

Abstract
The bacterial second messenger cyclic di-GMP (c-di-GMP) stimulates inflammation by initiating innate immune cell recruitment and triggering the release of proinflammatory cytokines and chemokines. These properties make c-di-GMP a promising candidate for use as a vaccine adjuvant, and numerous studies have demonstrated that administration of purified c-di-GMP with different antigens increases protection against infection in animal models. Here, we have developed a novel approach to produce c-di-GMP inside host cells as an adjuvant to exploit a host-pathogen interaction and initiate an innate immune response. We have demonstrated that c-di-GMP can be synthesized in vivo by transducing a diguanylate cyclase (DGC) gene into mammalian cells using an adenovirus serotype 5 (Ad5) vector. Expression of DGC led to the production of c-di-GMP in vitro and in vivo, and this was able to alter proinflammatory gene expression in murine tissues and increase the secretion of numerous cytokines and chemokines when administered to animals. Furthermore, coexpression of DGC modestly increased T-cell responses to a Clostridium difficile antigen expressed from an adenovirus vaccine, although no significant differences in antibody titers were observed. This adenovirus c-di-GMP delivery system offers a novel method to administer c-di-GMP as an adjuvant to stimulate innate immunity during vaccination.
INTRODUCTION
Cyclic di-GMP (c-di-GMP) is a bacterium-specific second messenger that controls a wide range of phenotypes, including motility, biofilm formation, and virulence (1). c-di-GMP was first discovered in 1987 by Benziman et al. (2) and since has been predicted to be utilized in >75% of all bacteria in representatives from every major bacterial phyla (3). Diguanylate cyclase (DGC) enzymes which contain conserved GGDEF domains synthesize c-di-GMP from two GTP molecules. In contrast, c-di-GMP is hydrolyzed by c-di-GMP-specific phosphodiesterase (PDE) enzymes which contain conserved EAL or HD-GYP domains (1). Bacteria typically contain numerous DGCs and PDEs within their genomes; for example, the marine bacterium Vibrio cholerae encodes 70 predicted c-di-GMP turnover domains (4).
Previous studies indicated that c-di-GMP is a potent stimulator of innate immunity in eukaryotic organisms. This occurs at least in part through the protein STING, which senses pathogen-derived nucleic acids in the cytoplasm and subsequently activates a signaling cascade to stimulate a type I interferon response (5, 6). Studies show that the presence of c-di-GMP can trigger the production of interleukin-2 (IL-2), IL-4, IL-5, IL-6, IL-8, IL-12p40, IL-17, IP-10, tumor necrosis factor alpha (TNF-α), keratinocyte-derived chemokine (KC), macrophage inflammatory protein 1α (MIP-1α), MIP-1β, MIP-2, monocyte chemotactic protein 1 (MCP-1), RANTES, beta interferon (IFN-β), and IFN-γ, stimulate the NLRP3 inflammasome pathway and promote the recruitment and activation of macrophages, NK cells, and αβ conventional T cells, and enhance dendritic cell (DC) maturation (6–14). Furthermore, in vivo studies have shown that coadministration of purified c-di-GMP with an antigen confers increased protection of animals in several different murine challenge models, including those utilizing Staphylococcus aureus, Klebsiella pneumoniae, and Streptococcus pneumoniae (10–12, 15).
Because c-di-GMP activates a robust immune response, there has been an ongoing focus on using c-di-GMP as an adjuvant to improve vaccine efficacy (16). Adjuvants are compounds administered alongside vaccine antigens for the purpose of enhancing the longevity and potency of the memory response or reducing the effective dose of the antigen without introducing toxic side effects. This is accomplished by stimulating the innate arm of the immune system, resulting in increased cytokine and chemokine production and upregulation of proinflammatory genes (17), which then enhances antigen recognition and response (18). The development of novel adjuvants may be critical to the success of vaccines targeting diseases for which vaccinations have previously failed such as those caused by Clostridium difficile and human immunodeficiency virus, malaria, and cancer. Despite the demand, currently there are few adjuvants approved for human use. The most commonly used adjuvants are aluminum salt (alum)-based; however, these adjuvants have drawbacks, including local reactions to administration, inadequate T-cell responses, and allergic IgE-type responses, and are ineffective with specific types of antigens (19). Other less commonly utilized adjuvants include oil and water emulsions, lipopolysaccharide derivatives, self-assembling viral nanoparticles, and cholera toxin B subunit (19). While each adjuvant offers different advantages and disadvantages, there is a large demand for novel adjuvants that can be paired with and improve vaccine antigens. Because c-di-GMP has effective immunostimulatory properties, it is a promising candidate as a novel adjuvant (16).
The studies examining the adjuvant properties of c-di-GMP utilized mucosal, intramuscular (IM), or systemic administration of chemically synthesized c-di-GMP (10–12, 15). While this method has been shown to effectively activate innate immunity, the mechanism by which c-di-GMP enters the cytoplasm of cells to stimulate STING is unknown as c-di-GMP has two phosphate groups that prevent it from easily passing through the cellular membrane. Consistent with this, it has been shown that c-di-GMP delivered with a liposome delivery system is more effective than c-di-GMP administered alone, suggesting that unmodified c-di-GMP does not efficiently enter host cells (20). Furthermore, chemical synthesis of pharmaceutical grade c-di-GMP in large quantities is costly. To bypass these limitations and provide another mechanism for delivery of c-di-GMP, we sought to synthesize c-di-GMP directly within host cells to stimulate innate immunity.
One vehicle that has been used for antigen and adjuvant delivery is replication-deficient adenovirus-based vectors (21). Adenovirus vectors transduce large fragments of DNA into a wide range of cells in order to synthesize proteins in vivo, and gene expression can be modulated and even localized to specific cell types. Unlike that for other types of viral delivery systems, the DNA delivered by adenovirus vectors does not integrate into the genome and thus the danger of insertional mutagenesis is circumvented (22). Adenovirus vectors have been shown to induce innate immunity, which is partially due to induction of the STING DNA recognition pathway (23). Additionally, adenovirus vectors can be produced cost efficiently in high abundance. Importantly, adenovirus vectors are currently being used in human clinical trials worldwide (24).
We investigated whether an adenovirus vector might be used to deliver a gene encoding a DGC into eukaryotic cells to synthesize c-di-GMP in vivo, manipulating this host-pathogen interaction to activate the innate arm of the immune system. We have constructed an adenovirus encoding the V. cholerae DGC VCA0956. We have shown that this DGC is capable of synthesizing c-di-GMP when virally transduced into cell culture lines. Furthermore, we have demonstrated that use of the VCA0956 adenovirus construct in a murine system results in c-di-GMP synthesis and subsequent upregulation of genes associated with innate immunity as well as increased secretion of numerous cytokines and chemokines. Finally, we have provided preliminary evidence that c-di-GMP produced by VCA0956 in vivo functions as an adjuvant by reducing the effective dose of a C. difficile toxin A (TA) antigen necessary to generate a T-cell response. Therefore, adenovirus vectors encoding DGCs offer a novel, alternative strategy to deliver c-di-GMP in vivo.
MATERIALS AND METHODS
All of the DNA manipulation and plasmid construction were performed as previously described (25). The VCA0956 gene was amplified from the Vibrio cholerae El tor strain C6706 using the DNA polymerase Phusion (New England BioLabs) and the oligonucleotides 5′-ATAGGTACCCCACCGTGATGACAACTGAAGATTTCA-3′ and 5′-ATACTCGAGTTAGAGCGGCATGACTCGAT-3′ (IDT). This product was then inserted into the plasmid pShuttle-CMV (cytomegalovirus) (26) by digestion with KpnI and XhoI (Fermentas) and ligated with a T4 DNA ligase (Invitrogen). Escherichia coli strain DH10B (Invitrogen) was used for harboring plasmid DNA, and sequence fidelity was confirmed by sequencing (Genewiz). The active site mutant allele was generated using the QuikChange Lightning site-directed mutagenesis kit (Agilent) with the primer 5′-TGACAGCTTATCGTTATGCCGCTGAAGAGTTTGCACTGAT-3′.
A first-generation, human adenovirus type 5 (Ad5) replication-deficient vector (deleted for the E1 and E3 genes) was used in this study (27). Recombination, viral propagation of the Ad5 vectors, and subsequent virus characterization were performed as previously described (27, 28). Viral particle (vp) numbers were determined by optical density measurement at 260 nm and validated as previously described (29). Construction of Ad5-null and Ad5-TA is described elsewhere (30, 31). All virus constructs were confirmed to be replication-competent adenovirus (RCA) negative using RCA PCR and direct sequencing methods (27), and the bacterial endotoxin content was found to be <0.15 endotoxin unit (EU)/ml (27). All procedures with recombinant adenovirus constructs were performed under BSL2 conditions.
All transfections of plasmid DNA into HeLa cells were performed with the TransIT-HeLaMonster transfection kit (Mirus) in 6-well plates with 2.5 μg plasmid DNA. For HeLa cell infections with adenovirus vectors, cells were infected with 2.0 × 109 viral particles (multiplicity of infection [MOI] of 500). Cell cultures were checked for confluence and morphology before and after transfection and for infection using microscopy. After 24 h of growth at 37°C in 5% CO2, the cells were dissociated using 300 μl 0.25% trypsin, and then cells were resuspended in 4 ml phosphate-buffered saline (PBS) and pelleted by centrifugation at 1,600 rpm at 4°C. Afterward, the cells were resuspended in 100 μl extraction buffer (40% acetonitrile, 40% methanol, and 0.1 N formic acid). The cell lysate was incubated at −20°C for 30 min and then centrifuged at the maximum speed for 10 min. The extraction buffer was removed from the pelleted debris and stored at −80°C until analysis.
Immediately prior to analysis, the extraction buffer was evaporated using a vacuum manifold, and the samples were rehydrated in 100 μl water. c-di-GMP was quantified using an Acquity ultraperformance liquid chromatography system (Waters) coupled with a Quattro Premier XE mass spectrometer (Waters) as previously described (32). The concentration of c-di-GMP was determined by generating an 8-point standard curve (1:2 dilutions) of chemically synthesized c-di-GMP (Biolog) ranging from 1.9 to 250 nM. The intracellular concentration was estimated by dividing the total molar amount of c-di-GMP extracted by the estimated total intracellular volume of HeLa cells extracted using cell counts, and size measurements were determined with a Countess automated cell counter (Life Technologies). The transfection efficiency was determined to be 18.2%, which was obtained by transfecting HeLa cells with a plasmid containing green fluorescent protein (GFP) under CMV promoter control and measuring the percentage of GFP-positive cells using flow cytometry. The infection efficiency of HeLa cells was determined to be 82.2% by infecting HeLa cells with Ad5-gfp (28) and quantifying the percentage of GFP-positive cells using flow cytometry.
Adult BALB/c wild-type (WT) male mice (6 to 8 weeks old) were used for all animal experiments (The Jackson Laboratory). For c-di-GMP quantification and innate studies, mice were anesthetized using isoflurane, and 2 × 1011 adenovirus vp per mouse (200 μl total volume, suspended in 1× sterile PBS) were administered intravenously (IV) via retro-orbital injection. After administration, mice were monitored every 6 h by lab personnel for mortality and other health parameters in accordance with the Michigan State University Environmental Health and Safety (EHS) and IACUC. After 24 h, the mice were sacrificed, and the spleen and the left lobe of the liver were isolated from each animal. Each tissue was placed in 500 μl PBS, and the tissue suspension was homogenized using an Omni tissue homogenizer (Omni International). Then 300 μl of homogenate was added to an equal volume of equilibrated phenol solution (Sigma). The homogenate-phenol solution was vortexed and centrifuged at 15,000 rpm for 10 min. The aqueous phase was removed and added to 500 μl chloroform. The mixture was vortexed and centrifuged at 15,000 rpm for 10 min. The aqueous phase was then removed and stored at −80°C until analysis.
Quantitative PCR was used to determine adenovirus abundance from DNA extracted from the liver tissue as previously described (33). Ad5 genome copy numbers were quantified using an ABI 7900HT fast real-time PCR system and SYBR green PCR master mix (Applied Biosystems) in a 15-μl reaction with a primer set for the Ad5 hexon gene that has been previously described (34). All PCRs were subjected to the following procedure: 95.0°C for 10 min, followed by 40 cycles of 95.0°C for 15 s and 60.0°C for 1 min. Standard curves to determine the number of viral genomes per liver cell were run in duplicate and consisted of 6 half-log dilutions using DNA extracted from purified Ad5 virus (27). As an internal control, liver DNA was quantified using primers spanning the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene (33), and standard curves were generated from the total genomic DNA. Melting curve analysis was performed to confirm the quality and specificity of the PCR (data not shown).
To determine the relative abundance of the specific liver-derived RNA transcript, reverse transcription (RT) was performed on RNA derived from the liver tissue using SuperScript III (Invitrogen) and random hexamers (Applied Biosystems) according to the manufacturer's instructions. RT reactions were diluted to a total volume of 60 μl, and 2 μl from each sample was used as the template for the subsequent PCRs. Quantitative PCRs were subsequently performed as described above using an ABI 7900HT fast real-time PCR system and SYBR green PCR master mix (Applied Biosystems) with primer sets that were previously described (27). The comparative threshold cycle (CT) method was used to determine relative gene expression using GAPDH to standardize expression levels across all samples. Relative expression changes were calculated by comparing the experimental levels of the liver transcript to the levels of the liver transcript derived from mock-treated animals.
IFN-β was quantified using the VeriKine mouse IFN beta enzyme-linked immunosorbent assay (ELISA) kit (PBL Assay Science) according to the manufacturer's instructions. Cytokine and chemokine concentrations were quantified from plasma samples using a Bio-Plex multiplex bead array system (Bio-Rad). At 6 and 24 h, blood samples were taken from mice using heparinized capillary tubes and EDTA-coated Microvettes (Sarstedt). The samples were centrifuged at 3,400 rpm for 10 min to isolate plasma. Samples were assayed for 12 independent cytokines and chemokines (IL-1α, IL-4, IL-6, IL-12-p40, IFN-γ, granulocyte colony-stimulating factor [G-CSF], eotaxin, KC, MCP-1, MIP-1α, MIP-1β, and RANTES) according to the manufacturer's instructions (Bio-Rad) via Luminex 100 technology (Luminex).
For adaptive immunity studies, mice were administered adenovirus ranging from 1 × 106 to 5 × 109 vp per mouse suspended in 25 μl PBS via IM injection into the tibialis anterior of the right hindlimb. To measure antigen-specific recall responses, mice were sacrificed, and the spleen was harvested after 14 days. Splenocytes were isolated and ex vivo stimulated with immunogenic peptides from the C. difficile TA library as previously described (31). Enzyme-linked immunosorbent spot assay (ELISpot) analysis was performed as previously described (31) using 96-well multiscreen high-protein binding Immobilon-P membrane plates (Millipore) and the Ready-Set-Go! IFN-γ mouse ELISpot kit (eBioscience). Spots were photographed and counted using an automated ELISpot reader system (Cellular Technology). To determine TA-specific IgG titers, ELISA-based titering was used on plasma samples taken from the mice 14 days postinfection (d.p.i.) as previously described (31).
All animal procedures were reviewed and approved by the Michigan State University EHS and IACUC. Care for the mice was provided in accordance with the U.S. PHS and AAALAC standards. Plasma and tissue samples were collected and handled in accordance with the Michigan State University IACUC.
RESULTS
Generating an adenovirus harboring a V. cholerae DGC.
c-di-GMP is an exciting new adjuvant that stimulates the innate immune system (16). These studies most frequently used chemically synthesized c-di-GMP. Because c-di-GMP is synthesized from GTP and GTP is abundant in the cytoplasm of eukaryotic organisms, we postulated that a DGC expressed under the control of a strong eukaryotic promoter/enhancer element would lead to c-di-GMP synthesis within the eukaryotic cell and subsequent enhancement of downstream innate immune responses. This approach offers a novel, alternative method to administer c-di-GMP as a vaccine adjuvant as opposed to direct delivery of the synthesized molecule. To identify a DGC that would produce c-di-GMP in the cytoplasm of a eukaryotic cell, we examined DGCs from V. cholerae, as V. cholerae is a well-studied model system for c-di-GMP signaling, and many V. cholerae DGCs have been shown to synthesize c-di-GMP in high concentrations (32). We selected the DGC VCA0956 due to the fact that it had no predicted N-terminal regulatory or transmembrane domains. Furthermore, VCA0956 has a canonical GGDEF domain and active site motif, and ectopic expression of VCA0956 has been shown to increase biofilm formation in both V. cholerae and Vibrio vulnificus (32, 35), repress motility in V. cholerae (36), and increase intracellular c-di-GMP in V. cholerae and Shewanella oneidensis (37–39).
To determine if VCA0956 is able to synthesize c-di-GMP in eukaryotic cytoplasm, we constructed a plasmid containing VCA0956 under the control of the constitutive CMV promoter/enhancer in the plasmid pShuttle-CMV. We also constructed a second vector containing the same VCA0956 allele with a mutation in the active site of the GGDEF domain (GGEEF → AAEEF). These plasmids were transfected into HeLa cells, and c-di-GMP levels were measured in cell lysates after 24 h using liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS). We found that eukaryotic cells transfected with the VCA0956 allele produced detectable levels of c-di-GMP (Fig. 1A). In contrast, no detectable c-di-GMP was observed in either cells transfected with the active site mutant allele or mock treatment controls (Fig. 1A). The estimated intracellular c-di-GMP concentrations of HeLa cells grown in 6-well dishes expressing VCA0956 are greater than the dissociation constant (Kd) range of the c-di-GMP binding protein STING (2.5 to 4.9 μM) (40, 41).Cell cultures were checked by microscopy, and we observed no discernible morphological differences between cells expressing VCA0956 and the controls. Furthermore, trypan blue staining indicated that treatment with VCA0956 did not appear to impact overall cell viability. Additionally, HeLa cells grown in T75 flasks, transfected with the VCA0956 plasmid, and measured 48 h later had less intracellular c-di-GMP, suggesting that c-di-GMP synthesis is transient (Fig. 1B). We speculate that c-di-GMP might be degraded in eukaryotic cells by nonspecific phosphodiesterase enzymes. We observed less c-di-GMP production in these experiments, which may be a function of decreased transfection efficiency in the flasks. Nevertheless, these results indicate that VCA0956 is capable of transiently synthesizing c-di-GMP in the cytoplasm of eukaryotic cells grown in vitro.
FIG 1.
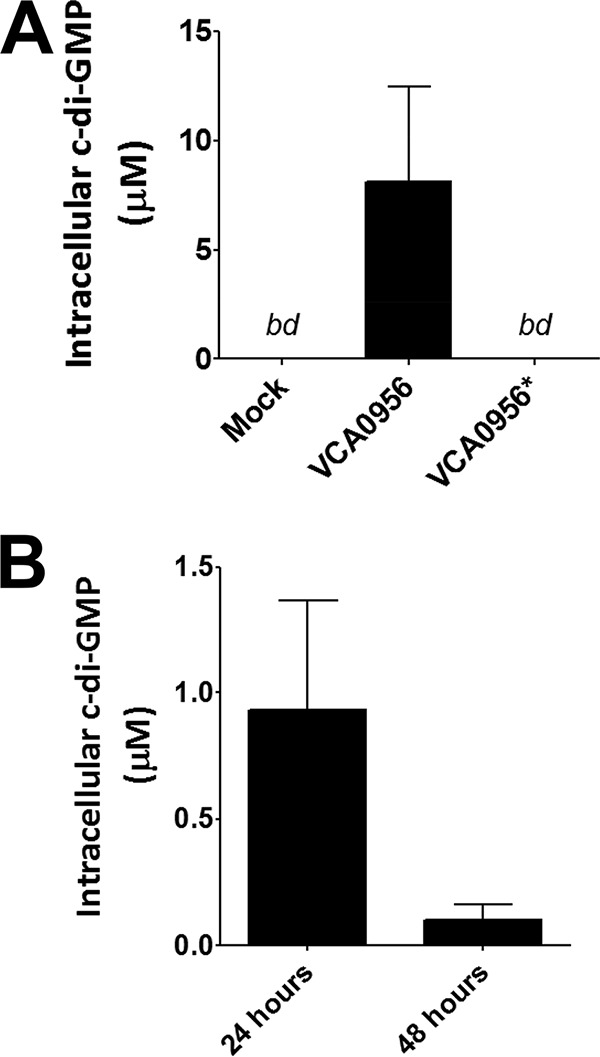
LC-MS/MS was used to quantify c-di-GMP in HeLa cells. (A) HeLa cells were transfected with plasmid vectors containing the VCA0956 allele or the active site mutant allele, VCA0956*. Bars represent the means from 5 independent cultures. (B) c-di-GMP in HeLa cells cultured in T75 flasks and transfected with plasmid vectors containing the VCA0956 allele at 24 and 48 h. Bars represent means from independent cell cultures (24 h, n = 3; 48 h, n = 2). bd, below detection.
The pShuttle-CMV-VCA0956 plasmid and its mutant allele counterpart were then used to construct and purify to high concentrations the respective recombinant Ad5-based vectors. To confirm that the VCA0956 Ad5 construct, herein referred to as Ad5-VCA0956, was able to produce c-di-GMP in eukaryotic cytoplasm, we infected HeLa cells (MOI of 500) with the Ad5-VCA0956 and Ad5-VCA0956 mutant allele (Ad5-VCA0956*) adenovirus vectors and measured c-di-GMP using LC-MS/MS after 24 h. The Ad5-null vector, an adenovirus construct carrying no transgene, was also included as a negative control. We found that cells infected with Ad5-VCA0956 produced high concentrations of c-di-GMP comparable to those with transfection of the pShuttle-CMV-VCA0956 plasmid, whereas cells infected with Ad5-VCA0956* or Ad5-null produced no detectable c-di-GMP (Fig. 2). Importantly, similar to VCA0956 plasmid transfections, infection with Ad5-VCA0956 had no noticeable impact on cell morphology or viability. These results demonstrate that an adenovirus vector can be used to deliver VCA0956 into HeLa cells to synthesize c-di-GMP.
FIG 2.
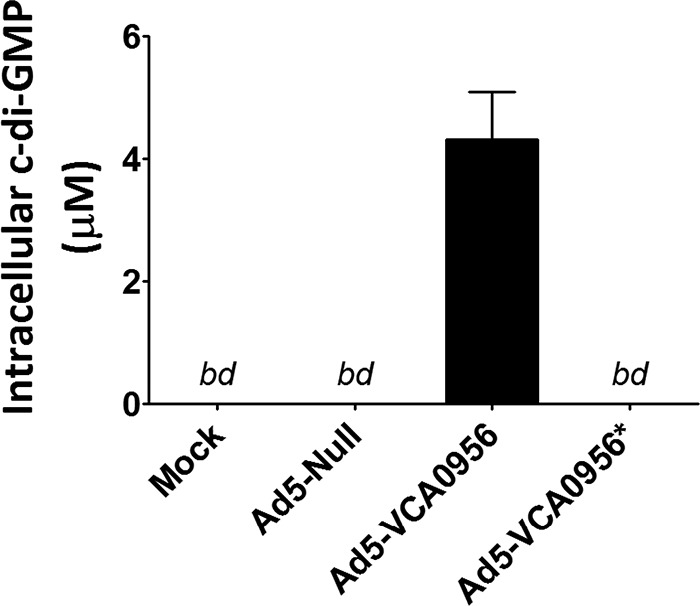
HeLa cells were infected with Ad5 vectors (MOI of 500). Bars represent the means from 3 independent cultures; error bars indicate standard deviations. bd, below detection.
Synthesis of c-di-GMP in vivo.
As the Ad5-VCA0956 vector is capable of producing c-di-GMP in HeLa cells in vitro, we next determined if our vector produces c-di-GMP in vivo in a murine model system. BALB/c mice (n = 3) were injected IV with the Ad5-null, Ad5-VCA0956, or Ad5-VCA0956* vectors, and quantitative PCRs were utilized to measure adenovirus genomes in the spleens and livers of injected mice at 24 h postinjection (h.p.i.). Using reverse transcription-quantitative PCR (qRT-PCR), we observed comparable Ad5 genome counts for each treatment in the liver and the spleen (Fig. 3A). Consistent with previous reports that the predominant tropism of adenovirus is in the liver (34, 42, 43), there were significantly more Ad5 genomes in the liver cells than in the spleen cells. We then measured c-di-GMP in both the liver and spleen using LC-MS/MS and found that the Ad5-VCA0956 vector produced detectable c-di-GMP in both tissues, whereas the Ad5-null and Ad5-VCA0956* vectors produced no detectable c-di-GMP (Fig. 3B). The concentration of c-di-GMP was consistent with the abundance of Ad5-VCA0956 genomes per cell, as the amount of c-di-GMP was significantly higher in the liver tissue than in the spleen. These data indicate that the Ad5-VCA0956 vector is capable of initiating c-di-GMP synthesis in a mouse.
FIG 3.
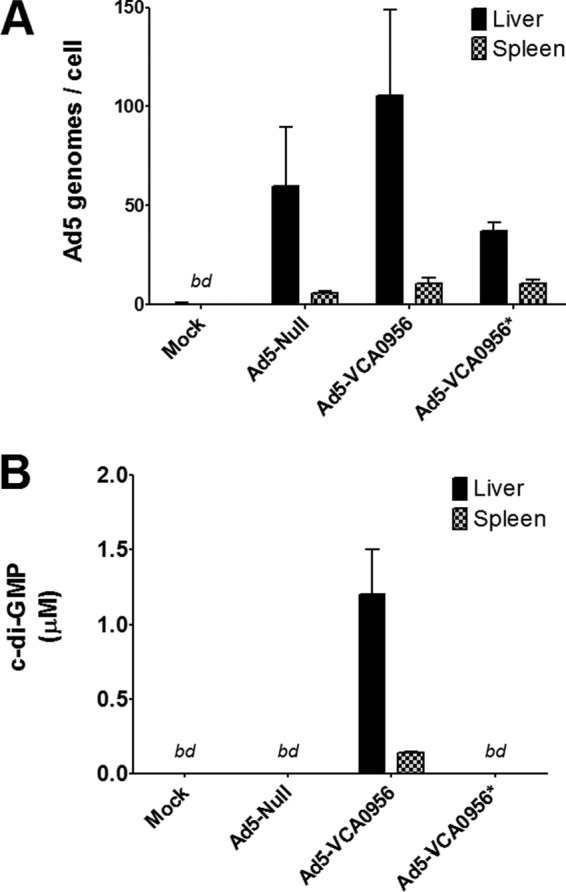
Infection of Ad5-VCA0956 in a murine system. (A) After 24 h, quantitative PCRs were used to quantify Ad5 genomes in liver cells or spleen cells. Data were normalized to an internal GADPH control. (B) LC-MS/MS was used to quantify c-di-GMP extracted from the liver or spleen. Bars represent the means from 3 independent mouse samples; error bars indicate standard deviations. bd, below detection.
c-di-GMP synthesized in vivo stimulates innate immunity in a mouse model.
It was previously shown that adenovirus vectors stimulate several proinflammatory innate immune response genes (21, 27, 33). To examine if Ad5-VCA0956 alters the profile of innate immune gene expression compared to the Ad5 vector alone, BALB/c mice (n = 3) were injected IV with Ad5-null, Ad5-VCA0956, and Ad5-VCA0956*, and qRT-PCRs were utilized to quantify the expression levels of several liver gene transcripts at 24 h.p.i. Infection with Ad5-VCA0956 had no observable effect on the health of the mice. We found that the Ad5-null treatment was able to stimulate 6 of the 12 markers examined (>2-fold) (ADAR, MCP-1, TLR2, IP10, Oas1a, and RIG1) (Fig. 4). These results are consistent with previous studies demonstrating that the adenovirus vector alone is capable of altering gene expression in the liver (26, 27). The expression of four genes was significantly (P < 0.05) higher in the Ad5-VCA0956 treatment than in the Ad5-VCA0956* treatment (Fig. 4A); these include the IFN-responsive gene ADAR, MCP-1, the Toll-like receptor (TLR) signaling pathway gene MyD88, and the pattern recognition receptor TLR2. It is worth noting that c-di-GMP sensing in the cytoplasm is thought to be independent of TLRs (10). Additionally, the expression of three genes was significantly (P < 0.05) repressed in the Ad5-VCA0956 treatment compared to that in the Ad5-VCA0956* treatment (Fig. 4B): the proinflammatory interleukin genes IL-18 and IL-1β and the interferon transcription factor IRF3. Interestingly, IRF3 has been shown to interact with STING to initiate a c-di-GMP-mediated host type I interferon response (5, 44, 45).
FIG 4.
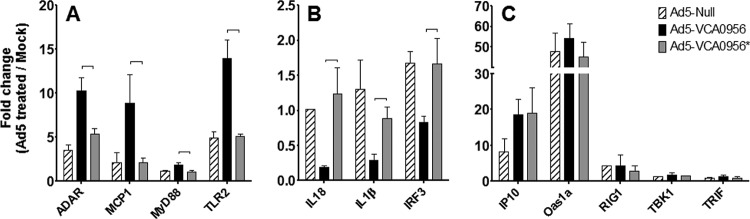
qRT-PCR of mouse liver gene transcripts 24 h after infection with Ad5 vectors. The data were normalized to an internal GADPH control. Fold change indicates each value normalized to values measured from mock-treated mice. Results are separated into liver gene expression increased by Ad5-VCA0956 (A), decreased by Ad5-VCA0956 (B), or unaffected by Ad5-VCA0956 (C). Bars represent the means from 3 independent mouse samples; error bars indicate standard deviations. Brackets indicate statistical significance, which was determined using a two-tailed Student's t test (P < 0.05).
In the cytoplasm, c-di-GMP interacts with STING to initiate a type I interferon response and activates IRF3, NF-κB, and the p38/Jun N-terminal protein kinase (JNK)/extracellular signal-regulated kinase (ERK) mitogen-activated protein (MAP) kinase signaling pathways, resulting in increased production of numerous cytokines and chemokines (5). To determine if Ad5-VCA0956 is capable of inducing type I interferons, we measured the concentration of IFN-β in the plasma of mice treated IV with Ad5-null, Ad5-VCA0956, or Ad5-VCA0956* at 6 h.p.i. and 24 h.p.i. We found that at 6 h.p.i., IFN-β concentrations were significantly higher in mice treated with Ad5-VCA0956 than in the other control mice (Fig. 5). At 24 h.p.i., IFN-β concentrations were undetectable in the control mice, whereas mice treated with Ad5-VCA0956 demonstrated IFN-β concentrations that were detectable, although lower than those at the 6 h.p.i. time point. These data indicate that Ad5-VCA0956 is capable of inducing a type I interferon response in mice.
FIG 5.
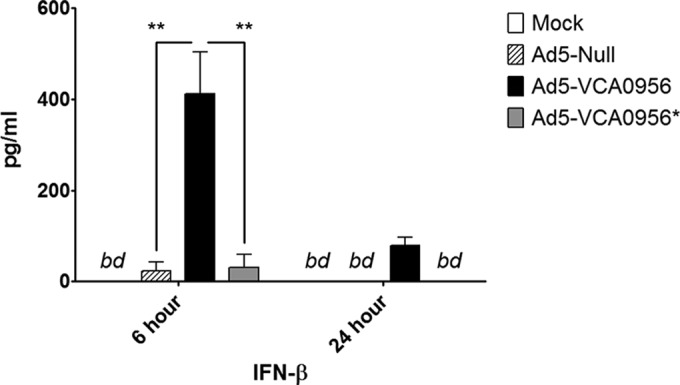
IFN-β concentrations in the plasma of mice infected with Ad5 vectors. Mice were infected with either Ad5-null, Ad5-VCA0956, or Ad5-VCA0956*. At 6 and 24 h, IFN-β was quantified from plasma samples. Brackets indicate statistical significance, which was determined using a one-way ANOVA test combined with a Bonferroni posttest (**, P < 0.01). Bars indicate the means from independent mouse plasma samples (n = 2: mock, Ad5-Null; n = 3: Ad5-VCA0956 and Ad5-VCA0956*), and error bars indicate standard deviations. bd, below detection.
In addition to IFN-β, we wanted to determine if other cytokines and chemokines were induced by Ad5-VCA0956. To this end, we directly quantified the abundance of cytokines and chemokines in the plasma of mice treated with Ad5-VCA0956 using a multiplexed assay system at 6 and 24 h.p.i. Consistent with prior studies showing that the adenovirus vector stimulates the secretion of proinflammatory cytokines and chemokines (27, 28), we observed 9 cytokines and chemokines that were modestly induced in the Ad5-null-treated mice compared to the naive mice (>3-fold) (IFN-γ, MCP-1, G-CSF, MIP-1α, IL-6, MIP-1β, IL-12p40, KC, and RANTES), and these differences were greatest at the 6-hour time point (Fig. 6). We found that 12 cytokines and chemokines, shown in Fig. 6, were significantly increased in the plasma of the Ad5-VCA0956-treated mice compared to those in the plasma of the control Ad5-VCA0956*-treated mice at one or both of the two time points. Furthermore, for the majority of cytokines and chemokines examined, the largest differences observed were at the 24-h time point, indicating that the effect of Ad5-VCA0956 is both more potent and longer lasting than that of the adenovirus vector alone. The inductions of most of these cytokines and chemokines are consistent with those in other studies examining the immunostimulatory effects of c-di-GMP (7, 9–13). Interestingly, we observed increases in IL-1α, G-CSF, and eotaxin levels in the Ad5-VCA0956-injected mice, which have not been previously reported to be induced by c-di-GMP. These data together indicate that the Ad5-VCA0956 vector is capable of inducing a robust innate response beyond that of the adenovirus vector alone in a murine model system.
FIG 6.

Plasma cytokine and chemokine levels in mice infected with Ad5 vectors. Mice were infected with either Ad5-null, Ad5-VCA0956, or Ad5-VCA0956*. At 6 and 24 h, cytokines and chemokines were quantified from plasma samples. Brackets indicate statistical significance, which was determined using a two-way ANOVA test combined with a Bonferroni posttest (*, P < 0.05, and **, P < 0.01). Bars indicate the means from independent mouse plasma samples (n = 2: mock, Ad5-null; n = 3: Ad5-VCA0956 and Ad5-VCA0956*), and error bars indicate standard deviations.
Ad5-VCA0956 lowers the effective dose for a T-cell response to a Clostridium difficile antigen.
The function of an adjuvant is to enhance the efficacy of a paired antigen by increasing the longevity and potency or reducing the effective dose. Our previous data show that Ad5-VCA0956 strongly upregulates inflammatory responses. To test if the Ad5-VCA0956 construct functions as a vaccine adjuvant, we determined if Ad5-VCA0956 could enhance the adaptive response to a C. difficile antigen. C. difficile, a Gram-positive spore-forming anaerobic bacteria, is the leading causative agent of nosocomial infections leading to diarrheal disease in the developed world. C. difficile-associated diarrhea (CDAD) represents nearly 1% of all hospital stays in the United States and can lead to septicemia, renal failure, and toxic megacolon (46). The incidence and mortality rates of C. difficile infections are rising in the United States, and the economic burden on the health care system is reported to be in the billions of dollars (46–50). Furthermore, to date there are no approved effective vaccines available for CDAD treatment or prevention (51).
We had previously developed an adenovirus vector that expresses the immunogenic portion of the C. difficile toxin A (Ad5-TA) and demonstrated that this vector protects mice from a toxin challenge by generating a humoral and T-cell response specific to toxin A in a murine model system (31). We hypothesized that supplementing this vaccine with the Ad5-VCA0956 adjuvant would enhance this humoral and T-cell response due to the strong innate immune stimulatory activity of VCA0956. We therefore vaccinated mice by IM injection with various concentrations of the Ad5-TA vector in combination with the Ad5-VCA0956 vector in equal ratios ranging from 1 × 106 to 5 × 109 viral particles (vp). After 2 weeks, we measured TA-specific IgG titers in the plasma of the vaccinated mice. At the 1 × 107 dose, we observed no significant changes in TA-specific IgG in the plasma of any of the treated mice compared to those in mice with the mock treatment, indicating that this dose of Ad5-TA and Ad5-VCA0956 is not sufficient to produce a robust IgG response in mice (Fig. 7A). In contrast, the 5 × 109 dose resulted in significantly increased TA-specific IgG for both Ad5-VCA0956 and Ad5-VCA0956*; however, the TA-specific IgG titers in the Ad5-VCA0956*-treated animals was modestly higher (2-way analysis of variance [ANOVA], P < 0.05) than those in animals treated with Ad5-VCA0956 (Fig. 7B), suggesting that higher doses of c-di-GMP have a negative impact on humoral immunity.
FIG 7.
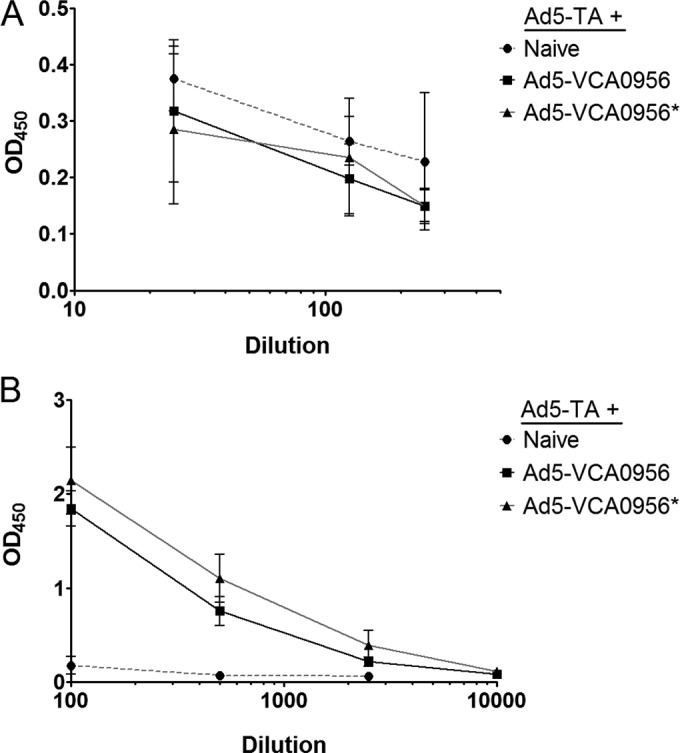
C. difficile TA-specific IgG from the plasma of mice vaccinated IM with 1 × 107 vp Ad5-TA and Ad5-VCA0956 (A) or 5 × 109 vp Ad5-TA and Ad5-VCA0956 (B) (both 14 d.p.i.) was quantified using an ELISA. The optical density at 450 nm (OD450) was measured at various plasma dilutions. Each point represents the mean of 6 independent mouse plasma samples, and error bars indicate standard deviations.
We also assessed TA-specific T-cell responses in the spleens of the naive and vaccinated animals using an IFN-γ ELISpot assay, utilizing the 15-mer peptide (VNGSRYYFDTDTAIA) that has been previously shown to elicit the secretion of IFN-γ in splenocytes of mice immunized with the Ad5-TA vector (31). We found that coinjection of equal amounts of the Ad5-TA and the mutant DGC allele vector Ad5-VCA0956* produced no induction of IFN-γ-secreting T cells over that of naive splenocytes at viral doses of 1 × 106 and 1 × 107 but did generate significant IFN-γ-producing T cells at 1 × 108 and 5 × 109 (Fig. 8). The numbers of spot-forming cells (SFCs) in the mice treated with Ad5-TA and Ad5-VCA0956* at the 5 × 109 dose were consistent with the numbers of SFCs in mice vaccinated with Ad5-TA alone (31). These data indicate that antigen-specific T-cell responses in splenocytes plateau at high levels of Ad5-TA independent of the addition of c-di-GMP. Although coinjection of 1 × 106 Ad5-TA with Ad5-VCA0956 did not produce increased IFN-γ levels, we observed significantly increased (P < 0.05) IFN-γ-producing T cells at a dose of 1 × 107 compared to cells derived from the DGC mutant-treated control (Fig. 8, black squares). However, the numbers of IFN-γ splenocytes did not reach those of the mice injected with higher concentrations of Ad5-TA and Ad5-VCA0956, suggesting only a modest improvement compared to the negative controls. The numbers of IFN-γ-producing T cells at injections of 1 × 108 and 5 × 109 Ad5-TA and Ad5-VCA0956 were similar to those in the DGC mutant control. No c-di-GMP was detected in the livers of mice infected with Ad5-VCA0956 at the 5 × 109 dose after 14 days, suggesting that even at high doses intramuscular administration of Ad5-VCA0956 does not lead to long-lasting c-di-GMP production at distal sites (data not shown). Thus, we conclude that although it does not increase a humoral response, c-di-GMP synthesized by Ad5-VCA0956 modestly lowers the effective dose to generate a T-cell response to Ad5-TA in a murine model system.
FIG 8.
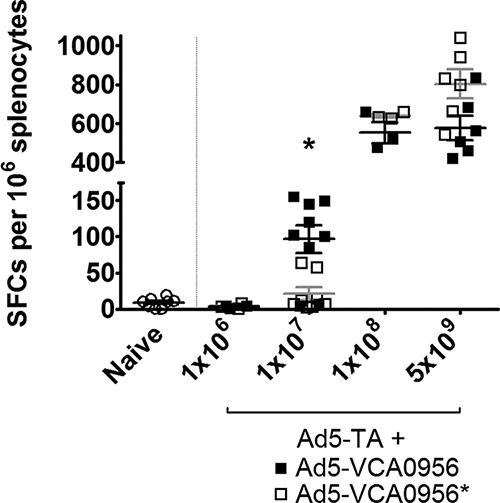
IFN-γ ELISpot analysis of mice vaccinated with Ad5-TA and Ad5 vectors. Mice were administered (IM) various doses of Ad-TA and either Ad-VCA0956 or Ad-VCA0956*. After 14 days, splenocytes were ex vivo stimulated with a C. difficile-specific peptide, and the number of IFN-γ-secreting splenocytes was determined using ELISpot. Each point represents an individual mouse. Lines indicate the mean of the replicates, and error bars indicate standard errors. *, statistically significant with a two-way ANOVA test combined with a Bonferroni posttest (P < 0.05).
DISCUSSION
With a current demand for novel vaccines that target difficult-to-treat diseases, it is crucial to have adjuvants to pair with these vaccines to optimize efficacy. Currently, there are a limited number of adjuvants available for clinical use, and there is a need for new adjuvants which can enhance the efficacy of vaccines to improve immunological protection (18, 52). Numerous studies have indicated that c-di-GMP is a promising novel adjuvant. Indeed, this second-messenger molecule has been shown to stimulate a robust type I interferon response and increase the secretion of numerous cytokines and chemokines to initiate a balanced Th1/Th2 response and to stimulate the inflammasome pathway and immune cell activation/recruitment (6–14). Our approach is novel in that it utilizes an adenovirus vector to deliver a c-di-GMP-producing enzyme DNA into cells, thereby synthesizing the adjuvant in vivo. Adenovirus vectors are promising in that they are cost-effective to produce and can efficiently deliver specific antigens or adjuvants into cells for in vivo production.
We have demonstrated that an adenovirus vector carrying a bacterial DGC is capable of synthesizing c-di-GMP in both human and mouse model systems. Similar to previous studies, we have demonstrated that c-di-GMP synthesized by Ad5-VCA0956 is able to induce a type I interferon response (Fig. 5). Furthermore, synthesis of c-di-GMP by Ad5-VCA0956 increases the secretion of numerous cytokines and chemokines (7, 9–14). Importantly, we have demonstrated that Ad5-VCA0956 induces an innate response beyond that of the adenovirus vector alone, which is capable of stimulating the STING system (23).These cytokines and chemokines induced by Ad5-VCA0956 include signals characteristic of both Th1-type (e.g., IFN-γ and IL-12) and Th2-type (e.g., IL-4 and IL-6) responses. Additionally, c-di-GMP production from Ad5-VCA0956 enhances activation of the innate immune system by activating TLR signaling (e.g., TLR2 and MyD88). It appears, however, that c-di-GMP synthesized in vivo negatively regulates the expression of inflammasome-dependent pathways in hepatocytes (IL-1β and IL-18) (Fig. 4). The significance of this finding is unclear, especially as it has been reported that c-di-GMP activates the NLRP3 inflammasome pathway (8). Importantly, we observed no signs of poor cell physiology or health in our cell cultures and animal models. Furthermore, our data indicate that the c-di-GMP synthesized by the Ad5-VCA0956 vector is transient and thus should enhance antigen recognition and response while minimizing any potentially unwanted long-term effects associated with administration, such as autoimmune activation (53). The mechanism by which c-di-GMP is being eliminated from cell cultures is unknown; we speculate that native eukaryotic phosphodiesterases are able to hydrolyze the second messenger.
We have also shown that c-di-GMP synthesized in vivo modestly reduces the effective antigen dose of Ad5-TA to produce a T-cell response to a vaccine antigen which targets the toxin of the human pathogen C. difficile. Reducing the dose required to initiate an adaptive immune response is of particular significance as high viral particle doses can lead to global toxicities, endothelial cell activation, and liver damage (33, 42, 54–56). Our data suggest that increased c-di-GMP did not enhance the humoral response, however, and we even observed modestly decreased antibody production against the C. difficile toxin. Whether these observations are specific to toxin A from C. difficile or more generally applicable to other antigens is under investigation.
While we have demonstrated that Ad5-VCA0956 is capable of in vivo c-di-GMP synthesis and has the potential to act as a vaccine adjuvant, further optimization is required to enhance this response. V. cholerae contains 40 predicted DGC alleles within its genome, and it has been shown that ectopic expression of these different DGCs results in different intracellular c-di-GMP concentrations (32). Hence, intracellular expression of other DGCs might produce different amounts of c-di-GMP in eukaryotic cells to optimize the intracellular concentration of c-di-GMP for different applications. Alternatively, other types of second messengers might be used to stimulate innate immunity. One example is to express a diadenylate cyclase to synthesize the related bacterial second messenger cyclic di-AMP (c-di-AMP) in vivo. c-di-AMP has similarly been shown to induce a robust innate immune response through STING-mediated recognition (57, 58). Another example is the dinucleotide cyclic GMP-AMP (cGAMP), a host second messenger produced in response to foreign DNA to activate a STING-dependent type I interferon response (59–62). As these second messengers stimulate a STING-mediated innate immune response, they are good alternative candidates for Ad-5-mediated in vivo synthesis. Different promoters might be used in lieu of the CMV promoter to produce localized or temporally controlled c-di-GMP production in the body. Finally, the kinetics of adjuvant production by DGCs and antigen expression might be key factors in stimulating increased adaptive responses.
Other research studies suggest that STING-dependent inflammation inhibits the development of cell-mediated immunity. Archer et al. recently showed that production of c-di-AMP by the intracellular bacterial pathogen Listeria monocytogenes inhibits cell-mediated immunity while inducing inflammatory cytokines in a STING-dependent manner (63). We did not observe a significant inhibition of either antibody production or IFN-γ-producing memory T cells. Whether these differences are due to the delivery route (L. monocytogenes versus Ad5 transduction), the levels of the signal, or other factors remains to be determined, but addressing this question has significant implications for using c-di-GMP or c-di-AMP as a vaccine adjuvant.
c-di-GMP has been shown to enhance protection against other pathogens, including S. aureus, K. pneumoniae, and S. pneumoniae (10–12, 15), indicating that c-di-GMP has broad antigen-adjuvant synergy. Although the results of this study imply that the c-di-GMP produced from adenovirus vectors may not enhance vaccines that rely on antibody production, such as those targeting bacterial toxins, the Ad5-VCA0956-stimulated c-di-GMP innate immune response might enhance protection of vaccines that drive cell-mediated immunity such as those targeting viral infections or cancers. Consistent with this idea, c-di-GMP has been shown to exhibit anticancer properties in a number of studies (20, 64, 65), which are thought to be mediated through stimulation of a type I interferon response as observed here. Miyabe et al. (20) showed that enhancing c-di-GMP entry into cancer cells using liposomes increased its efficacy; adenovirus delivery of DGCs to tumors might function similarly by driving synthesis of c-di-GMP in cancer cells. One advantage of using adenovirus over general administration for this purpose is that modified adenovirus vectors have been constructed to target specific tissue types (66), and c-di-GMP might be directly delivered to tumor cells or other tissues.
ACKNOWLEDGMENTS
This work was supported by the NIH (grants U19AI090872, R01AR056981, and R21AI105499), the Region V Great Lakes RCE (NIH award 2-U54-AI-057153), the Osteopathic Heritage Foundation, and the MSU Foundation. We also acknowledge support from the Rudolph Hugh Fellowship, the Russell B. DuVall Scholarship, and the Marvin Hensley Fellowship to B.J.K. and assistance from the MSU mass spectrometry facility to quantify c-di-GMP.
Footnotes
Published ahead of print 17 September 2014
REFERENCES
- 1. Römling U, Galperin MY, Gomelsky M. 2013. Cyclic di-GMP: the first 25 years of a universal bacterial second messenger. Microbiol. Mol. Biol. Rev. 77:1–52. 10.1128/MMBR.00043-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ross P, Weinhouse H, Aloni Y, Michaeli D, Weinbergerohana P, Mayer R, Braun S, Devroom E, Vandermarel GA, Vanboom JH, Benziman M. 1987. Regulation of cellulose synthesis in Acetobacter xylinum by cyclic diguanylic acid. Nature 325:279–281. 10.1038/325279a0. [DOI] [PubMed] [Google Scholar]
- 3. Seshasayee ASN, Fraser GM, Luscombe NM. 2010. Comparative genomics of cyclic-di-GMP signaling in bacteria: post-translational regulation and catalytic activity. Nucleic Acids Res. 38:5970–5981. 10.1093/nar/gkq382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Galperin MY, Nikolskaya AN, Koonin EV. 2001. Novel domains of the prokaryotic two-component signal transduction systems. FEMS Microbiol. Lett. 203:11–21. 10.1111/j.1574-6968.2001.tb10814.x. [DOI] [PubMed] [Google Scholar]
- 5. McWhirter SM, Barbalat R, Monroe KM, Fontana MF, Hyodo M, Joncker NT, Ishii KJ, Akira S, Colonna M, Chen ZJJ, Fitzgerald KA, Hayakawa Y, Vance RE. 2009. A host type I interferon response is induced by cytosolic sensing of the bacterial second messenger cyclic-di-GMP. J. Exp. Med. 206:1899–1911. 10.1084/jem.20082874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sauer JD, Sotelo-Troha K, von Moltke J, Monroe KM, Rae CS, Brubaker SW, Hyodo M, Hayakawa Y, Woodward JJ, Portnoy DA, Vance RE. 2011. The N-ethyl-N-nitrosourea-induced Goldenticket mouse mutant reveals an essential function of Sting in the in vivo interferon response to listeria monocytogenes and cyclic dinucleotides. Infect. Immun. 79:688–694. 10.1128/IAI.00999-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ebensen T, Schulze K, Riese P, Link C, Morr M, Guzman CA. 2007. The bacterial second messenger cyclic diGMP exhibits potent adjuvant properties. Vaccine 25:1464–1469. 10.1016/j.vaccine.2006.10.033. [DOI] [PubMed] [Google Scholar]
- 8. Abdul-Sater AA, Tattoli I, Jin L, Grajkowski A, Levi A, Koller BH, Allen IC, Beaucage SL, Fitzgerald KA, Ting JPY, Cambier JC, Girardin SE, Schindler C. 2013. Cyclic-di-GMP and cyclic-di-AMP activate the NLRP3 inflammasome. EMBO Rep. 14:900–906. 10.1038/embor.2013.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ebensen T, Schulze K, Riese P, Morr M, Guzman CA. 2007. The bacterial second messenger cdiGMP exhibits promising activity as a mucosal adjuvant. Clin. Vaccine Immunol. 14:952–958. 10.1128/CVI.00119-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Karaolis DKR, Means TK, Yang D, Takahashi M, Yoshimura T, Muraille E, Philpott D, Schroeder JT, Hyodo M, Hayakawa Y, Talbot BG, Brouillette E, Malouin F. 2007. Bacterial c-di-GMP is an immunostimulatory molecule. J. Immunol. 178:2171–2181. 10.4049/jimmunol.178.4.2171. [DOI] [PubMed] [Google Scholar]
- 11. Karaolis DKR, Newstead MW, Zeng XY, Hyodo M, Hayakawa Y, Bhan U, Liang H, Standiford TJ. 2007. Cyclic di-GMP stimulates protective innate immunity in bacterial pneumonia. Infect. Immun. 75:4942–4950. 10.1128/IAI.01762-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yan HB, KuoLee R, Tram K, Qiu HY, Zhang JB, Patel GB, Chen WX. 2009. 3′,5′-Cyclic diguanylic acid elicits mucosal immunity against bacterial infection. Biochem. Biophys. Res. Commun. 387:581–584. 10.1016/j.bbrc.2009.07.061. [DOI] [PubMed] [Google Scholar]
- 13. Gray PM, Forrest G, Wisniewski T, Porter G, Freed DC, DeMartino JA, Zaller DM, Guo ZQ, Leone J, Fu TM, Vora KA. 2012. Evidence for cyclic diguanylate as a vaccine adjuvant with novel immunostimulatory activities. Cell Immunol. 278:113–119. 10.1016/j.cellimm.2012.07.006. [DOI] [PubMed] [Google Scholar]
- 14. Blaauboer SM, Gabrielle VD, Jin L. 2014. MPYS/STING-mediated TNF-α, not type I IFN, is essential for the mucosal adjuvant activity of (3′-5′)-cyclic-di-guanosine-monophosphate in vivo. J. Immunol. 192:492–502. 10.4049/jimmunol.1301812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ogunniyi AD, Paton JC, Kirby AC, McCullers JA, Cook J, Hyodo M, Hayakawa Y, Karaolis DKR. 2008. C-di-GMP is an effective immunomodulator and vaccine adjuvant against pneumococcal infection. Vaccine 26:4676–4685. 10.1016/j.vaccine.2008.06.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen W, Kuolee R, Yan HB. 2010. The potential of 3′,5′-cyclic diguanylic acid (c-di-GMP) as an effective vaccine adjuvant. Vaccine 28:3080–3085. 10.1016/j.vaccine.2010.02.081. [DOI] [PubMed] [Google Scholar]
- 17. Mosca F, Tritto E, Muzzi A, Monaci E, Bagnoli F, Iavarone C, O'Hagan D, Rappuoli R, De Gregorio E. 2008. Molecular and cellular signatures of human vaccine adjuvants. Proc. Natl. Acad. Sci. U. S. A. 105:10501–10506. 10.1073/pnas.0804699105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Coffman RL, Sher A, Seder RA. 2010. Vaccine adjuvants: putting innate immunity to work. Immunity 33:492–503. 10.1016/j.immuni.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gupta RK. 1998. Aluminum compounds as vaccine adjuvants. Adv. Drug Deliv. Rev. 32:155–172. 10.1016/S0169-409X(98)00008-8. [DOI] [PubMed] [Google Scholar]
- 20. Miyabe H, Hyodo M, Nakamura T, Sato Y, Hayakawa Y, Harashima H. 2014. A new adjuvant delivery system ‘cyclic di-GMP/YSK05 liposome' for cancer immunotherapy. J. Control. Release 184:20–27. 10.1016/j.jconrel.2014.04.004. [DOI] [PubMed] [Google Scholar]
- 21. Hartman ZC, Appledorn DM, Amalfitano A. 2008. Adenovirus vector induced innate immune responses: impact upon efficacy and toxicity in gene therapy and vaccine applications. Virus Res. 132:1–14. 10.1016/j.virusres.2007.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Aldhamen YA, Seregin SS, Amalfitano A. 2011. Immune recognition of gene transfer vectors: focus on adenovirus as a paradigm. Front. Immun. 2:40. 10.3389/fimmu.2011.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lam E, Stein S, Falck-Pedersen E. 2014. Adenovirus detection by the cGAS/STING/TBK1 DNA sensing cascade. J. Virol. 88:974–981. 10.1128/JVI.02702-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fukazawa T, Matsuoka J, Yamatsuji T, Maeda Y, Durbin ML, Naomoto Y. 2010. Adenovirus-mediated cancer gene therapy and virotherapy (review). Int. J. Mol. Med. 25:3–10. 10.3892/ijmm_00000306. [DOI] [PubMed] [Google Scholar]
- 25. Sambrook J, Russell DW. 2001. Molecular cloning—a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 26. Seregin SS, Aldhamen YA, Appledorn DM, Zehnder J, Voss T, Godbehere S, Amalfitano A. 2011. Use of DAF-displaying adenovirus vectors reduces induction of transgene- and vector-specific adaptive immune responses in mice. Hum. Gene Ther. 22:1083–1094. 10.1089/hum.2010.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Seregin SS, Aldhamen YA, Appledorn DM, Schuldt NJ, McBride AJ, Bujold M, Godbehere SS, Amalfitano A. 2009. CR1/2 is an important suppressor of adenovirus-induced innate immune responses and is required for induction of neutralizing antibodies. Gene Ther. 16:1245–1259. 10.1038/gt.2009.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Seregin SS, Aldhamen YA, Appledorn DM, Hartman ZC, Schuldt NJ, Scott J, Godbehere S, Jiang HX, Frank MM, Amalfitano A. 2010. Adenovirus capsid-display of the retro-oriented human complement inhibitor DAF reduces Ad vector-triggered immune responses in vitro and in vivo. Blood 116:1669–1677. 10.1182/blood-2010-03-276949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Amalfitano A, Hauser MA, Hu H, Serra D, Begy CR, Chamberlain JS. 1998. Production and characterization of improved adenovirus vectors with the E1, E2b, and E3 genes deleted. J. Virol. 72:926–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Morgan J, Ng P, Graham F. 2002. Construction of first-generation adenoviral vectors, p 389–414 In Morgan J.R. (ed), Gene therapy protocols, vol 69 Springer, New York, NY. [DOI] [PubMed] [Google Scholar]
- 31. Seregin SS, Aldhamen YA, Rastall DPW, Godbehere S, Amalfitano A. 2012. Adenovirus-based vaccination against Clostridium difficile toxin A allows for rapid humoral immunity and complete protection from toxin A lethal challenge in mice. Vaccine 30:1492–1501. 10.1016/j.vaccine.2011.12.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Massie JP, Reynolds EL, Koestler BJ, Cong J-P, Agostoni M, Waters CM. 2012. Quantification of high-specificity cyclic diguanylate signaling. Proc. Natl. Acad. Sci. U. S. A. 109:12746–12751. 10.1073/pnas.1115663109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Seregin SS, Appledorn DM, McBride AJ, Schuldt NJ, Aldhamen YA, Voss T, Wei JP, Bujold M, Nance W, Godbehere S, Amalfitano A. 2009. Transient pretreatment with glucocorticoid ablates innate toxicity of systemically delivered adenoviral vectors without reducing efficacy. Mol. Ther. 17:685–696. 10.1038/mt.2008.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Appledorn DM, Kiang A, McBride A, Jiang H, Seregin S, Scott JM, Stringer R, Kousa Y, Hoban M, Frank MM, Amalfitano A. 2008. Wild-type adenoviruses from groups A-F evoke unique innate immune responses, of which HAd3 and SAd23 are partially complement dependent. Gene Ther. 15:885–901. 10.1038/gt.2008.18. [DOI] [PubMed] [Google Scholar]
- 35. Nakhamchik A, Wilde C, Rowe-Magnus DA. 2008. Cyclic-di-GMP regulates extracellular polysaccharide production, biofilm formation, and rugose colony development by Vibrio vulnificus. Appl. Environ. Microbiol. 74:4199–4209. 10.1128/AEM.00176-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hunter JL, Severin GB, Koestler BJ, Waters CM. 2014. The Vibrio cholerae diguanylate cyclase VCA0965 has an AGDEF active site and synthesizes cyclic di-GMP. BMC Microbiol. 14:22. 10.1186/1471-2180-14-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Koestler BJ, Waters CM. 2013. Exploring environmental control of cyclic di-GMP signaling in Vibrio cholerae by using the ex vivo lysate cyclic di-GMP assay (TELCA). Appl. Environ. Microbiol. 79:5233–5241. 10.1128/AEM.01596-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tamayo R, Schild S, Pratt JT, Camili A. 2008. Role of cyclic di-GMP during El tor biotype Vibrio cholerae infection: characterization of the in vivo-induced cyclic di-GMP phosphodiesterase CdpA. Infect. Immun. 76:1617–1627. 10.1128/IAI.01337-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Thormann KM, Duttler S, Saville RM, Hyodo M, Shukla S, Hayakawa Y, Spormann AM. 2006. Control of formation and cellular detachment from Shewanella oneidensis MR-1 biofilms by cyclic di-GMP. J. Bacteriol. 188:2681–2691. 10.1128/JB.188.7.2681-2691.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, Hayakawa Y, Vance RE. 2011. STING is a direct innate immune sensor of cyclic di-GMP. Nature 478:515–518. 10.1038/nature10429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yin Q, Tian Y, Kabaleeswaran V, Jiang X, Tu D, Eck MJ, Chen ZJ, Wu H. 2012. Cyclic di-GMP sensing via the innate immune signaling protein STING. Mol. Cell 46:735–745. 10.1016/j.molcel.2012.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Everett RS, Hodges BL, Ding EY, Xu F, Serra D, Amalfitano A. 2003. Liver toxicities typically induced by first-generation adenoviral vectors can be reduced by use of E1, E2b-deleted adenoviral vectors. Hum. Gene Ther. 14:1715–1726. 10.1089/104303403322611737. [DOI] [PubMed] [Google Scholar]
- 43. Nakamura T, Sato K, Hamada H. 2003. Reduction of natural adenovirus tropism to the liver by both ablation of fiber-coxsackievirus and adenovirus receptor interaction and use of replaceable short fiber. J. Virol. 77:2512–2521. 10.1128/JVI.77.4.2512-2521.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tanaka Y, Chen ZJ. 2012. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci. Signal. 5:ra20. 10.1126/scisignal.2002521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. de Almeida LA, Carvalho NB, Oliveira FS, Lacerda TLS, Vasconcelos AC, Nogueira L, Bafica A, Silva AM, Oliveira SC. 2011. MyD88 and STING signaling pathways are required for IRF3-mediated IFN-β induction in response to Brucella abortus infection. PLoS One 6:e23135. 10.1371/journal.pone.0023135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lucado J, Gould C, Elixhauser A. 2012. Clostridium difficile infections (CDI) in hospital stays, 2009. HCUP statistical brief no. 124. US Department of Health and Human Services, Agency for Healthcare Research and Quality, Rockville, MD. [PubMed] [Google Scholar]
- 47. Morris AM, Jobe BA, Stoney M, Sheppard BC, Deveney CW, Deveney KE. 2002. Clostridium difficile colitis: an increasingly aggressive iatrogenic disease? Arch. Surg. 137:1096–1100. 10.1001/archsurg.137.10.1096. [DOI] [PubMed] [Google Scholar]
- 48. Redelings MD, Sorvillo F, Mascola L. 2007. Increase in Clostridium difficile-related mortality rates, United States, 1999-2004. Emerg. Infect. Dis. 13:1417–1419. 10.3201/eid1309.061116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kyne L, Hamel MB, Polavaram R, Kelly CP. 2002. Health care costs and mortality associated with nosocomial diarrhea due to Clostridium difficile. Clin. Infect. Dis. 34:346–353. 10.1086/338260. [DOI] [PubMed] [Google Scholar]
- 50. Dubberke ER, Wertheimer AI. 2009. Review of current literature on the economic burden of Clostridium difficile infection. Infect. Control Hosp. Epidemiol. 30:57–66. 10.1086/592981. [DOI] [PubMed] [Google Scholar]
- 51. Aslam S, Hamill RJ, Musher DM. 2005. Treatment of Clostridium difficile-associated disease: old therapies and new strategies. Lancet Infect. Dis. 5:549–557. 10.1016/S1473-3099(05)70215-2. [DOI] [PubMed] [Google Scholar]
- 52. Reed SG, Bertholet S, Coler RN, Friede M. 2009. New horizons in adjuvants for vaccine development. Trends Immunol. 30:23–32. 10.1016/j.it.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 53. Zandman-Goddard G, Shoenfeld Y. 2005. Infections and SLE. Autoimmunity 38:473–485. 10.1080/08916930500285352. [DOI] [PubMed] [Google Scholar]
- 54. Wolins N, Lozier J, Eggerman TL, Jones E, Aguilar-Córdova E, Vostal JG. 2003. Intravenous administration of replication-incompetent adenovirus to rhesus monkeys induces thrombocytopenia by increasing in vivo platelet clearance. Br. J. Haematol. 123:903–905. 10.1046/j.1365-2141.2003.04719.x. [DOI] [PubMed] [Google Scholar]
- 55. Appledorn DM, McBride A, Seregin S, Scott JM, Schuldt N, Kiang A, Godbehere S, Amalfitano A. 2008. Complex interactions with several arms of the complement system dictate innate and humoral immunity to adenoviral vectors. Gene Ther. 15:1606–1617. 10.1038/gt.2008.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Schiedner G, Hertel S, Kochanek S. 2000. Efficient transformation of primary human amniocytes by E1 functions of Ad5: generation of new cell lines for adenoviral vector production. Hum. Gene Ther. 11:2105–2116. 10.1089/104303400750001417. [DOI] [PubMed] [Google Scholar]
- 57. Barker JR, Koestler BJ, Carpenter VK, Burdette DL, Waters CM, Vance RE, Valdivia RH. 2013. STING-dependent recognition of cyclic di-AMP mediates type I interferon responses during Chlamydia trachomatis infection. mBio 4:e00018-13. 10.1128/mBio.00018-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Woodward JJ, Iavarone AT, Portnoy DA. 2010. c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type i interferon response. Science 328:1703–1705. 10.1126/science.1189801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sun L, Wu J, Du F, Chen X, Chen ZJ. 2013. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339:786–791. 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wu J, Sun L, Chen X, Du F, Shi H, Chen C, Chen ZJ. 2013. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339:826–830. 10.1126/science.1229963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Gao D, Wu J, Wu Y-T, Du F, Aroh C, Yan N, Sun L, Chen ZJ. 2013. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science 341:903–906. 10.1126/science.1240933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Li X-D, Wu J, Gao D, Wang H, Sun L, Chen ZJ. 2013. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science 341:1390–1394. 10.1126/science.1244040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Archer KA, Durack J, Portnoy DA. 2014. STING-dependent type I IFN production inhibits cell-mediated immunity to Listeria monocytogenes. PLoS Pathog. 10:e1003861. 10.1371/journal.ppat.1003861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chandra D, Quispe-Tintaya W, Jahangir A, Asafu-Adjei D, Ramos I, Sintim HO, Zhou J, Hayakawa Y, Karaolis DK, Gravekamp C. 2014. STING ligand c-di-GMP improves cancer vaccination against metastatic breast cancer. Cancer Immunol. Res. 2:901–910. 10.1158/2326-6066.CIR-13-0123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Karaolis DKR, Cheng K, Lipsky M, Elnabawi A, Catalano J, Hyodo M, Hayakawa Y, Raufman J-P. 2005. 3′,5′-Cyclic diguanylic acid (c-di-GMP) inhibits basal and growth factor-stimulated human colon cancer cell proliferation. Biochem. Biophys. Res. Commun. 329:40–45. 10.1016/j.bbrc.2005.01.093. [DOI] [PubMed] [Google Scholar]
- 66. Reetz J, Herchenröder O, Pützer BM. 2014. Peptide-based technologies to alter adenoviral vector tropism: ways and means for systemic treatment of cancer. Viruses. 6:1540–1563. 10.3390/v6041540. [DOI] [PMC free article] [PubMed] [Google Scholar]