

Abstract
Salmonella pathogenicity islands 1 and 2 (SPI-1 and SPI-2) have essential roles in the pathogenesis of Salmonella enterica. Previously, we reported transcriptional cross talk between SPI-1 and SPI-2 when the SPI-1 regulator HilD induces expression of the SsrA/B two-component system, the central positive regulator of SPI-2, during the growth of Salmonella to late stationary phase in LB rich medium. Here, we further define the mechanism of the HilD-mediated expression of ssrAB. Expression analysis of cat transcriptional fusions containing different regions of ssrAB revealed the presence of negative regulatory sequences located downstream of the ssrAB promoter. In the absence of these negative cis elements, ssrAB was expressed in a HilD-independent manner and was no longer repressed by the global regulator H-NS. Consistently, when the activity of H-NS was inactivated, the expression of ssrAB also became independent of HilD. Furthermore, electrophoretic mobility shift assays showed that both HilD and H-NS bind to the ssrAB region containing the repressing sequences. Moreover, HilD was able to displace H-NS bound to this region, whereas H-NS did not displace HilD. Our results support a model indicating that HilD displaces H-NS from a region downstream of the promoter of ssrAB by binding to sites overlapping or close to those sites bound by H-NS, which leads to the expression of ssrAB. Although the role of HilD as an antagonist of H-NS has been reported before for other genes, this is the first study showing that HilD is able to effectively displace H-NS from the promoter of one of its target genes.
INTRODUCTION
Acquisition of DNA fragments by horizontal transfer and subsequent adaptation of regulatory mechanisms to control the spatiotemporal expression of the newly acquired genes have been pivotal events in the evolution of Salmonella pathogenicity (1, 2). Salmonella pathogenicity islands 1 and 2 (SPI-1 and SPI-2) are chromosomal regions composed of 39 and 44 genes, respectively, which were acquired at different evolutionary times and have crucial roles in the pathogenesis of Salmonella (2–4). SPI-1 genes are required for Salmonella invasion of the intestinal epithelium, leading to enteritis, whereas SPI-2 genes are needed mainly for Salmonella survival and replication within macrophages and consequent establishment of systemic disease (2–5). SPI-2 genes also induce a nonproliferative intracellular lifestyle by restraining growth inside phagocytes and nonphagocytic cells and contribute to the development of intestinal inflammatory and diarrheal disease, as they enhance Salmonella's transepithelial passage, as well as foster growth inside phagocytes present in the lamina propria (6–12). In agreement with their pathogenicity roles, SPI-1 genes are expressed when bacteria are in the intestinal lumen or associated with the epithelium or with extruding enterocytes, as well as in a subpopulation of bacteria hyperreplicating in the cytosol of epithelial cells, whereas SPI-2 genes are expressed when bacteria are inside macrophages and also when they are in the intestinal lumen prior to penetrating the intestine, as well as in the lamina propria and in the underlying mucosa (13–18). In vitro, the SPI-1 and SPI-2 genes are expressed during early and late stationary phase, respectively, when Salmonella is grown in nutrient-rich media, such as Luria-Bertani (LB) medium (19–22). Additionally, the SPI-2 genes, but not the SPI-1 genes, are expressed when Salmonella is grown in acidic minimal media containing low concentrations of phosphate, calcium, and magnesium (16, 20, 22).
The AraC-like transcriptional regulator HilD, encoded within SPI-1, positively regulates the expression of the SPI-1 genes in a cascade fashion, mainly by directly inducing the expression of hilA, which encodes the master regulator of SPI-1 (2, 5, 23–26). HilD also directly controls the expression of the SPI-1 genes hilD, hilC, and invF, as well as other acquired and ancestral genes located outside SPI-1, such as rtsA, flhDC, siiA, lpxR, ytfK, STM14_1282, and STM14_2342 (2, 25–31). Previously, we demonstrated that HilD directly induces the expression of the ssrAB operon, which is located in SPI-2 and codes for the SsrA/B two-component system, the central positive regulator of SPI-2, thus establishing a transcriptional cross talk between SPI-1 and SPI-2 (21). Interestingly, HilD is required for the expression of ssrAB when Salmonella is grown to late stationary phase in LB medium but not when it is grown in minimal media (21). Instead, when Salmonella is grown in minimal media, the expression of ssrAB and thus the SPI-2 genes is self-regulated by SsrA/B as well as by other regulators, such as the two-component systems OmpR/EnvZ and PhoP/Q and the MarR-like regulator SlyA (32–38). Therefore, in response to distinct growth conditions, at least two different pathways induce the expression of the ssrAB genes in vitro, which may be consistent with the fact that the SPI-2 genes can be expressed in different compartments inside Salmonella hosts (18).
A myriad of other Salmonella-specific and global regulators have been involved in the expression of the SPI-1 and SPI-2 genes, most of them acting on the expression of hilD, hilA, or ssrAB (2, 5, 25, 38–41). H-NS is a constitutively expressed, abundant nucleus-associated protein that represses the expression of both the SPI-1 and SPI-2 genes, as well as many other Salmonella genes, by binding to AT-rich sequences commonly present in acquired DNA (39, 42, 43). Moreover, H-NS has played an important role in the evolution of Salmonella pathogenicity by preventing uncontrolled expression of genes present in acquired DNA that may be deleterious to bacterial fitness (42–44). Therefore, H-NS is considered a genome sentinel (45). Regulatory proteins have been adapted to counteract H-NS-mediated repression at specific promoters, thereby enabling the regulated expression of newly acquired genes (46, 47).
Several studies strongly support the idea that HilD induces the expression of hilA and rtsA by counteracting the repression exerted by H-NS on the promoters of these genes (23, 24, 27, 48, 49). Furthermore, it has been shown that H-NS directly represses ssrAB and that HilD is required for the expression of ssrAB but only if H-NS is present (21, 42, 43, 50). This suggests that HilD induces the expression of ssrAB also by counteracting H-NS-mediated repression of its promoter. However, how HilD counteracts the H-NS-mediated repression of ssrAB, or even of other target genes, had not yet been determined.
In this work, by combining genetic and biochemical strategies, we determined that H-NS represses the expression of ssrAB by initially binding to sequences located downstream of the promoter of this operon. Furthermore, our results revealed that in the absence of the downstream region bound by H-NS or when H-NS is inactivated, the expression of ssrAB becomes independent of HilD. Moreover, we found that HilD is able to displace H-NS from ssrAB by binding to sites overlapping or close to those sites bound by H-NS, which leads to the expression of ssrAB. Our results elucidate the mechanism by which HilD and H-NS regulate the expression of ssrAB and show for the first time that HilD is able to displace H-NS from one of its target genes.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Bacterial strains used in this work are listed in Table S1 in the supplemental material. Bacterial cultures were grown at 37°C in LB medium containing 1% tryptone, 0.5% yeast agar, and 1% NaCl at pH 7.5 or in N minimal medium (N-MM) containing 5 mM KCl, 7.5 mM (NH4)2SO4, 0.5 mM K2SO4, 1 mM KH2PO4, 100 mM Tris-HCl (pH 7.5), 10 μM MgCl2, 0.5% glycerol, and 0.1% Casamino Acids. When necessary, media were supplemented with ampicillin (200 μg/ml), streptomycin (100 μg/ml), tetracycline (10 μg/ml), or kanamycin (20 μg/ml). Cultures for chloramphenicol acetyltransferase (CAT) assays were performed as we described previously (21, 41). Culture samples were taken after 10 or 16 h of growth in LB medium or N-MM, respectively, on the basis of our previous work to optimize the expression of SPI-2 genes under these growth conditions (21, 41).
Construction of plasmids.
Plasmids and primers used in this study are listed in Tables S1 and S2 in the supplemental material, respectively. To construct the plasmids containing the ssrAB transcriptional fusions, different segments of the regulatory region of ssrAB were amplified by PCR with the primer pairs SsaBF/SsrBR-S6E, SsrAB-2A/SsrBR-S6E, SsrAB-2B/SsrBR-S6E, SsrAB-55/SsrBR-S6E, SsaBF/SsrAB+336, SsaBF/SsrAB+240, SsaBF/SsrAB-1A, SsaBF/SsrAB+69, and SsaBF/SsrAB-1B. The PCR products were digested with BglII and SalI restriction enzymes and then cloned into the BamHI and SalI sites of the vector pKK232-8, which carries a promoterless cat gene (Amersham Pharmacia LKB Biotechnology), generating plasmids pssrAB-cat−302/+478, pssrAB-cat−208, pssrAB-cat−106, pssrAB-cat−55, pssrAB-cat+336, pssrAB-cat+240, pssrAB-cat+119, pssrAB-cat+69, and pssrAB-cat+10, respectively.
CAT assays.
The CAT assays and protein quantification to calculate CAT-specific activities were performed as described previously (51).
Expression and purification of MBP-HilD and H-NS–FH.
Maltose-binding protein (MBP)–HilD and H-NS–FLAG-His6 (H-NS–FH) were expressed in Escherichia coli BL21/DE3 containing pMAL-HilD1 or pBAD-H-NS–FH and purified by using an amylose column or a HiTrap Ni2+-chelating column, respectively, as described previously (21, 52).
EMSAs.
DNA fragments 1A to 1E, which have overlapping ends and, all together, span the region of ssrAB from positions −302 to +478 (−302/+478 region), were amplified by PCR with primer pairs SsaBF/SsrAB-11aR, SsrAB-11bF/SsrAB-11bR, SsrAB-11cF/SsrAB-11cR, SsrAB-11dF/SsrAB-11dR, and SsrAB-11eF/SsrBR-6SE, respectively, using plasmid pssrAB-cat−302/+478 as the template. The DNA fragment containing the regulatory region of sigD, used as a negative control, was amplified by PCR from chromosomal DNA with the primer pair SigD-BHIF/SigD-Rv1 and Salmonella enterica serovar Typhimurium SL1344. PCR products were purified using the QIAquick PCR purification kit (Qiagen). Binding reactions were performed by mixing ∼100 ng of each PCR product with increasing concentrations of purified MBP-HilD or H-NS–FH in binding buffer containing 10 mM Tris-HCl (pH 8), 50 mM KCl, 1 mM dithiothreitol (DTT), 0.5 mM EDTA, 5% glycerol, and 10 μg ml−1 bovine serum albumin (BSA), in a total volume of 20 μl. Protein-DNA binding reaction mixtures were incubated at room temperature for 20 min and then electrophoretically separated in 6% nondenaturing polyacrylamide gels in 0.5× Tris-borate-EDTA buffer at room temperature. For competitive electrophoretic mobility shift assays (EMSAs), we used the DNA fragment containing the −302/+478 region of ssrAB, which was amplified by PCR with the primer pair SsaBF/SsrBR-S6E. This fragment was first incubated with 0.6 μM H-NS–FH or 0.5 μM MBP-HilD for 15 min and then incubated with increasing concentrations of MBP-HilD or H-NS–FH, respectively, for an additional 20 min. These protein-DNA binding reaction mixtures were electrophoretically separated in 6% nondenaturing polyacrylamide gels as described above. The DNA fragments were stained with ethidium bromide and visualized with an Alpha-Imager UV transilluminator (Alpha Innotech Corp.).
Western blotting.
MBP-HilD or H-NS–FH present in the protein-DNA complexes of the EMSAs were transferred from the polyacrylamide gels to 0.45-μm-pore-size nitrocellulose membranes (Bio-Rad), using a semidry transfer apparatus (Bio-Rad). Membranes obtained from separate gels were blocked with 5% nonfat milk and incubated with anti-FLAG M2 (Sigma) or anti-MBP (Sigma) monoclonal antibodies at a 1:1,000 or 1:3,000 dilution, respectively. Horseradish peroxidase-conjugated anti-mouse antibody (Pierce), at a dilution of 1:10,000, was used as the secondary antibody. Bands on the blotted membranes were developed by incubation with the Western Lightning chemiluminescence reagent plus (Perkin-Elmer) and exposed to Kodak X-Omat films.
RESULTS
cis elements required for the regulation of ssrAB by HilD.
In a previous study, we showed that HilD is required for the expression of ssrAB; furthermore, we demonstrated that HilD binds to a DNA fragment spanning the region from −302 to +478 of ssrAB (−302/+478 region) (21). To determine whether this region contains all the cis elements required for the HilD-mediated regulation of ssrAB, a transcriptional fusion of the −302/+478 region to the cat gene was constructed in plasmid pKK232-8, an expression reporter system that we have successfully used in S. Typhimurium (21, 41, 53). The CAT-specific activity directed by the plasmid carrying this transcriptional fusion, ssrB-cat−302/+478 (Fig. 1), was determined in wild-type (WT) S. Typhimurium strain SL1344 and its derivative ΔhilD mutant grown in LB medium at 37°C, conditions that favor the HilD-mediated expression of ssrAB (21, 41). The expression of the ssrB-cat−302/+478 fusion was reduced in the ΔhilD mutant but was restored to WT levels in the presence of a plasmid expressing HilD (Fig. 2), indicating that the −302/+478 region contains all the cis sequences necessary for the HilD-mediated expression of ssrAB.
FIG 1.

Schematic representation of the ssrAB loci and the ssrAB-cat transcriptional fusions used in this work. The transcriptional start site (+1) of ssrAB is indicated by a bent arrow. The positions spanning the region between ssaB and ssrAB are shown. The transcriptional fusions are represented as lines with a short black arrow at their 3′ ends, indicating the respective ssrAB region (lines) and the cat reporter gene (arrows). The positions spanning the ssrAB fragment carried by each fusion, as well as the name of each fusion, are shown. Fusions are grouped with regard to 5′ or 3′ deletions with respect to the ssrAB-cat−302/+478 fusion. All positions are indicated with respect to the transcriptional start site of ssrAB.
FIG 2.
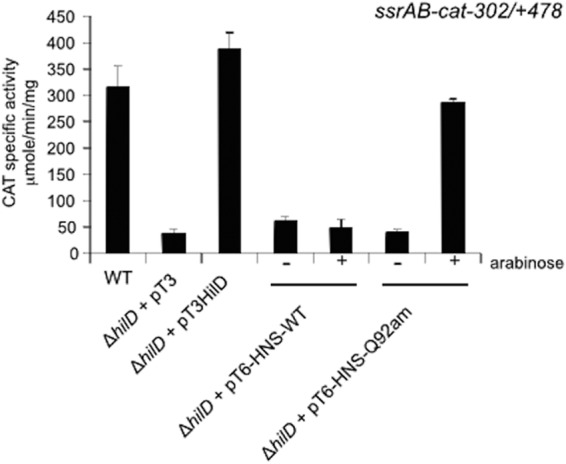
HilD antagonizes the H-NS-mediated repression exerted on ssrAB. Expression of the ssrAB-cat−302/+478 transcriptional fusion, carried by plasmid pssrAB-cat−302/+478, was tested in the WT S. Typhimurium strain and its isogenic ΔhilD mutant containing the vector pMPM-T3 or the plasmid pT3-HilD1, which expresses HilD, as well as in the ΔhilD mutant containing the plasmid pT6-HNS-WT or pT6-HNS-Q92am, which express WT H-NS or a dominant negative C-terminally truncated form of H-NS (H-NSQ92am), respectively, from an arabinose-inducible promoter. CAT-specific activity was determined from samples collected from bacterial cultures grown for 10 h in LB medium at 37°C. Expression of WT H-NS or H-NSQ92am from plasmids pT6-HNS-WT and pT6-HNS-Q92am, respectively, was induced by adding 0.1% l-arabinose to the medium (+arabinose). The data are the averages of results of three independent experiments performed in duplicate. Bars represent the standard deviations.
To further identify sequences within the −302/+478 region required for the regulation of ssrAB by HilD, a series of ssrAB-cat fusions carrying different 5′ or 3′ deletions was constructed (Fig. 1). The 5′-deletion fusions have a common 3′ end located at position +478 and are numbered according to the position of their 5′ end, whereas the 3′-deletion fusions have a common 5′ end located at position −302 and are numbered according to the position of their 3′ end (Fig. 1). The CAT-specific activity directed by the plasmids carrying these ssrAB-cat transcriptional fusions was determined in the WT S. Typhimurium strain and its derivative ΔhilD mutant grown in LB medium at 37°C.
The ssrAB-cat−208 and ssrAB-cat−106 5′-deletion fusions showed an expression pattern similar to that shown by the ssrAB-cat−302/+478 fusion; both showed comparable levels of expression in the WT strain and decreased expression in the ΔhilD mutant (Fig. 3A), indicating that the sequence upstream of position −106 is not required for the expression of ssrAB under the conditions tested. In contrast, the ssrAB-cat−55 fusion showed reduced CAT activity in the WT strain with respect to that shown by the ssrAB-cat−302/+478, ssrAB-cat−208, and ssrAB-cat−106 fusions; however, its expression was further reduced in the ΔhilD mutant compared to that in the WT strain (Fig. 3A). These results indicate that the sequence between positions −106 and −55 has a positive regulatory role for the expression of ssrAB but that it is not essential for the HilD-mediated regulation of this operon.
FIG 3.

Analysis of the cis-acting sequences required for the HilD-mediated regulation of ssrAB. Expression of the ssrAB-cat−302/+478, ssrAB-cat−208, ssrAB-cat−106, ssrAB-cat−55, ssrAB-cat+336, ssrAB-cat+240, ssrAB-cat+119, ssrAB-cat+69, and ssrAB-cat+10 transcriptional fusions contained in plasmids pssrAB-cat−302/+478, pssrAB-cat−208, pssrAB-cat−106, pssrAB-cat−55, pssrAB-cat+336, pssrAB-cat+240, pssrAB-cat+119, pssrAB-cat+69, and pssrAB-cat+10, respectively, was tested in the WT S. Typhimurium strain and its isogenic ΔhilD mutant. CAT-specific activity was determined from samples collected from bacterial cultures grown in LB medium (A and B) or N-MM (C and D) at 37°C for 10 h or 16 h, respectively. The data are the averages of results from three independent experiments performed in duplicate. Bars represent the standard deviations.
The ssrAB-cat+336 and ssrAB-cat+240 3′-deletion fusions were similarly expressed in the WT strain, and their expression was reduced 3- and 2-fold, respectively, in the ΔhilD mutant, whereas the expression of the ssrAB-cat+119, ssrAB-cat+69, and ssrAB-cat+10 fusions was unaffected in the ΔhilD mutant (Fig. 3B). Thus, these results indicate that the region between positions +119 and +336 contains negative regulatory sequences that are required to maintain the HilD-mediated regulation of ssrAB. Interestingly, the expression level shown by the ssrAB-cat+336 fusion was higher than that shown by the ssrAB-cat−302/+478 fusion (Fig. 3A and B), indicating that the region between positions +336 and +478 also contains negative regulatory sequences.
In agreement with our previous results indicating that HilD is required for the expression of SsrB when Salmonella is grown in LB medium but not in minimal media (21), the absence of HilD did not affect the expression of the ssrAB-cat fusions when the strains were grown in N-MM (Fig. 3C and D).
Together, these results delimit the region containing the cis elements required for the HilD-mediated regulation of ssrAB, which spans positions −55 to +240. Additionally, these results indicate that the region between positions +119 and +478 contains negative regulatory sequences and that, in the absence of this region, the expression of ssrAB becomes independent of HilD.
cis elements required for the repression of ssrAB by H-NS.
H-NS represses ssrAB as well as many other Salmonella genes (21, 39, 42, 43, 50). A Salmonella Δhns mutant shows severe growth defects and seems to be viable only after acquiring secondary mutations (43, 44); however, the effect of H-NS in a WT Salmonella strain can be analyzed by using an H-NS mutant that does not have binding activity but still forms heterodimers with WT monomers and thus acts as a dominant negative mutant (21, 50). By using this strategy, we previously demonstrated that HilD is required for the expression of SsrB in the presence of H-NS but not when it is inactivated (21). Additionally, we have shown that H-NS binds to a DNA fragment spanning the region from −302 to +478 of ssrAB (21). To determine if the −302/+478 region contains all the cis elements required for the H-NS-mediated repression of ssrAB, we analyzed the expression of the ssrB-cat−302/+478 fusion in the WT strain and its derivative ΔhilD mutant containing the plasmid pT6-HNS-WT or pT6-HNS-Q92am, which expresses WT H-NS or a C-terminally truncated H-NS derivative corresponding to the N-terminal dimerization domain (H-NSQ92am), respectively, from an arabinose-inducible promoter. The strains containing the respective plasmids were grown in LB medium at 37°C in the presence or absence of 0.1% arabinose, which induces the expression of the WT and mutant H-NS proteins. As shown in Fig. 2, induction of the dominant negative H-NSQ92am domain, but not of WT H-NS, restored the expression of the ssrB-cat−302/+478 fusion in the ΔhilD mutant. This indicates that the −302/+478 region contains the cis elements necessary for the repression of ssrAB by H-NS.
To further localize the regulatory sequences required for the repression of ssrAB by H-NS, we analyzed the effect of dominant negative H-NSQ92am on the expression of the different ssrAB-cat fusions in the absence of HilD. Induction of H-NSQ92am derepressed the ssrAB-cat−302/+478, ssrAB-cat−208, ssrAB-cat−106, and ssrAB-cat−55 fusions in the ΔhilD mutant to levels similar to those shown in the WT strain (Fig. 3A and 4A). Notably, this phenomenon was seen even for the ssrAB-cat−55 fusion, which showed expression levels under all conditions tested that were lower than those shown by the ssrAB-cat−302/+478, ssrAB-cat−208, and ssrAB-cat−106 fusions (Fig. 3A and C and 4A). Thus, these results indicate that the sequence upstream of position −55 is not required for the H-NS-mediated repression of ssrAB.
FIG 4.

Analysis of the cis-acting sequences required for the H-NS-mediated repression of ssrAB. Expression of the ssrAB-cat−302/+478, ssrAB-cat−208, ssrAB-cat−106, ssrAB-cat−55, ssrAB-cat+336, ssrAB-cat+240, ssrAB-cat+119, ssrAB-cat+69, and ssrAB-cat+10 transcriptional fusions contained in plasmids pssrAB-cat−302/+478, pssrAB-cat−208, pssrAB-cat−106, pssrAB-cat−55, pssrAB-cat+336, pssrAB-cat+240, pssrAB-cat+119, pssrAB-cat+69, and pssrAB-cat+10, respectively, was tested in the S. Typhimurium ΔhilD mutant containing the plasmid pT6-HNS-Q92am, which expresses a dominant negative C-terminally truncated form of H-NS (H-NSQ92am) from an arabinose-inducible promoter. (A and B) Results for the 5′- and 3′-deletion fusions, respectively. CAT-specific activity was determined from samples collected from bacterial cultures grown for 10 h in LB medium at 37°C. Expression of H-NSQ92am from pT6-HNS-Q92am was induced by adding 0.1% l-arabinose to the medium (+ arabinose). The data are the averages of results from three independent experiments performed in duplicate. Bars represent the standard deviations.
The expression analysis of the ssrAB-cat+336, ssrAB-cat+240, ssrAB-cat+119, ssrAB-cat+69, and ssrAB-cat+10 fusions in the ΔhilD mutant revealed that consecutive deletions of the +240/+336 and +119/+240 regions result in an evident gradual increase in the expression of ssrAB (Fig. 3B and 4B). Moreover, induction of H-NSQ92am derepressed the ssrAB-cat+336 and ssrAB-cat+240 fusions in the ΔhilD mutant to levels similar to those shown in the WT strain (Fig. 3B and 4B) but did not affect the expression of ssrAB-cat+119, ssrAB-cat+69, and ssrAB-cat+10 fusions (Fig. 4B), which indicates that when H-NS is inactivated, the +119/+336 region is not able to mediate repression of ssrAB. In contrast, even in the presence of H-NSQ92am, the ssrAB-cat+336 fusion showed a level of expression higher than that reached by the ssrAB-cat−302/+478 fusion (Fig. 4A and B), indicating that the +336/+478 region mediates H-NS-independent repression of ssrAB.
Together, these results demonstrate that the region between positions +119 and +336 contains multiple cis elements required for the H-NS-mediated repression of ssrAB. In addition, they show that in the absence of these negative cis elements, or when H-NS is inactivated, the expression of ssrAB becomes independent of HilD, further supporting the role of HilD as an antagonist of H-NS-mediated repression of ssrAB.
Binding sites for HilD and H-NS are located close to each other in ssrAB.
Our data from the expression analysis showed that HilD and H-NS regulate the expression of ssrAB mainly by acting on the −55/+240 and +119/+336 regions, respectively. To further dissect the cis sequences required for the HilD/H-NS-mediated regulation of ssrAB, we analyzed the interaction of HilD and H-NS to different segments of the −302/+478 region by electrophoretic mobility shift assays (EMSAs). Purified MBP-HilD (maltose-binding protein–HilD) and H-NS–FH (H-NS-FLAG-His6) proteins were used to perform nonradioactive EMSAs with five ∼200-bp DNA fragments (termed 1A to 1E), which overlap each other by ∼50 bp at their ends and thus all together span the −302/+478 region (Fig. 5A). As a negative control in these binding assays, we used a fragment containing the regulatory region of sigD, a gene that is not directly regulated by HilD or H-NS (21). As shown in Fig. 5B, both the MBP-HilD and H-NS–FH proteins shifted specifically DNA fragments 1B, 1C, and 1D but did not shift DNA fragments 1A and 1E or the negative control, sigD. Considering the positions of the overlapping ends of these DNA fragments, these results indicate that HilD and H-NS bind to the region spanning positions −111 to +287 of ssrAB (Fig. 5A). To determine the specific sites bound by HilD in this region, we performed DNase I protection assays; however, we were unable to obtain specific protected sites in these assays. Instead, HilD protection extended through the entire −111/+287 region (data not shown). Despite this, since MBP-HilD and H-NS–FH bound DNA fragments 1B and 1D, which do not overlap, our results indicate that both HilD and H-NS bind to at least two different sites, located in the regions spanning positions −111 to +37 and +136 to +287 (Fig. 5A).
FIG 5.

HilD and H-NS bind to the same regions of ssrAB. (A) Schematic representation of the ssrAB loci and the five ∼200-bp overlapping DNA fragments (1A to 1E) spanning the −302/+478 region that were used in the EMSAs. Important positions, with respect to the transcriptional start site of ssrAB, are indicated. The two different regions (−111/+37 and +136/+287) bound by HilD and H-NS, deduced from the EMSAs with the five overlapping DNA fragments, are indicated by two gray-filled boxes inside the dashed-line box representing the entire region (−111/+287) bound by these proteins. (B) EMSAs. The ssrAB DNA fragments 1A to 1E were incubated with increasing concentrations of purified MBP-HilD (0, 0.2, 0.3, 0.4, and 0.5 μM) or H-NS–FH (0, 0.3, 0.4, 0.5, and 0.6 μM). A fragment containing the regulatory region of sigD, a gene that is not regulated directly by HilD or H-NS, was used as a negative control. The DNA-protein complexes, which are indicated by an asterisk, were resolved in a nondenaturing 6% polyacrylamide gel and stained with ethidium bromide.
HilD displaces H-NS from ssrAB.
Our results from EMSAs suggested that HilD and H-NS bind to sites located close to each other in ssrAB. Therefore, HilD may antagonize the repressor activity of H-NS by affecting its interaction with ssrAB. To investigate this possibility, we performed competitive EMSAs to examine the effect of HilD on H-NS bound to the −302/+478 region of ssrAB. A DNA fragment spanning this region was first incubated with H-NS, and then increasing amounts of HilD were added. The DNA–H-NS complexes and the DNA-HilD complexes were detected by staining the DNA fragments, as well as by immunoblotting with anti-FLAG antibodies for H-NS–FH and with anti-MBP antibodies for MBP-HilD. As shown in Fig. 6A, increasing amounts of MBP-HilD shifted the DNA–H-NS complex to a slower-migrating complex similar to that formed by MBP-HilD; furthermore, the Western blots indicated that increasing amounts of the DNA-HilD complexes correlate with decreasing amounts of H-NS–FH bound to the DNA fragments. Similar assays were performed by first incubating the ssrAB fragment with MBP-HilD and then adding to the reaction mixture increasing amounts of H-NS–FH, which showed that H-NS is not able to shift the DNA-HilD complexes or to affect the amounts of MBP-HilD bound to the DNA fragments (Fig. 6B). Interestingly, small amounts of H-NS were detected in the DNA-HilD complexes generated by incubating first with H-NS–FH and then with MBP-HilD (Fig. 6A), as well as in those generated by incubating first with MBP-HilD and then with H-NS–FH (Fig. 6B), which may indicate that HilD displaces H-NS from most, but not all, of its binding sites in ssrAB.
FIG 6.

HilD displaces H-NS from ssrAB. Shown are the results of competitive EMSAs between H-NS, which was added first, and HilD (A) or between HilD, which was added first, and H-NS (B) in the −302/+478 region of ssrAB. The upper panels show the ethidium bromide-stained gels, and the middle and lower panels show the immunoblot detection of H-NS–FH and MBP-HilD from the DNA-protein complexes, respectively, obtained from different membranes. (A) H-NS–FH was added at 0.6 μM (lanes 3 to 7), and MBP-HilD was added at 0.2, 0.3, 0.4, and 0.5 μM (lanes 4 to 7, respectively). No protein was added in lane 1, and MBP-HilD was added at 0.5 μM in lane 2. (B) MBP-HilD was added at 0.5 μM (lanes 3 to 7), and H-NS–FH was added at 0.3, 0.4, 0.5, and 0.6 μM (lanes 4 to 7). No protein was added in lane 1, and H-NS–FH was added at 0.6 μM in lane 2. Similar results were obtained from three different experiments.
Together, our data demonstrate that HilD effectively displaces H-NS bound to those sites that mediate the repression of ssrAB.
DISCUSSION
In this study, we elucidated the mechanism by which HilD and H-NS regulate the expression of ssrAB and thus the SPI-2 genes (Fig. 7).
FIG 7.

Model for the HilD- and H-NS-mediated regulation of ssrAB. H-NS represses ssrAB by binding to multiple sites located in a region spanning the promoter and downstream sequence, which may form an H-NS filament complex that blocks the access of the RNA polymerase to the ssrAB promoter. HilD binds to sites overlapping or close to most of the sites bound by H-NS and thus displaces H-NS from most of its binding sites, which disrupts the H-NS nucleoprotein complex, thus leading to the expression of ssrAB. The regions required for the regulation of ssrAB by HilD and H-NS, defined in this study by expression and binding assays, are shown.
Our expression analysis revealed the presence of negative regulatory sequences located downstream of the promoter of ssrAB (Fig. 3 and 4). In the absence of the +119/+336 region, the expression of ssrAB was no longer repressed by H-NS (Fig. 4), whereas deletion of the sequence upstream of the position −55 did not affect the H-NS-mediated repression of ssrAB (Fig. 3 and 4). Furthermore, we showed that H-NS binds to the region spanning positions −111 to +287 of ssrAB (Fig. 5). Taken together, these results indicate that H-NS represses the expression of ssrAB by acting on the −55/+287 region. This region has a very high AT content (69.3%) with respect to that of the entire S. Typhimurium genome (47.8%), consistent with the fact that H-NS binds to and acts on AT-rich sequences (42, 43).
H-NS represses gene expression by forming nucleoprotein complexes, which trap or exclude the RNA polymerase at or from promoters, mainly by two mechanisms (45, 46, 54–57). By the first mechanism, H-NS binds to two sites located further apart, upstream and downstream of the promoter, and then induces a DNA–H-NS–DNA bridge that forms a repressor nucleoprotein stem-loop. By the second mechanism, H-NS initially binds to one or more high-affinity sites that are close together, termed nucleation sites, which leads to its interaction with lower-affinity sites and thus to its polymerization along DNA, to form a repressor nucleoprotein filament.
Our results showed that the sequence upstream of position −55 is not required for the H-NS-mediated repression of ssrAB (Fig. 3 and 4) but that consecutive deletion of the +240/+336 and +119/+240 regions results in a gradual release of the H-NS-mediated repression of ssrAB (Fig. 4). Furthermore, our EMSAs revealed that H-NS binds to at least two different sites along the −111/+287 region of ssrAB (Fig. 5). Taken together, these results favor the model in which H-NS represses the expression of ssrAB initially by binding to two nucleation sites and then by forming a nucleoprotein filament on the promoter (Fig. 7).
Our results revealed that deletion of the +336/+478 region increases around 6-fold the expression of ssrAB, which is evident when Salmonella is grown either in LB medium (Fig. 3A and B) or in N-MM (Fig. 3C and D) and even when H-NS is inactivated (Fig. 4A and B). Thus, these results indicate that the +336/+478 region mediates H-NS-independent repression of ssrAB. There are no reports indicating the action of any trans or cis factor on this region. Furthermore, preliminary bioinformatics analyses of this region showed no obvious putative binding sites for any transcriptional repressor of a known binding consensus sequence or a sequence forming an intrinsic transcriptional terminator. Therefore, how the +336/+478 region negatively affects the expression of ssrAB remains to be determined.
Several DNA-binding proteins have been shown to relieve the transcriptional-silencing activity of H-NS in different bacteria (47). These proteins are able to displace or remodel the H-NS nucleoprotein complexes by binding to sites overlapping or close to those nucleation sites for H-NS, which might arise simply as a result of the DNA-distorting effect exerted by most transcriptional factors at their DNA-binding sites and the relatively weak DNA-binding activity of H-NS (45–47, 54, 55, 58). Interestingly, most of the AraC-like regulators that control virulence gene expression, such as HilD, ToxT, VirF, CfaD, RegA, and Rns, act as H-NS antagonists, and consistently, they bind to AT-rich sites (47, 59).
Our results indicate that the −55/+240 region contains the cis elements required for positive HilD-mediated regulation of ssrAB (Fig. 3). In addition, we found that when the activity of H-NS is inactivated (Fig. 2 and 4) or when the +119/+336 region is deleted (Fig. 3 and 4), full expression of ssrAB becomes independent of HilD. In agreement with these results, we previously showed that full expression of the chromosomally encoded SsrA and SsrB proteins requires HilD in the presence of H-NS but not when it is inactivated (21). Furthermore, in the present study, we determined that HilD binds to at least two different sites located in the same region bound by H-NS, −111 and +287 (Fig. 5). Moreover, by competitive EMSAs we demonstrated that HilD is able to displace H-NS from ssrAB, whereas H-NS does not affect HilD binding to ssrAB (Fig. 6). Interestingly, our results suggest that HilD displaces H-NS from most, but not all, of its binding sites in ssrAB (Fig. 6), which is consistent with our genetic and binding results indicating the presence of several H-NS binding sites in ssrAB. Thus, some of these H-NS binding sites may be unproductive for the repression of ssrAB, or another regulator may act along with HilD to counteract a double mechanism of H-NS-mediated repression of ssrAB.
All together, these results indicate that HilD induces the expression of ssrAB by displacing the H-NS nucleoprotein complex from the promoter of this operon (Fig. 7).
HilD directly regulates at least 11 other genes; however, evidence that HilD acts as an antagonist of H-NS exists only for hilA and rtsA (23, 24, 26–31, 48, 49, 60). How HilD counteracts the H-NS-mediated repression of hilA and rtsA and whether HilD acts as an antagonist of H-NS with its other target genes remain to be determined.
HilD-binding sites have been determined upstream of the promoters of hilA, hilC, hilD, rtsA, and flhDC; these sites present a high AT content and show a high degree of degeneracy between them (24, 26, 27, 31). However, HilD regulates invF by binding to regions located both upstream and downstream of the HilC/D-dependent invF promoter (28, 60). Furthermore, our results indicate that HilD induces the expression of ssrAB by binding to at least two different AT-rich regions: one located on or close to the promoter and the other located far downstream (Fig. 3 and 5). These data indicate that HilD can act at different positions with respect to the promoters, which is consistent with its role as an antagonist of H-NS.
The high degree of degeneracy between the known HilD-binding sites makes it difficult to predict the specific binding sites of HilD in the −111/+287 region of ssrAB. However, at least 10 copies of the CnaTTnnT motif (predominant, conserved, and degenerate bases are indicated by uppercase and lowercase letters and the letter n, respectively), somewhat conserved in two different HilD-binding consensus sequences previously reported (27, 31), can be found in this region (data not shown). Site-directed mutagenesis of this predicted motif in the −111/+287 region may help to better define the binding sites of HilD in ssrAB.
HilD, HilC, and RtsA are homologous AraC-like regulators that bind to very similar DNA sites; therefore, when these regulators are overexpressed, they are able to self-activate their own expression and to activate the expression of one another, as well as to induce the expression of most genes belonging to the HilD regulon; however, HilD is considered to be dominant, as deletion of hilD, but not of hilC or rtsA, drastically reduces the expression of the targets of HilD (23, 24, 26–30, 61–64). Our results indicate that the expression of ssrAB is not affected in a ΔhilC (21) or ΔrtsA mutant, but a plasmid expressing HilC can restore the expression of ssrAB in the ΔhilD mutant (unpublished data). Thus, it is possible that the feed forward loop constituted by HilD, HilC, and RtsA can regulate the expression of ssrAB.
Previously, we showed that OmpR (the response regulator of the OmpR/EnvZ two-component system), in addition to HilD, is required for the expression of SsrA/B when Salmonella is grown in LB medium; moreover, OmpR is required for the expression of SsrA/B even when H-NS is inactivated (21). Therefore, the expression of ssrAB seems to require the coordinated actions of HilD, which antagonizes repression by H-NS, and OmpR, which probably mediates the interaction of the RNA polymerase with the promoter of this operon. The S. Typhimurium ugtL and pagC genes are regulated by a similar mechanism, involving the coordinated actions of SlyA, which antagonizes H-NS-mediated repression of these genes, and PhoP (the response regulator of the PhoP/PhoQ two-component system), which acts as a conventional transcriptional activator (65).
Our data from this study show that the sequence between positions −106 and −55 has a positive regulatory role for the expression of ssrAB, but this sequence was not required for the HilD- and H-NS-mediated regulation of this operon (Fig. 3 and 4). Since OmpR binds to the −83/+6 region (32, 33), it is possible that the sequence spanning positions −106 to −55 is involved in the positive regulation of ssrAB by OmpR.
Interestingly, HilD is required for the expression of ssrAB when Salmonella is grown in LB medium but not when it is grown in minimal media, such as N-MM (Fig. 3) (21). In minimal media, OmpR, PhoP, SsrB, and SlyA have been involved in the expression of ssrAB (32–38). It is known that SlyA induces gene expression mainly by acting as an antagonist of H-NS (47). Therefore, SlyA might replace HilD to counteract H-NS-mediated repression of ssrAB when Salmonella is grown in minimal media. To determine how HilD, OmpR, and SlyA coordinate to regulate the expression of ssrAB in response to different growth conditions is a matter of our current and future studies.
The results from this study further expand the current knowledge about the mechanisms regulating the expression of the SPI-2 genes, which have an essential role in Salmonella virulence.
Supplementary Material
ACKNOWLEDGMENTS
We thank Angie Hinz for critical reading of the manuscript.
This work was supported by grants from the Dirección General de Asuntos del Personal Académico de la UNAM (IN205512) and from the Consejo Nacional de Ciencia y Tecnología (179071) to V.H.B. L.C.M. was supported by a predoctoral fellowship from CONACYT (169380).
Footnotes
Published ahead of print 18 August 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.01799-14.
REFERENCES
- 1.Schmidt H, Hensel M. 2004. Pathogenicity islands in bacterial pathogenesis. Clin. Microbiol. Rev. 17:14–56. 10.1128/CMR.17.1.14-56.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fàbrega A, Vila J. 2013. Salmonella enterica serovar Typhimurium skills to succeed in the host: virulence and regulation. Clin. Microbiol. Rev. 26:308–341. 10.1128/CMR.00066-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hansen-Wester I, Hensel M. 2001. Salmonella pathogenicity islands encoding type III secretion systems. Microbes Infect. 3:549–559. 10.1016/S1286-4579(01)01411-3. [DOI] [PubMed] [Google Scholar]
- 4.Haraga A, Ohlson MB, Miller SI. 2008. Salmonellae interplay with host cells. Nat. Rev. Microbiol. 6:53–66. 10.1038/nrmicro1788. [DOI] [PubMed] [Google Scholar]
- 5.Moest TP, Meresse S. 2013. Salmonella T3SSs: successful mission of the secret(ion) agents. Curr. Opin. Microbiol. 16:38–44. 10.1016/j.mib.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 6.Bispham J, Tripathi BN, Watson PR, Wallis TS. 2001. Salmonella pathogenicity island 2 influences both systemic salmonellosis and Salmonella-induced enteritis in calves. Infect. Immun. 69:367–377. 10.1128/IAI.69.1.367-377.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coburn B, Li Y, Owen D, Vallance BA, Finlay BB. 2005. Salmonella enterica serovar Typhimurium pathogenicity island 2 is necessary for complete virulence in a mouse model of infectious enterocolitis. Infect. Immun. 73:3219–3227. 10.1128/IAI.73.6.3219-3227.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coombes BK, Coburn BA, Potter AA, Gomis S, Mirakhur K, Li Y, Finlay BB. 2005. Analysis of the contribution of Salmonella pathogenicity islands 1 and 2 to enteric disease progression using a novel bovine ileal loop model and a murine model of infectious enterocolitis. Infect. Immun. 73:7161–7169. 10.1128/IAI.73.11.7161-7169.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hapfelmeier S, Stecher B, Barthel M, Kremer M, Muller AJ, Heikenwalder M, Stallmach T, Hensel M, Pfeffer K, Akira S, Hardt WD. 2005. The Salmonella pathogenicity island (SPI)-2 and SPI-1 type III secretion systems allow Salmonella serovar typhimurium to trigger colitis via MyD88-dependent and MyD88-independent mechanisms. J. Immunol. 174:1675–1685. 10.4049/jimmunol.174.3.1675. [DOI] [PubMed] [Google Scholar]
- 10.Grant AJ, Morgan FJ, McKinley TJ, Foster GL, Maskell DJ, Mastroeni P. 2012. Attenuated Salmonella Typhimurium lacking the pathogenicity island-2 type 3 secretion system grow to high bacterial numbers inside phagocytes in mice. PLoS Pathog. 8:e1003070. 10.1371/journal.ppat.1003070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muller AJ, Kaiser P, Dittmar KE, Weber TC, Haueter S, Endt K, Songhet P, Zellweger C, Kremer M, Fehling HJ, Hardt WD. 2012. Salmonella gut invasion involves TTSS-2-dependent epithelial traversal, basolateral exit, and uptake by epithelium-sampling lamina propria phagocytes. Cell Host Microbe 11:19–32. 10.1016/j.chom.2011.11.013. [DOI] [PubMed] [Google Scholar]
- 12.Nuñez-Hernández C, Alonso A, Pucciarelli MG, Casadesús J, García-del Portillo F. 2014. Dormant intracellular Salmonella enterica serovar Typhimurium discriminates among Salmonella pathogenicity island 2 effectors to persist inside fibroblasts. Infect. Immun. 82:221–232. 10.1128/IAI.01304-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown NF, Vallance BA, Coombes BK, Valdez Y, Coburn BA, Finlay BB. 2005. Salmonella pathogenicity island 2 is expressed prior to penetrating the intestine. PLoS Pathog. 1:e32. 10.1371/journal.ppat.0010032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Knodler LA, Vallance BA, Celli J, Winfree S, Hansen B, Montero M, Steele-Mortimer O. 2010. Dissemination of invasive Salmonella via bacterial-induced extrusion of mucosal epithelia. Proc. Natl. Acad. Sci. U. S. A. 107:17733–17738. 10.1073/pnas.1006098107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cirillo DM, Valdivia RH, Monack DM, Falkow S. 1998. Macrophage-dependent induction of the Salmonella pathogenicity island 2 type III secretion system and its role in intracellular survival. Mol. Microbiol. 30:175–188. 10.1046/j.1365-2958.1998.01048.x. [DOI] [PubMed] [Google Scholar]
- 16.Deiwick J, Nikolaus T, Erdogan S, Hensel M. 1999. Environmental regulation of Salmonella pathogenicity island 2 gene expression. Mol. Microbiol. 31:1759–1773. 10.1046/j.1365-2958.1999.01312.x. [DOI] [PubMed] [Google Scholar]
- 17.Eriksson S, Lucchini S, Thompson A, Rhen M, Hinton JC. 2003. Unravelling the biology of macrophage infection by gene expression profiling of intracellular Salmonella enterica. Mol. Microbiol. 47:103–118. 10.1046/j.1365-2958.2003.03313.x. [DOI] [PubMed] [Google Scholar]
- 18.Laughlin RC, Knodler LA, Barhoumi R, Payne HR, Wu J, Gomez G, Pugh R, Lawhon SD, Baumler AJ, Steele-Mortimer O, Adams LG. 2014. Spatial segregation of virulence gene expression during acute enteric infection with Salmonella enterica serovar Typhimurium. mBio 5(1):e00946–13. 10.1128/mBio.00946-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lundberg U, Vinatzer U, Berdnik D, von Gabain A, Baccarini M. 1999. Growth phase-regulated induction of Salmonella-induced macrophage apoptosis correlates with transient expression of SPI-1 genes. J. Bacteriol. 181:3433–3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miao EA, Miller SI. 2000. A conserved amino acid sequence directing intracellular type III secretion by Salmonella typhimurium. Proc. Natl. Acad. Sci. U. S. A. 97:7539–7544. 10.1073/pnas.97.13.7539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bustamante VH, Martínez LC, Santana FJ, Knodler LA, Steele-Mortimer O, Puente JL. 2008. HilD-mediated transcriptional cross-talk between SPI-1 and SPI-2. Proc. Natl. Acad. Sci. U. S. A. 105:14591–14596. 10.1073/pnas.0801205105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kroger C, Colgan A, Srikumar S, Handler K, Sivasankaran SK, Hammarlof DL, Canals R, Grissom JE, Conway T, Hokamp K, Hinton JC. 2013. An infection-relevant transcriptomic compendium for Salmonella enterica serovar Typhimurium. Cell Host Microbe 14:683–695. 10.1016/j.chom.2013.11.010. [DOI] [PubMed] [Google Scholar]
- 23.Schechter LM, Damrauer SM, Lee CA. 1999. Two AraC/XylS family members can independently counteract the effect of repressing sequences upstream of the hilA promoter. Mol. Microbiol. 32:629–642. 10.1046/j.1365-2958.1999.01381.x. [DOI] [PubMed] [Google Scholar]
- 24.Schechter LM, Lee CA. 2001. AraC/XylS family members, HilC and HilD, directly bind and derepress the Salmonella typhimurium hilA promoter. Mol. Microbiol. 40:1289–1299. 10.1046/j.1365-2958.2001.02462.x. [DOI] [PubMed] [Google Scholar]
- 25.Golubeva YA, Sadik AY, Ellermeier JR, Slauch JM. 2012. Integrating global regulatory input into the Salmonella pathogenicity island 1 type III secretion system. Genetics 190:79–90. 10.1534/genetics.111.132779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Olekhnovich IN, Kadner RJ. 2002. DNA-binding activities of the HilC and HilD virulence regulatory proteins of Salmonella enterica serovar Typhimurium. J. Bacteriol. 184:4148–4160. 10.1128/JB.184.15.4148-4160.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Olekhnovich IN, Kadner RJ. 2007. Role of nucleoid-associated proteins Hha and H-NS in expression of Salmonella enterica activators HilD, HilC, and RtsA required for cell invasion. J. Bacteriol. 189:6882–6890. 10.1128/JB.00905-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Akbar S, Schechter LM, Lostroh CP, Lee CA. 2003. AraC/XylS family members, HilD and HilC, directly activate virulence gene expression independently of HilA in Salmonella typhimurium. Mol. Microbiol. 47:715–728. 10.1046/j.1365-2958.2003.03322.x. [DOI] [PubMed] [Google Scholar]
- 29.Ellermeier CD, Ellermeier JR, Slauch JM. 2005. HilD, HilC and RtsA constitute a feed forward loop that controls expression of the SPI1 type three secretion system regulator HilA in Salmonella enterica serovar Typhimurium. Mol. Microbiol. 57:691–705. 10.1111/j.1365-2958.2005.04737.x. [DOI] [PubMed] [Google Scholar]
- 30.Petrone BL, Stringer AM, Wade JT. 2014. Identification of HilD-regulated genes in Salmonella enterica serovar Typhimurium. J. Bacteriol. 196:1094–1101. 10.1128/JB.01449-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Singer HM, Kuhne C, Deditius JA, Hughes KT, Erhardt M. 2014. The Salmonella Spi1 virulence regulatory protein HilD directly activates transcription of the flagellar master operon flhDC. J. Bacteriol. 196:1448–1457. 10.1128/JB.01438-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee AK, Detweiler CS, Falkow S. 2000. OmpR regulates the two-component system SsrA-SsrB in Salmonella pathogenicity island 2. J. Bacteriol. 182:771–781. 10.1128/JB.182.3.771-781.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Feng X, Oropeza R, Kenney LJ. 2003. Dual regulation by phospho-OmpR of ssrA/B gene expression in Salmonella pathogenicity island 2. Mol. Microbiol. 48:1131–1143. 10.1046/j.1365-2958.2003.03502.x. [DOI] [PubMed] [Google Scholar]
- 34.Feng X, Walthers D, Oropeza R, Kenney LJ. 2004. The response regulator SsrB activates transcription and binds to a region overlapping OmpR binding sites at Salmonella pathogenicity island 2. Mol. Microbiol. 54:823–835. 10.1111/j.1365-2958.2004.04317.x. [DOI] [PubMed] [Google Scholar]
- 35.Bijlsma JJ, Groisman EA. 2005. The PhoP/PhoQ system controls the intramacrophage type three secretion system of Salmonella enterica. Mol. Microbiol. 57:85–96. 10.1111/j.1365-2958.2005.04668.x. [DOI] [PubMed] [Google Scholar]
- 36.Linehan SA, Rytkonen A, Yu XJ, Liu M, Holden DW. 2005. SlyA regulates function of Salmonella pathogenicity island 2 (SPI-2) and expression of SPI-2-associated genes. Infect. Immun. 73:4354–4362. 10.1128/IAI.73.7.4354-4362.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Okada N, Oi Y, Takeda-Shitaka M, Kanou K, Umeyama H, Haneda T, Miki T, Hosoya S, Danbara H. 2007. Identification of amino acid residues of Salmonella SlyA that are critical for transcriptional regulation. Microbiology 153:548–560. 10.1099/mic.0.29259-0. [DOI] [PubMed] [Google Scholar]
- 38.Fass E, Groisman EA. 2009. Control of Salmonella pathogenicity island-2 gene expression. Curr. Opin. Microbiol. 12:199–204. 10.1016/j.mib.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baños RC, Vivero A, Aznar S, García J, Pons M, Madrid C, Juárez A. 2009. Differential regulation of horizontally acquired and core genome genes by the bacterial modulator H-NS. PLoS Genet. 5:e1000513. 10.1371/journal.pgen.1000513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Queiroz MH, Madrid C, Paytubi S, Balsalobre C, Juárez A. 2011. Integration host factor alleviates H-NS silencing of the Salmonella enterica serovar Typhimurium master regulator of SPI1, hilA. Microbiology 157:2504–2514. 10.1099/mic.0.049197-0. [DOI] [PubMed] [Google Scholar]
- 41.Martínez LC, Yakhnin H, Camacho MI, Georgellis D, Babitzke P, Puente JL, Bustamante VH. 2011. Integration of a complex regulatory cascade involving the SirA/BarA and Csr global regulatory systems that controls expression of the Salmonella SPI-1 and SPI-2 virulence regulons through HilD. Mol. Microbiol. 80:1637–1656. 10.1111/j.1365-2958.2011.07674.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lucchini S, Rowley G, Goldberg MD, Hurd D, Harrison M, Hinton JC. 2006. H-NS mediates the silencing of laterally acquired genes in bacteria. PLoS Pathog. 2:e81. 10.1371/journal.ppat.0020081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Navarre WW, Porwollik S, Wang Y, McClelland M, Rosen H, Libby SJ, Fang FC. 2006. Selective silencing of foreign DNA with low GC content by the H-NS protein in Salmonella. Science 313:236–238. 10.1126/science.1128794. [DOI] [PubMed] [Google Scholar]
- 44.Sturm A, Heinemann M, Arnoldini M, Benecke A, Ackermann M, Benz M, Dormann J, Hardt WD. 2011. The cost of virulence: retarded growth of Salmonella Typhimurium cells expressing type III secretion system 1. PLoS Pathog. 7:e1002143. 10.1371/journal.ppat.1002143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dorman CJ. 2007. H-NS, the genome sentinel. Nat. Rev. Microbiol. 5:157–161. 10.1038/nrmicro1598. [DOI] [PubMed] [Google Scholar]
- 46.Navarre WW, McClelland M, Libby SJ, Fang FC. 2007. Silencing of xenogeneic DNA by H-NS-facilitation of lateral gene transfer in bacteria by a defense system that recognizes foreign DNA. Genes Dev. 21:1456–1471. 10.1101/gad.1543107. [DOI] [PubMed] [Google Scholar]
- 47.Stoebel DM, Free A, Dorman CJ. 2008. Anti-silencing: overcoming H-NS-mediated repression of transcription in Gram-negative enteric bacteria. Microbiology 154:2533–2545. 10.1099/mic.0.2008/020693-0. [DOI] [PubMed] [Google Scholar]
- 48.Schechter LM, Jain S, Akbar S, Lee CA. 2003. The small nucleoid-binding proteins H-NS, HU, and Fis affect hilA expression in Salmonella enterica serovar Typhimurium. Infect. Immun. 71:5432–5435. 10.1128/IAI.71.9.5432-5435.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Olekhnovich IN, Kadner RJ. 2006. Crucial roles of both flanking sequences in silencing of the hilA promoter in Salmonella enterica. J. Mol. Biol. 357:373–386. 10.1016/j.jmb.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 50.Duong N, Osborne S, Bustamante VH, Tomljenovic AM, Puente JL, Coombes BK. 2007. Thermosensing coordinates a cis-regulatory module for transcriptional activation of the intracellular virulence system in Salmonella enterica serovar Typhimurium. J. Biol. Chem. 282:34077–34084. 10.1074/jbc.M707352200. [DOI] [PubMed] [Google Scholar]
- 51.Puente JL, Bieber D, Ramer SW, Murray W, Schoolnik GK. 1996. The bundle-forming pili of enteropathogenic Escherichia coli: transcriptional regulation by environmental signals. Mol. Microbiol. 20:87–100. 10.1111/j.1365-2958.1996.tb02491.x. [DOI] [PubMed] [Google Scholar]
- 52.Barba J, Bustamante VH, Flores-Valdez MA, Deng W, Finlay BB, Puente JL. 2005. A positive regulatory loop controls expression of the locus of enterocyte effacement-encoded regulators Ler and GrlA. J. Bacteriol. 187:7918–7930. 10.1128/JB.187.23.7918-7930.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Martínez LC, Martínez-Flores I, Salgado H, Fernández-Mora M, Medina-Rivera A, Puente JL, Collado-Vides J, Bustamante VH. 2014. In silico identification and experimental characterization of regulatory elements controlling the expression of the Salmonella csrB and csrC genes. J. Bacteriol. 196:325–336. 10.1128/JB.00806-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fang FC, Rimsky S. 2008. New insights into transcriptional regulation by H-NS. Curr. Opin. Microbiol. 11:113–120. 10.1016/j.mib.2008.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dillon SC, Dorman CJ. 2010. Bacterial nucleoid-associated proteins, nucleoid structure and gene expression. Nat. Rev. Microbiol. 8:185–195. 10.1038/nrmicro2261. [DOI] [PubMed] [Google Scholar]
- 56.Ali SS, Xia B, Liu J, Navarre WW. 2012. Silencing of foreign DNA in bacteria. Curr. Opin. Microbiol. 15:175–181. 10.1016/j.mib.2011.12.014. [DOI] [PubMed] [Google Scholar]
- 57.Lim CJ, Lee SY, Kenney LJ, Yan J. 2012. Nucleoprotein filament formation is the structural basis for bacterial protein H-NS gene silencing. Sci. Rep. 2:509. 10.1038/srep00509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dorman CJ, Kane KA. 2009. DNA bridging and antibridging: a role for bacterial nucleoid-associated proteins in regulating the expression of laterally acquired genes. FEMS Microbiol. Rev. 33:587–592. 10.1111/j.1574-6976.2008.00155.x. [DOI] [PubMed] [Google Scholar]
- 59.Yang J, Tauschek M, Robins-Browne RM. 2011. Control of bacterial virulence by AraC-like regulators that respond to chemical signals. Trends Microbiol. 19:128–135. 10.1016/j.tim.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 60.Lim S, Lee B, Kim M, Kim D, Yoon H, Yong K, Kang DH, Ryu S. 2012. Analysis of HilC/D-dependent invF promoter expression under different culture conditions. Microb. Pathog. 52:359–366. 10.1016/j.micpath.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 61.Lucas RL, Lee CA. 2001. Roles of hilC and hilD in regulation of hilA expression in Salmonella enterica serovar Typhimurium. J. Bacteriol. 183:2733–2745. 10.1128/JB.183.9.2733-2745.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ellermeier CD, Slauch JM. 2003. RtsA and RtsB coordinately regulate expression of the invasion and flagellar genes in Salmonella enterica serovar Typhimurium. J. Bacteriol. 185:5096–5108. 10.1128/JB.185.17.5096-5108.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ellermeier CD, Slauch JM. 2004. RtsA coordinately regulates DsbA and the Salmonella pathogenicity island 1 type III secretion system. J. Bacteriol. 186:68–79. 10.1371/journal.ppat.1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Saini S, Ellermeier JR, Slauch JM, Rao CV. 2010. The role of coupled positive feedback in the expression of the SPI1 type three secretion system in Salmonella. PLoS Pathog. 6:e1001025. 10.1371/journal.ppat.1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Perez JC, Latifi T, Groisman EA. 2008. Overcoming H-NS-mediated transcriptional silencing of horizontally acquired genes by the PhoP and SlyA proteins in Salmonella enterica. J. Biol. Chem. 283:10773–10783. 10.1074/jbc.M709843200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.