

Abstract
Combinations of glycopeptides and β-lactams exert synergistic antibacterial activity, but the evolutionary mechanisms driving resistance to both antibiotics remain largely unexplored. By repeated subculturing with increasing vancomycin (VAN) and cefuroxime (CEF) concentrations, we isolated an evolved strain of the model bacterium Bacillus subtilis with reduced susceptibility to both antibiotics. Whole-genome sequencing revealed point mutations in genes encoding the major σ factor of RNA polymerase (sigA), a cell shape-determining protein (mreB), and the ρ termination factor (rho). Genetic-reconstruction experiments demonstrated that the G-to-C substitution at position 336 encoded by sigA (sigAG336C), in the domain that recognizes the −35 promoter region, is sufficient to reduce susceptibility to VAN and works cooperatively with the rhoG56C substitution to increase CEF resistance. Transcriptome analyses revealed that the sigAG336C substitution has wide-ranging effects, including elevated expression of the general stress σ factor (σB) regulon, which is required for CEF resistance, and decreased expression of the glpTQ genes, which leads to fosfomycin (FOS) resistance. Our findings suggest that mutations in the core transcriptional machinery may facilitate the evolution of resistance to multiple cell wall antibiotics.
INTRODUCTION
The evolution of multidrug resistance among pathogenic bacteria is an increasing threat to global public health because the rate of resistance evolution far outpaces that of development of new antibiotics (1–3). Combinations of antibiotics have emerged as a promising strategy to enhance antibiotic effectiveness (4, 5), and they are commonly classified as additive (no interaction), synergistic (greater than the additive effect), or antagonistic (less than the additive effect) (6, 7). The synergistic interactions between glycopeptides and β-lactams have been demonstrated, especially against staphylococci with reduced susceptibilities to vancomycin (VAN) (8, 9). Glycopeptide antibiotics, such as VAN, inhibit cell wall biosynthesis by binding tightly to the d-Ala-d-Ala terminus of the peptidoglycan pentapeptide, thereby blocking the transpeptidation and transglycosylation reactions in peptidoglycan assembly (10). The β-lactam antibiotics, including cephalosporins, such as cefuroxime (CEF), inhibit cell wall cross-linking by inactivating specific transpeptidases known as penicillin-binding proteins (PBPs) (11, 12). Although synergistic combinations of drugs have clinical benefits, in some cases, resistance to drug combinations may evolve at an even higher rate than resistance to individual drugs (13).
Whole-genome resequencing has enormous resolving power for identifying the genetic changes underlying adaptation. Thus, it has been widely used to explore the evolution of antibiotic resistance that results from the accumulation of random mutations (14–17). Thousands of bacterial genome sequences are available in the public domain and can be used as reference genomes for comparison (18).
Mutations affecting bacterial RNA polymerase (RNAP) arise during many selections (2, 19–23), suggesting that alterations to RNAP can facilitate adaptation. Bacterial RNAP is an essential, highly conserved, multisubunit enzyme that is responsible for transcription (24). It consists of the core (α2ββ′ω) enzyme, a dissociable σ factor (24), and, in Gram-positive bacteria, an additional δ subunit that enhances transcriptional specificity (25, 26). For transcription initiation, the core enzyme must bind one of various σ factors, each of which recognizes a specific promoter sequence (27). Bacteria have one housekeeping σ factor (σ70 in Escherichia coli; σA in Bacillus subtilis) and several alternative σ factors that are activated in response to either environmental or developmental signals. Thus, the replacement of one σ factor by another redirects transcription of different subsets of genes (28–30).
The transcription termination factor Rho is required for one of two bacterial termination mechanisms, Rho dependent and intrinsic (31, 32). Rho is broadly distributed among the Bacteria and highly conserved, but some bacterial species do not have a homolog (33, 34). Rho is essential in enterobacteria, such as E. coli (35), but dispensable in the Gram-positive bacteria B. subtilis and Staphylococcus aureus (33, 36). In B. subtilis, the cellular level of Rho is less than 5% that of E. coli, and initially, rho itself and the trp operon were the only genes known to be regulated by Rho-dependent termination (37, 38). In addition, Gram-positive bacteria, except for Micrococcus luteus, are resistant to the Rho-inhibiting antibiotic bicyclomycin, suggesting that Rho is usually nonessential in these bacteria (33, 39).
Here, we describe mutations resulting from the selection of B. subtilis for reduced susceptibility to VAN and CEF. Whole-genome resequencing and genetic-reconstruction studies demonstrate that single-nucleotide substitutions within sigA and rho are responsible for the observed effects. These results highlight the ability of mutations affecting the core transcriptional machinery to mediate adaptation to cell envelope antibiotics.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The bacterial strains, plasmids, and primers used in this study are listed in Table 1. Null mutant strains were constructed by using long-flanking homology PCR (LFH-PCR) as described previously (40). Cells were routinely cultured at 37°C in lysogeny broth (LB) medium or minimal medium supplemented with either 2% glucose or 2% glycerol-3-phosphate (glycerol-3-P). Minimal medium contained 40 mM potassium morpholinepropanesulfonate (MOPS) (adjusted to pH 7.4 with KOH), 2 mM potassium phosphate buffer (pH 7.0), glucose (2% [wt/vol]), (NH4)2SO4 (2 g/liter), MgSO4 · 7H2O (0.2 g/liter), trisodium citrate · 2H2O (1 g/liter), potassium glutamate (1 g/liter), tryptophan (10 mg/liter), 3 nM (NH4)6Mo7O24, 400 nM H3BO3, 100 μM FeCl3, 30 nM CoCl2, 10 nM CuSO4, 10 nM ZnSO4, and 80 nM MnCl2. For B. subtilis, the following antibiotics were used for selection: spectinomycin (100 μg/ml), kanamycin (15 μg/ml), chloramphenicol (10 μg/ml), or macrolide-lincosamide-streptogramin B (containing 1 μg/ml erythromycin and 25 μg/ml lincomycin).
TABLE 1.
Strains, plasmids, and primers
| Strain, plasmid, or primer | Genotype, description, or sequence (5′→3′)a | Source or referenceb |
|---|---|---|
| Strains | ||
| W168 | trpC2 | BGSC no. 1A1 |
| HB13513 | W168 sigAG336C mreBL201S rhoG56C | Evolved isolate |
| HB13679 | W168 sigAWT-mls | LFH PCR with W168 |
| HB13680 | W168 sigAG336C-mls | LFH PCR with W168 |
| HB13681 | W168 kan-mreBWT | LFH PCR with W168 |
| HB13682 | W168 kan-mreBL201S | LFH PCR with W168 |
| HB13683 | W168 spc-rhoWT | LFH PCR with W168 |
| HB13684 | W168 spc-rhoG56C | LFH PCR with W168 |
| HB13690 | W168 sigAWT-mls spc-rhoG56C | LFH PCR with HB13684 |
| HB13691 | W168 sigAG336C-mls spc-rhoG56C | LFH PCR with HB13684 |
| HB13685 | W168 rho::spc | LFH PCR with W168 |
| HB13686 | W168 penP::kan | LFH PCR with W168 |
| HB13689 | W168 rho::spc penP::kan | HB13686 chr DNA with HB13685 |
| HB0008 | CU1065 fosB::cat | 63 |
| HB13698 | W168 fosB::cat sigAWT-mls | HB0008 chr DNA with HB13679 |
| HB13699 | W168 fosB::cat sigAG336C-mls | HB0008 chr DNA with HB13680 |
| HB11302 | CU1065 bshC::cat | Laboratory stock |
| HB13700 | W168 bshC::cat sigAWT-mls | HB11302 chr DNA with HB13679 |
| HB13701 | W168 bshC::cat sigAG336C-mls | HB11302 chr DNA with HB13680 |
| HB13551 | W168 sigB::cat | 72 |
| HB13709 | W168 sigB::cat sigAWT-mls | HB13551 chr DNA with HB13679 |
| HB13710 | W168 sigB::cat sigAG336C-mls | HB13551 chr DNA with HB13680 |
| HB13713 | W168 sigAWT-mls amyE::Pspac(hy)-glpTQ (cat) | pYH006 with HB13679 |
| HB13714 | W168 sigAG336C-mls amyE::Pspac(hy)-glpTQ (cat) | pYH006 with HB13680 |
| Plasmids | ||
| pPL82 | IPTG-inducible expression vector (amyE integration) | 43 |
| pYH006 | Pspac(hy)-glpTQ in pPL82 | This study |
| Primers (no. [name]) | ||
| 6074 (sigA up-F) | CGCCTACGCTCAAAAGATTG | |
| 6075 [sigA up-R (mls)] | GAGGGTTGCCAGAGTTAAAGGATCTTCAGTAAAATAAAGGCATATTATCCA | |
| 6076 [sigA do-F (mls)] | CGATTATGTCTTTTGCGCAGTCGGCTGCAAATGAACATTGTGGTG | |
| 6077 (sigA do-R) | CACTTGTCATCACAACTTTTCTCAA | |
| 6078 (mreB up-F) | ACAATGAGAGCTCTTCGCCA | |
| 6079 [mreB up-R (kan)] | CCTATCACCTCAAATGGTTCGCTGGCAAAAATACCCTAAAGGGAAAA | |
| 6080 [mreB do-F (kan)] | CGAGCGCCTACGAGGAATTTGTATCGAAAGGAAAAAGGATATTTGTAACACTT | |
| 6081 (mreB do-R) | TGGATGTGCTCCAGTGCTTT | |
| 6082 (rho up-F) | GTCGCAGTTGACCCTCTTGA | |
| 6083 [rho up-R (spc)] | CGTTACGTTATTAGCGAGCCAGTCAAGGTTTTGACACGGAATTGA | |
| 6084 [rho do-F (spc)] | CAATAAACCCTTGCCCTCGCTACGTCAATTCCGTGTCAAAACCTT | |
| 6085 (rho do-R) | AGCTCTGCTTCAGGCTGGTT | |
| 6086 (rho up-F) for KO | GCGTTGATATCCTGTTCGGA | |
| 6087 [rho up-R (spc)] for KO | CGTTACGTTATTAGCGAGCCAGTCCATAAAAACACCACGCTTTTCA | |
| 6088 [rho do-F (spc)] for KO | CAATAAACCCTTGCCCTCGCTACGACCATCCTTGCTACGGCTCT | |
| 6089 (rho do-R) for KO | GTACATAGGCTTCGCTCCCC | |
| 6090 (penP up-F) | GATACTGCAGGCCCCTTTTC | |
| 6091 [penP up-R (kan)] | CCTATCACCTCAAATGGTTCGCTGCTATGGATTTTGCTTCGGCA | |
| 6092 [penP do-F (kan)] | CGAGCGCCTACGAGGAATTTGTATCGGTGGCAATTCTGTCAAACCG | |
| 6093 (penP do-R) | CAAAAGCTCCAGGAAAAGAAGA | |
| 6094 (glpT RT-F) | TCAACGGATGGTTTCAAGGA | |
| 6095 (glpT RT-R) | GCCGTATCGCACGAAATAAA | |
| 5397 (16S rRNA-8F) | AGAGTTTGATCCTGGCTCAG | |
| 5398 (16S rRNA-519R) | GTATTACCGCGGCTGCTGG | |
| 6205 (glpTQ Pspac-F) | GCGCCCCGGGCACAAACAGCAAAGGGGGA | |
| 6206 (glpTQ Pspac-R) | GCGCTCTAGATTTTTGCTTTTAATAACCCTTTTT |
Restriction sites are underlined, and the sequences complementary to antibiotic resistance cassettes for LFH PCR are in boldface.
chr, chromosomal.
Selection of antibiotic resistance.
The MICs for vancomycin and cefuroxime in combination were determined using a Bioscreen C microbial-growth analyzer. Overnight cultures of B. subtilis W168 were subcultured by 1:100 dilution in 5 ml of Mueller-Hinton (MH) medium (Difco) containing sub-MICs of the combination of the antibiotics (vancomycin, 0.1 μg/ml; cefuroxime, 2 μg/ml). Then, 24-h cultures were inoculated 1:100 into 5 ml fresh medium containing both antibiotics, with the concentrations incrementally increasing for 10 days. Bacterial glycerol stocks were frozen at −80°C every day. The day 6 cultures were streaked on an LB agar plate, and the susceptibilities of 10 colonies to vancomycin and cefuroxime were compared. Finally, a representative colony (HB13513) was selected for whole-genome resequencing.
Whole-genome resequencing.
B. subtilis wild-type and HB13513 mutant strains were grown in LB medium to an optical density at 600 nm (OD600) of 0.4, and their genomic DNAs were purified using the Qiagen DNeasy blood and tissue kit. The quantity and purity of DNA were determined using a NanoDrop spectrophotometer (Nanodrop Technology, Inc., Wilmington, DE), and DNA was sequenced using an Illumina HiSeq 2000 at the Cornell University Life Sciences Core Laboratories Center. To identify single-nucleotide polymorphisms (SNPs), the resulting genomic sequence data were assembled with MOSAIC, using the reference sequence (41) under GenBank accession number ABQK00000000.
Genetic reconstruction.
Each point mutation was moved into the wild-type chromosome using LFH PCR as described previously (2). The upstream and downstream fragments were amplified from either the wild type or HB13513 using four primers for each SNP (Table 1) and then joined to an antibiotic resistance cassette (mls, kan, or spc), which was introduced as a selectable marker after the stop codon or before the promoter region. The final PCR products were transformed into the wild-type B. subtilis strain, and the correct transfer of the point mutation was verified by DNA sequencing.
Plasmid construction.
Molecular techniques were performed as described by Sambrook and Russell (42). Plasmid pPL82 was used for the expression of glpTQ under the control of the isopropyl-β-d-thiogalactopyranoside (IPTG)-inducible promoter Pspac(hy) (43). To construct pYH006 (pPL82-glpTQ), the promoterless glpTQ operon was amplified from B. subtilis wild-type chromosomal DNA by PCR using the primers 6205 (glpTQ Pspac-F)/6206 (glpTQ Pspac-R). Construct integrity was verified by DNA sequencing. The resulting plasmid was then integrated by double-crossover homologous recombination into the amyE locus.
Disk diffusion assays.
Disk diffusion assays were performed as described previously (2). Briefly, strains were grown in LB at 37°C with shaking to an OD600 of 0.4. A 100-μl aliquot of these cultures was added to 4 ml of 0.7% MH soft agar (kept at 45°C) and directly poured onto MH plates (containing 15 ml of 1.5% MH agar). After 30 min incubation at room temperature, the plates were dried for 20 min in a laminar airflow hood. Filter paper disks containing the antibiotics to be tested were placed on top of the MH agar, and the plates were incubated at 37°C for 16 to 18 h. The diameters of the inhibition zones were measured after subtraction of the diameter of the filter paper disk (6.5 mm). The following antibiotics and quantities were used in the disk diffusion assays: cefuroxime, 50 μg; ceftazidime, 30 μg; ceftriaxone, 30 μg; ampicillin, 50 μg; penicillin G, 50 μg; piperacillin, 100 μg; oxacillin, 1 μg; and fosfomycin (FOS), 100 μg or 500 μg.
MIC assays.
The Etest assay was performed similarly to the disk diffusion assay. Etest strips (bioMérieux) impregnated with VAN or CEF (at concentrations ranging from 0.016 to 256 μg/ml) were applied to MH agar plates, and the plates were incubated at 37°C for 16 to 18 h. The MIC was determined from the scale at the intersection of bacterial growth with the Etest strip. For the growth inhibition assay, strains were grown to an OD600 of 0.4 and then diluted 1:400 in MH broth. Aliquots (200 μl) of the diluted cultures were dispensed in a Bioscreen 100-well microtiter plate, and a range of at least 9 antibiotic concentrations close to the MIC were added to each well. Growth was measured spectrophotometrically (OD600) every 30 min for 24 h using a Bioscreen C microbial-growth analyzer at 37°C with continuous shaking. The MIC was defined as the lowest concentration of antibiotic that completely inhibited growth (OD600 < 0.2) at the 16-h or 24-h time point.
RNA preparation and microarray analyses.
Total RNA was isolated from strains HB13679 (sigAWT-mls) and HB13680 (sigAG336C mls [where sigAG336C represents the G-to-C substitution at position 336 of sigA]) grown in LB medium to an OD600 of 0.4, using the RNeasy minikit (Qiagen) followed by DNase treatment with Turbo DNA free (Ambion). The quantity and purity of RNA were determined using a NanoDrop spectrophotometer. Two microarray analyses were performed in biological triplicate with a dye swap. cDNA labeling and microarray analysis were performed as described previously (44). The GenePix Pro software package (version 6.0) was used for image processing and analysis. The normalized microarray data sets were filtered to remove those genes that were not expressed at levels significantly above background under either condition (sum of mean fluorescence intensities, <20). In addition, the mean and standard deviation of the fluorescence intensities were computed for each gene, and those for which the standard deviation was greater than the mean value were ignored. The fold change was calculated by using the average signal intensities for HB13680 divided by those for HB13679.
RT-PCR assay.
For reverse transcription (RT)-PCR, total RNA (1 μg) was reverse transcribed into cDNA by using random hexamers and a TaqMan reverse transcription kit (Roche). The cDNA was then amplified by PCR using gene-specific primer sets (Table 1). The reaction mixture was denatured (95°C for 3 min) and then subjected to 20 thermal cycles (95°C for 30 s, 54°C for 30 s, and 72°C for 1 min) and a final extension (72°C for 10 min). Primer pair 6094 (glpT RT-F)/6095 (glpT RT-R) was used to detect the glpT transcript. 16S rRNA was used as a normalization control. The PCR products were separated on a 1.5% agarose gel, stained with ethidium bromide, and visualized.
Microarray data accession number.
The microarray data set is available in the NCBI GEO database under accession number GSE55202.
RESULTS AND DISCUSSION
Development of resistance to the combination of VAN and CEF in B. subtilis.
We combined experimental evolution and whole-genome sequencing to define a pathway enabling B. subtilis to develop increased resistance to the combination of VAN and CEF. The MICs of VAN and CEF in MH medium for B. subtilis W168 are 0.25 μg/ml and 6 μg/ml, respectively. When either antibiotic was present at 0.5× MIC, the observed MIC for the other was reduced by ∼2-fold, indicative of synergism. To select for increased resistance against this combination of cell wall antibiotics, wild-type cells were continuously subcultured for 10 days with increasing concentrations of both antibiotics. Cells were taken every day before subculturing for frozen bacterial stocks, and their growth rates were measured spectrophotometrically using a Bioscreen C growth analyzer at 37°C. As the cells grew very slowly from day 7 (Fig. 1A), we focused on the day 6 strain. We first isolated a single colony of the evolved strain (HB13513) by streaking it onto an LB agar plate and determined the MICs of VAN and CEF by using an Etest assay. Strain HB13513 displayed an ∼8-fold-increased MIC of CEF, together with a modest (≤2-fold) increase in the MIC of VAN (Fig. 1B). These data suggest that B. subtilis HB13513 has undergone significant evolution of CEF resistance. Despite extended exposure to VAN, only a modest decrease in VAN susceptibility was observed in this B. subtilis isolate.
FIG 1.

Experimental evolution of increased resistance to the combination of VAN and CEF in B. subtilis. (A) Growth rates of the evolved strains. Wild-type B. subtilis W168 (BGSC 1A1) cells were grown by repeated subculturing with selection for increasing resistance to both VAN and CEF. (B) Determination of MICs of VAN and CEF for the evolved isolate after 6 days of culturing. Etest strips (bioMérieux) with VAN or CEF (concentrations, 0.016 to 256 μg/ml) were applied to MH agar plates, and then the MIC values were read as the point of intersection or by interpolation after 18 h of incubation at 37°C. (C) Sanger sequencing for the confirmation of SNPs (as originally identified by Illumina-based whole-genome resequencing). Strain HB13513 has four point mutations in three genes. The mutated residues are indicated by asterisks, and the altered amino acids are listed at the bottom of each panel. (D) Determinations of the VAN and CEF MICs for evolved strains. The MICs of VAN and CEF were determined by the growth inhibition assay and Etest assay, respectively. (E) Sequential fixation of mutations during the evolution of resistance to the combination of VAN and CEF.
Identification of mutations that drive the evolution of reduced antibiotic susceptibility.
To identify the genomic basis of these changes in susceptibility to VAN and CEF, we used whole-genome resequencing of HB13513 and its parental wild type to reveal four SNPs that distinguish the two strains. These four SNPs affect three genes (sigA, mreB, and rho) and lead to single amino acid changes in each encoded protein. These mutations were confirmed by Sanger DNA sequencing (Fig. 1C).
When antibiotic susceptibility was evaluated by the growth inhibition assay or Etest assay, the MICs of VAN and CEF gradually increased over time, as expected (Fig. 1D). To determine the order of fixation of these mutations, we sequenced these loci in each sequential daily culture. As shown in Fig. 1E, evolution proceeded through the sequential fixation of mutations in the rho, sigA, and mreB genes. The first mutation arising (rhoG56C) correlates with an ∼4-fold increase in CEF resistance, with another significant increase associated with the next two mutations at days 4 and 5 (which were also correlated with an increase in the MIC of VAN).
The sigAG336C substitution is responsible for both altered growth and reduced VAN susceptibility.
To determine which of these mutations confer the reduced susceptibility to VAN and/or CEF, we moved each point mutation into the parental wild-type strain, as described previously (2). An antibiotic resistance cassette was introduced as a selectable marker either after the stop codon or before the promoter region of each gene (Fig. 2A). These genetic-reconstruction experiments demonstrated that the altered growth phenotype (longer lag phase) results from the sigAG336C substitution (Fig. 2B). This is consistent with the appearance of this mutation in the day 4 culture (Fig. 1E), which also had a longer lag phase (Fig. 1A). Our results provide an example of the general trend that antibiotic resistance mutations affecting essential genes often increase fitness in the presence of antibiotic but result in decreased fitness (including reductions in growth and virulence) in the absence of antibiotic (45).
FIG 2.

Identification of the genetic determinants for slow growth and reduced VAN susceptibility. (A) Genetic reconstruction of the substitutions in the wild-type chromosomal DNA. The antibiotic resistance cassette (mls, kan, or spc) was placed as a selectable marker after the stop codon or before the promoter region. The SNPs, indicated by asterisks, were moved into the parental wild-type strain by transformation of the LFH-PCR product. (B) Determinants of the slow-growth phenotype in the HB13513 mutant. Growth assays were performed in LB medium in the absence of antibiotic. (C) Determinants of reduced VAN susceptibility in the HB13513 mutant. Growth assays were performed in MH medium in the absence or presence of VAN. The data are representative of at least three independent experiments.
To identify genetic determinants that affect susceptibility to VAN, we compared the growth rates of the reconstructed strains in the presence of VAN using a Bioscreen C growth analyzer. As shown in Fig. 2C, the sigAG336C substitution is largely responsible for the reduced VAN susceptibility, although the mreBL201S substitution also has a slight effect. These results suggest that the sigAG336C substitution is sufficient for reduced VAN susceptibility (comparable to that noted in strain HB13513) (Fig. 2C), but this change also results in overall reduced fitness (Fig. 2B).
Although the sigAG336C substitution occurred at day 4 and the mreBL201S substitution occurred at day 5 (Fig. 1E), the cultures from days 2 and 3 were also slightly less susceptible to VAN (Fig. 1D). This may be due to induction of an adaptive-tolerance response. For example, VAN is known to be an inducer of several cell envelope stress responses, including the σM regulon (30, 46) and the LiaRS two-component system (47).
The sigAG336C and rhoG56C alleles together reduce susceptibility to CEF.
A similar genetic-reconstruction strategy was used to identify the determinants of reduced CEF susceptibility in HB13513. These studies demonstrate that both sigAG336C and rhoG56C reduce CEF susceptibility, and the combination is sufficient to confer a 9-fold increase in the MIC (Fig. 3). In contrast, the mreBL201S substitution was not directly involved in CEF resistance (Fig. 3A). Notably, the rhoG56C substitution led to an over 4-fold increase in the CEF MIC in the Etest assays (Fig. 3B). These results suggest that the reduced susceptibility to CEF in the HB13513 strain is attributable to the effects of both sigAG336C and rhoG56C.
FIG 3.

Identification of the genetic determinants for decreased CEF susceptibility. (A) Determination of the CEF susceptibilities of the reconstructed strains. Note that in the course of introducing each of these mutations into the parent strain (by selection for the linked antibiotic resistance marker [Fig. 2A]), recombination can occur either proximal to the SNP (leaving a wild-type allele in the chromosome) or distal to the SNP (leading to a strain with the SNP in the chromosome). The former integrants, which provide a control to rule out effects from the integrated antibiotic cassette, are designated by a superscript WT, and the latter are identified by the corresponding amino acid change. Disk diffusion assays were performed on MH agar plates with a filter paper disk containing 50 μg CEF. Each bar represents the average zone of inhibition, expressed as the total diameter minus the diameter of the filter paper disk (6.5 mm). Three independent experiments were performed for each strain, and the standard deviations are indicated by error bars. (B) Determination of MICs for CEF using an Etest assay. The Etest strips (bioMérieux) with CEF concentrations of 0.016 to 256 μg/ml were applied to MH agar plates, and the MIC was read by interpolation after 18 h of incubation at 37°C.
A rho-null mutant displays reduced susceptibility to β-lactam antibiotics.
We next sought to determine how the rhoG56C mutation might affect Rho function. The active form of Rho is a ring-shaped homohexamer with RNA-binding, ATP hydrolysis, translocase, and RNA/DNA helicase activities (32, 48). Each monomer has two distinct functional domains: an N-terminal RNA-binding domain (RNA-BD) and a C-terminal ATP-binding domain (ATP-BD) (49), but the secondary RNA-binding site (SBS) also exists in the ATP-BD (50).
Analysis of conserved motifs in B. subtilis Rho using the NCBI Conserved Domain database (51) revealed that the rhoG56C substitution lies close to the N-terminal RNA-binding domain (Fig. 4A). Since the 6 RNA-binding domains of the Rho hexamer together compose the primary RNA-binding site (52, 53), we hypothesized that this substitution might impair RNA binding to Rho and therefore would be a loss-of-function mutation. Indeed, the MIC of CEF for a rho-null mutant was exactly the same as that for the reconstructed rhoG56C strain (Fig. 4B). Bicyclomycin is an antibiotic that inhibits Rho function by interacting with its secondary RNA-binding site (54). Although B. subtilis is not sensitive to bicyclomycin (since Rho is not essential), bicyclomycin dramatically reduced CEF susceptibility (Fig. 4C), indicating that bicyclomycin works antagonistically with respect to β-lactams. Moreover, the rho-null mutant displayed reduced sensitivity to other broad-spectrum β-lactams, especially cephalosporins (Fig. 4D).
FIG 4.

A rho-null mutation or bicyclomycin reduces β-lactam susceptibility. (A) Schematic representation of the location of the rhoG56C substitution. The conserved motifs were identified using the NCBI Conserved Domain database (http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi). (B) Effect of the rho gene deletion on the sensitivity of B. subtilis to CEF. MICs were determined using an Etest assay. (C) Effect of bicyclomycin inhibition on the sensitivity of B. subtilis to CEF. Treatment with the Rho inhibitor bicyclomycin increases the CEF MIC in a concentration-dependent manner. MICs were determined by the growth inhibition assay in MH broth. The data are reported as median values from three independent experiments. The MIC endpoint was established after 16 h of incubation. (D) Effect of the rho gene deletion on the sensitivity of B. subtilis to broad-spectrum β-lactams. Disk diffusion assays were performed on MH agar plates. Three independent experiments were performed for each strain, and the standard deviations are indicated by error bars. (E) Effect of the penP gene deletion on CEF resistance in the rho-null mutant. A representative data set from the disk diffusion assay is shown.
In B. subtilis, global transcriptomics approaches have recently revealed that a rho-null mutant results in numerous extended mRNAs (up to 12 kb), thereby affecting the expression of dozens of downstream genes that collectively comprise ∼2% of the genome (55). Interestingly, penP, which encodes a putative β-lactamase, is among the genes with 3′-extended mRNAs in a rho-null mutant. We therefore hypothesized that a rho-null mutation might affect the levels or translation of penP mRNA, possibly accounting for the observed CEF resistance. However, penP is not required for β-lactam resistance in the rho-null mutant (Fig. 4E).
As β-lactams target the PBPs, we also examined the impact of pbp gene deletions on the reduced CEF susceptibility in the rho-null mutant. Previously, we used a Bocillin-FL competition assay to demonstrate that CEF targets PBP1 (ponA), PBP2b (pbpB), PBP2c (pbpF), and PBP4 (pbpD) (56). To determine if increased expression of one or more of these PBPs might contribute to elevated CEF resistance, we measured the resistance of rho-null strains additionally lacking one or more PBPs in a zone-of-inhibition assay. However, none of the deletions tested led to a significant change in CEF resistance in the rho-null mutant. For example, rho-null mutant strains carrying an additional mutation(s) in ponA, pbpD, pbpF, pbpG, or dacA were at least as resistant to CEF as the rho-null mutant, and even a quadruple mutant (rho, pbpD, pbpF, and pbpG) was as resistant as the rho single mutant (data not shown). Thus, we conclude that inactivation of rho is a new genetic mechanism affecting β-lactam susceptibility in B. subtilis, but our results do not explain why this mutation confers resistance.
The sigAG336C substitution affects sensitivity to multiple antibiotics.
We next sought to determine how the sigAG336C mutation alters antibiotic susceptibility. The sigAG336C substitution is located in the helix-turn-helix (HTH) motif in region 4 that recognizes the −35 promoter element (Fig. 5A). Since sigA is essential, we hypothesized that this is an altered-function mutation likely affecting either core binding, promoter recognition, or possibly interaction with one or more regulatory proteins (57). We used disk diffusion assays to determine whether the sigAG336C substitution affected susceptibility to other cell wall antibiotics, including fosfomycin, bacitracin, and d-cycloserine. Indeed, the sigAG336C substitution made B. subtilis highly resistant to FOS, whereas the substitutions in mreB and rho did not (Fig. 5B). Our results demonstrate that the sigAG336C substitution alone leads to reductions in susceptibility to CEF, VAN, and FOS.
FIG 5.

The sigAG336C substitution drives the acquisition of FOS resistance. (A) Schematic representation of the location of the sigAG336C substitution. The conserved motifs were identified by using the NCBI Conserved Domain database. (B) Determination of the FOS susceptibilities of the reconstructed strains. Disk diffusion assays were performed on MH agar plates with a filter paper disk containing FOS. The asterisks indicate no inhibition. The error bars indicate standard deviations. (C) Microarray transcriptional analysis of strains HB13679 (sigAWT-mls) and HB13680 (sigAG336C-mls) under nonstress conditions. RNA was extracted from cells grown in LB medium to an OD600 of 0.4. The fluorescence intensity data are representative of two independent experiments, each of which was performed with dye swaps on each of three biological replicates. Each point represents the gene expression value for a single gene, with a subset of genes indicated by larger symbols as noted (see the text).
The sigAG336C substitution alters the transcriptome.
We used microarray-based gene expression profiling to better understand the genetic basis for resistance to antibiotics in the sigAG336C mutant. This mutation clearly leads to genome-wide changes in gene expression between the mutant (HB13680) and wild-type (HB13679) strains even under nonstress conditions (Fig. 5C). Significantly, in the sigAG336C mutant (HB13680), 28 genes of the σB general stress response regulon (58) were upregulated >2-fold, and other operons (including glpTQ) were strongly (>4-fold) downregulated (Fig. 5C). Since the mRNA level for sigB was not increased in the sigAG336C mutant, the increased expression of the σB regulon is unlikely to be due to an increase in the σB protein level. One possible explanation is that the mutant σA has reduced stability or binding affinity to core, thereby enabling σB to compete more readily for RNA polymerase core enzyme. Alternatively, the mutant σA may alter the expression of proteins involved in the posttranslational control of σB activity.
The sigAG336C substitution mediates CEF resistance by increased expression of the σB regulon.
To determine whether σB is involved in the enhanced resistance to CEF, we introduced a null allele of sigB into the sigAG336C mutant and performed disk diffusion assays. In contrast to the wild type, introduction of the sigAG336C mutation did not lead to an increase in CEF resistance in the sigB-null mutant background. Indeed, the opposite was observed: the sigB sigAG336C double mutant was more sensitive to CEF than the sigB single mutant, as judged by the diameter of growth inhibition (Fig. 6). However, after overnight growth, there was weak growth in the sigAG336C mutant strain relatively close to the disk, consistent with some protective effect (Fig. 6, upper right). Nevertheless, these results suggest that sigB is critical for the ability of the sigAG336C mutation to increase CEF resistance, consistent with the elevated expression of the σB regulon observed in the mutant strain (Fig. 5C). There are ∼200 genes in the σB regulon (59–62), and the identity of the gene(s) that mediates reduced susceptibility to CEF is still unknown.
FIG 6.
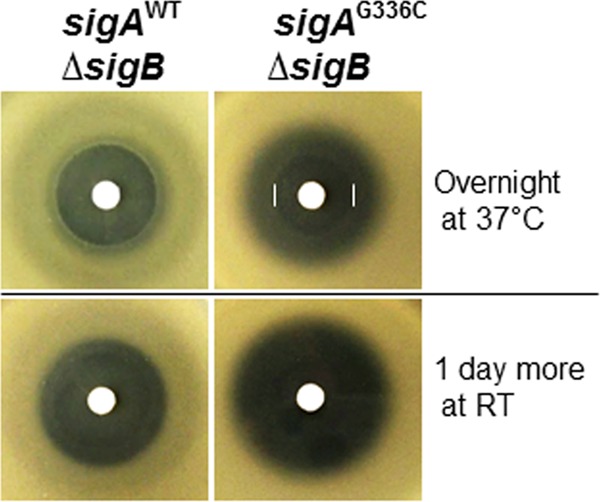
Reduced CEF susceptibility in the sigAG336C mutant is dependent on σB. The sigAG336C allele no longer decreases CEF susceptibility in strains carrying a sigB deletion. Disk diffusion assays were performed on MH agar plates with a filter paper disk containing 50 μg CEF. Three independent experiments were performed for each strain, and one representative experiment is shown. Note that the sigAG336C allele allows weak growth relatively close to the CEF disk, as visualized after overnight growth (upper right, white bars), but the overall diameters of the zones of growth inhibition are larger in the strains carrying the sigAG336C and sigB alleles (right) than in the strains carrying the sigB-null mutant (left). RT, room temperature.
The sigAG336C substitution mediates FOS resistance by decreased expression of glpTQ.
We next sought to define the mechanism by which the sigAG336C substitution confers resistance to FOS, which, unlike CEF and VAN, acts on the very early cytosolic steps of peptidoglycan biogenesis. We have shown previously that the σW-regulated fosB gene is a major FOS resistance determinant (63), and a bshC-null mutant lacking bacillithiol (BSH), a cofactor for the FosB bacillithiol transferase, is also highly FOS sensitive (64). Although cells lacking FosB or BshC are very sensitive to FOS, they still displayed an increase in FOS resistance upon introduction of the sigAG336C allele (Fig. 7A). We conclude that the sigAG336C allele can act independently of FosB. Motivated by our characterization of the transcriptional changes resulting from the sigAG336C substitution (Fig. 5C), we next asked whether σB might be involved in FOS resistance. In contrast with the results with CEF, disruption of sigB had no significant effect on the ability of the sigAG336C substitution to confer FOS resistance (data not shown).
FIG 7.

FOS resistance in the sigAG336C mutant is largely due to decreased glpTQ transcription. (A) The sigAG336C substitution can still confer FOS resistance in strains lacking fosB or bshC. Disk diffusion assays were performed on MH agar plates with a filter paper disk containing FOS. The asterisks indicate no inhibition. Each bar represents the average zone of inhibition, expressed as the total diameter minus the diameter of the filter paper disk (6.5 mm). Three independent experiments were performed for each strain, and the standard deviations are indicated by error bars. (B) RT-PCR analysis of glpT transcription. RNA was extracted from cells grown under the same conditions as for panel A. 16S rRNA was used as a loading control. (C) Growth comparison of strains HB13679 (sigAWT-mls) and HB13680 (sigAG336C-mls). Liquid growth assays were performed at 37°C in minimal medium supplemented with 2% glucose or 2% glycerol-3-P. The data are representative of at least three independent experiments. (D) IPTG induction of glpTQ restores FOS sensitivity in the sigAG336C mutant. Disk diffusion assays were performed on MH agar plates with or without 0.1 mM IPTG. The asterisk indicates no inhibition. Three independent experiments were performed for each strain. The error bars indicate standard deviations.
Mutations in glpT are known to be one mechanism for FOS resistance (65, 66), because GlpT is a glycerol-3-phosphate permease via which FOS enters the cell (67). The observed decrease in the transcription level of glpT seen in the microarray (Fig. 5C) was confirmed by RT-PCR analysis (Fig. 7B). This decreased expression is functionally significant, since the sigAG336C mutant displayed a growth defect in minimal medium containing 2% glycerol-3-P, suggesting that it has lower levels of GlpT (Fig. 7C). To test whether this decreased expression of glpTQ was responsible for FOS resistance, the glpTQ operon was expressed under the control of the IPTG-inducible Pspac(hy) promoter after integration into the amyE locus of the sigAG336C mutant. Indeed, IPTG induction of glpTQ restored FOS sensitivity (Fig. 7D). These results suggest that the sigAG336C substitution decreases the expression of glpTQ and that this decrease is sufficient to confer FOS resistance.
The molecular basis for the decreased expression of the glpTQ operon in the sigAG336C mutant is unclear. We noticed that the residue affected by this mutation (G336) is close to residues previously implicated in contacting activator proteins. Specifically, amino acids near and within the first helix of the HTH motif in region 4 of the E. coli σ70 mediate contact with the response regulator PhoB (68). Therefore, we speculated that SigAG336C may be defective for interaction with PhoP (the equivalent of the E. coli PhoB) or other, similar regulators. However, almost all PhoP-regulated genes are essentially unchanged in the sigAG336C mutant (Fig. 5C and data not shown), suggesting that PhoPR activity is not affected by SigAG336C. It is also known that only the glpQ gene of the glpTQ operon is under the control of PhoPR (69), and glpT has a SigA-type promoter (70). Thus, it is likely that transcription of the glpTQ operon is reduced directly due to a change in the promoter activation function of SigAG336C.
Conclusions.
Our data highlight the ability of mutational changes in the core transcriptional machinery to drive the evolution of decreased sensitivity to antibiotics in B. subtilis. To date, mutations in rpoB (β subunit) and rpoC (β′ subunit) have been correlated with the emergence of changes in antibiotic resistance for B. subtilis and several human pathogens (2, 19, 20, 71). While the mechanistic bases for these changes are often unclear, we have shown previously that an rpoC mutation can decrease antibiotic susceptibility by increasing the activities of three extracytoplasmic function (ECF) σ factors (σM, σW, and σX) (72).
Here, we show that sigAG336C is sufficient to reduce susceptibility to VAN and FOS and that it contributes (with rhoG56C) to decreased CEF susceptibility. In contrast with rpoB and rpoC, mutations in sigA and rho have not, to our knowledge, been previously linked to the emergence of antibiotic resistance in bacteria. Indeed, there are relatively few examples where altered-function mutations in a primary (essential) σ factor have emerged in random genetic selections (as opposed to experiments explicitly designed to recover such mutations). We are aware of three examples that include mutations leading to (i) an A-signaling defect in Myxococcus xanthus (73), (ii) suppression of defects associated with an inability to synthesize ppGpp in E. coli (74), and (iii) increased conversion of aminoimidazole ribotide to hydroxymethyl pyrimidine in Salmonella enterica (75). Here, we extend this list to include changes in antibiotic susceptibility. We also demonstrate that a rho-null mutation displays broad effects on susceptibility to β-lactam antibiotics in B. subtilis, but the mechanistic basis of this effect is presently unclear. Changes in Rho in E. coli have been previously shown to lead to widespread changes in the fitness landscape (76). Our results suggest that the Rho inhibitor bicyclomycin (which is used in agriculture as a feed additive) may increase the phenotypic resistance of Gram-positive bacteria to β-lactams. In sum, our data suggest that changes in the activity of the primary σ factor σA and the termination factor Rho may contribute to the evolution of antibiotic resistance.
ACKNOWLEDGMENTS
This work was supported by a grant from the National Institutes of Health (GM047446). Y.H.L. was partly supported by a research grant from Dongseo University (20140078).
Footnotes
Published ahead of print 11 August 2014
REFERENCES
- 1.Taubes G. 2008. The bacteria fight back. Science 321:356–361. 10.1126/science.321.5887.356. [DOI] [PubMed] [Google Scholar]
- 2.Chopra I. 2013. The 2012 Garrod lecture: discovery of antibacterial drugs in the 21st century. J. Antimicrob. Chemother. 68:496–505. 10.1093/jac/dks436. [DOI] [PubMed] [Google Scholar]
- 3.Norrby SR, Nord CE, Finch R. 2005. Lack of development of new antimicrobial drugs: a potential serious threat to public health. Lancet Infect. Dis. 5:115–119. 10.1016/S1473-3099(05)01283-1. [DOI] [PubMed] [Google Scholar]
- 4.Berenbaum MC. 1989. What is synergy? Pharmacol. Rev. 41:93–141. [PubMed] [Google Scholar]
- 5.Eggimann P, Revelly JP. 2006. Should antibiotic combinations be used to treat ventilator-associated pneumonia? Semin. Respir. Crit. Care Med. 27:68–81. 10.1055/s-2006-933675. [DOI] [PubMed] [Google Scholar]
- 6.Yeh PJ, Hegreness MJ, Aiden AP, Kishony R. 2009. Drug interactions and the evolution of antibiotic resistance. Nat. Rev. Microbiol. 7:460–466. 10.1038/nrmicro2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yeh P, Tschumi AI, Kishony R. 2006. Functional classification of drugs by properties of their pairwise interactions. Nat. Genet. 38:489–494. 10.1038/ng1755. [DOI] [PubMed] [Google Scholar]
- 8.Climo MW, Patron RL, Archer GL. 1999. Combinations of vancomycin and beta-lactams are synergistic against staphylococci with reduced susceptibilities to vancomycin. Antimicrob. Agents Chemother. 43:1747–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perichon B, Courvalin P. 2006. Synergism between beta-lactams and glycopeptides against VanA-type methicillin-resistant Staphylococcus aureus and heterologous expression of the vanA operon. Antimicrob. Agents Chemother. 50:3622–3630. 10.1128/AAC.00410-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reynolds PE. 1989. Structure, biochemistry and mechanism of action of glycopeptide antibiotics. Eur. J. Clin. Microbiol. Infect. Dis. 8:943–950. 10.1007/BF01967563. [DOI] [PubMed] [Google Scholar]
- 11.Sauvage E, Kerff F, Terrak M, Ayala JA, Charlier P. 2008. The penicillin-binding proteins: structure and role in peptidoglycan biosynthesis. FEMS Microbiol. Rev. 32:234–258. 10.1111/j.1574-6976.2008.00105.x. [DOI] [PubMed] [Google Scholar]
- 12.Waxman DJ, Strominger JL. 1983. Penicillin-binding proteins and the mechanism of action of beta-lactam antibiotics. Annu. Rev. Biochem. 52:825–869. 10.1146/annurev.bi.52.070183.004141. [DOI] [PubMed] [Google Scholar]
- 13.Hegreness M, Shoresh N, Damian D, Hartl D, Kishony R. 2008. Accelerated evolution of resistance in multidrug environments. Proc. Natl. Acad. Sci. U. S. A. 105:13977–13981. 10.1073/pnas.0805965105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hachmann AB, Sevim E, Gaballa A, Popham DL, Antelmann H, Helmann JD. 2011. Reduction in membrane phosphatidylglycerol content leads to daptomycin resistance in Bacillus subtilis. Antimicrob. Agents Chemother. 55:4326–4337. 10.1128/AAC.01819-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tomasz A. 2013. The use of whole genome sequencing to solve an epidemiological puzzle. EMBO Mol. Med. 5:486–487. 10.1002/emmm.201302622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mwangi MM, Wu SW, Zhou Y, Sieradzki K, de Lencastre H, Richardson P, Bruce D, Rubin E, Myers E, Siggia ED, Tomasz A. 2007. Tracking the in vivo evolution of multidrug resistance in Staphylococcus aureus by whole-genome sequencing. Proc. Natl. Acad. Sci. U. S. A. 104:9451–9456. 10.1073/pnas.0609839104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hegreness M, Kishony R. 2007. Analysis of genetic systems using experimental evolution and whole-genome sequencing. Genome Biol. 8:201. 10.1186/gb-2007-8-1-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Edwards DJ, Holt KE. 2013. Beginner's guide to comparative bacterial genome analysis using next-generation sequence data. Microb. Inform Exp. 3:2. 10.1186/2042-5783-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cui L, Isii T, Fukuda M, Ochiai T, Neoh HM, Camargo IL, Watanabe Y, Shoji M, Hishinuma T, Hiramatsu K. 2010. An RpoB mutation confers dual heteroresistance to daptomycin and vancomycin in Staphylococcus aureus. Antimicrob. Agents Chemother. 54:5222–5233. 10.1128/AAC.00437-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Friedman L, Alder JD, Silverman JA. 2006. Genetic changes that correlate with reduced susceptibility to daptomycin in Staphylococcus aureus. Antimicrob. Agents Chemother. 50:2137–2145. 10.1128/AAC.00039-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Watanabe Y, Cui L, Katayama Y, Kozue K, Hiramatsu K. 2011. Impact of rpoB mutations on reduced vancomycin susceptibility in Staphylococcus aureus. J. Clin. Microbiol. 49:2680–2684. 10.1128/JCM.02144-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Conrad TM, Frazier M, Joyce AR, Cho BK, Knight EM, Lewis NE, Landick R, Palsson BO. 2010. RNA polymerase mutants found through adaptive evolution reprogram Escherichia coli for optimal growth in minimal media. Proc. Natl. Acad. Sci. U. S. A. 107:20500–20505. 10.1073/pnas.0911253107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brinkman CL, Bumgarner R, Kittichotirat W, Dunman PM, Kuechenmeister LJ, Weaver KE. 2013. Characterization of the effects of an rpoC mutation that confers resistance to the Fst peptide toxin-antitoxin system toxin. J. Bacteriol. 195:156–166. 10.1128/JB.01597-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murakami KS, Darst SA. 2003. Bacterial RNA polymerases: the wholo story. Curr. Opin. Struct. Biol. 13:31–39. 10.1016/S0959-440X(02)00005-2. [DOI] [PubMed] [Google Scholar]
- 25.Juang YL, Helmann JD. 1994. The delta subunit of Bacillus subtilis RNA polymerase. An allosteric effector of the initiation and core-recycling phases of transcription. J. Mol. Biol. 239:1–14. [DOI] [PubMed] [Google Scholar]
- 26.Lopez de Saro FJ, Woody AY, Helmann JD. 1995. Structural analysis of the Bacillus subtilis delta factor: a protein polyanion which displaces RNA from RNA polymerase. J. Mol. Biol. 252:189–202. 10.1006/jmbi.1995.0487. [DOI] [PubMed] [Google Scholar]
- 27.Borukhov S, Severinov K. 2002. Role of the RNA polymerase sigma subunit in transcription initiation. Res. Microbiol. 153:557–562. 10.1016/S0923-2508(02)01368-2. [DOI] [PubMed] [Google Scholar]
- 28.Gruber TM, Gross CA. 2003. Multiple sigma subunits and the partitioning of bacterial transcription space. Annu. Rev. Microbiol. 57:441–466. 10.1146/annurev.micro.57.030502.090913. [DOI] [PubMed] [Google Scholar]
- 29.Paget MS, Helmann JD. 2003. The sigma70 family of sigma factors. Genome Biol. 4:203. 10.1186/gb-2003-4-1-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cao M, Wang T, Ye R, Helmann JD. 2002. Antibiotics that inhibit cell wall biosynthesis induce expression of the Bacillus subtilis sigma(W) and sigma(M) regulons. Mol. Microbiol. 45:1267–1276. 10.1046/j.1365-2958.2002.03050.x. [DOI] [PubMed] [Google Scholar]
- 31.Peters JM, Vangeloff AD, Landick R. 2011. Bacterial transcription terminators: the RNA 3′-end chronicles. J. Mol. Biol. 412:793–813. 10.1016/j.jmb.2011.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Richardson JP. 2002. Rho-dependent termination and ATPases in transcript termination. Biochim. Biophys. Acta 1577:251–260. 10.1016/S0167-4781(02)00456-6. [DOI] [PubMed] [Google Scholar]
- 33.Washburn RS, Marra A, Bryant AP, Rosenberg M, Gentry DR. 2001. rho is not essential for viability or virulence in Staphylococcus aureus. Antimicrob. Agents Chemother. 45:1099–1103. 10.1128/AAC.45.4.1099-1103.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Opperman T, Richardson JP. 1994. Phylogenetic analysis of sequences from diverse bacteria with homology to the Escherichia coli rho gene. J. Bacteriol. 176:5033–5043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Das A, Court D, Adhya S. 1976. Isolation and characterization of conditional lethal mutants of Escherichia coli defective in transcription termination factor rho. Proc. Natl. Acad. Sci. U. S. A. 73:1959–1963. 10.1073/pnas.73.6.1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Quirk PG, Dunkley EA, Jr, Lee P, Krulwich TA. 1993. Identification of a putative Bacillus subtilis rho gene. J. Bacteriol. 175:647–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yakhnin H, Babiarz JE, Yakhnin AV, Babitzke P. 2001. Expression of the Bacillus subtilis trpEDCFBA operon is influenced by translational coupling and Rho termination factor. J. Bacteriol. 183:5918–5926. 10.1128/JB.183.20.5918-5926.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ingham CJ, Dennis J, Furneaux PA. 1999. Autogenous regulation of transcription termination factor Rho and the requirement for Nus factors in Bacillus subtilis. Mol. Microbiol. 31:651–663. 10.1046/j.1365-2958.1999.01205.x. [DOI] [PubMed] [Google Scholar]
- 39.Nowatzke WL, Keller E, Koch G, Richardson JP. 1997. Transcription termination factor Rho is essential for Micrococcus luteus. J. Bacteriol. 179:5238–5240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mascher T, Margulis NG, Wang T, Ye RW, Helmann JD. 2003. Cell wall stress responses in Bacillus subtilis: the regulatory network of the bacitracin stimulon. Mol. Microbiol. 50:1591–1604. 10.1046/j.1365-2958.2003.03786.x. [DOI] [PubMed] [Google Scholar]
- 41.Srivatsan A, Han Y, Peng J, Tehranchi AK, Gibbs R, Wang JD, Chen R. 2008. High-precision, whole-genome sequencing of laboratory strains facilitates genetic studies. PLoS Genet. 4:e1000139. 10.1371/journal.pgen.1000139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 43.Quisel JD, Burkholder WF, Grossman AD. 2001. In vivo effects of sporulation kinases on mutant Spo0A proteins in Bacillus subtilis. J. Bacteriol. 183:6573–6578. 10.1128/JB.183.22.6573-6578.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hachmann AB, Angert ER, Helmann JD. 2009. Genetic analysis of factors affecting susceptibility of Bacillus subtilis to daptomycin. Antimicrob. Agents Chemother. 53:1598–1609. 10.1128/AAC.01329-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Andersson DI, Levin BR. 1999. The biological cost of antibiotic resistance. Curr. Opin. Microbiol. 2:489–493. 10.1016/S1369-5274(99)00005-3. [DOI] [PubMed] [Google Scholar]
- 46.Eiamphungporn W, Helmann JD. 2008. The Bacillus subtilis sigma(M) regulon and its contribution to cell envelope stress responses. Mol. Microbiol. 67:830–848. 10.1111/j.1365-2958.2007.06090.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mascher T, Zimmer SL, Smith TA, Helmann JD. 2004. Antibiotic-inducible promoter regulated by the cell envelope stress-sensing two-component system LiaRS of Bacillus subtilis. Antimicrob. Agents Chemother. 48:2888–2896. 10.1128/AAC.48.8.2888-2896.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Skordalakes E, Berger JM. 2006. Structural insights into RNA-dependent ring closure and ATPase activation by the Rho termination factor. Cell 127:553–564. 10.1016/j.cell.2006.08.051. [DOI] [PubMed] [Google Scholar]
- 49.Bear DG, Andrews CL, Singer JD, Morgan WD, Grant RA, von Hippel PH, Platt T. 1985. Escherichia coli transcription termination factor Rho has a two-domain structure in its activated form. Proc. Natl. Acad. Sci. U. S. A. 82:1911–1915. 10.1073/pnas.82.7.1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wei RR, Richardson JP. 2001. Identification of an RNA-binding site in the ATP binding domain of Escherichia coli Rho by H2O2/Fe-EDTA cleavage protection studies. J. Biol. Chem. 276:28380–28387. 10.1074/jbc.M102444200. [DOI] [PubMed] [Google Scholar]
- 51.Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Lu F, Marchler GH, Mullokandov M, Omelchenko MV, Robertson CL, Song JS, Thanki N, Yamashita RA, Zhang D, Zhang N, Zheng C, Bryant SH. 2011. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 39:D225–D229. 10.1093/nar/gkq1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Modrak D, Richardson JP. 1994. The RNA-binding domain of transcription termination factor rho: isolation, characterization, and determination of sequence limits. Biochemistry 33:8292–8299. 10.1021/bi00193a016. [DOI] [PubMed] [Google Scholar]
- 53.Skordalakes E, Berger JM. 2003. Structure of the Rho transcription terminator: mechanism of mRNA recognition and helicase loading. Cell 114:135–146. 10.1016/S0092-8674(03)00512-9. [DOI] [PubMed] [Google Scholar]
- 54.Magyar A, Zhang X, Kohn H, Widger WR. 1996. The antibiotic bicyclomycin affects the secondary RNA binding site of Escherichia coli transcription termination factor Rho. J. Biol. Chem. 271:25369–25374. 10.1074/jbc.271.41.25369. [DOI] [PubMed] [Google Scholar]
- 55.Nicolas P, Mader U, Dervyn E, Rochat T, Leduc A, Pigeonneau N, Bidnenko E, Marchadier E, Hoebeke M, Aymerich S, Becher D, Bisicchia P, Botella E, Delumeau O, Doherty G, Denham EL, Fogg MJ, Fromion V, Goelzer A, Hansen A, Hartig E, Harwood CR, Homuth G, Jarmer H, Jules M, Klipp E, Le Chat L, Lecointe F, Lewis P, Liebermeister W, March A, Mars RA, Nannapaneni P, Noone D, Pohl S, Rinn B, Rugheimer F, Sappa PK, Samson F, Schaffer M, Schwikowski B, Steil L, Stulke J, Wiegert T, Devine KM, Wilkinson AJ, van Dijl JM, Hecker M, Volker U, Bessieres P, Noirot P. 2012. Condition-dependent transcriptome reveals high-level regulatory architecture in Bacillus subtilis. Science 335:1103–1106. 10.1126/science.1206848. [DOI] [PubMed] [Google Scholar]
- 56.Luo Y, Helmann JD. 2012. Analysis of the role of Bacillus subtilis sigma(M) in beta-lactam resistance reveals an essential role for c-di-AMP in peptidoglycan homeostasis. Mol. Microbiol. 83:623–639. 10.1111/j.1365-2958.2011.07953.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gruber TM, Markov D, Sharp MM, Young BA, Lu CZ, Zhong HJ, Artsimovitch I, Geszvain KM, Arthur TM, Burgess RR, Landick R, Severinov K, Gross CA. 2001. Binding of the initiation factor sigma(70) to core RNA polymerase is a multistep process. Mol. Cell 8:21–31. 10.1016/S1097-2765(01)00292-1. [DOI] [PubMed] [Google Scholar]
- 58.Hecker M, Pane-Farre J, Volker U. 2007. SigB-dependent general stress response in Bacillus subtilis and related gram-positive bacteria. Annu. Rev. Microbiol. 61:215–236. 10.1146/annurev.micro.61.080706.093445. [DOI] [PubMed] [Google Scholar]
- 59.Helmann JD, Wu MF, Kobel PA, Gamo FJ, Wilson M, Morshedi MM, Navre M, Paddon C. 2001. Global transcriptional response of Bacillus subtilis to heat shock. J. Bacteriol. 183:7318–7328. 10.1128/JB.183.24.7318-7328.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nannapaneni P, Hertwig F, Depke M, Hecker M, Mader U, Volker U, Steil L, van Hijum SA. 2012. Defining the structure of the general stress regulon of Bacillus subtilis using targeted microarray analysis and random forest classification. Microbiology 158:696–707. 10.1099/mic.0.055434-0. [DOI] [PubMed] [Google Scholar]
- 61.Petersohn A, Brigulla M, Haas S, Hoheisel JD, Volker U, Hecker M. 2001. Global analysis of the general stress response of Bacillus subtilis. J. Bacteriol. 183:5617–5631. 10.1128/JB.183.19.5617-5631.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Price CW, Fawcett P, Ceremonie H, Su N, Murphy CK, Youngman P. 2001. Genome-wide analysis of the general stress response in Bacillus subtilis. Mol. Microbiol. 41:757–774. 10.1046/j.1365-2958.2001.02534.x. [DOI] [PubMed] [Google Scholar]
- 63.Cao M, Bernat BA, Wang Z, Armstrong RN, Helmann JD. 2001. FosB, a cysteine-dependent fosfomycin resistance protein under the control of sigma(W), an extracytoplasmic-function sigma factor in Bacillus subtilis. J. Bacteriol. 183:2380–2383. 10.1128/JB.183.7.2380-2383.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gaballa A, Newton GL, Antelmann H, Parsonage D, Upton H, Rawat M, Claiborne A, Fahey RC, Helmann JD. 2010. Biosynthesis and functions of bacillithiol, a major low-molecular-weight thiol in Bacilli. Proc. Natl. Acad. Sci. U. S. A. 107:6482–6486. 10.1073/pnas.1000928107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bordi C, Butcher BG, Shi Q, Hachmann AB, Peters JE, Helmann JD. 2008. In vitro mutagenesis of Bacillus subtilis by using a modified Tn7 transposon with an outward-facing inducible promoter. Appl. Environ. Microbiol. 74:3419–3425. 10.1128/AEM.00476-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lindgren V. 1978. Mapping of a genetic locus that affects glycerol 3-phosphate transport in Bacillus subtilis. J. Bacteriol. 133:667–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Santoro A, Cappello AR, Madeo M, Martello E, Iacopetta D, Dolce V. 2011. Interaction of fosfomycin with the glycerol 3-phosphate transporter of Escherichia coli. Biochim. Biophys. Acta 1810:1323–1329. 10.1016/j.bbagen.2011.07.006. [DOI] [PubMed] [Google Scholar]
- 68.Makino K, Amemura M, Kim SK, Nakata A, Shinagawa H. 1993. Role of the sigma 70 subunit of RNA polymerase in transcriptional activation by activator protein PhoB in Escherichia coli. Genes Dev. 7:149–160. 10.1101/gad.7.1.149. [DOI] [PubMed] [Google Scholar]
- 69.Antelmann H, Scharf C, Hecker M. 2000. Phosphate starvation-inducible proteins of Bacillus subtilis: proteomics and transcriptional analysis. J. Bacteriol. 182:4478–4490. 10.1128/JB.182.16.4478-4490.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nilsson RP, Beijer L, Rutberg B. 1994. The glpT and glpQ genes of the glycerol regulon in Bacillus subtilis. Microbiology 140:723–730. 10.1099/00221287-140-4-723. [DOI] [PubMed] [Google Scholar]
- 71.Howden BP, Peleg AY, Stinear TP. 2014. The evolution of vancomycin intermediate Staphylococcus aureus (VISA) and heterogenous-VISA. Infect. Genet. Evol. 21:575–582. 10.1016/j.meegid.2013.03.047. [DOI] [PubMed] [Google Scholar]
- 72.Lee YH, Nam KH, Helmann JD. 2013. A mutation of the RNA polymerase beta' subunit (rpoC) confers cephalosporin resistance in Bacillus subtilis. Antimicrob. Agents Chemother. 57:56–65. 10.1128/AAC.01449-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Davis JM, Mayor J, Plamann L. 1995. A missense mutation in rpoD results in an A-signalling defect in Myxococcus xanthus. Mol. Microbiol. 18:943–952. 10.1111/j.1365-2958.1995.18050943.x. [DOI] [PubMed] [Google Scholar]
- 74.Hernandez VJ, Cashel M. 1995. Changes in conserved region 3 of Escherichia coli sigma 70 mediate ppGpp-dependent functions in vivo. J. Mol. Biol. 252:536–549. 10.1006/jmbi.1995.0518. [DOI] [PubMed] [Google Scholar]
- 75.Dougherty MJ, Downs DM. 2004. A mutant allele of rpoD results in increased conversion of aminoimidazole ribotide to hydroxymethyl pyrimidine in Salmonella enterica. J. Bacteriol. 186:4034–4037. 10.1128/JB.186.12.4034-4037.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Freddolino PL, Goodarzi H, Tvazoie S. 2012. Fitness landscape transformation through a single amino acid change in the Rho terminator. PLoS genetics 8:e1002744. 10.1371/journal.pgen.1002744. [DOI] [PMC free article] [PubMed] [Google Scholar]