin of any potential tumor phenotype (defects in involution, hyperproliferation of luminal/HR+ cells, both, or other reasons), as well as which of the miRNA targets are involved in it. Further research will be necessary to fully define any potential role of miR-424(322)/503 in breast tumorigenesis.
Overall, our studies have demonstrated that CDC25A is targeted by miR-424(322)/503 in mammary epithelial cells and have uncovered the upregulation of this microRNA cluster as part of the multilayered mechanism involved in the efficient downregulation of CDC25A during TGF-β-mediated cytostasis (Fig. 9).
FIG 9.

Mechanistic model for miR-424/503 during TGF-β-mediated cytostasis in mammary epithelial cells. (A) General overview of the proposed molecular mechanistic model. When stimulated with TGF-β, mammary epithelial cells activate a signal transduction cascade triggered by SMAD proteins that aim to inhibit cell cycle progression by arresting cells in G1 phase. Induced expression of antiproliferative p15INK4B and the repression of c-Myc and CDC25A are well-known key factors associated with cell cycle arrest in response to TGF-β. We describe the upregulation of miR-424(322) and miR-503 by TGF-β as a novel mechanism that cooperates with transcriptional repression and proteasome-mediated degradation to reduce the expression level of CDC25A. (B) Proposed miR-424/503 functional activity at a cellular level. Under normal conditions, luminal/HR+ cells can foster proliferation of surrounding epithelial cells (ER−) through the secretion of locally acting growth factors. However, luminal/HR+ cells are themselves restrained from proliferation through an autocrine TGF-β cytostatic program that involves downregulation of CDC25A in part due to the increased expression of miR-424 and -503. In their absence, the inhibitory function of TGF-β is reduced, and the resulting increased CDC25A protein levels promote hyperproliferation of luminal/HR+ cells.
ACKNOWLEDGMENTS
This work was supported by Programa Beatriu de Pinós (BP-DGR 2011).
We thank Andi Molin and Emily Vasiliou for technical assistance.
Footnotes
Published ahead of print 29 September 2014
REFERENCES
- 1.Sundqvist A, Ten Dijke P, van Dam H. 2012. Key signaling nodes in mammary gland development and cancer: Smad signal integration in epithelial cell plasticity. Breast Cancer Res. 14:204. 10.1186/bcr3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Howard BA, Gusterson BA. 2000. Human breast development. J. Mammary Gland Biol. Neoplasia 5:119–137. 10.1023/A:1026487120779. [DOI] [PubMed] [Google Scholar]
- 3.Macias H, Hinck L. 2012. Mammary gland development. Wiley Interdiscip. Rev. Dev. Biol. 1:533–557. 10.1002/wdev.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Massague J. 2008. TGFbeta in cancer. Cell 134:215–230. 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sandhu C, Garbe J, Bhattacharya N, Daksis J, Pan CH, Yaswen P, Koh J, Slingerland JM, Stampfer MR. 1997. Transforming growth factor beta stabilizes p15INK4B protein, increases p15INK4B-cdk4 complexes, and inhibits cyclin D1-cdk4 association in human mammary epithelial cells. Mol. Cell. Biol. 17:2458–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iavarone A, Massague J. 1997. Repression of the CDK activator Cdc25A and cell-cycle arrest by cytokine TGF-beta in cells lacking the CDK inhibitor p15. Nature 387:417–422. 10.1038/387417a0. [DOI] [PubMed] [Google Scholar]
- 7.Chen CR, Kang Y, Massague J. 2001. Defective repression of c-myc in breast cancer cells: A loss at the core of the transforming growth factor beta growth arrest program. Proc. Natl. Acad. Sci. U. S. A. 98:992–999. 10.1073/pnas.98.3.992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim WY, Sharpless NE. 2006. The regulation of INK4/ARF in cancer and aging. Cell 127:265–275. 10.1016/j.cell.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 9.Boutros R, Lobjois V, Ducommun B. 2007. CDC25 phosphatases in cancer cells: key players? Good targets? Nat. Rev. Cancer 7:495–507. 10.1038/nrc2169. [DOI] [PubMed] [Google Scholar]
- 10.Iavarone A, Massague J. 1999. E2F and histone deacetylase mediate transforming growth factor beta repression of cdc25A during keratinocyte cell cycle arrest. Mol. Cell. Biol. 19:916–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ray D, Terao Y, Nimbalkar D, Chu LH, Donzelli M, Tsutsui T, Zou X, Ghosh AK, Varga J, Draetta GF, Kiyokawa H. 2005. Transforming growth factor beta facilitates beta-TrCP-mediated degradation of Cdc25A in a Smad3-dependent manner. Mol. Cell. Biol. 25:3338–3347. 10.1128/MCB.25.8.3338-3347.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Caporali A, Meloni M, Vollenkle C, Bonci D, Sala-Newby GB, Addis R, Spinetti G, Losa S, Masson R, Baker AH, Agami R, le Sage C, Condorelli G, Madeddu P, Martelli F, Emanueli C. 2011. Deregulation of microRNA-503 contributes to diabetes mellitus-induced impairment of endothelial function and reparative angiogenesis after limb ischemia. Circulation 123:282–291. 10.1161/CIRCULATIONAHA.110.952325. [DOI] [PubMed] [Google Scholar]
- 13.Zhao H, Wang J, Gao L, Wang R, Liu X, Gao Z, Tao Z, Xu C, Song J, Ji X, Luo Y. 2013. MiRNA-424 protects against permanent focal cerebral ischemia injury in mice involving suppressing microglia activation. Stroke 44:1706–1713. 10.1161/STROKEAHA.111.000504. [DOI] [PubMed] [Google Scholar]
- 14.Sarkar S, Dey BK, Dutta A. 2010. MiR-322/424 and -503 are induced during muscle differentiation and promote cell cycle quiescence and differentiation by down-regulation of Cdc25A. Mol. Biol. Cell 21:2138–2149. 10.1091/mbc.E10-01-0062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Llobet-Navas D, Rodriguez-Barrueco R, Castro V, Ugalde AP, Sumazin P, Jacob-Sendler D, Demircan B, Castillo-Martin M, Putcha P, Marshall N, Villagrasa P, Chan J, Sanchez-Garcia F, Pe'er D, Rabadan R, Iavarone A, Cordon-Cardo C, Califano A, Lopez-Otin C, Ezhkova E, Silva JM. 2014. The miR-424(322)/503 cluster orchestrates remodeling of the epithelium in the involuting mammary gland. Genes Dev. 28:765–782. 10.1101/gad.237404.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clarke RB, Howell A, Potten CS, Anderson E. 1997. Dissociation between steroid receptor expression and cell proliferation in the human breast. Cancer Res. 57:4987–4991. [PubMed] [Google Scholar]
- 17.Grimm SL, Rosen JM. 2006. Stop! In the name of transforming growth factor-beta: keeping estrogen receptor-alpha-positive mammary epithelial cells from proliferating. Breast Cancer Res. 8:106. 10.1186/bcr1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ewan KB, Oketch-Rabah HA, Ravani SA, Shyamala G, Moses HL, Barcellos-Hoff MH. 2005. Proliferation of estrogen receptor-alpha-positive mammary epithelial cells is restrained by transforming growth factor-beta1 in adult mice. Am. J. Pathol. 167:409–417. 10.1016/S0002-9440(10)62985-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salmon P, Kindler V, Ducrey O, Chapuis B, Zubler RH, Trono D. 2000. High-level transgene expression in human hematopoietic progenitors and differentiated blood lineages after transduction with improved lentiviral vectors. Blood 96:3392–3398. [PubMed] [Google Scholar]
- 20.Tang F, Hajkova P, Barton SC, Lao K, Surani MA. 2006. MicroRNA expression profiling of single whole embryonic stem cells. Nucleic Acids Res. 34:e9. 10.1093/nar/gnj009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee DH, Nguyen JT, Barbisin M, Xu NL, Mahuvakar VR, Andersen MR, Lao KQ, Livak KJ, Guegler KJ. 2005. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 33:e179. 10.1093/nar/gni178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shehata M, Teschendorff A, Sharp G, Novcic N, Russell A, Avril S, Prater M, Eirew P, Caldas C, Watson CJ, Stingl J. 2012. Phenotypic and functional characterization of the luminal cell hierarchy of the mammary gland. Breast Cancer Res. 14:R134. 10.1186/bcr3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M, Jr, Jungkamp AC, Munschauer M, Ulrich A, Wardle GS, Dewell S, Zavolan M, Tuschl T. 2010. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 141:129–141. 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu Q, Fu H, Sun F, Zhang H, Tie Y, Zhu J, Xing R, Sun Z, Zheng X. 2008. miR-16 family induces cell cycle arrest by regulating multiple cell cycle genes. Nucleic Acids Res. 36:5391–5404. 10.1093/nar/gkn522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Finnerty JR, Wang WX, Hebert SS, Wilfred BR, Mao G, Nelson PT. 2010. The miR-15/107 group of microRNA genes: evolutionary biology, cellular functions, and roles in human diseases. J. Mol. Biol. 402:491–509. 10.1016/j.jmb.2010.07.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. 2007. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol. Cell 27:91–105. 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lewis BP, Burge CB, Bartel DP. 2005. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120:15–20. 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 28.Shirdel EA, Xie W, Mak TW, Jurisica I. 2011. NAViGaTing the micronome—using multiple microRNA prediction databases to identify signalling pathway-associated microRNAs. PLoS One 6:e17429. 10.1371/journal.pone.0017429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karakas B, Weeraratna A, Abukhdeir A, Blair BG, Konishi H, Arena S, Becker K, Wood W, III, Argani P, De Marzo AM, Bachman KE, Park BH. 2006. Interleukin-1 alpha mediates the growth proliferative effects of transforming growth factor-beta in p21 null MCF-10A human mammary epithelial cells. Oncogene 25:5561–5569. 10.1038/sj.onc.1209540. [DOI] [PubMed] [Google Scholar]
- 30.Debnath J, Muthuswamy SK, Brugge JS. 2003. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods 30:256–268. 10.1016/S1046-2023(03)00032-X. [DOI] [PubMed] [Google Scholar]
- 31.Nilsson I, Hoffmann I. 2000. Cell cycle regulation by the Cdc25 phosphatase family. Prog. Cell Cycle Res. 4:107–114. 10.1007/978-1-4615-4253-7_10. [DOI] [PubMed] [Google Scholar]
- 32.Nguyen AV, Pollard JW. 2000. Transforming growth factor beta3 induces cell death during the first stage of mammary gland involution. Development 127:3107–3118. [DOI] [PubMed] [Google Scholar]
- 33.Gorska AE, Jensen RA, Shyr Y, Aakre ME, Bhowmick NA, Moses HL. 2003. Transgenic mice expressing a dominant-negative mutant type II transforming growth factor-beta receptor exhibit impaired mammary development and enhanced mammary tumor formation. Am. J. Pathol. 163:1539–1549. 10.1016/S0002-9440(10)63510-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bouquet F, Pal A, Pilones KA, Demaria S, Hann B, Akhurst RJ, Babb JS, Lonning SM, DeWyngaert JK, Formenti SC, Barcellos-Hoff MH. 2011. TGFbeta1 inhibition increases the radiosensitivity of breast cancer cells in vitro and promotes tumor control by radiation in vivo. Clin. Cancer Res. 17:6754–6765. 10.1158/1078-0432.CCR-11-0544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Melisi D, Ishiyama S, Sclabas GM, Fleming JB, Xia Q, Tortora G, Abbruzzese JL, Chiao PJ. 2008. LY2109761, a novel transforming growth factor beta receptor type I and type II dual inhibitor, as a therapeutic approach to suppressing pancreatic cancer metastasis. Mol. Cancer Ther. 7:829–840. 10.1158/1535-7163.MCT-07-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnson K, Kirkpatrick H, Comer A, Hoffmann FM, Laughon A. 1999. Interaction of Smad complexes with tripartite DNA-binding sites. J. Biol. Chem. 274:20709–20716. [DOI] [PubMed] [Google Scholar]
- 37.Ameres SL, Zamore PD. 2013. Diversifying microRNA sequence and function. Nat. Rev. Mol. Cell. Biol. 14:475–488. 10.1038/nrm3611. [DOI] [PubMed] [Google Scholar]
- 38.Guo H, Ingolia NT, Weissman JS, Bartel DP. 2010. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466:835–840. 10.1038/nature09267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aqeilan RI, Calin GA, Croce CM. 2010. miR-15a and miR-16-1 in cancer: discovery, function and future perspectives. Cell Death Differ. 17:215–220. [DOI] [PubMed] [Google Scholar]
- 40.Aqeilan RI, Calin GA, Croce CM. 2010. miR-15a and miR-16-1 in cancer: discovery, function and future perspectives. Cell Death Differ. 17:215–220. 10.1038/cdd.2009.69. [DOI] [PubMed] [Google Scholar]
- 41.Dvinge H, Git A, Graf S, Salmon-Divon M, Curtis C, Sottoriva A, Zhao Y, Hirst M, Armisen J, Miska EA, Chin SF, Provenzano E, Turashvili G, Green A, Ellis I, Aparicio S, Caldas C. 2013. The shaping and functional consequences of the microRNA landscape in breast cancer. Nature 497:378–382. 10.1038/nature12108. [DOI] [PubMed] [Google Scholar]
- 42.Ray D, Terao Y, Fuhrken PG, Ma ZQ, DeMayo FJ, Christov K, Heerema NA, Franks R, Tsai SY, Papoutsakis ET, Kiyokawa H. 2007. Deregulated CDC25A expression promotes mammary tumorigenesis with genomic instability. Cancer Res. 67:984–991. 10.1158/0008-5472.CAN-06-3927. [DOI] [PubMed] [Google Scholar]