

Abstract
The mechanisms involved in the virulence of Yersinia pestis, the plague pathogen, are not fully understood. In previous research, we found that a Y. pestis mutant lacking the HicB3 (YPO3369) putative orphan antitoxin was attenuated for virulence in a murine model of bubonic plague. Toxin-antitoxin systems (TASs) are widespread in prokaryotes. Most bacterial species possess many TASs of several types. In type II TASs, the toxin protein is bound and neutralized by its cognate antitoxin protein in the cytoplasm. Here we identify the hicA3 gene encoding the toxin neutralized by HicB3 and show that HicA3-HicB3 constitutes a new functional type II TAS in Y. pestis. Using biochemical and mutagenesis-based approaches, we demonstrate that the HicA3 toxin is an RNase with a catalytic histidine residue. HicB3 has two functions: it sequesters and neutralizes HicA3 by blocking its active site, and it represses transcription of the hicA3B3 operon. Gel shift assays and reporter fusion experiments indicate that the HicB3 antitoxin binds to two operators in the hicA3B3 promoter region. We solved the X-ray structures of HicB3 and the HicA3-HicB3 complex; thus, we present the first crystal structure of a TA complex from the HicAB family. HicB3 forms a tetramer that can bind two HicA3 toxin molecules. HicA3 is monomeric and folds as a double-stranded-RNA-binding domain. The HicB3 N-terminal domain occludes the HicA3 active site, whereas its C-terminal domain folds as a ribbon-helix-helix DNA-binding motif.
INTRODUCTION
The Gram-negative enterobacterium Yersinia pestis is the causal agent of plague, a disease that is usually transmitted via a flea bite or (more rarely) via the inhalation of aerosols (1). Flea-borne plague leads to bubonic plague or (to a lesser extent) primary septicemic plague, whereas aerosol transmission produces pneumonia (2). To better understand the mechanisms responsible for disease production, we previously screened a library of Y. pestis deletion mutants for attenuated virulence in a rat model of bubonic plague (3). Each mutant in the library lacked one or more of the genes determined by comparative transcriptome analysis to be upregulated in vivo (4). One of the virulence-attenuated mutants lacked the uncharacterized ypo3369 gene (3). Although it had been suggested that ypo3369 (also referred to as hicB3) encoded an antitoxin from a toxin-antitoxin system (TAS) (5), the associated toxin gene had yet to be identified.
Toxin-antitoxin systems were originally defined as two-component modules encoded by bicistronic operons in a wide range of bacteria (6), with one gene encoding a toxic protein and the other encoding a specific antitoxin. Although most toxins are RNases (7), some can target membranes (8), DNA gyrase (9), or ribosomes (10) or can phosphorylate proteins (11, 12). Overall, the activities of the toxin interfere with replication or translation and thus lead to growth arrest or even cell death (13). The toxin gene may be located upstream or downstream of the antitoxin gene. Three different classes of TASs have been defined according to the biochemical nature of the antitoxin. In type I TASs, the antitoxin is a noncoding RNA that is able to hybridize with the toxin mRNA and block its translation or target it for degradation (14). In type II TASs, the antitoxin is a small protein that binds to and neutralizes a toxic protein (i.e., through protein-protein interactions). The type II antitoxin is usually also a DNA-binding protein that can block the promoter region of the TA operon (15). In type III TASs, the antitoxin is an RNA that directly binds to and neutralizes the toxin protein (16, 17). Most recently, three-component modules have been described and included in the list of type II TASs (18, 19). The third component in these systems is a repressor that regulates the transcription of the operon.
In Y. pestis, a total of 10 putative type II TAS loci have been identified on the chromosome of the virulent CO92 strain (5, 20, 21). Five of these systems belong to the HigBA family, two others to the HicAB family, and one each to the MqsRA, Phd-Doc, and RelBE families (5). However, Goulard et al. showed that only three toxin candidates (HicA1, HigB2, and RelE) were indeed toxic when overexpressed in Y. pestis (5). Two orphan antitoxin genes (hicB3 and relB2) had also been identified in the CO92 genome (5).
Starting from the candidate virulence gene hicB3 (ypo3369), we used genetic, biochemical, and structural approaches to discover and characterize a new TAS in Y. pestis: HicA3-HicB3. We also report the first crystal structure of a toxin-antitoxin complex from the HicA-HicB family.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
The strains and plasmids used in this study are listed in Table 1. Y. pestis strain KIM6+ was used to analyze in vitro phenotypes, because it lacks the pCD1 virulence plasmid present in strain CO92 (22). The sequence of the hicA3-hicB3 locus is exactly the same in CO92 and KIM6+. Strains were cultivated in LB broth or on LB agar plates (at 37°C for Escherichia coli or 28°C for Y. pestis). Antibiotics and other chemicals were used at the following final concentrations: ampicillin (Ap), 200 μg ml−1; kanamycin (Km), 25 μg ml−1; trimethoprim (Tp), 25 μg ml−1; isopropyl-β-d-thiogalactopyranoside (IPTG), 24 μg ml−1; Irgasan (Irg), 1 μg ml−1; sucrose, 5% (wt/vol); 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (Xgal), 40 μg ml−1. Arabinose (Ara; 0.5 or 1 mM) or IPTG (1 mM) was added to the cultures to induce Para or Plac, respectively.
TABLE 1.
Strains and plasmids
| Strain or plasmid | Relevant propertya | Source or reference |
|---|---|---|
| Strains | ||
| E. coli | ||
| BL21(DE3) | lon ompT; used for protein production | 52 |
| CC118 λpir | ΔlacX74 recA1; used for construction of reporter plasmids bearing pir-dependent R6K replication origin | 53 |
| DH5α | recA1 endA1 Δ(argF-lac)U169 ϕ80dlacZΔM15; used for cloning | 54 |
| MG1655 | K-12 WT strain | 55 |
| S17-1 λpir | Donor strain in conjugation | 56 |
| Y. pestis | ||
| CO92 | Virulent strain | 57 |
| CO92 ΔhicA3B3::Tp | hicA3B3 deleted and replaced with the FRT-Tpr-FRT cassette from pEP1087 | This work |
| KIM6+ | Lacks the pYV (also called pCD) virulence plasmid; attenuated strain | 22 |
| YPEP430 | KIM6+ ΔlacZ | This work |
| YSBT26 | KIM6+ ΔhicB3::Tp; Tpr | This work |
| YSBT34 | KIM6+ ΔhicA3B3::Tp; Tpr | This work |
| YSBT54 | KIM6+ ΔlacZ ΔhicB3::Tp; derived from YPEP430; Tpr | This work |
| YSBT55 | KIM6+ ΔlacZ ΔhicA3B3::Tp; derived from YPEP430; Tpr | This work |
| YSBT59 | KIM6+ ΔlacZ PhicA3::lacZ; chromosomal insertion of pSBT30 into YPEP430; Kmr | This work |
| YSBT61 | KIM6+ ΔlacZ PhicA3::lacZ ΔhicA3B3::Tp; chromosomal insertion of pSBT30 into YSBT55; Kmr Tpr | This work |
| YSBT62 | KIM6+ ΔlacZ hicB3::lacZ; chromosomal insertion of pSBT36 into YPEP430; Kmr | This work |
| YSBT151 | KIM6+ ΔlacZ hicB3::lacZ; chromosomal insertion of pSBT172 into YPEP430; Kmr | This work |
| YSBT152 | KIM6+ ΔlacZ hicB3::lacZ ΔhicB3::Tp; chromosomal insertion of pSBT172 into YSBT54; Kmr Tpr | This work |
| YSBT157 | KIM6+ ΔlacZ hicB3::lacZ ΔhicB3-FRT; derived from YSBT152 by deletion of the Tpr cassette; Kmr | This work |
| YSBT170 | KIM6+ ΔlacZ ΔPhicA3::Tp-term hicB3::lacZ; Tpr cassette with transcription terminator; Kmr | This work |
| YSBT172 | KIM6+ ΔlacZ ΔPhicA3::Tp-term hicB3::lacZ ΔhicB3-FRT; Tpr cassette with transcription terminator; Kmr | This work |
| YSBT173 | KIM6+ ΔlacZ ΔPhicA3-FRT hicB3::lacZ; derived from YSB170 by deletion of the Tpr cassette; Kmr | This work |
| YSBT175 | KIM6+ ΔlacZ ΔPhicA3-FRT hicB3::lacZ ΔhicB3-FRT; derived from YSBT172 by deletion of the Tpr cassette; Kmr | This work |
| Plasmids | ||
| pAK-Not | Expression vector, Plac promoter, ori ColE1; Cmr | 58 |
| pAKK-Not | pSBT41 derivative with deletion of the NotI fragment bearing hicB3; Plac promoter; Kmr | This work |
| pBAD30 | Expression vector, Para promoter, ori p15A; Apr | 59 |
| pCRII | Cloning vector, Plac promoter; Apr Kmr | Invitrogen |
| pEP1013 | Red recombinase vector, pKD46 derivative bearing sacB; Apr | 3 |
| pEP1087 | Template for the FRT-Tpr-FRT cassette amplification; ori R6K; Apr Tpr | 3 |
| pEP1164 | pCRII bearing the hicA3 promoter region (208-bp insert) | This work |
| pEP1165 | pCRII bearing hicA3-hicB3 cloned opposite Plac (939-bp insert) | This work |
| pEP1216 | Template for the Tpr-terminator cassette amplification; ori R6K; Apr Tpr | This work |
| pEP1319 | pEP1320 bearing hicB3 cotranscribed with dfrB; ori R6K; Tpr | This work |
| pEP1320 | Cloning vector bearing dfrB, ori R6K; Tpr | This work |
| pEP1336 | lacZ reporter plasmid bearing mutated PhicA3 MU1-BS2 DNA fragment; Apr | This work |
| pEP1339 | lacZ reporter plasmid bearing WT PhicA3 BS1-BS2 DNA fragment; Apr | This work |
| pEP1350 | lacZ reporter plasmid bearing mutated PhicA3 BS1-MU2 DNA fragment; Apr | This work |
| pEP1352 | lacZ reporter plasmid bearing mutated PhicA3 MU1-MU2′ DNA fragment; Apr | This work |
| pET24a+ | C-terminal 6-histidine tag expression vector, T7 promoter; Kmr | Novagen |
| pFLP2 | FLP recombinase expression vector; Apr | 24 |
| pSBT7 | pCRII bearing hicB3 under the control of Plac (151 bp upstream from the hicB3 start site and 39 bp downstream from the hicB3 stop site) | This work |
| pSBT10 | pBAD30 bearing hicA3 and its SD sequence as an EcoRI-SalI insert | This work |
| pSBT13 | pCRII bearing hicB3 flanked by NotI sites | This work |
| pSBT18 | pAK-Not bearing hicB3 under the control of Plac; pSBT13 NotI fragment insertion; Cmr | This work |
| pSBT30 | pVIK112 bearing an EcoRI fragment from pEP1164; PhicA3-lacZ fusion; Kmr | This work |
| pSBT36 | pVIK112 bearing an EcoRI-EcoRV fragment from pSBT7 cloned into EcoRI and SmaI | This work |
| pSBT41 | pSBT18 derivative in which the Cmr cassette is replaced by the Kmr cassette from pUC4K | This work |
| pSBT71 | pET24a+ bearing hicA3B3 as an NdeI-XhoI insert to produce HicA3 and HicB3-6His | This work |
| pSBT73 | pCRII bearing hicB3 with NdeI and XhoI flanking sites | This work |
| pSBT74 | pET24a+ bearing hicB3 cloned as an NdeI-XhoI fragment from pSBT73, to produce HicB3-6His | This work |
| pSBT113 | pCRII bearing hicA3-H28A with NdeI and XhoI flanking sites | This work |
| pSBT116 | pET24a+ bearing the hicA3-H28A NdeI-XhoI fragment from pSBT113, to produce HicA3-H28A–6His | This work |
| pSBT172 | pVIK112 bearing the hicA3-hicB3 intergenic region (253 bp upstream and 99 bp downstream from the hicB3 ATG) | This work |
| pSBT174 | pCRII bearing the 5′ end of hicB3 (codons 1 to 93) with flanking NotI sites | This work |
| pSBT230 | pCRII bearing hicA3-H28A with flanking EcoRI and SalI sites | This work |
| pSBT231 | pCRII bearing hicB1 with flanking NotI and SalI sites | This work |
| pSBT232 | pCRII bearing hicB2 with flanking NotI and SalI sites | This work |
| pSBT237 | pBAD30 bearing the hicA3-H28A EcoRI-SalI fragment of pSBT230 cloned under the control of Para | This work |
| pSBT238 | pAKK-Not bearing the hicB1 NotI-SalI fragment of pSBT231 cloned under the control of Plac | This work |
| pSBT239 | pAKK-Not bearing the hicB2 NotI-SalI fragment of pSBT232 cloned under the control of Plac | This work |
| pUC4K | Source of Kmr cassette; Apr Kmr | Amersham |
| pVIK112 | ori R6K suicide vector bearing the promoterless lacZ reporter gene; Kmr | 60 |
Ap, ampicillin; Km, kanamycin; Tp, trimethoprim; Cm, chloramphenicol; Plac, lactose operon promoter; Para, arabinose operon promoter; SD, Shine-Dalgarno.
Y. pestis mutant construction.
Mutants were constructed using the Red recombinase technique (23) and pEP1013 (3). Antibiotic-labeled PCR products were generated using pEP1087 or pEP1216 templates. Primers are listed in Table S1 in the supplemental material.
To delete chromosomal antibiotic resistance cassettes flanked by FLP recombination target (FRT) sites, pFLP2 (24) was electroporated into some constructs. Transformants were selected and were checked for loss of the cassette. To generate Y. pestis KIM6+ lacZ reporter strains, the suicide plasmids pSBT30, pSBT36, and pSBT172 were introduced into YPEP430 and its ΔhicA3B3 or ΔhicB3 derivatives by conjugation. Transconjugants were selected on LB-Km-Irg-Xgal plates. Correct integration of the transcriptional fusion at the chromosomal hicA3B3 locus was assessed by PCR. For all Y. pestis mutants, the presence of the instable chromosomal pgm locus was verified by streaking onto Congo red plates (25). Conservation of the endogenous plasmids was checked using multiplex PCR with primer pairs Ymt1/Ymt2 and Pla1/Pla2 for KIM6+ derivatives and the additional YopH3/YopH4 primer pair for CO92 constructs.
Growth assays.
E. coli MG1655 bearing both pSBT10 and pSBT41 was grown overnight at 37°C in LB-Ap-Km medium and was used to inoculate three cultures at an optical density at 600 nm (OD600) of 0.05. In the first culture, 0.5 mM Ara was added after 120 min of growth. In the second culture, 0.5 mM Ara was added after 120 min of growth, and 0.5 mM IPTG was added after 195 min. No inducer was added to the third culture. OD600 was measured every 30 min after induction.
Y. pestis KIM6+(pSBT10) and KIM6+(pBAD30) were grown overnight at 28°C in LB-Ap medium and were used to inoculate cultures at an OD600 of 0.05. After 180 min of growth, 1 mM Ara was added to the cultures. OD600 was measured every hour after induction.
Site-directed mutagenesis.
The megaprimer PCR method adapted from reference 26 was used to replace the hicA3 His28 codon with an alanine codon. Two 100-bp megaprimers were obtained by PCR amplification of Y. pestis KIM6+ genomic DNA using the forward external primer 3369aNde or HicA3RI, the mutated internal reverse primer HicA3H28A, and Pfu polymerase (Stratagene). In a second round of PCR, purified megaprimers were used with the reverse external primer 3369aXho or HicA3Sal to amplify hicA3H28A from the hicA3B3-bearing PCR product obtained with primer pair 3368F1/3369R1. The resulting NdeI-XhoI and EcoRI-SalI fragments were further cloned into pET24a+ and pBAD30 to yield pSBT116 and pSBT237, respectively.
Protein production and purification.
E. coli BL21(DE3) transformants were grown at 37°C until the OD600 reached 0.5. Protein overexpression was induced by the addition of 0.5 mM IPTG. After 3 h, cells were harvested, resuspended in buffer A (50 mM Tris [pH 8], 300 mM NaCl), and lysed using a French press. His-tagged proteins were purified from cleared lysates on a HisPur Ni-nitrilotriacetic acid (NTA) column (Thermo Scientific). Proteins were eluted with 5 ml of buffer A–300 mM imidazole. Further purification was carried out on a Superdex 75 size exclusion column (GE Healthcare) equilibrated with buffer A. Fractions of interest were pooled and were concentrated using an Amicon Ultra-4 centrifugal filter unit (molecular weight cutoff, 3,000 [3K] or 10K; Merck Millipore). For circular dichroism (CD) measurements, buffer A was replaced with NaP buffer (150 mM NaH2PO4–Na2HPO4 [pH 7.2]) by dialysis.
For crystallization experiments, cells were disrupted by sonication, and proteins were purified on a Ni-NTA agarose column (Qiagen) via elution with 100 mM, 200 mM, and 300 mM imidazole in buffer A with 5 mM Tris-(2-carboxyethyl)phosphine (TCEP). Proteins were then injected onto a HiTrap Heparin HP column (GE Healthcare) and were eluted with an NaCl gradient. Fractions of interest were concentrated and were injected onto a Superdex 75 column equilibrated with buffer A with 5 mM TCEP. These fractions were pooled and were concentrated using a Vivaspin 20 centrifugal concentrator (molecular weight cutoff, 5K; GE Healthcare). Selenomethionine (SeMet)-labeled HicB3 was prepared as described in reference 27 and was purified in the same way as the native protein.
HicA3 was purified from the HicA3-HicB3-6His complex via the following “water shock” procedure, which we found serendipitously. The complex was purified as described above except that after the first concentration step, fractions containing the HicA3-HicB3-6His complex were injected onto a Superdex 75 column equilibrated with water. Proteins that eluted as a single peak in the dead volume were then reconcentrated and reinjected onto a Superdex 75 column equilibrated with buffer A plus 5 mM TCEP. The proteins then eluted as two peaks: the first corresponded to a HicA3-depleted HicA3-HicB3-6His complex, and the second corresponded to HicA3 alone. Analytical size exclusion chromatography (SEC) revealed that (i) water-shocked purified HicA3 was dimeric and (ii) HicA3-H28A–6His purified in buffer A was monomeric. The misfolded, water-shocked protein was dialyzed against NaP buffer, denatured in 8 M guanidium chloride, and then dialyzed stepwise against NaP buffer. Finally, HicA3 was purified on a Superdex 75 size exclusion column and was concentrated using an Amicon Ultra-4 centrifugal filter unit (molecular weight cutoff, 3K). The same denaturation/renaturation protocol was applied to HicA3-H28A–6His. The circular dichroism spectra of renatured HicA3 and HicA3-H28A–6His were identical to that of native HicA3-H28A–6His.
Protein concentrations were determined by absorbance at 280 nm using a NanoVue Plus spectrophotometer (GE Healthcare) or a Bradford assay (Bio-Rad). The secondary-structure contents of the proteins were checked by CD, and the integrity of the protein sequences was checked with mass spectrometry.
5′ RACE.
The hicA3 and hicB3 transcription start sites (TSSs) were mapped using rapid amplification of 5′ cDNA ends (5′ RACE) according to the method described in reference 28. Briefly, total RNA was extracted from a 2-ml Y. pestis culture at an OD600 of 1 by using an RNeasy minikit (Qiagen). The RNA concentration was measured with a NanoVue Plus spectrophotometer. A 100-μl reaction volume containing RNA (6 μg), 20 U RNaseOUT (Invitrogen), and 10 U of tobacco acid pyrophosphatase (TAP; Epicentre) in TAP buffer was incubated for 30 min at 37°C. Control RNA (with no TAP treatment) was incubated under the same conditions. The 38-nucleotide (nt) RACE RNA adapter (500 pmol) was added to the tubes prior to phenol-chloroform extraction and ethanol precipitation. Pellets were dissolved in 13 μl water, denatured at 90°C for 5 min, and then quick-chilled on ice. The RACE adapter was ligated overnight at 17°C in a 20-μl reaction volume containing 10 U T4 RNA ligase (Epicentre), 5 μM ATP, 10% dimethyl sulfoxide (DMSO), and 0.4 U RNaseOUT in T4 RNA ligase buffer. Primer RlacZ (2 pmol) was added to RNA prior to phenol-chloroform extraction and ethanol precipitation. Pellets were dissolved in 20 μl of water, and 10 μl was used for reverse transcription with SuperScript III reverse transcriptase (Invitrogen). The cDNA was then amplified by PCR with primers specific for the RNA adapter (B6) and the target mRNA (3369aR1 or 3369R2). PCR products were purified from a 2% agarose gel, cloned into pCRII (Invitrogen), and sequenced. The absence of DNA contamination in the RNA preparation was assessed by PCR.
Gel shift assays.
DNA fragments containing either the hicA3 upstream region (365 bp) or part of the ymt gene (517 bp; the control fragment) were amplified by PCR using primer pair 3368F1/3369aR1 or Ypmt1/Ypmt2, respectively. PCR products were purified using the NucleoSpin Gel and PCR Clean-up kit (Macherey-Nagel). Reaction mixtures (20 μl) containing 50 ng of each DNA fragment and 0, 50, 100, or 150 ng of HicB3-6His or the HicA3-HicB3-6His complex in gel shift buffer (10 mM Tris [pH 7.5], 50 mM NaCl, 0.5 mM dithiothreitol [DTT], 1 mM MgCl2, and 2.5% glycerol) were incubated for 20 min at room temperature (RT) and were loaded onto a 6% acrylamide–TBE (Tris-borate EDTA) gel (89 mM Tris-borate [pH 8], 2 mM EDTA). After migration, DNA was visualized by ethidium bromide staining.
For gel shift assays with smaller DNA fragments, pairs of complementary oligonucleotides (66-mers or 80-mers) (see Table S1 in the supplemental material) were annealed by boiling for 5 min in a water bath and slow cooling of the bath to RT. The DNA fragments (1.25 pmol) were incubated with 2.3 pmol of HicB3-6His (corresponding to 150 ng) in a 20-μl final volume of gel shift buffer. Samples were run in a 7% acrylamide-TBE gel.
RNase activity assay.
An 11-kb RNA transcript containing part of the hepatitis C virus subgenomic replicon (29) was used as a substrate. In a 10-μl final volume of DNase I buffer (Ambion), 0.4 pmol of this RNA was incubated for 30 min at 37°C with 50 pmol of HicA3, HicA3-H28A–6His, HicA3-HicB3-6His, or HicB3-6His. Next, 2 μl of Gel Loading Buffer II (Ambion) was added to each tube, and samples were run in a 1% agarose-TBE gel. The RNA was visualized by ethidium bromide staining.
Virulence assay.
Groups of 8- to 9-week-old female OF1 mice (Charles River, France) were inoculated intradermally with ∼10 cells of Y. pestis CO92 or its ΔhicA3B3 derivative, as described previously (30). Survival was monitored daily for 15 days after inoculation.
Crystallization and crystal structure resolution.
All crystallizations were performed according to the vapor diffusion method at 293K. Crystals of selenium-labeled HicB3 (HicB3-SeMet) were obtained from a 1:1 mixture of 13.5 mg/ml protein in 50 mM Tris (pH 8)–0.3 M sodium chloride–5 mM TCEP buffer and a reservoir solution of 25% polyethylene glycol 3000–0.1 M morpholineethanesulfonic acid (MES) (pH 6.5).
For the HicA3B3 complex, we obtained crystals only in the presence of the subtilisin A protease. In this protocol, 58 μl of HicA3B3 solution (39 mg/ml in 50 mM Tris [pH 8]–0.3 M sodium chloride–5 mM TCEP) was mixed with 2 μl of a 0.5-mg/ml subtilisin A solution. The mixture was immediately used in the crystallization trials. The best conditions were obtained in 2.4 M disodium malonate solution.
Crystals were flash-frozen in liquid nitrogen in a two-step soaking protocol by using 15% and 30% ethylene glycol as a cryoprotectant for HicB3-SeMet and glycerol for HicA3B3. Diffraction data were collected at 100K on the Proxima 1 beamline at the Soleil synchrotron (Gif-sur-Yvette, France), using a Pilatus detector. The images were integrated with the XDS program and were processed using the Collaborative Computational Project Number 4 (CCP4) suite of programs (31). The initial models were completed and adjusted with the COOT program and were then refined using the REFMAC, PHENIX, and BUSTER programs.
The positions of the selenium atoms were determined using the automated procedure implemented in the SHELXD program at an optimal resolution of 4.4 Å and were refined using PHASER. Noncrystallographic symmetry and density modification were performed using PARROT. Automatic model building was performed using BUCCANEER. SHELXD, PARROT, PHASER, and BUCCANEER were all implemented in the CCP4 suite.
The structure of the HicA3B3 complex was solved by applying the molecular replacement method with PHASER. Both the N-terminal domain of the HicB3 structure (HicB3-Nt; 83 amino acids [aa]) (our work) and the TTHA1913 structure (PDB code 1WHZ) were used as search models. The experimental map was improved by solvent modification using the DM program. The resulting map was of very good quality, and ARP/wARP automatically built most of the protein model (291 of the 298 residues). The crystal structure at a resolution of 2.12 Å was refined to crystallographic R and Rfree factors of 18 and 21.8%, respectively (for the statistics, see Table S2 in the supplemental material). The refined structure consists of residues 1 to 85 for chains A and C (HicB3-Nt) and residues 1 to 66 for chains B and D (HicA3).
Protein structure accession numbers.
The PDB accession codes for the structures determined in this study are 4P7D for HicB3 and 4P78 for HicA3-HicB3.
RESULTS
HicA3-HicB3 is a new TAS in Y. pestis.
We reported previously that Y. pestis lacking ypo3369 is attenuated for virulence (3). ypo3369 is referred to as hicB3 by another research group, since its 135-aa product presents homology with the HicB antitoxin in the Escherichia coli HicA-HicB TAS (5). We hypothesized that the loss of virulence by the ypo3369 mutant resulted from a growth defect caused by the absence of toxin neutralization. In silico analysis revealed an open reading frame upstream of hicB3; it putatively encoded a 66-aa protein sharing 26% and 44% identity with the E. coli HicA (EcHicA) and Y. pestis HicA1 toxins, respectively (see Fig. S1 in the supplemental material). We called this gene hicA3. To establish whether or not the HicA3-HicB3 system was a bona fide TAS, we monitored the growth of E. coli MG1655 containing two plasmids: one harbored hicA3 under the control of the arabinose-inducible promoter Para, and the other harbored hicB3 under the control of the IPTG-inducible promoter Plac (Fig. 1A). Addition of arabinose to the culture medium induced growth arrest, whereas subsequent IPTG addition restored bacterial growth; this result suggests that HicA3 overproduction is bacteriostatic and that HicB3 is able to neutralize this toxicity. In contrast, overproduction of HicA3 with HicB1 or HicB2 (the other two Y. pestis HicB family antitoxins [5]) was bacteriostatic (Fig. 1B). Thus, neither protein is able to neutralize HicA3.
FIG 1.

Growth curves in LB broth. (A) E. coli MG1655 bearing both pSBT10 (Para-hicA3) and pSBT41 (Plac-hicB3) was grown in three flasks in parallel. Arrows indicate the addition of inducers; 0.5 mM arabinose (Ara) was added to two cultures (filled and open circles), and 1 mM IPTG was added to one culture (filled circles). Shaded circles correspond to growth in the absence of inducers. (B) E. coli DH5α bearing pSBT10 (Para-hicA3) and either pSBT41 (Plac-hicB3), pSBT238 (Plac-hicB1), pSBT239 (Plac-hicB2), or pAKK (empty plasmid). Inducers were added to each of the four cultures. (C) Y. pestis KIM6+ bearing pBAD30 (empty plasmid) or pSBT10 (Para-hicA3). After 180 min, 1 mM Ara was added to both cultures. (D) Y. pestis KIM6+ and ΔhicB3 mutants. Plasmid pSBT7 (pHicB3) bears the hicB3 gene. Each curve is representative of the results of at least three independent experiments.
We next evaluated the toxicity of HicA3 in Y. pestis. The KIM6+ strain transformed with the Para-hicA3 plasmid was grown in LB medium; upon the addition of arabinose, hicA3 induction triggered bacteriostasis (Fig. 1C). We also constructed KIM6+ ΔhicB3 and ΔhicA3B3 mutants and compared their respective growth rates. In the absence of hicB3, the presence of hicA3 conferred a slow-growth phenotype that was complemented by a hicB3-bearing plasmid (Fig. 1D). In contrast, the deletion of both hicA3 and hicB3 did not affect the growth rate of Y. pestis—confirming that the toxic effect required HicA3 and that HicB3 was an antitoxin (Fig. 1D). Taken as a whole, our data indicate that hicA3 and hicB3 together constitute a new two-component type II TAS.
hicA3B3 is an operon and is repressed by HicB3.
A 174-bp intergenic region separates hicA3 and hicB3 on the CO92 chromosome (Fig. 2A), suggesting that hicB3 could be transcribed independently of hicA3. DNA fragments containing the putative hicA3 promoter (PhicA3) or encompassing part or all of the intergenic region were cloned and transcriptionally fused to the lacZ reporter gene (Fig. 2A). A high level of β-galactosidase activity on an Xgal plate was detected only for E. coli expressing lacZ under the control of PhicA3 (Fig. 2B). This observation suggested that (i) a promoter is present upstream of hicA3 and (ii) there is no constitutive promoter in the intergenic region.
FIG 2.

(A) Genomic organization of the Y. pestis CO92 hicA3-hicB3 locus. This organization is strictly conserved for the KIM6+ chromosome. The DNA fragments cloned into pVIK112 to yield PhicA3-lacZ and hicB3-lacZ reporter fusions are depicted here: the pSBT30 insert encompasses the sequence extending 117 bp upstream and 90 bp downstream of the hicA3 start codon, while the pSBT36 and pSBT172 inserts encompass the sequence extending 151 or 253 bp upstream of the hicB3 start codon and 99 bp downstream. (B) LacZ phenotype of E. coli CC118 λpir bearing pVIK112 (empty plasmid), pSBT30, pSBT36, or pSBT172 on Xgal plates. Transformants were patched onto LB-Km-Xgal plates and were incubated at 37°C for 24 h. The intensity of the blue color reflects the β-galactosidase activity level.
Toxin-antitoxin operon promoters are usually repressed by the antitoxin or the TA complex (15). To establish whether HicB3 could repress PhicA3, the PhicA3-lacZ reporter fusion was introduced into the chromosome of Y. pestis ΔlacZ strains lacking or not lacking hicA3B3 (Fig. 3A). On Xgal plates, the parental strain bearing PhicA3-lacZ was LacZ−, whereas the ΔhicA3B3 mutant carrying the same reporter fusion was LacZ+ (Fig. 3A). When a plasmid bearing a wild-type (WT) copy of hicB3 or carrying the hicA3B3 operon was introduced into the ΔhicA3B3 PhicA3-lacZ strain, the LacZ− phenotype was restored (Fig. 3A). These observations suggested that PhicA3 is repressed by HicB3.
FIG 3.

Schematic representation of the hicA3B3 region in the different Y. pestis KIM6+ ΔlacZ reporter strains and the associated LacZ phenotypes on Xgal plates. P indicates the hicA3 promoter. Strains were patched onto LB-Km-Xgal plates and were incubated at 28°C for 48 h. (A) hicA3′-lacZ fusions generated by chromosomal integration of pSBT30. YSBT61 transformed with pSBT7 (pHicB3) or pEP1165 (pHicA3B3) turned LacZ−, while the pCRII (empty-plasmid) transformant remained LacZ+. (B) hicB3′-lacZ fusions generated by chromosomal integration of pSBT172. YSBT151 has a Lac+/− phenotype, whereas YSBT157 has a Lac+ phenotype. YSBT151 and YSBT157 were also transformed with the plasmids mentioned above: pHicB3 and pHicA3B3 generate full repression on both strains (LacZ− phenotype), while the empty plasmid does not. Derivatives of YSBT151 and YSB157 bearing an 88-bp deletion around PhicA3 (ΔP) do not exhibit any β-galactosidase activity.
To evaluate hicB3 expression and regulation in Y. pestis, we introduced the hicB3-lacZ transcriptional fusion into the chromosomes of ΔlacZ strains lacking or not lacking hicB3 (Fig. 3B). Although both the parental and ΔhicB3 strains expressed hicB3-lacZ, the expression level of the fusion was higher in the ΔhicB3 background, which agreed with the observed derepression of PhicA3 in the absence of HicB3. When the reporter strains were transformed with a plasmid bearing hicB3, the chromosomal hicB3-lacZ fusion was fully repressed in both strains (Fig. 3B). This observation indicated that either (i) hicB3 is transcribed mainly from the PhicA3 promoter or (ii) any alternative hicB3 promoters are also repressed by HicB3. In order to distinguish between these two possibilities, we deleted 88 bp within the PhicA3 promoter region upstream of the hicB3-lacZ fusion on the chromosomes of the HicB3+ and HicB3− isogenic strains (Fig. 3B). No hicB3-lacZ expression was detected in the absence of the PhicA3 promoter—even in the strain lacking the HicB3 repressor (Fig. 3B). Overall, these data indicate that hicB3 is transcribed mainly from PhicA3 and that the activity of the hicB3-lacZ fusion detected in the HicB3+ strain resulted from transcriptional read-through from PhicA3.
We then used rapid amplification of 5′ cDNA ends (5′ RACE) to identify the transcription start sites (TSSs) for the hicA3 and hicB3 genes. Total RNA purified from the ΔhicA3B3 PhicA3-lacZ reporter strain (in which PhicA3 is fully active) and the hicB3-lacZ reporter strain was used to localize the TSSs of hicA3 and hicB3, respectively. For hicA3, a single TSS was identified 23 bp upstream of the HicA3 initiation codon (see Fig. S2A in the supplemental material), from which we deduced the −10 (TATGAT) and −35 (TTGACT) boxes of the PhicA3 promoter (Fig. 4A). For hicB3, the longest mRNA was initiated at the hicA3 TSS—confirming that hicA3 and hicB3 form an operon. Several smaller mRNAs initiating between positions +144 and +365 relative to the hicA3 TSS were also detected (see Fig. S2B). In contrast to the longest mRNA, most of these mRNAs were unaffected by TAP treatment, suggesting that they were monophosphorylated and therefore were not primary transcripts. In silico analysis of the +144-to-+365 region failed to reveal any other promoter candidates. Overall, the data suggest that these mRNAs are truncated forms of hicA3B3 mRNA.
FIG 4.

(A) Nucleotide sequence of the hicA3 upstream region. The −10 and −35 promoter sequences, the ribosome binding site (SD), and the HicA3 initiation codon (ATG) are underlined. The BS1 and BS2 palindromic sequences are indicated by converging arrows. Letters in boldface correspond to bases conserved in the repeat. The TSS (+1) is indicated by an arrow. (B) Gel shift assay of DNA fragments bearing PhicA3 (365 bp) or part of ymt (control sequence; 517 bp) incubated with HicB3-6His or HicA3-HicB3-6His for 20 min at RT. (C) Gel shift assay of DNA fragments BS1-BS2, MU1-BS2, BS1-MU2, and MU1-MU2′ (1.25 pmol), incubated in the presence (+) or absence (−) of 2.3 pmol of HicB3-6His for 20 min at RT. (D) Promoter activities of the BS1-BS2, MU1-BS2, BS1-MU2, and MU1-MU2′ DNA fragments cloned upstream of lacZ. The four recombinant plasmids were introduced into E. coli CC118 λpir carrying a compatible plasmid bearing the hicB3 gene (HicB3+) or an empty vector (no HicB3). Transformants were patched onto LB-Ap-Tp-Xgal plates.
HicB3 binds a dyad symmetry DNA motif.
We performed gel shift experiments to establish whether HicB3 and/or the HicA3-HicB3 complex binds to the PhicA3 region in vitro. The purified six-histidine-tagged HicB3 protein (HicB3-6His) or the purified HicA3-HicB3-6His complex was incubated with a 365-bp PCR product bearing PhicA3 or with a control DNA fragment taken from outside the hicA3B3 region (Fig. 4B). Both HicB3-6His and the HicA3-HicB3-6His complex were able to bind the DNA fragment encompassing PhicA3 but not the control fragment.
Sequence analysis of the PhicA3 region revealed the presence of two 15-bp inverted repeats, corresponding to the dyad symmetry consensus T(G/A)GGT(A/G)TNA(C/T)ACC(T/C)A (Fig. 4A). We named these palindromes BS1 (bases −57 to −42 relative to the hicA3 TSS) and BS2 (bases −17 to −2) and tested their ability to bind purified HicB3-6His in vitro. We used four different DNA fragments containing the −62-to-+5 region and bearing either intact BS1 and BS2 sequences or sequences with base substitutions in BS1 (MU1-BS2 fragment), BS2 (BS1-MU2 fragment), or both (MU1-MU2′ fragment) in gel shift assays. The substitutions were chosen so as to affect neither the −35 and −10 boxes of PhicA3 nor the TSS (see Fig. S3 in the supplemental material). HicB3-6His binding was observed with the DNA fragments bearing either BS1 or BS2 but not when both sites were mutated (Fig. 4C). In the MU1-MU2′ fragment, substitutions in MU2′ affected only the second half of the consensus—showing that the dyad symmetry of the sequence is required for HicB3 binding in vitro.
To evaluate the role of the HicB3 binding sites in the regulation of PhicA3 in vivo, the BS1-BS2, MU1-BS2, BS1-MU2, and MU1-MU2′ DNA fragments were cloned upstream of the lacZ reporter gene. The four reporter plasmids conferred a LacZ+ phenotype on E. coli on Xgal plates, indicating that each insert contained an active promoter (Fig. 4D). When a second compatible plasmid bearing the hicB3 gene was introduced into these four strains, the WT BS1-BS2 and mutated MU1-BS2 promoters were fully repressed (giving a LacZ− phenotype); in contrast, the mutated BS1-MU2 promoter remained active but at a lower level (LacZ+/− phenotype), while the mutated MU1-MU2′ promoter remained fully active (LacZ+ phenotype) (Fig. 4D). These in vivo data show that although HicB3 is able to bind both the BS1 and BS2 sites, the main operator of PhicA3 is BS2, which overlaps with the −10 box.
HicA3 is an RNase.
We sought to purify HicA3 and thus study its activity. We were initially unable to overproduce HicA3-6His alone (because of its high toxicity in E. coli) or to purify HicA3 after denaturation of the HicA3-HicB3 complex (presumably because of its very high affinity for HicB3). We serendipitously found a “water shock” and renaturation procedure (described in Materials and Methods) that enabled us to purify HicA3. Since the E. coli HicA toxin had been shown to degrade mRNA (21), we tested the RNase activity of HicA3 on an in vitro-transcribed viral mRNA substrate. This mRNA was hydrolyzed by HicA3 but not by HicB3-6His or the HicA3-HicB3-6His complex (Fig. 5). Although the HicA3-HicB3-6His complex lacks RNase activity, it is able to bind RNA, as indicated by the RNA shift observed. HicA3 was also able to degrade two other in vitro-transcribed mRNAs (data not shown), suggesting that it can target various mRNAs in vivo.
FIG 5.
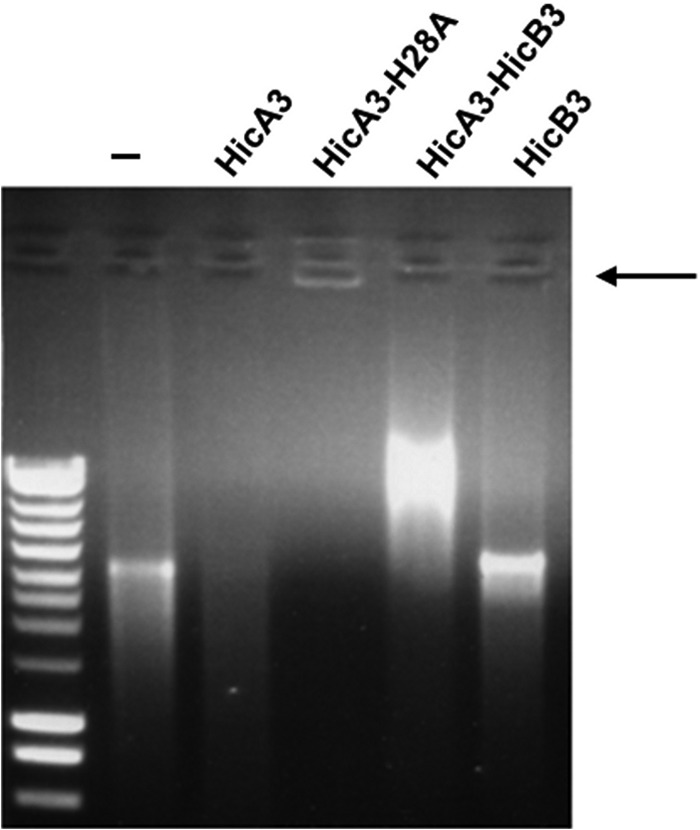
Test of HicA3, HicA3-H28A–6His, HicA3-HicB3-6His, and HicB3-6His for RNase activity. The arrow indicates the stacking of the mRNA–HicA3-H28A–6His complex in the well.
We hypothesized that the histidine 28 (His28) residue of HicA3 is involved in RNase activity, because it is conserved in the E. coli HicA and Y. pestis HicA1 toxins (see Fig. S1 in the supplemental material). Indeed, when we replaced His28 with alanine (H28A) via site-directed mutagenesis, the resulting overproduction of HicA3-H28A–6His was not toxic to E. coli (data not shown). Furthermore, the purified protein was inactive in vitro but was able to aggregate mRNA, as indicated by the stacking of the substrate in the well (Fig. 5).
HicA3B3 is not required for virulence in a bubonic plague model.
Taken as a whole, our data show that HicB3 is the antitoxin for the HicA3 toxin and is also a transcriptional repressor. Thus, the attenuated virulence of the ΔhicB3 mutant described in our previous work (3) could result from either (i) the inability of the mutant to grow efficiently in vivo when HicA3 is not neutralized or (ii) the regulation of virulence genes by HicB3 in addition to its role as an antitoxin. To distinguish between these hypotheses, we deleted the whole hicA3B3 operon from the CO92 chromosome. In contrast to the attenuated ΔhicB3 mutant, the ΔhicA3B3 mutant was fully virulent in the murine model of bubonic plague (Fig. 6). Thus, HicB3 is not required for virulence in the absence of the HicA3 toxin.
FIG 6.
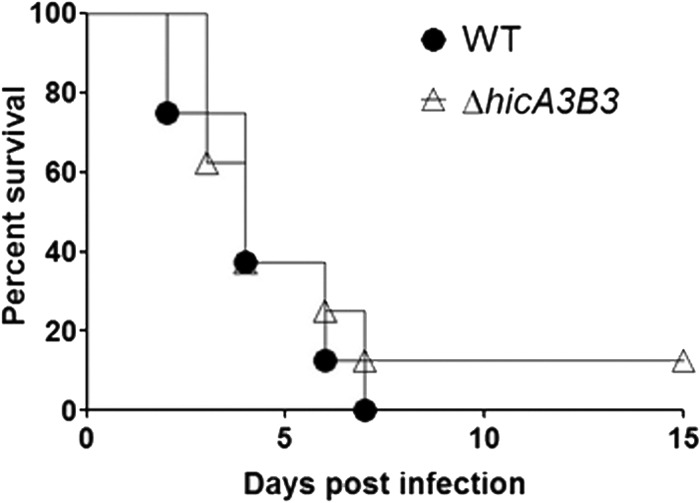
Survival rate (expressed as a percentage) of OF1 mice injected intradermally with 10 CFU of wild-type Y. pestis CO92 (filled circles) or CO92 ΔhicA3B3 (open triangles).
The HicB3 antitoxin is a tetramer.
We solved the X-ray crystal structure of HicB3 at a resolution of 2.12 Å (see Table S2 in the supplemental material). HicB3 forms a tetramer, the symmetry of which can best be described as a dimer of dimers. The HicB3 monomer consists of two domains connected by a linker (residues 85 to 92) containing a short helical α4 stretch (Fig. 7A). The N-terminal (Nt) domain adopts an antiparallel β1β2β3α1α2α3β4 fold. The long α1 helix lies in the cradle formed by the bend in the β-sheet, while the two short α2 and α3 helices flank the other face of the β-sheet. The C-terminal (Ct) β5α5α6 domain (residues 93 to 135) forms a ribbon-helix-helix (RHH) motif. HicB3 dimerizes through this domain (Fig. 7A); the β5 strands from two RHH motifs form a central antiparallel β-sheet, and the pairs of helices form a helical bundle. In one subunit, the α5 helix of the RHH motif packs against the α1 helix of the N-terminal domain of the opposite subunit. In the other subunit, the α5 helix interacts with the linker situated between the α1 and α2 helices. The two dimers bind through their N-terminal domains to form a ring-type tetramer (Fig. 7B and C). This interface is stabilized by the packing of hydrophobic patches (Ile2, Ile78, and Phe81 from one subunit, and Phe31, Ile34, Tyr66, Ile67, Ile78, and Leu79 from the other).
FIG 7.

Crystal structures. (A) Schematic presentation of the HicB3 dimer. α-Helices, β-strands, and N- and C-terminal residues for one monomer are labeled. Dimerization occurs via the β5 strand of the C-terminal RHH domain in each subunit. (B) Perpendicular views of the HicB3 tetramer. Two HicB3 dimers interact via their N-terminal domains to form a tetramer. (C) View of two interacting N-terminal domains in the tetramer. (D) HicA3–HicB3-Nt tetrameric complex. The two HicA3 subunits are shown in red, and the two HicB3 subunits are shown in pink and blue. Position 85 indicates the last residue of HicB3-Nt.
We used size exclusion chromatography coupled to multiangle laser light scattering (SEC-MALS) measurements to analyze HicB3. The monodisperse sample in SEC corresponds to a molecular mass of 63.4 kDa, close to the value of 65.2 kDa expected for a tetramer (see Fig. S4A in the supplemental material). These observations were confirmed by analytical ultracentrifugation measurements showing that 95% of the HicB3 species in solution form a globular homotetramer (see Fig. S4C).
The HicA3-HicB3 complex primarily forms a heterohexamer.
We were able to obtain crystals for the HicA3-HicB3 complex only when subtilisin A was added during the crystallization process. We collected a complete diffraction data set at a resolution of 2.12 Å (see Table S2 in the supplemental material). The protease had cleaved off the C-terminal domain of HicB3 because no electron density was present beyond residue 85. The asymmetric crystal unit obtained under these conditions contains two copies of HicA3 and two copies of the N-terminal domain of HicB3 (HicB3-Nt), which thus form a heterotetramer (referred to below as HicA3–HicB3-Nt). HicB3-Nt has the same structure in the complex and in the unbound HicB3 protein (root mean square deviation for 85 superposed residues, 1.4 Å). The HicA3–HicB3-Nt complex is elongated, with a HicA3 subunit binding to each end of the HicB3-Nt dimer interface (Fig. 7D). HicA3 adopts an α1β1β2β3α2 fold characteristic of a double-stranded RNA (dsRNA)-binding domain. The HicA3 α2 helix packs against the β-sheet of HicB3. The β-sheets of HicA3 and HicB3 are juxtaposed in the complex but do not form a continuous β-sheet. The α1 helix of HicB3 covers one face of the HicA3 β-sheet. The interface is stabilized by both hydrophobic and polar interactions (10 hydrogen bonds and 5 salt bridges). The His28 residue required for HicA3 RNase activity is situated at the N-terminal end of the β2 strand and is completely buried at the interface with HicB3, suggesting that HicB3 neutralizes HicA3 by blocking its active site. Overall, 28% of the available surface area of HicA3 is masked by complex formation.
When using SEC-MALS to determine the stoichiometry of the HicA3-HicB3 complex (in the absence of subtilisin processing), we measured a molecular mass of about 78.6 kDa, close to the value of 79.8 kDa calculated for a hexamer of two HicA3 units and four HicB3 units (see Fig. S4B in the supplemental material). The analytical ultracentrifugation data are compatible with the presence of 90% of the molecules in solution as a 2:4 heterohexamer (see Fig. S4D in the supplemental material). Interestingly, the superposition of the structures of the HicA3–HicB3-Nt heterotetramer and the HicB3 homotetramer revealed a steric clash between the end of the β-sheet in the two HicA3 subunits and the end of the last α-helix (α6) in HicB3 subunits 2 and 4 (Fig. 8A). This observed steric hindrance is thus consistent with the formation of a heterohexamer composed of two subunits of HicA3 bound to opposing subunits of a HicB3 tetramer (Fig. 8B).
FIG 8.
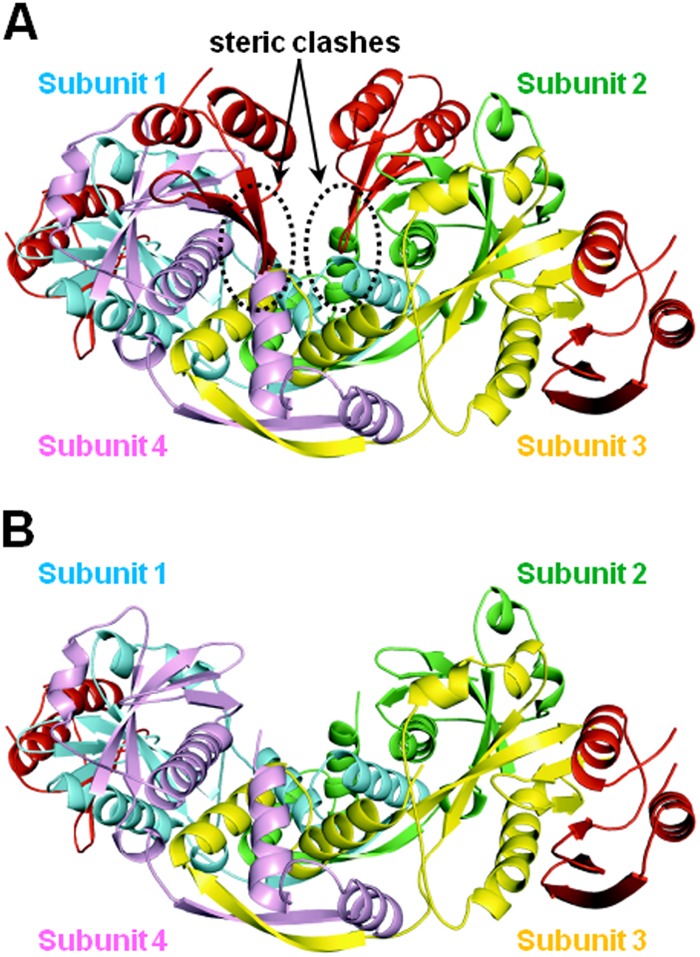
In silico models of the HicA3-HicB3 complexes. The HicA3 subunits are shown in red, and the four HicB3 subunits are shown in green, blue, pink, and yellow. (A) The putative hetero-octameric complex. The steric clashes between two HicA3 subunits and the C-terminal domains of HicB3 subunits 2 and 4 are circled. (B) The proposed heterohexameric complex.
DISCUSSION
We have identified HicA3 and characterized HicA3B3, a novel, functional type II TAS in Y. pestis. HicA3 is a 66-aa monomeric RNase. The HicB3 antitoxin has two functions: it neutralizes HicA3 through direct binding and represses hicA3B3 transcription. Type II two-component TASs are highly modular and can be classified into 11 families as a function of their structure or mode of action (for a recent review, see reference 32). The X-ray structures of TA complexes have been solved for seven families. The present report is the first for a HicAB family complex. In the various TASs, some toxins are monomeric and others are homodimeric (33–36). Antitoxins are thought to be dimeric and bind DNA via an N- or C-terminal dimerization domain folding as an RHH, helix-turn-helix (HTH), PhD-like-, or AbrB-like domain (37). The antitoxin dimer binds one, two, or four toxin monomers (33, 36, 38). HicB3 assembles as a dimer of dimers. Each tetramer possesses two RHH DNA-binding folds and is able to receive two HicA3 molecules. The HicA3B3 complex is therefore the first example of a tetrameric antitoxin that binds two toxin monomers. In the HicA3-HicB3 complex, the HicB3 N-terminal domain binds to one side of the toxin and significantly occludes the catalytic His28. We also show that both the HicB3 tetramer and the HicA3B3 complex bind to 15-bp operators flanking PhicA3 in vitro and repress PhicA3 in vivo. As observed for other RHH transcription regulators (39, 40), DNA binding is probably mediated via insertion of the two HicB3 RHH domains into the major groove of the DNA double helix, with each ribbon interacting with one TRGGTRT half-site. In silico analysis using the Regulatory Sequence Analysis Tools website (http://www.rsat.eu) (41) did not reveal any other occurrences of the operator sequence in the Y. pestis CO92 genome—suggesting that hicA3B3 is the only operon regulated by HicB3.
In canonical, well-studied TASs, such as RelBE and Phd-Doc, interaction with DNA consists primarily of the binding of the antitoxin to operator sequences. The toxin acts as either a corepressor or a derepressor, depending on the toxin/antitoxin ratio (42). At low toxin concentrations, toxin binding enhances the affinity of the antitoxin for the operator. At high toxin concentrations, affinity for the operator decreases. This “conditional cooperativity” mechanism relies on allosteric modification of the antitoxin upon toxin binding (43, 44). In contrast to canonical type II TASs, the dimeric E. coli MqsA antitoxin (a member of the HTH repressor family) does not exhibit conditional cooperativity (45). MqsA is fully folded and binds DNA alone. The MqsR toxin is not a corepressor, since the MqsRA complex is unable to bind DNA. However, the toxin destabilizes the MqsA-DNA repression complex via allosteric modification (45). Our data suggest that HicA3 may not have a corepressor function, since (i) the HicB3 tetramer is already fully folded in the absence of HicA3 and (ii) HicB3 and the HicA3B3 complex bind DNA in vitro to the same extent. However, we observed that PhicA3 repression can be alleviated by overexpression of the nontoxic HicA3-H28A protein in vivo (data not shown). This finding suggests that excess HicA3 destabilizes the ternary HicA3-HicB3-operator complex and titrates out the HicB3 repressor. Further research will be required to establish whether HicA3 is solely a derepressor (like MqsA [45]) or both a corepressor and a derepressor.
X-ray crystallography revealed that HicA3 has a dsRNA-binding fold, which suggests that the toxin can cleave mRNA in the vicinity of double-stranded regions. This fits with the observation that E. coli HicA degrades both mRNA and transfer-mRNA (tmRNA) (21). Targeted mutagenesis of HicA3 highlighted His28 as required for RNase activity. While this article was in preparation, Butt et al. reported the solution structure of the Burkholderia pseudomallei HicA toxin (BpsHicA) and showed that BpsHicA requires Gly22 and His24 for RNase activity (46). These residues correspond to Gly26 and His28 in HicA3. BpsHicA and HicA3 share the same folding structure.
The fact that we were able to construct a Y. pestis ΔhicB3 mutant indicates that the expression of chromosomal hicA3 in the absence of the HicB3 antitoxin is not lethal; the mutant grows slowly, but it does grow. This observation suggests that the amount of HicA3 RNase produced from hicA3 mRNA is not highly toxic to Y. pestis. Hence, either (i) the cleavage rate of HicA3 is too low to produce bacteriostasis, (ii) HicA3 is produced in very low quantities (even though PhicA3 is derepressed), or (iii) HicA3 targets are not essential for growth. These hypotheses will be evaluated in future studies. A comparative analysis of the cleavage rates of the HicA3, HicA1, EcHicA, and BpsHicA RNases and mutant HicA3 proteins may help to characterize the catalytic mechanism of these RNases.
A few TASs have been shown to be involved in virulence in Salmonella enterica (47), Haemophilus influenzae (48), and Mycobacterium tuberculosis (49). We suggested previously that hicB3 may be involved in plague pathogenesis (3). Our present results show that a Y. pestis ΔhicA3B3 mutant is fully virulent; hence, the loss of virulence of the ΔhicB3 mutant is due to inefficient in vivo growth caused by the activity of free HicA3 RNase, not to the lack of HicB3 as a regulator. Y. pestis encodes three other active type II TASs and seven putative ones (5) that could compensate for the loss of HicA3B3. The other complete HicAB system (HicA1B1) was a possible candidate for this role. However, we found that a ΔhicA1B1 ΔhicA3B3 double mutant was fully virulent in the murine plague model (unpublished data). It is worth noting that in other species, deletion of three to five TA operons was required before a change in phenotype could be observed (50, 51). Although our data indicate that HicA3B3 is not important for Y. pestis virulence in the rodent, one cannot rule out the possibility that the system is required in other environments (e.g., in the flea or for survival in the soil). The B. pseudomallei HicAB sytem has recently been shown to play a role in persister cell formation following exposure to ciprofloxacin (46). Future experiments should evaluate the role of the HicA1B1 and HicA3B3 systems in Y. pestis persistence.
Supplementary Material
ACKNOWLEDGMENTS
We thank Laure Marceau for sequencing, Hervé Drobecq for mass spectrometry analysis, Julien Herrou for suggesting the use of subtilisin to crystallize the complex, and Rayan Farhat for the gift of viral mRNA. We thank the BSL-3 Facility staff of the Institut Pasteur de Lille, Karine Blondeau and Seiki Achiedo for the production of SeMet-labeled HicB3, and the staff at Proxima 1 for help with synchrotron data collection and processing (and particularly Pierre Legrand for helpful discussions). We are grateful to Françoise Jacob-Dubuisson, Alain Baulard, and Michel Simonet for critical reading of the manuscript.
Work in the laboratory of F.S. was funded by INSERM, CNRS, Institut Pasteur de Lille, and Université Lille Nord de France, Région Nord-Pas-de-Calais Arcir Emergence grant 07230045 (to F.S.), and ANR grant 07-MIME-017-01 IVOTIMP (to F.S.). Work in the laboratory of H.V.T. was funded by the French Infrastructure for Integrated Structural Biology (FRISBI) and ANR grant 10-INSB-05-01. The analytical ultracentrifugation measurements were performed at the Imagif facility (Gif-sur-Yvette, France). S.B.-T. received a doctoral studentship from the Ministère de l'Enseignement Supérieur et de la Recherche.
Footnotes
Published ahead of print 11 August 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.01932-14.
REFERENCES
- 1.Butler T. 2013. Plague gives surprises in the first decade of the 21st century in the United States and worldwide. Am. J. Trop. Med. Hyg. 89:788–793. 10.4269/ajtmh.13-0191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lathem WW, Crosby SD, Miller VL, Goldman WE. 2005. Progression of primary pneumonic plague: a mouse model of infection, pathology, and bacterial transcriptional activity. Proc. Natl. Acad. Sci. U. S. A. 102:17786–17791. 10.1073/pnas.0506840102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pradel E, Lemaître N, Merchez M, Ricard I, Reboul A, Dewitte A, Sebbane F. 2014. New insights into how Yersinia pestis adapts to its mammalian host during bubonic plague. PLoS Pathog. 10:e1004029. 10.1371/journal.ppat.1004029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sebbane F, Lemaître N, Sturdevant DE, Rebeil R, Virtaneva K, Porcella SF, Hinnebusch BJ. 2006. Adaptive response of Yersinia pestis to extracellular effectors of innate immunity during bubonic plague. Proc. Natl. Acad. Sci. U. S. A. 103:11766–11771. 10.1073/pnas.0601182103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goulard C, Langrand S, Carniel E, Chauvaux S. 2010. The Yersinia pestis chromosome encodes active addiction toxins. J. Bacteriol. 192:3669–3677. 10.1128/JB.00336-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yamaguchi Y, Park J-H, Inouye M. 2011. Toxin-antitoxin systems in bacteria and archaea. Annu. Rev. Genet. 45:61–79. 10.1146/annurev-genet-110410-132412. [DOI] [PubMed] [Google Scholar]
- 7.Cook GM, Robson JR, Frampton RA, McKenzie J, Przybilski R, Fineran PC, Arcus VL. 2013. Ribonucleases in bacterial toxin–antitoxin systems. Biochim. Biophys. Acta 1829:523–531. 10.1016/j.bbagrm.2013.02.007. [DOI] [PubMed] [Google Scholar]
- 8.Unoson C, Wagner EGH. 2008. A small SOS-induced toxin is targeted against the inner membrane in Escherichia coli. Mol. Microbiol. 70:258–270. 10.1111/j.1365-2958.2008.06416.x. [DOI] [PubMed] [Google Scholar]
- 9.Bernard P, Kézdy KE, Van Melderen L, Steyaert J, Wyns L, Pato ML, Higgins PN, Couturier M. 1993. The F plasmid CcdB protein induces efficient ATP-dependent DNA cleavage by gyrase. J. Mol. Biol. 234:534–541. 10.1006/jmbi.1993.1609. [DOI] [PubMed] [Google Scholar]
- 10.Zhang Y, Inouye M. 2011. RatA (YfjG), an Escherichia coli toxin, inhibits 70S ribosome association to block translation initiation. Mol. Microbiol. 79:1418–1429. 10.1111/j.1365-2958.2010.07506.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Correia FF, D'Onofrio A, Rejtar T, Li L, Karger BL, Makarova K, Koonin EV, Lewis K. 2006. Kinase activity of overexpressed HipA is required for growth arrest and multidrug tolerance in Escherichia coli. J. Bacteriol. 188:8360–8367. 10.1128/JB.01237-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Germain E, Castro-Roa D, Zenkin N, Gerdes K. 2013. Molecular mechanism of bacterial persistence by HipA. Mol. Cell 52:248–254. 10.1016/j.molcel.2013.08.045. [DOI] [PubMed] [Google Scholar]
- 13.Yamaguchi Y, Inouye M. 2011. Regulation of growth and death in Escherichia coli by toxin–antitoxin systems. Nat. Rev. Microbiol. 9:779–790. 10.1038/nrmicro2651. [DOI] [PubMed] [Google Scholar]
- 14.Fozo EM, Hemm MR, Storz G. 2008. Small toxic proteins and the antisense RNAs that repress them. Microbiol. Mol. Biol. Rev. 72:579–589. 10.1128/MMBR.00025-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hayes F, Van Melderen L. 2011. Toxins-antitoxins: diversity, evolution and function. Crit. Rev. Biochem. Mol. Biol. 46:386–408. 10.3109/10409238.2011.600437. [DOI] [PubMed] [Google Scholar]
- 16.Fineran PC, Blower TR, Foulds IJ, Humphreys DP, Lilley KS, Salmond GP. 2009. The phage abortive infection system, ToxIN, functions as a protein–RNA toxin–antitoxin pair. Proc. Natl. Acad. Sci. U. S. A. 106:894–899. 10.1073/pnas.0808832106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Samson JE, Bélanger M, Moineau S. 2013. Effect of the abortive infection mechanism and type III toxin/antitoxin system AbiQ on the lytic cycle of Lactococcus lactis phages. J. Bacteriol. 195:3947–3956. 10.1128/JB.00296-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zielenkiewicz U, Cegłowski P. 2005. The toxin-antitoxin system of the streptococcal plasmid pSM19035. J. Bacteriol. 187:6094–6105. 10.1128/JB.187.17.6094-6105.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hallez R, Geeraerts D, Sterckx Y, Mine N, Loris R, Van Melderen L. 2010. New toxins homologous to ParE belonging to three-component toxin-antitoxin systems in Escherichia coli O157:H7. Mol. Microbiol. 76:719–732. 10.1111/j.1365-2958.2010.07129.x. [DOI] [PubMed] [Google Scholar]
- 20.Pandey DP, Gerdes K. 2005. Toxin–antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes. Nucleic Acids Res. 33:966–976. 10.1093/nar/gki201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jørgensen MG, Pandey DP, Jaskolska M, Gerdes K. 2009. HicA of Escherichia coli defines a novel family of translation-independent mRNA interferases in bacteria and archaea. J. Bacteriol. 191:1191–1199. 10.1128/JB.01013-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sikkema DJ, Brubaker RR. 1987. Resistance to pesticin, storage of iron, and invasion of HeLa cells by yersiniae. Infect. Immun. 55:572–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645. 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP. 1998. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212:77–86. 10.1016/S0378-1119(98)00130-9. [DOI] [PubMed] [Google Scholar]
- 25.Fetherston JD, Schuetze P, Perry RD. 1992. Loss of the pigmentation phenotype in Yersinia pestis is due to the spontaneous deletion of 102 kb of chromosomal DNA which is flanked by a repetitive element. Mol. Microbiol. 6:2693–2704. 10.1111/j.1365-2958.1992.tb01446.x. [DOI] [PubMed] [Google Scholar]
- 26.Burke E, Barik S. 2003. Megaprimer PCR, p 525–531 In Bartlett JMS, Stirling D. (ed), PCR protocols. Humana Press, Totowa, NJ. 10.1385/1-59259-384-4:525. [DOI] [Google Scholar]
- 27.Quevillon-Cheruel S, Collinet B, Trésaugues L, Minard P, Henckes G, Aufrère R, Blondeau K, Zhou C-Z, Liger D, Bettache N, Poupon A, Aboulfath I, Leulliot N, Janin J, van Tilbeurgh H. 2007. Cloning, production, and purification of proteins for a medium-scale structural genomics project, p 21–37 In Walker JM, Doublíe S. (ed), Macromolecular crystallography protocols. Humana Press, Totowa, NJ. 10.1007/978-1-59745-209-0_2. [DOI] [PubMed] [Google Scholar]
- 28.Argaman L, Hershberg R, Vogel J, Bejerano G, Wagner EGH, Margalit H, Altuvia S. 2001. Novel small RNA-encoding genes in the intergenic regions of Escherichia coli. Curr. Biol. 11:941–950. 10.1016/S0960-9822(01)00270-6. [DOI] [PubMed] [Google Scholar]
- 29.Lohmann V, Körner F, Koch J-O, Herian U, Theilmann L, Bartenschlager R. 1999. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285:110–113. 10.1126/science.285.5424.110. [DOI] [PubMed] [Google Scholar]
- 30.Lemaître N, Ricard I, Pradel E, Foligné B, Courcol R, Simonet M, Sebbane F. 2012. Efficacy of ciprofloxacin-gentamicin combination therapy in murine bubonic plague. PLoS One 7:e52503. 10.1371/journal.pone.0052503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. 2011. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67:235–242. 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park SJ, Son WS, Lee B-J. 2013. Structural overview of toxin–antitoxin systems in infectious bacteria: a target for developing antimicrobial agents. Biochim. Biophys. Acta 1834:1155–1167. 10.1016/j.bbapap.2013.02.027. [DOI] [PubMed] [Google Scholar]
- 33.Gazit E, Sauer RT. 1999. The Doc toxin and Phd antidote proteins of the bacteriophage P1 plasmid addiction system form a heterotrimeric complex. J. Biol. Chem. 274:16813–16818. 10.1074/jbc.274.24.16813. [DOI] [PubMed] [Google Scholar]
- 34.Loris R, Dao-Thi M-H, Bahassi EM, Van Melderen L, Poortmans F, Liddington R, Couturier M, Wyns L. 1999. Crystal structure of CcdB, a topoisomerase poison from E. coli. J. Mol. Biol. 285:1667–1677. 10.1006/jmbi.1998.2395. [DOI] [PubMed] [Google Scholar]
- 35.Overgaard M, Borch J, Gerdes K. 2009. RelB and RelE of Escherichia coli form a tight complex that represses transcription via the ribbon–helix–helix motif in RelB. J. Mol. Biol. 394:183–196. 10.1016/j.jmb.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kamada K, Hanaoka F, Burley SK. 2003. Crystal structure of the MazE/MazF complex: molecular bases of antidote-toxin recognition. Mol. Cell 11:875–884. 10.1016/S1097-2765(03)00097-2. [DOI] [PubMed] [Google Scholar]
- 37.Anantharaman V, Aravind L. 2003. New connections in the prokaryotic toxin-antitoxin network: relationship with the eukaryotic nonsense-mediated RNA decay system. Genome Biol. 4:R81. 10.1186/gb-2003-4-12-r81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kamphuis M, Chiara Monti M, van den Heuvel HR, Lopez-Villarejo J, Diaz-Orejas R, Boelens R. 2007. Structure and function of bacterial Kid-Kis and related toxin-antitoxin systems. Protein Pept. Lett. 14:113–124. 10.2174/092986607779816096. [DOI] [PubMed] [Google Scholar]
- 39.Brown BM, Bowie JU, Sauer RT. 1990. Arc repressor is tetrameric when bound to operator DNA. Biochemistry (Mosc.) 29:11189–11195. 10.1021/bi00503a006. [DOI] [PubMed] [Google Scholar]
- 40.Bøggild A, Sofos N, Andersen KR, Feddersen A, Easter AD, Passmore LA, Brodersen DE. 2012. The crystal structure of the intact E. coli RelBE toxin-antitoxin complex provides the structural basis for conditional cooperativity. Structure 20:1641–1648. 10.1016/j.str.2012.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van Helden J, André B, Collado-Vides J. 2000. A web site for the computational analysis of yeast regulatory sequences. Yeast 16:177–187. 10.1002/(SICI)1097-0061(20000130)16:2<177::AID-YEA516>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 42.Overgaard M, Borch J, Jørgensen MG, Gerdes K. 2008. Messenger RNA interferase RelE controls relBE transcription by conditional cooperativity. Mol. Microbiol. 69:841–857. 10.1111/j.1365-2958.2008.06313.x. [DOI] [PubMed] [Google Scholar]
- 43.Garcia-Pino A, Balasubramanian S, Wyns L, Gazit E, De Greve H, Magnuson RD, Charlier D, van Nuland NAJ, Loris R. 2010. Allostery and intrinsic disorder mediate transcription regulation by conditional cooperativity. Cell 142:101–111. 10.1016/j.cell.2010.05.039. [DOI] [PubMed] [Google Scholar]
- 44.Winther KS, Gerdes K. 2012. Regulation of enteric vapBC transcription: induction by VapC toxin dimer-breaking. Nucleic Acids Res. 40:4347–4357. 10.1093/nar/gks029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brown BL, Lord DM, Grigoriu S, Peti W, Page R. 2013. The Escherichia coli toxin MqsR destabilizes the transcriptional repression complex formed between the antitoxin MqsA and the mqsRA operon promoter. J. Biol. Chem. 288:1286–1294. 10.1074/jbc.M112.421008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Butt A, Higman VA, Williams C, Crump MP, Hemsley CM, Harmer N, Titball RW. 2014. The HicA toxin from Burkholderia pseudomallei has a role in persister cell formation. Biochem. J. 459:333–344. 10.1042/BJ20140073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.De la Cruz MA, Zhao W, Farenc C, Gimenez G, Raoult D, Cambillau C, Gorvel J-P, Méresse S. 2013. A toxin-antitoxin module of Salmonella promotes virulence in mice. PLoS Pathog. 9:e1003827. 10.1371/journal.ppat.1003827. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 48.Ren D, Walker AN, Daines DA. 2012. Toxin-antitoxin loci vapBC-1 and vapXD contribute to survival and virulence in nontypeable Haemophilus influenzae. BMC Microbiol. 12:263. 10.1186/1471-2180-12-263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ramage HR, Connolly LE, Cox JS. 2009. Comprehensive functional analysis of Mycobacterium tuberculosis toxin-antitoxin systems: implications for pathogenesis, stress responses, and evolution. PLoS Genet. 5:e1000767. 10.1371/journal.pgen.1000767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim Y, Wang X, Ma Q, Zhang X-S, Wood TK. 2009. Toxin-antitoxin systems in Escherichia coli influence biofilm formation through YjgK (TabA) and fimbriae. J. Bacteriol. 191:1258–1267. 10.1128/JB.01465-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Frampton R, Aggio RBM, Villas-Bôas SG, Arcus VL, Cook GM. 2012. Toxin-antitoxin systems of Mycobacterium smegmatis are essential for cell survival. J. Biol. Chem. 287:5340–5356. 10.1074/jbc.M111.286856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Studier FW, Moffatt BA. 1986. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 189:113–130. 10.1016/0022-2836(86)90385-2. [DOI] [PubMed] [Google Scholar]
- 53.Herrero M, de Lorenzo V, Timmis KN. 1990. Transposon vectors containing non-antibiotic resistance selection markers for cloning and stable chromosomal insertion of foreign genes in gram-negative bacteria. J. Bacteriol. 172:6557–6567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Grant SG, Jessee J, Bloom FR, Hanahan D. 1990. Differential plasmid rescue from transgenic mouse DNAs into Escherichia coli methylation-restriction mutants. Proc. Natl. Acad. Sci. U. S. A. 87:4645–4649. 10.1073/pnas.87.12.4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guyer MS, Reed RR, Steitz JA, Low KB. 1981. Identification of a sex-factor-affinity site in E. coli as gamma delta. Cold Spring Harb. Symp. Quant. Biol. 45(Part 1):135–140. 10.1101/SQB.1981.045.01.022. [DOI] [PubMed] [Google Scholar]
- 56.Simon R, Priefer U, Pühler A. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram negative bacteria. Nat. Biotechnol. 1:784–791. 10.1038/nbt1183-784. [DOI] [Google Scholar]
- 57.Doll JM, Zeitz PS, Ettestad P, Bucholtz AL, Davis T, Gage K. 1994. Cat-transmitted fatal pneumonic plague in a person who traveled from Colorado to Arizona. Am. J. Trop. Med. Hyg. 51:109–114. [DOI] [PubMed] [Google Scholar]
- 58.Veiga E, de Lorenzo V, Fernández LA. 1999. Probing secretion and translocation of a β-autotransporter using a reporter single-chain Fv as a cognate passenger domain. Mol. Microbiol. 33:1232–1243. 10.1046/j.1365-2958.1999.01571.x. [DOI] [PubMed] [Google Scholar]
- 59.Guzman L-M, Belin D, Carson MJ, Beckwith JON. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 177:4121–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kalogeraki VS, Winans SC. 1997. Suicide plasmids containing promoterless reporter genes can simultaneously disrupt and create fusions to target genes of diverse bacteria. Gene 188:69–75. 10.1016/S0378-1119(96)00778-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.