

Abstract
For most phages, holins control the timing of host lysis. During the morphogenesis period of the infection cycle, canonical holins accumulate harmlessly in the cytoplasmic membrane until they suddenly trigger to form lethal lesions called holes. The holes can be visualized by cryo-electron microscopy and tomography as micrometer-scale interruptions in the membrane. To explore the fine structure of the holes formed by the lambda holin, S105, a cysteine-scanning accessibility study was performed. A collection of S105 alleles encoding holins with a single Cys residue in different positions was developed and characterized for lytic function. Based on the ability of 4-acetamido-4′-((iodoacetyl) amino) stilbene-2,2′-disulfonic acid, disodium salt (IASD), to modify these Cys residues, one face of transmembrane domain 1 (TMD1) and TMD3 was judged to face the lumen of the S105 hole. In both cases, the lumen-accessible face was found to correspond to the more hydrophilic face of the two TMDs. Judging by the efficiency of IASD modification, it was concluded that the bulk of the S105 protein molecules were involved in facing the lumen. These results are consistent with a model in which the perimeters of the S105 holes are lined by the holin molecules present at the time of lysis. Moreover, the findings that TMD1 and TMD3 face the lumen, coupled with previous results showing TMD2-TMD2 contacts in the S105 dimer, support a model in which membrane depolarization drives the transition of S105 from homotypic to heterotypic oligomeric interactions.
INTRODUCTION
Host lysis at the end of the bacteriophage infection cycle is one of the most common cellular fates in the biosphere. For most phages, the holin controls the timing of lysis and thus the length and fecundity of the infection cycle. The best-studied holin is the S105 protein of phage lambda, one of two products of the lambda S gene (Fig. 1 and 2), the other being the S107 antiholin (1–4). Throughout the morphogenesis period of the infection cycle, S105 accumulates in the membrane without detectable effect on cell physiology or membrane integrity. Suddenly, at an allele-specific time, the S105 population is said to “trigger” to form lethal membrane lesions. Triggering is detectable by a sudden halt in culture growth and respiration, collapse of the membrane potential, massive ion leakage into the medium, and loss of viability. Moreover, triggering can be imposed prematurely by causing a sudden reduction in the membrane potential, using energy poisons like 2,4-dinitrophenol (DNP) and cyanide (5). The physiological effects observed for the cell are due to the formation of micrometer-scale membrane lesions, or holes, approximately 1 to 3 per cell and averaging >340 nm in diameter (6), the largest membrane lesions described in biology. The formation of these massive holes allows folded, fully active cytoplasmic endolysins to be released nonspecifically into the periplasm, resulting in rapid degradation of the peptidoglycan (2, 7). The term “hole” was chosen to distinguish these lesions from channels and pores that have been characterized in bacterial membranes (8).
FIG 1.
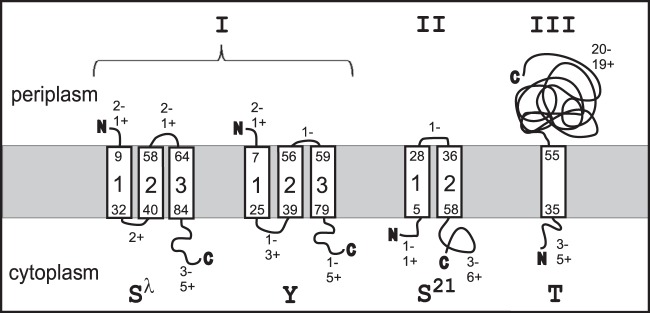
Three topology classes of the holins. Shown are the membrane topologies of λ S105, P2 Y, phage 21 S21, and T4 T. Both the borders of TMDs and the numbers of charges in the loops are indicated.
FIG 2.

Primary structure of S105. The positions of single-cysteine substitutions are indicated above the sequence. In each case, the allele also contains the Cys51Ser substitution, indicated by the asterisk. The three TMDs are boxed (17). The antiholin S107 has the same primary structure, except for two additional residues, Met-Lys, at the N terminus. Plus and minus signs indicate charged amino acid residues.
The finding that the holes were of such unprecedented size explained previous observations that S105 holes were nonspecific with respect to heterogeneous endolysins and were permissive for ∼0.5-MDa endolysin–β-galactosidase chimeras (7). Similar nonspecificity and permissiveness for the large endolysin chimeras had been noted for other holins, the Y holin from coliphage P2 and the T holin from coliphage T4 (9, 10). Recently, cryo-electron microscopy (cryo-EM) studies revealed that both of these holins formed similar micrometer-scale interruptions in the inner membrane (IM) (11). Y, like S105, is a class I holin (three transmembrane domains [TMDs], with N in and C out), although they share no detectable sequence similarity (Fig. 1). However, T is a class III holin, with a single TMD (N in and C out) (Fig. 1). The fact that these unrelated holins all formed the micrometer-scale lesions suggested that this terminal phenotype is a general feature of holin function and thus the most common cytocidal membrane lesion in the biosphere.
Other than the massive size, nothing is known about the structure of the hole formed by the canonical holins like S105, Y, and T. The number of S105, Y, and T proteins present at the time of hole formation is consistent with the notion that most, if not all, of the available holin is involved in forming the hole perimeter, based on the assumption that at least one of the TMDs in each holin molecule is part of the hole wall (6). Here, we present the results of a cysteine-scanning accessibility study for S105 assembled into the micrometer-scale holes. The results are discussed in terms of a general model for hole formation and thus for the temporal regulation of the phage infection cycle.
MATERIALS AND METHODS
Materials, strains, phages, plasmids, and growth media.
The Escherichia coli strain XL1-Blue, the lysis-defective thermoinducible prophage λΔSR, and the lysis-proficient thermoinducible prophage λS105 (expressing S105) have been described previously (12–15). In general, S105 was expressed from plasmid pS105, which carries the phage lambda lysis gene cassette (S105RRzRz1) under its native promoter, pR′. The other lysis genes were inactivated by nonsense mutations, so that only S105 was expressed. The bacterial strains, phages, and plasmids used in this work are listed in Table 1. Media, growth conditions, and thermal induction of the λ lysis genes from a prophage and/or plasmid have been described previously (5, 14, 16, 17). Bacterial cultures were grown in standard LB medium supplemented with ampicillin (100 μg/ml) and chloramphenicol (10 μg/ml) for the maintenance of plasmids and prophages, respectively.
TABLE 1.
Strains, phages, and plasmids
| Strain, phage, or plasmid | Genotype and/or featuresa | Source or reference |
|---|---|---|
| Strains | ||
| E. coli MC4100 | K-12 F− araD139 Δ(argF-lac)U169 ΔfhuA rpsL150 relA1 flbB5301 deoC1 ptsF25 rbsR | Laboratory stock |
| E. coli XL1-Blue | K-12 recA endA1 gyrA96 thi1 hsdR17 supE44 relA1 (F′ proAB lacIqZΔM15::Tn10 [Tetr]) | Stratagene |
| Phages | ||
| λ Cmr Δ(SR) | stf::cat::tfa cI857 Δ(SR); replacement of stf and tfa genes (λ nt 19996–22220) with the cat gene (33); Δ(SR); deletion of λ nt 45136–45815 (13) | Laboratory stock |
| λ-S105 | cI857 SM1L; expresses S105 only | Laboratory stock |
| Plasmids | ||
| pS105 | λ lysis cassette under pR′ on the pBR322 backbone; SM1L allele; expresses S105 only | 18 |
| pS105C51S | Same as pS105 but with C51S mutation | 17 |
| pS105C51S,A52V | Same as pS105C51S with addition of A52V mutation | This study |
nt, nucleotides.
Standard DNA manipulations, PCR, site-directed mutagenesis, and DNA sequencing.
Isolation of plasmid DNA, DNA amplification by PCR, DNA transformation, and DNA sequencing were performed as previously described (17). Primers were obtained from Integrated DNA Technologies, Coralville, IA, and were used without further purification. Single-cysteine substitutions were made in the corresponding plasmids by site-directed mutagenesis using the QuikChange kit from Stratagene as described previously (17). pS105 was used as the template for all primers, except the cysteine mutation primers (excluding A52V For/Rev). pS105C51S was used as the template for all of the cysteine mutation primers. The DNA sequences of all constructs were verified by automated fluorescence sequencing performed at Eton Bioscience Inc., San Diego, CA.
TCA precipitation.
Culture aliquots of 1 or 5 ml were added to 111 μl or 555 μl, respectively, of cold 6.1 N-trichloroacetic acid (TCA) and then placed on ice for 30 min. The precipitate was collected by centrifugation (15,000 rpm in a tabletop microcentrifuge or 3,000 rpm in a clinical centrifuge) and washed once with acetone, resuspending the pellet completely. The pellets were air dried and resuspended in SDS-PAGE loading buffer. Proteins were separated on SDS-16.5% PAGE with a 4% stacking gel. Western blotting and immunodetection with anti-S antibodies were performed as previously described (5).
Cysteine modification.
Cultures were grown to an A550 of 0.4, induced by a thermal shift to 42°C for 15 min (14, 18), and aerated until the time of holin triggering or, in the case of nonlethal S105 alleles, for 50 min (16). Cells corresponding to 0.25 A550 unit were collected by centrifugation, washed twice with 1 ml Tris-buffered saline (TBS) buffer (25 mM Tris-HCl, 150 mM NaCl, pH 7.2), and then resuspended in 0.25 ml of TBS. Each sample was divided into two 125-μl aliquots. To one, 10 mM 4-acetamido-4′-((iodoacetyl) amino) stilbene-2,2′-disulfonic acid, disodium salt (IASD) (Invitrogen), was added, and to the other, an equivalent amount of water was added. After 30 min at room temperature, 50 mM l-cysteine was added to quench any unreacted IASD. After 10 min, the cells were diluted by the addition of 0.75 ml TBS, collected by centrifugation, washed twice with 1 ml TBS, resuspended in 100 μl PB (50 mM phosphate buffer, pH 7), and extracted with 750 μl chloroform-methanol-water (1:4:1). After incubation on ice for 30 min, the denatured and delipidated proteins were collected by centrifugation at 13,000 × g for 5 min at 4°C. The protein pellets were washed once with 400 μl 95% methanol and resuspended in 100 μl PEGylation buffer (10 M urea-1% SDS-1 mM EDTA-0.6 M Tris, pH 7; adapted from the work of Lu and Deutsch [19]). Fifty microliters of each sample was transferred to a clean tube, treated with 0.2 mM polyethylene glycol (PEG)-maleimide (Creative Biochem) for 30 min at room temperature, and then precipitated with 1 ml cold ethanol. After being kept overnight at 20°C, proteins were collected by centrifugation at 13,000 × g for 15 min at 4°C. The pellets were air dried and resuspended in sample loading buffer for analysis by SDS-PAGE (20). For experiments involving depolarization of the membrane, 1 mM DNP was added to the culture at the time of harvesting and used to supplement TBS in each step until quenching with l-cysteine.
Western blotting.
SDS-PAGE and Western blotting were performed as described previously (21); 16.5% Tris-Tricine gels were used to separate protein samples. An antibody raised in rabbit against the S105 C-terminal peptide was used as the primary antibody to detect S105 protein variants (22). Horseradish peroxidase-conjugated goat anti-rabbit secondary antibody was from Pierce.
RESULTS
Cys-scanning mutagenesis of S105.
S105 has a single Cys residue in position 51 in the middle of TMD2. This position tolerates a Ser substitution without loss of lytic function; in fact, the C51S (S105C51S) allele exhibits significantly early triggering (23). Using the S105C51S allele as the parent, a collection of Cys substitution alleles had been created previously for studies aimed at determining the membrane topology and genetic analysis of S105 (17, 18). However, some of these alleles were found to be nonlytic, making them unsuitable for probing the lumenal surfaces of the S105 holes. Using helical projections as a guide, site-directed mutagenesis of S105C51S was performed until there were multiple single-Cys substitutions along each face of all three TMDs, in addition to the N-terminal periplasmic domain and the cytoplasmic C-terminal tail domain, all of which retained lytic function (Fig. 2 and Table 2). In order to have an isogenic nonlytic control, each Cys substitution was also introduced into S105C51S, A52V. In the wild-type (wt) and C51S context, the A52V change abrogates S105 hole formation and results in a holin protein that accumulates as a harmless, stable dimer (18). In each case, the resultant triple mutant, carrying C51S, A52V, and the particular Cys substitution, retained the nonlytic phenotype (data not shown). Table 2 shows a compilation of both the preexisting and new single-Cys alleles in the C51S context, along with their triggering times. All the alleles exhibited normal accumulation of protein after induction, irrespective of lytic functionality (Fig. 3). Neither the functional nor the nonfunctional alleles exhibited detectable intermolecular disulfide bond formation in samples taken by TCA precipitation from induced cultures (data not shown). It should be noted that the nonfunctionality of some of the Cys substitutions reinforces the extraordinary phenotypic sensitivity of the S105 protein, and holins in general, to conservative substitutions throughout the TMDs (3, 18).
TABLE 2.
Amino acid changes in lytically functional single-Cys alleles and triggering times of the alleles
| Amino acid position | Amino acid residue or changea | Triggering time (min)b |
|---|---|---|
| 7 | H7C | 55 |
| TMD1 positions | ||
| 12 | A12C | 35 |
| 13 | I13C | 30 |
| 14 | L14C | 35 |
| 15 | A15C | 20 |
| 16 | A16C | 20 |
| 17 | K17C | 75 |
| 20 | G20C | 30 |
| 21 | I21C | 20 |
| 23 | A23C | 75 |
| 24 | I24C | 35 |
| 27 | F27C | 75 |
| 28 | A28C | 20 |
| TMD2 positions | ||
| 45 | V45C | 30 |
| 49 | T48C | 60 |
| 51 | C51 (parental) | 35 |
| 53 | I53C | 45 |
| 54 | I54C | 65 |
| 55 | A55C | 45 |
| 57 | F57C | 50 |
| TMD3 positions | ||
| 68 | S68C | 55 |
| 69 | S69C | 30 |
| 71 | L71C | 70 |
| 72 | A72C | 50 |
| 74 | I74C | 55 |
| 75 | T75C | 40 |
| 77 | V77C | 45 |
| 78 | F78C | 40 |
| 79 | I79C | 70 |
| 80 | G80C | 55 |
| 81 | Y81C | 75 |
| 95 | A95C | 30 |
| 108 | Stop108C | 30 |
All changes are missense changes, and all are in the context of the C51S substitution.
Triggering time in minutes after induction.
FIG 3.
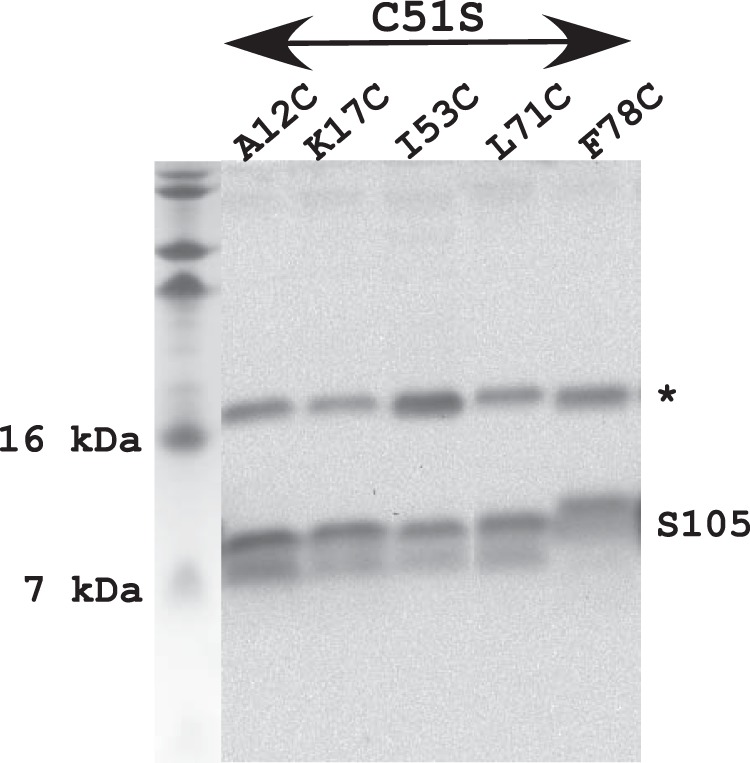
S105 Cys substitution variants accumulate normally. Lanes (from left): molecular mass marker, A12C variant, K17C variant, I53C variant, L71C variant, and F78C variant. The asterisk indicates a nonspecific band used as a loading control.
Analyzing the S hole by cysteine accessibility.
To probe the structure of the holin in the membrane, we used a variation of cysteine-scanning accessibility (17, 24). This approach involves treating whole cells expressing a particular S105 single-cysteine substitution allele with the membrane-impermeant thiol reagent IASD. After quenching the Cys modification reaction, cells are solubilized with SDS-urea, and the extent of IASD modification is assessed by derivatizing the remaining free cysteines with a high-molecular-mass (5-kDa) derivative of methoxy-PEG-maleimide. We first examined the accessibility of cysteines located in either the periplasmic loop or the cytoplasmic domain of S105C51S. As can be seen in Fig. 4A, the His7 position at the N terminus of S105C51S shows protection from PEGylation by IASD treatment, irrespective of being in the lytic (S105C51S) or nonlytic (S105C51S, A52V) context, as expected for the periplasmic location. In contrast, the protection of the cytoplasmic C-terminal positions (Ala94 and Cys108) is efficient only for the lytic S105C51S version but not for S105C51S, A52V, as expected, since IASD should pass freely through the micrometer-scale S105 holes (25). Moreover, depolarization of the membrane with DNP does not improve IASD labeling of the C-terminal positions in the A52V context, as expected, since this mutant is incapable of either premature or spontaneous triggering (data not shown). We conclude that the nonlytic A52V variant does not impair the impermeability of the cytoplasmic membrane to IASD. In addition, positions all around TMD1 (Leu14, Ala15, Ala23, and Ile24), TMD2 (Thr49, Cys51, lle53, lle54, and Ala55), and TMD3 (positions Ser68, Ile74, Phe78, and Tyr81) exhibit no IASD labeling in the context of this nonlytic allele. These data also confirm the previous mapping of the three TMDs (17).
FIG 4.

IASD protection analysis of S105. (A) Immunoblot analysis, as described in Materials and Methods. ‘‘PEG'' indicate the positions of the PEGylated species. IASD protection is indicated by an IASD-dependent decrease in the PEGylated species. (B) IASD-protected positions in TMD1, -2, and -3 map to the most hydrophilic face. Helical projections of all three TMDs are shown, with hydrophilic residues represented as circles, hydrophobic residues as diamonds, acidic residues as triangles, and basic residues as pentagons. Hydrophobicity is also color coded: the most hydrophobic residue is green, and the green becomes lighter with decreasing hydrophobicity, with zero hydrophobicity coded as yellow. The charged residues are light blue. The blue and red boxes indicate positions modified and not modified by IASD, respectively. The blue arcs indicate the lumen faces of TMD1 and TMD3.
The most hydrophilic faces of TMD1 and -3 line the S105 hole.
Since IASD penetrates the lesion formed by S105, it was practical to use the IASD modification method to determine which faces of the three S105 TMDs line the lumen of the hole. For TMD1, positions Ala12, Leu14, Ala15, and Ala23 all showed IASD protection from PEGylation to the same extent as the N-terminal periplasmic residues, whereas positions Ile13, Ala16, Lys17, Gly20, Ile21, Ile24, Phe27, and Ala28 exhibited no protection, despite the lytic functionality of each derivative (Fig. 4A). In helical projection, the IASD-accessible and -inaccessible positions form continuous arcs roughly equivalent to opposite faces of TMD1 (blue arcs in Fig. 4B). Similar clustering into continuous arcs of accessible and protected positions was observed for TMD3. In contrast, none of the seven positions tested in TMD2 exhibited IASD accessibility, irrespective of lytic function (Fig. 4A).
The simplest interpretation of these findings is that the S105 hole is lined by S105 molecules in which one face of TMD1 and one face of TMD3 are oriented toward the aqueous lumen, with the opposite faces and all of TMD2 embedded in the lipid or masked by a protein-protein interface. Moreover, although thiol accessibility is only a semiquantitative methodology, due to position effects of thiol reactivity to both IASD and PEGylation reagents, nevertheless, the extent of PEGylation and the difference in PEGylation between the IASD-treated and untreated samples were nearly identical for the putative lumen-facing positions and the periplasmically localized N-terminal position (His7). This indicates that nearly all of the S105 molecules in the cell are actually involved in the formation of hole walls.
DISCUSSION
The results presented here address for the first time the molecular structure of the fatal membrane lesion, or hole, formed by the lambda holin, S105. There are several important lessons. The first is that essentially all of the ∼103 holin molecules end up facing the aqueous lumen of the hole; in other words, the S105 molecules line the walls of the holes. Second, two of the three TMDs of S105, TMD1 and TMD3, face the lumen of the hole. These conclusions enable us to leverage previous findings from cryo-electron microscopy and tomography, where it was directly observed that, after triggering, S105 forms large (micrometer-scale) interruptions in the cytoplasmic membrane (6). The images could not distinguish between the simplest alternatives for the nature of the interruptions, which would be either large aqueous channels or massive two-dimensional S105 aggregates, or perhaps intermediate “Swiss cheese” structures with multiple channels embedded within the aggregates. Moreover, there was no information about how much of the holin population actually participated in hole formation. The results described here strongly favor the simplest and most satisfying model, where all the holins end up as units of the hole wall. The results are quantitatively consistent with this perspective: 1 to 3 holes with an average 340-nm diameter per cell would mean 1 to 3 μm of hole wall perimeter. An S105 holin with two TMDs facing the lumen would occupy ∼2 nm of wall perimeter, which is consistent with the ∼103 S105 molecules per cell estimated from comparative Western blotting. A corollary is that no host protein would be required in the final structure of the hole, a notion supported by cross-linking studies showing that S forms regular oligomers and, parenthetically, by the fact that S105 also forms lethal holes in yeast and mammalian cells (18, 26, 27).
These are the first data that address the structure of the holes and may provide a basis for modeling a pathway to hole formation. Cross-linking studies indicate that the homotypic interface in the homodimer that dominates the untriggered membrane involves the face of TMD2 occupied by Cys51. If it is assumed that the hydrophilic faces of TMD1 and TMD3 that eventually face the lumen of the hole must be sequestered from the lipid in the homodimer, the simplest model would have these faces sequestered intramolecularly (Fig. 5). The current understanding of holin function is that massive aggregation occurs after the holin, or rather the holin homodimer, reaches a critical (two-dimensional) concentration, akin to the transition to purple membrane formation when bacteriorhodopsin is induced (4, 28). One effect of this aggregation could be to bring the intramolecularly sequestered hydrophilic surfaces into adjacency, which might make a transition to intermolecular sequestration feasible. Hydration of these intermolecular interfaces by water molecules from either the periplasm or the cytoplasm could provide the driving force for a concerted reorganization of the aggregate into the macroscopic holes. The model in Fig. 5 makes specific predictions about heterotypic intermolecular and intramolecular interfaces in the final hole; these predictions can be tested by cysteine cross-linking studies, as was done for the TMD-TMD interactions of the LacY permease (29).
FIG 5.
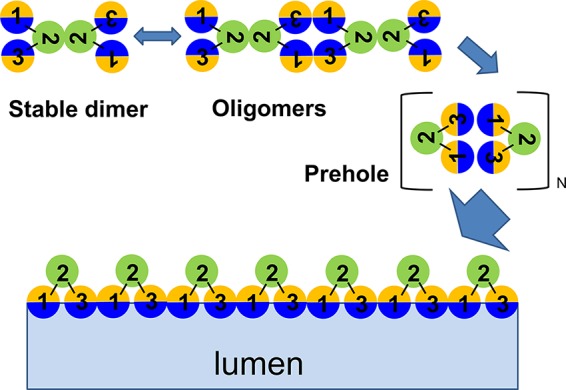
General model of the S105 hole formation pathway. A top-down view of a region of the cytoplasmic membrane is shown; each circle represents a helical TMD. At least four steps, described in the text, are required for hole formation: first, stable dimerization of λ S; second, oligomerization; third, raft formation and helical-face rearrangements; fourth, TMD1 and TMD3 lining the lumen of the hole. The three TMDs (green, TMD2; sectored, TMD1 and TMD3) in a single S molecule are outlined. TMD1 and TMD3 contain two interacting faces, hydrophilic (the face that is also accessible to IASD) and hydrophobic, represented by blue and orange, respectively.
This perspective is reinforced by parallels with the current model for the pathway to pinholin-mediated lysis. Pinholins are a distinct class of holins that mimic nearly every aspect of the canonical holin function, including accumulating as a harmless dimer and then undergoing triggering. The prototype pinholin, S2168, has class II topology (2 TMDs; N in and C in) (Fig. 1). For S2168, the product of triggering is not a few micrometer-scale holes, as in the case of the canonical holins, but instead, ∼103 heptameric “pinholes,” in each of which a single TMD, TMD2, lines a lumen estimated to be ∼2 nm in diameter (30). As shown here for TMD1 and TMD3 of S105, Cys-scanning accessibility studies indicated that in the case of S2168 as well, the most hydrophilic face of TMD2 faces the lumen. Genetic analysis also suggests that during the pathway to triggering, this face is sequestered, first intramolecularly and then intermolecularly (31, 32).
For both canonical holins and pinholins, the membrane potential plays a determinative role in the lysis pathway. Obviously, in the case of either the formation of a few micrometer-scale holes by the canonical holins or of ∼103 pinholes by the pinholin, the cytoplasmic membrane would be completely depolarized. However, the key physiological feature of the holin lysis timing program is that artificial depolarization of the membrane causes premature triggering. By extrapolation, we have suggested that in the normal pathway, a spontaneous depolarization event provides the free energy for the massive quaternary rearrangements needed for hole formation (7, 28). In the current model, the notion is that once a critical concentration of holin dimer is reached, large two-dimensional aggregates, designated rafts, are nucleated and form rapidly (4, 7, 28). These raft prehole structures are proposed to be dominated by intimated TMD-TMD helical packing and are thus depleted in lipid, providing the basis for a local collapse of the membrane potential. It seems likely that the primary effect of the loss of polarization may be on the orientation of the TMD helices in the bilayer, perhaps favoring a transition to the intermolecular sequestration of the hydrophilic faces or the rearrangement into the hole wall arrangement (Fig. 5).
Finally, taken together, the results presented here and the previous analysis of the pinholin pathway suggest that it may be feasible to predict the lumenal faces of other holins. Recently, another holin, P2 Y, although lacking detectable similarity to S105, has been shown to adapt the same topology and follow the same general pathway, including triggering to form micrometer-scale holes (9). It also has two TMDs that have faces with widely disparate hydrophobic characters, but in this case, they are TMD2 and TMD3 rather than TMD1 and TMD3, as in S105. Experiments are under way to test this prediction by cysteine-scanning accessibility.
ACKNOWLEDGMENTS
We thank the Young laboratory members, past and present, for their helpful criticisms and suggestions.
This work was supported by Public Health Service grant GM27099 to R.Y.
Footnotes
Published ahead of print 4 August 2014
REFERENCES
- 1.Wang IN, Smith DL, Young R. 2000. Holins: the protein clocks of bacteriophage infections. Annu. Rev. Microbiol. 54:799–825. 10.1146/annurev.micro.54.1.799. [DOI] [PubMed] [Google Scholar]
- 2.Young R, Wang IN. 2006. Phage lysis, p 104–126 In Calendar R. (ed), The bacteriophages, 2nd ed. Oxford University Press, Oxford, United Kingdom. [Google Scholar]
- 3.Young R. 2002. Bacteriophage holins: deadly diversity. J. Mol. Microbiol. Biotechnol. 4:21–36. [PubMed] [Google Scholar]
- 4.Young R. 2013. Phage lysis: do we have the hole story yet? Curr. Opin. Microbiol. 16:790–797. 10.1016/j.mib.2013.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gründling A, Manson MD, Young R. 2001. Holins kill without warning. Proc. Natl. Acad. Sci. U. S. A. 98:9348–9352. 10.1073/pnas.151247598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dewey JS, Savva CG, White RL, Vitha S, Holzenburg A, Young R. 2010. Micron-scale holes terminate the phage infection cycle. Proc. Natl. Acad. Sci. U. S. A. 107:2219–2223. 10.1073/pnas.0914030107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang IN, Deaton JF, Young R. 2003. Sizing the holin lesion with an endolysin-β-galactosidase fusion. J. Bacteriol. 185:779–787. 10.1128/JB.185.3.779-787.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Young R. 1992. Bacteriophage lysis: mechanism and regulation. Microbiol. Rev. 56:430–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.To KH, Dewey J, Weaver J, Park T, Young R. 2013. Functional analysis of a class I holin, p2 y. J. Bacteriol. 195:1346–1355. 10.1128/JB.01986-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ramanculov E, Young R. 2001. Functional analysis of the phage T4 holin in a lambda context. Mol. Genet. Genomics 265:345–353. 10.1007/s004380000422. [DOI] [PubMed] [Google Scholar]
- 11.Savva CG, Dewey JS, Moussa SH, To KH, Holzenburg A, Young R. 2014. Stable micron-scale holes are a general feature of canonical holins. Mol. Microbiol. 91:57–65. 10.1111/mmi.12439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gründling A, Smith DL, Bläsi U, Young R. 2000. Dimerization between the holin and holin inhibitor of phage lambda. J. Bacteriol. 182:6075–6081. 10.1128/JB.182.21.6075-6081.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Raab R, Neal G, Sohaskey C, Smith J, Young R. 1988. Dominance in lambda S mutations and evidence for translational control. J. Mol. Biol. 199:95–105. 10.1016/0022-2836(88)90381-6. [DOI] [PubMed] [Google Scholar]
- 14.Smith DL. 1998. Purification and biochemical characterization of the bacteriophage lambda holin. Ph.D. dissertationTexas A&M University, College Station, TX. [Google Scholar]
- 15.Smith DL, Young R. 1998. Oligohistidine tag mutagenesis of the lambda holin gene. J. Bacteriol. 180:4199–4211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang CY, Nam K, Young R. 1995. S gene expression and the timing of lysis by bacteriophage lambda. J. Bacteriol. 177:3283–3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gründling A, Blasi U, Young R. 2000. Biochemical and genetic evidence for three transmembrane domains in the class I holin, lambda S. J. Biol. Chem. 275:769–776. 10.1074/jbc.275.2.769. [DOI] [PubMed] [Google Scholar]
- 18.Gründling A, Blasi U, Young R. 2000. Genetic and biochemical analysis of dimer and oligomer interactions of the lambda S holin. J. Bacteriol. 182:6082–6090. 10.1128/JB.182.21.6082-6090.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu J, Deutsch C. 2001. Pegylation: a method for assessing topological accessibilities in Kv1.3. Biochemistry 40:13288–13301. 10.1021/bi0107647. [DOI] [PubMed] [Google Scholar]
- 20.Tran TA, Struck DK, Young R. 2007. The T4 RI antiholin has an N-terminal signal anchor release domain that targets it for degradation by DegP. J. Bacteriol. 189:7618–7625. 10.1128/JB.00854-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park T, Struck DK, Deaton JF, Young R. 2006. Topological dynamics of holins in programmed bacterial lysis. Proc. Natl. Acad. Sci. U. S. A. 103:19713–19718. 10.1073/pnas.0600943103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barenboim M, Chang CY, dib Hajj F, Young R. 1999. Characterization of the dual start motif of a class II holin gene. Mol. Microbiol. 32:715–727. 10.1046/j.1365-2958.1999.01385.x. [DOI] [PubMed] [Google Scholar]
- 23.Johnson-Boaz R, Chang CY, Young R. 1994. A dominant mutation in the bacteriophage lambda S gene causes premature lysis and an absolute defective plating phenotype. Mol. Microbiol. 13:495–504. 10.1111/j.1365-2958.1994.tb00444.x. [DOI] [PubMed] [Google Scholar]
- 24.Sun J, Kaback HR. 1997. Proximity of periplasmic loops in the lactose permease of Escherichia coli determined by site-directed cross-linking. Biochemistry 36:11959–11965. 10.1021/bi971172k. [DOI] [PubMed] [Google Scholar]
- 25.Akabas MH, Stauffer DA, Xu M, Karlin A. 1992. Acetylcholine receptor channel structure probed in cysteine-substitution mutants. Science 258:307–310. 10.1126/science.1384130. [DOI] [PubMed] [Google Scholar]
- 26.Garrett J, Bruno C, Young R. 1990. Lysis protein S of phage lambda functions in Saccharomyces cerevisiae. J. Bacteriol. 172:7275–7277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Agu CA, Klein R, Lengler J, Schilcher F, Gregor W, Peterbauer T, Blasi U, Salmons B, Gunzburg WH, Hohenadl C. 2007. Bacteriophage-encoded toxins: the lambda-holin protein causes caspase-independent non-apoptotic cell death of eukaryotic cells. Cell Microbiol. 9:1753–1765. 10.1111/j.1462-5822.2007.00911.x. [DOI] [PubMed] [Google Scholar]
- 28.White R, Chiba S, Pang T, Dewey JS, Savva CG, Holzenburg A, Pogliano K, Young R. 2011. Holin triggering in real time. Proc. Natl. Acad. Sci. U. S. A. 108:798–803. 10.1073/pnas.1011921108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaback HR, Sahin-Toth M, Weinglass AB. 2001. The kamikaze approach to membrane transport. Nat. Rev. Mol. Cell. Biol. 2:610–620. 10.1038/35085077. [DOI] [PubMed] [Google Scholar]
- 30.Pang T, Savva CG, Fleming KG, Struck DK, Young R. 2009. Structure of the lethal phage pinhole. Proc. Natl. Acad. Sci. U. S. A. 106:18966–18971. 10.1073/pnas.0907941106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pang T, Park T, Young R. 2010. Mutational analysis of the S21 pinholin. Mol. Microbiol. 76:68–77. 10.1111/j.1365-2958.2010.07080.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pang T, Fleming TC, Pogliano K, Young R. 2013. Visualization of pinholin lesions in vivo. Proc. Natl. Acad. Sci. U. S. A. 110:E2054–E2063. 10.1073/pnas.1222283110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schweizer HP. 1990. The pUC18CM plasmids: a chloramphenicol resistance gene cassette for site-directed insertion and deletion mutagenesis in Escherichia coli. Biotechniques 8:612–613, 616. [PubMed] [Google Scholar]