

Abstract
A newly recognized Serratia species, termed South African Caenorhabditis briggsae isolate (SCBI), is both a mutualist of the nematode Caenorhabditis briggsae KT0001 and a pathogen of lepidopteran insects. Serratia sp. strain SCBI displays high proteolytic activity, and because secreted proteases are known virulence factors for many pathogens, the purpose of this study was to identify genes essential for extracellular protease activity in Serratia sp. strain SCBI and to determine what role proteases play in insect pathogenesis and cytotoxicity. A bank of 2,100 transposon mutants was generated, and six SCBI mutants with defective proteolytic activity were identified. These mutants were also defective in cytotoxicity. The mutants were found defective in genes encoding the following proteins: alkaline metalloprotease secretion protein AprE, a BglB family transcriptional antiterminator, an inosine/xanthosine triphosphatase, GidA, a methyl-accepting chemotaxis protein, and a PIN domain protein. Gene expression analysis on these six mutants showed significant downregulation in mRNA levels of several different types of predicted protease genes. In addition, transcriptome sequencing (RNA-seq) analysis provided insight into how inactivation of AprE, GidA, and a PIN domain protein influences motility and virulence, as well as protease activity. Using quantitative reverse transcription-PCR (qRT-PCR) to further characterize expression of predicted protease genes in wild-type Serratia sp. SCBI, the highest mRNA levels for the alkaline metalloprotease genes (termed prtA1 to prtA4) occurred following the death of an insect host, while two serine protease and two metalloprotease genes had their highest mRNA levels during active infection. Overall, these results indicate that proteolytic activity is essential for cytotoxicity in Serratia sp. SCBI and that its regulation appears to be highly complex.
INTRODUCTION
Members of the genus Serratia are found widespread around the globe and are well-known for their roles as insect pathogens (1, 2). A newly recognized Serratia species, termed South African Caenorhabditis briggsae isolate (SCBI), was identified following its isolation from the nematode C. briggsae KT0001 (3). These C. briggsae KT0001 nematodes were recovered from soil samples through Galleria mellonella bait traps in three provinces in South Africa (3). While Serratia sp. strain SCBI is nonpathogenic to Caenorhabditis nematodes, these bacteria are lethal to G. mellonella and the tobacco hornworm, Manduca sexta (4). When injected into the hemocoel of either species in numbers less than 1,000 CFU, larvae die within 72 h.
A hallmark of Serratia spp. is their ability to produce and secrete a variety of enzymes into the external milieu. Expression and secretion of these exoenzymes, which includes proteases, lipases, DNases, and chitinases, are usually growth phase dependent, with activities not seen until late-exponential or stationary-phase growth (5–8). In addition, expression of these exoenzymes is largely regulated by the substrate upon which they degrade (9–11). The plethora of extracellular proteins produced by Serratia spp. allow for invasion and colonization of a wide number of habitats, thereby contributing directly or indirectly to virulence against a broad host range. In particular, protease activity has been recognized as a virulence factor in the opportunistic pathogen Serratia marcescens, with proteases actively contributing to both insect and human pathogenesis (12, 13).
S. marcescens is capable of secreting multiple kinds of proteases, yet the majority of activity is due to a 56-kDa metalloprotease termed PrtA, or serralysin (14, 15). Secreted by a typical ABC transport system termed LipBCD (16, 17), PrtA causes a variety of pathogenic effects. When cultured, keratitis-causing Serratia spp. produce up to 10× more proteases and cause more-severe lesions than isolates that exhibit less proteolytic activity. The severity of these infections is directly correlated with PrtA levels (18). On a molecular level, PrtA enhances vascular permeability through activation of the Hageman factor/kallikrein-kinin system (19–21). PrtA degrades various protease inhibitors and crucial components of the mammalian host complement system in human plasma, reducing the ability of the host to clear pathogens (15, 22–24). PrtA also destroys immunoglobulin (IgG and IgA) by hydrolyzing the heavy chains of these immunoglobulins near the hinge region (15, 25). In human lung squamous cell carcinoma EBC-1 cells, PrtA induces an inflammatory response through the activation of a protease-activated receptor 2, inducing interleukin-6 and interleukin-8 expression (26).
Protease activity in Serratia has also been linked with invasion and destruction of various mammalian cell lines. Incubation of fibroblast cells with purified S. marcescens PrtA results in the destruction of more than 50% of cells within 1 h (15). Mutant strains lacking the 56-kDa metalloprotease are no longer cytotoxic toward HeLa cells (27). Proteases found in Serratia proteamaculans and Serratia grimesii also have cytotoxic properties. S. proteamaculans strain 94 produces a 32-kDa thermostable protealysin that is able of cleaving filamentous actin and matrix metalloprotease MMP2 in human larynx carcinoma HEp-2 cells (28–30). Additionally, S. proteamaculans strain 94 is able to infect HEp-2 cells and was retained within approximately 10% of cells. This was the first finding that any S. proteamaculans strain was capable of eukaryotic cell invasion. Similarly, S. grimesii produces grimelysin, a novel metalloprotease, which has specific actin-hydrolyzing activity and mediates HEp-2 cell invasion (31).
While the genes responsible for protease activity and secretion have been elucidated in S. marcescens, the mechanisms regulating the expression of PrtA and other proteases are not well understood. There is very little knowledge of the transcription factors involved in the regulation of protease production. However, lipB (the ATP-binding component of the LipBCD transporter) expression is under the control of the swr quorum-sensing system in S. marcescens (32). In addition, the catabolite regulation protein (CRP) of S. marcescens is an indirect regulator of PrtA, and its inactivation results in increased proteolytic activity (33). CRP acts as a global regulator, and its activity is influenced by the intracellular cyclic AMP (cAMP) concentration, which in turn is regulated by the level of intracellular glucose. Besides affecting protease activity, the role of cAMP-CRP has been linked to chitinase and phospholipase activities, as well as pilus and flagellum production (33, 34).
Similar to other Serratia spp., Serratia sp. strain SCBI has protease, lipase, DNase, alkaline phosphatase, and chitinase activities, and is cytotoxic toward the mammalian Buffalo green monkey kidney (BGMK) cell line (4). The Serratia sp. strain SCBI genome contains a high number of protease genes, including four PrtA homologues, at least two extracellular serine proteases, and two or more extracellular metalloproteases (F. Abebe-Akele, L. S. Tisa, V. S. Cooper, P. J. Hatcher, E. Abebe, and W. K. Thomas, submitted for publication). The goal of this study was to determine whether or not protease activity was important in insect pathogenesis and cytotoxicity in Serratia sp. SCBI. The first step was to identify mutants defective in proteolytic activity by screening a miniHimar RB1 transposon mutant library on skim milk agar plates. Six mutants were identified, the point of transposon insertion was determined, and these mutants were assayed for cytotoxicity, virulence, motility, and hemolysis. For several of these mutants with significantly altered virulence, their transcriptomes were analyzed by transcriptome sequencing (RNA-seq). To gain further insight into whether protease activity may play a role in insect pathogenesis and cytotoxicity, quantitative reverse transcription-PCR (qRT-PCR) was utilized to ascertain the conditions under which expression of eight predicted protease genes was optimal in Serratia sp. SCBI.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
A complete list of all bacterial strains and plasmids used in this study is given in Table 1. Bacteria were grown overnight at 37°C in LB medium (1% tryptone, 0.5% yeast extract, 1% NaCl) with appropriate antibiotics when required.
TABLE 1.
Bacterial strains and plasmids used in this study
| Bacterial strain or plasmid | Description or purpose | Reference or source |
|---|---|---|
| Serratia sp. strains | ||
| SCBI | Wild type | 3 |
| 16-D2 | Transposon mutant with disrupted aprE | This study |
| 22-A7 | Transposon mutant with disrupted bglB | This study |
| 22-B9 | Transposon mutant with disrupted inosine/xanthosine triphosphatase | This study |
| 22-C7 | Transposon mutant with disrupted gidA | This study |
| 22-D7 | Transposon mutant with disrupted MCP | This study |
| 22-F6 | Transposon mutant with disrupted PIN protein | This study |
| E. coli strains | ||
| S17-1 λpir | Tpr Smr recA thi pro hsdS(r− m+) RP4::2-Tc::Mu::Km Tn7 λpir; donor strain for transposon mutagenesis | 64 |
| DH5α λpir | supE44 ΔlacU169 (ϕlacZΔM15) recA1 endA1 hsdR17 thi-1 gyrA96 relA1; λpir phage lysogen; host strain for gene complementation | 65 |
| Plasmids | ||
| pMiniHimar RB1 | Transposon mutagenesis | 35 |
| pCR2.1-TOPO | TOPO cloning | Invitrogen |
| pBAD33-Cm | Expression vector | 37 |
| pBAD33-AprE | pBAD33 vector containing cloned aprE plus tolC | This study |
| pBAD33-GidA | pBAD33 vector containing cloned gidA | This study |
| pBAD33-MCP | pBAD33 vector containing cloned gene (SCBI_0077) coding for MCP | This study |
| pBAD33-PIN | pBAD33 vector containing cloned gene (SCBI_0044) coding for the PIN protein | This study |
Cell culture growth conditions.
Buffalo green monkey kidney (BGMK) cells were grown and maintained at 37°C in minimum essential medium (Flow Laboratories) supplemented with medium 199 (Sigma), 10% fetal bovine serum (FBS), 16 mM HEPES buffer, 12 mM NaHCO3, 2 mM l-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin, pH 7.3.
Construction of Serratia sp. strain SCBI transposon mutant library.
Serratia sp. strain SCBI was mutagenized by the use of the MiniHimar transposon RB1 (35). Both the donor (Escherichia coli S17-1 λpir/pMiniHimar RB1) and recipient (Serratia sp. SCBI) were grown overnight in LB medium containing appropriate antibiotics (30 μg/ml kanamycin for the donor and 10 μg/ml polymyxin B sulfate for the recipient). One milliliter of each culture was centrifuged at 10,000 × g for 2 min, and the pellet was washed twice with phosphate-buffered saline (PBS) before 20 μl of a 1:1 cell mixture was spotted onto the center of an LB agar plate and incubated at 37°C for 24 h. Following incubation, the cell mixture was resuspended in 1 ml PBS, and transconjugants were selected by overnight growth of 100-μl aliquots from 10−5 and 10−6 dilutions on LB agar containing 100 μg/ml kanamycin and 30 μg/ml polymyxin B sulfate at 37°C. A total of 2,100 isolated transconjugant colonies were selected and grown at 37°C overnight in LB medium containing 40 μg/ml kanamycin and 10 μg/ml polymyxin B sulfate in 96-well plates before addition of an equal volume of 60% glycerol and storage at −80°C for future use.
Molecular analysis of transposon mutants.
Genomic DNA (gDNA) from each clone was prepared by the standard chloroform-isoamyl alcohol extraction technique. The gDNA was digested with NsiI (New England BioLabs), which does not cut the transposon, followed by ligation with T4 ligase (New England BioLabs). The ligated DNA was electroporated into E. coli DH5α λpir cells, and transformants were selected on LB plates containing 25 μg/ml kanamycin. The transposon-carrying plasmids were isolated and sequenced using the transposon-specific primers Himar1 (5′-CAT TTA ATA CTA GCG ACG CCA TCT-3′) and 615 (5′-TCG GGT ATC GCT CTT GAA GGG-3′). Sequences were compared to the Serratia sp. SCBI genome using BLASTN.
Protease assay.
Proteolytic activity was determined by a liquid azocasein assay described previously (4). Briefly, cultures were grown overnight at 28°C, which is the optimal temperature for protease activity in Serratia sp. SCBI (4), diluted to an optical density at 600 nm (OD600) of 2.0, and centrifuged for 5 min at 16,060 × g. An aliquot of 500 μl of the resulting supernatant was mixed with 500 μl of azocasein solution (2% azocasein, 50 mM Tris-HCl) and incubated for 1 h at 37°C with gentle agitation. An aliquot of 500 μl of a 10% trichloroacetic acid solution was added to each sample, shaken vigorously, and centrifuged for 10 min at 12,000 × g. The A450 of the resulting supernatant was determined on a spectrophotometer to measure the amount of released substrate. The following equation was used to calculate the units of enzymatic activity (U) for the protease assay: U = ΔAbs/g substrate/ml/h where Abs is absorbance. Activity was standardized as units per 109 cells. Cell numbers were determined by the OD600 reading of the culture prior to centrifugation. An OD600 of 1.0 represents 2 × 109 cells/ml.
To test the inhibitory effect of protease inhibitors, 50 mM EDTA and/or 1 mM phenylmethanesulfonyl fluoride (PMSF) were added to appropriate azocasein-supernatant samples.
Screening for mutants deficient in protease activity.
Individual colonies of transposon-induced mutants of Serratia sp. strain SCBI were screened for protease by the use of a skim milk plate assay. Skim milk agar (1% tryptone, 0.5% yeast extract, 1% NaCl, 3% powdered skim milk) was poured into oversized petri dishes (150 by 15 mm; Fisher Scientific, Canada). Using a 96-well replicator, the transposants stored at −80°C were transferred into new 96-well microtiter plates containing fresh LB medium containing 25 μg/ml kanamycin and incubated overnight at 37°C. The freshly grown overnight culture was replica plated directly into skim milk medium, and the plates were incubated for 48 h at 28°C. Mutants with loss or reduction in protease activity were confirmed by the use of a liquid azocasein assay as described above.
Insect viability assay.
Serratia sp. SCBI miniHimar RB1 mutants defective in protease activity were analyzed for their ability to kill third-instar M. sexta larvae using methods described previously (4). Briefly, overnight bacterial cultures of mutants were initially diluted to an OD600 of 1.0 in LB medium. The bacterial culture was further diluted severalfold to obtain an average CFU value of 2 × 103 for each mutant. For each diluted sample, 10 μl was directly injected into the insect hemocoel by use of a sterilized Hamilton syringe. Wild-type Serratia sp. SCBI and E. coli EPI300 were used as controls. For all of the samples, including the controls, 10 larvae were used. The larvae were held individually with food for up to 14 days at 37°C under a 16-h light/8-h dark cycle. This temperature (37°C) provides the optimal rate of killing by Serratia sp. SCBI without any significant impact on larval health (4). The larvae were monitored for insect mortality or delays in insect development during that time period. The insect viability assay was repeated a total of three times (in total, 30 larva were injected per bacterial culture; 10 larva each replicate). Mortality was measured as the percentage of larvae killed by the bacteria relative to the total number of larvae injected per sample. From these data, the LT50 values were calculated and defined as the average time required for 50% of the population to die from infection.
Cytotoxicity assay.
Cytotoxic activity of bacterial culture supernatant was determined on BGMK cells as described previously (4). Briefly, cell viability assays were carried out in 96-well flat-bottom plastic microplates. BGMK cells were seeded at a density of 5 × 103 cells per well and incubated at 37°C in growth medium overnight. Bacteria were grown overnight in BGMK growth medium at 28°C and centrifuged for 10 min at 16,060 × g, and the resulting supernatant was filter sterilized. The supernatant samples were assayed for protein concentration using the bicinchoninic acid (BCA) assay (Promega) and diluted to 2 mg/ml total protein. Growth medium was removed from the BGMK cells and replaced with 2 mg/ml bacterial supernatant, and BGMK cells were incubated for 24 h at 37°C. For the positive control, 2% SDS was used, while the growth medium was used as a negative control. After 24 h, the liquid medium was removed from all wells, and 100 μl of BGMK medium containing 0.25 mg/ml 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was added to each well. The cells were incubated for 4 h at 37°C during which time yellow MTT was reduced to purple formazan in the mitochondria of living BGMK cells. The colored formazan product was dissolved in 150 μl dimethyl sulfoxide (DMSO) for 15 min at 37°C with gentle shaking. Each sample was assayed in triplicate. The conversion of MTT to formazan was quantified as a change in A562. The percent viability was determined by dividing the average A562 for a given sample by the average of the negative-control samples.
Hemolysis assay.
Rates of hemolytic activity were measured by the use of a liquid assay described previously (4). Serratia sp. SCBI demonstrates the same rate of hemolytic activity at 28°C and 37°C; therefore, this assay can be performed at either temperature. Briefly, sheep red blood cells (SRBCs) were washed three times in phosphate-buffered saline (PBS) and suspended to a final concentration of 10% (vol/vol) in PBS. Overnight bacterial cultures were diluted to an OD600 of 0.005 (an estimated 4.0 × 106 CFU) in LB medium. For each culture condition, 100 μl of the diluted bacterial culture was removed and mixed with 1 ml washed SRBCs. The mixture was incubated at 28°C or 37°C for 4 h with gentle agitation. Every hour, 125-μl samples were removed and centrifuged at 16,060 × g for 1 min to removed unlysed SRBCs. The resulting supernatant was diluted 1:10 in distilled water (dH2O), and the A405 was determined with a spectrophotometer to measure the amount of released hemoglobin. For a positive control, 1 ml of SRBCs was lysed by addition of 100 μl of 20% sodium dodecyl sulfate (SDS) and incubated at the test temperature of 28°C for 15 min. For negative controls, 100 μl of LB medium was added to 1 ml washed SRBCs incubated at the test temperature of 28°C, and samples were treated as described above. The following equation was used to calculate hemolytic units (HU): HU = [(A405 of sample – A405 of negative control)/A405 of positive control] × 100.
Motility assay.
Swimming and swarming motility was assayed as described previously (4, 36). For swimming, the plates were incubated for 8 h at 37°C, the optimal temperature for swimming motility (4), and the swim ring diameter was measured. For swarming, the plates were incubated for 18 to 42 h at the test temperature (22°C or 28°C), and the swarm ring diameter was measured. The optimal rate of swarming by Serratia sp. SCBI is 28°C (4), and therefore, this temperature was used for the majority of swarm assays. However, the test temperature of 22°C was necessary to assay swarming motility by mutant 16-D2 due to the increased speed at which it moved at 28°C.
Statistical analysis.
Data were analyzed by one-way analysis of variance using JMP 10 software (SDS Institute, Inc.). Student's t test provided comparisons of means.
Genetic complementation.
For genetic complementation analysis, the wild-type genes were amplified with primers listed in the supplemental material (see Table S1 in the supplemental material), a template of 100 ng Serratia sp. SCBI gDNA, and OneTaq hot start DNA polymerase (New England BioLabs). The PCR program was as follows: (i) an initial denaturation step of 30 s at 94°C; (ii) 30 cycles, with 1 cycle consisting of 30 s at 94°C, 30 s at 50°C, and 3 min at 68°C; (iii) a final extension step of 10 min at 68°C. PCR products were cleaned up using the QIAquick PCR purification kit (Qiagen) according to the manufacturer's instructions. Approximately 100 ng of cleaned-up PCR product was cloned into vector pCR2.1-TOPO using the TOPO TA Cloning kit (Invitrogen) following the manufacturer's instructions. Invitrogen primers M13 Forward (−20) and M13 Reverse were used to determine the orientation of gene insertion. Depending on the orientation of the insertion in pCR2.1-TOPO, plasmid DNA was cut either with XbaI and SacI or with XbaI and HindIII (New England BioLabs). Digested DNA was ligated into pBAD33Cm (37), which had been cut with the same restriction enzymes. The vector was subsequently renamed pBAD33-AprE, pBAD33-GidA, pBAD33-MCP, or pBAD33-PIN, depending on which gene(s) it carried. The pBAD33-AprE construction also contained the tolC gene. These constructs were electroporated into E. coli DH5α λpir, and selection was made on LB plates containing 25 μg/ml chloramphenicol. Plasmids were extracted from successfully transformed E. coli DH5α λpir and confirmed by restriction analysis and sequencing. Confirmed constructs were introduced into both the appropriate mutant and wild-type Serratia sp. strain SCBI and were selected on LB plates containing 150 μg/ml chloramphenicol.
RNA extraction and cDNA synthesis.
To analyze gene expression during stationary growth phase, the cultures were grown at 22, 28, or 37°C for 18 h, diluted to an OD600 of 0.6, centrifuged at 16,060 × g for 5 min, and treated with RNAprotect. To analyze gene expression during exponential growth phase, the cultures were grown at 22, 28, or 37°C until they reached an OD600 of 0.6 (∼3 h), centrifuged at 16,060 × g for 5 min, and treated with RNAprotect. To analyze expression from bacterial cells during hemolysis, the protocol for the hemolysis assay (described above) was followed with the modification that following 3 h of incubation, a 2-ml aliquot (SRBCs plus bacterial cells) was collected, centrifuged at 16,060 × g for 5 min, and treated with RNAprotect. To obtain cells during infection of M. sexta, the insect viability assay protocol (described above) was followed; however, after 24 h of incubation at 37°C (while larvae were still alive) or after 48 h of incubation (soon after larvae have died), 100 μl of hemolymph was collected by cutting a proleg of the larva or removing the head, being careful not to cut the gut. The hemolymph was mixed with 100 μl PBS containing 20 mg/ml l-cysteine, passed through a Qiashredder homogenizer (Qiagen), and treated with RNAprotect. All samples were frozen overnight at −80°C. RNA extraction was performed using the RNeasy minikit (Qiagen) following the manufacturer's instructions, followed by treatment with DNase (New England BioLabs). Four micrograms of RNA was transcribed into cDNA using GoScript reverse transcriptase (Promega) following the manufacturer's instructions, quantified with a Qubit 2.0 fluorometer (Invitrogen), and diluted to 10 ng/μl.
Analysis of gene expression with quantitative RT-PCR.
Amplification and detection of gene expression were performed using the Stratagene Mx3000P QPCR system (Agilent Technologies). The primers used for these experiments are listed in the supplemental material (see Table S1 in the supplemental material). Each primer sequence was blasted against the Serratia sp. SCBI genome to ensure specificity to the target gene. Standard curves were generated using Serratia sp. SCBI genomic DNA and each primer set to test primer efficiency before use. The rplU gene was used as the normalizer for all qRT-PCR experiments. The RT-PCRs were done using 50 ng template cDNA, SYBR green PCR master mix (Applied Biosystems), and primer mix (0.3 μM) in a 25-μl reaction mixture. The following thermal cycler parameters were used: (i) 15 min at 95°C; (ii) 40 cycles, with 1 cycle consisting of 15 s at 95°C and 30 s at 60°C; and (iii) 1 thermal disassociation cycle of 1 min at 95°C and 30 s at 55°C for 30 s and incremental increases in temperature to 95°C for 30 s. Reactions were performed in triplicate, and the comparative threshold cycle method was used to quantify gene expression.
RNA-seq sample preparation.
To analyze gene expression during stationary growth phase, cultures were grown at 28°C for 18 h, diluted to an OD600 of 0.6, centrifuged at 16,060 × g for 5 min, and treated with RNAprotect. All samples were frozen overnight at −80°C. RNA extraction was performed using the RNeasy minikit (Qiagen) following the manufacturer's instructions, followed by treatment with DNase (New England BioLabs). RNA quality was evaluated on a BioAnalyzer RNA nano chip (Agilent). rRNA was removed using the MICROBExpress bacterial mRNA enrichment kit (Ambion) following the manufacturer's instructions. RNA cleanup was done using the MEGAclear kit (Ambion) following the manufacturer's instructions. cDNA libraries were prepared using the TruSeq RNA sample preparation kit (Illumina) following the manufacturer's instructions. The cDNA library was verified for appropriate fragment size (200 to 300 bp) on a BioAnalyzer DNA chip (Agilent). Samples were amplified onto flow cells using an Illumina cBot and sequenced on an Illumina HiSeq2000.
RNA-seq data analysis.
Sequencing reads were processed using CLC Genomics Workbench version 6.5.1 (CLC bio) using the default settings for prokaryotic genomes. Reads were transformed and normalized using square root transformation and quantile normalization, respectively. Reads were mapped to a version of the Serratia sp. SCBI genome that had all rRNA genes removed with the limit for removal of low-quality sequences set at 0.05 and a maximum mismatch of 2 nucleotides allowed. Statistical analysis was done on proportions using false discovery rate (FDR) correction to compare data sets.
Accession numbers.
The genome sequence and its annotations are available at NCBI under GenBank accession numbers CP003424 and CP003425. RNA-seq information is available in the NCBI Gene Expression Omnibus database under accession number GSE60476.
RESULTS
Identification of six transposon mutants with defective protease activity.
A Serratia sp. SCBI miniHimar RB1 mutant library (∼2,100 mutants) was generated and screened for protease activity on skim milk agar plates. The production of clearing zones on this medium is indicative of the presence of proteases capable of degrading casein, and Serratia sp. SCBI shows strong activity on skim milk agar (4). Six mutants that either failed to produce a clearing zone or had a clearing zone smaller than that of wild-type Serratia sp. SCBI were identified. The mutants were confirmed with an azocasein assay and shown to maintain a loss or reduction of proteolytic activity (Fig. 1). Mutant 16-D2 showed no protease activity at all, whereas mutants 22-A7, 22-B9, 22-C7, 22-D7, and 22-F6 all had a significant reduction in activity (P < 0.05).
FIG 1.
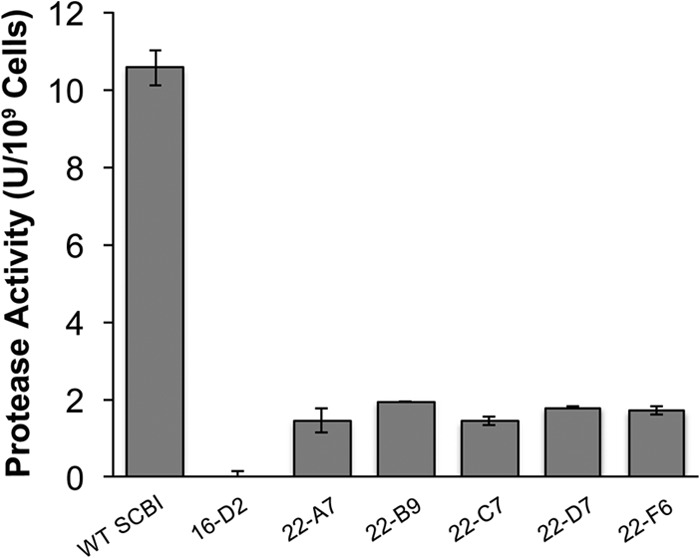
Six Serratia sp. SCBI transposon mutants with defective protease activity were identified. Proteolytic activity was determined using an azocasein assay, which was performed with strains grown for 18 h at 28°C. Values are means ± standard deviations (error bars) from at least two independent experiments. WT, wild type.
Molecular characterization of the attenuated mutants.
Rescue cloning successfully identified the gene inactivated by transposon insertion in all six mutants (see Fig. S1 in the supplemental material). No mutant had a hit in a protease gene. Mutant 16-D2 was inactivated in the aprE gene (SCBI_3625), which encodes a predicted membrane fusion protein important in alkaline protease secretion (see Fig. S1B). Homologous to the S. marcescens lipC gene, aprE is the second gene in a three-gene operon that appears to encode an ABC secretion system. The predicted genes in this operon are an ATP-binding protein, aprE, and a TolC protein. Mutant 22-A7 had an insertion in a bglB (SCBI_3572) gene, which is predicted to encode an antiterminator (see Fig. S1C). This BglB antiterminator appears to be the second gene in a two-gene operon, with both genes predicted to code for BglB proteins. Mutant 22-B9 had an insertion 12 bp downstream of the start codon in a predicted inosine/xanthosine triphosphatase gene (SCBI_0601) (see Fig. S1D). Mutant 22-C7 had an insertion in gidA (SCBI_4598), which is a predicted tRNA uridine 5-carboxymethyl-aminomethyl modification enzyme (see Fig. S1E). Mutant 22-D7 had an insertion in SCBI_0077, which encodes a predicted methyl-accepting chemotaxis protein II (see Fig. S1F). Mutant 22-F6 had an insertion at the 177-bp position in the SCBI_0044 gene, which codes for a predicted nucleic acid-binding protein that contains a PIN domain (see Fig. S1G). This PIN domain protein appears to be the second gene in a two-gene operon that also contains a predicted copG gene.
Genetic complementation of Serratia sp. SCBI mutants.
The genetically complemented mutants and controls were assayed for protease activity using both the liquid azocasein assay and skim milk agar plates (Table 2). The introduction of the vector without an insert did not significantly change the phenotypes of parental wild-type Serratia sp. SCBI or any of the mutants tested. Genetic complementation of mutants 16-D2, 22-C7, 22-D7, and 22-F6 restored wild-type levels of proteolytic activity. These results indicate that these genes were responsible for these phenotypic effects. Introduction of bglB and the inosine/xanthosine triphosphatase genes were unable to complement the defect in mutants 22-A7 and 22-B9.
TABLE 2.
Genetic complementation of mutants 16-D2, 22-C7, 22-D7, and 22-F6 restored wild-type phenotypes
| Straina | Radius (mm) of the clearing zone on skim milk agarb,e | Protease activity azocasein assay (U/109 cells)c,e | Insect pathogenesis LT50 value (no. of days)d,e |
|---|---|---|---|
| WT Serratia sp. strain SCBI | 8.1 ± 0.2 | 11.42 ± 0.57 | 2.96 |
| WT SCBI + pBAD33 | 7.7 ± 0.3 | 10.86 ± 0.43 | 3.08 |
| WT SCBI + pBAD33-AprE | 8.3 ± 0.2 | 11.59 ± 0.98 | 3.08 |
| WT SCBI + pBAD33-GidA | 8.2 ± 0.1 | 11.98 ± 0.47 | 2.87 |
| WT SCBI + pBAD33-MCP | 8.1 ± 0.2 | 11.31 ± 0.67 | 2.96 |
| WT SCBI + pBAD33-PIN | 8.3 ± 0.2 | 10.90 ± 0.47 | 3.21 |
| Mutant 16-D2 | 0.5 ± 0.0* | 0.25 ± 0.05* | 1.48* |
| Mutant 16-D2 + pBAD33 | 0.5 ± 0.1* | 0.38 ± 0.06* | 1.61* |
| Mutant 16-D2 + pBAD33-AprE | 7.1 ± 0.4 | 10.12 ± 1.76 | 2.75 |
| Mutant 22-C7 | 1.0 ± 0.1* | 1.24 ± 0.27* | 8.35* |
| Mutant 22-C7 + pBAD33 | 1.0 ± 0.2* | 1.36 ± 0.57* | 8.49* |
| Mutant 22-C7 + pBAD33-GidA | 6.9 ± 0.4* | 9.21 ± 2.13 | 3.54 |
| Mutant 22-D7 | 1.0 ± 0.2* | 1.52 ± 0.04* | 5.29* |
| Mutant 22-D7 + pBAD33 | 1.1 ± 0.1* | 1.59 ± 0.05* | 5.05* |
| Mutant 22-D7 + pBAD33-MCP | 7.5 ± 0.3 | 9.74 ± 2.03 | 3.67 |
| Mutant 22-F6 | 1.2 ± 0.2* | 1.69 ± 0.02* | 5.22* |
| Mutant 22-F6 + pBAD33 | 1.2 ± 0.2* | 1.56 ± 0.01* | 4.90* |
| Mutant 22-F6 + pBAD33-PIN | 7.8 ± 0.5 | 10.35 ± 1.67 | 3.21 |
Wild-type (WT) or mutant bacterial strain alone or carrying plasmid pBAD33 or derivatives of pBAD33.
Skim milk agar plates were spot inoculated with strains that had been grown for 18 h at 28°C in LB medium. After 48 h of incubation, the radius of the clearing zones were measured. These values were expressed in millimeters and are the averages of four measurements. The standard deviations are from at least two independent experiments.
Proteolytic activity was determined using an azocasein assay and performed with strains grown for 18 h at 28°C in LB medium. Values are averages of three measurements ± standard deviations from at least two independent experiments.
LT50 values represent the average time (in days) for 50% of the M. sexta larval population to die.
Values that are significantly different (P < 0.05) from the value for wild-type Serratia sp. SCBI are shown with an asterisk.
Loss of proteolytic activity results in loss of cytotoxicity toward BGMK cells.
Since protease activity has been identified as a cytotoxic factor for S. marcescens (27), the six Serratia sp. SCBI protease mutants were tested for their cytotoxic effects against BGMK cells. Following 24 h of incubation at 37°C, an average of 45% of BGMK cells were viable when exposed to wild-type Serratia sp. SCBI supernatant (Fig. 2A). In contrast, the protease mutants showed minimal cytotoxic effects (P < 0.05). Between 93% and 97% of BGMK cells were still viable in wells exposed to supernatant from mutants 16-D2, 22-A7, 22-B9, 22-C7, 22-D7, and 22-F6. BGMK cells incubated in normal growth medium was used as a control and represented 100% viability.
FIG 2.
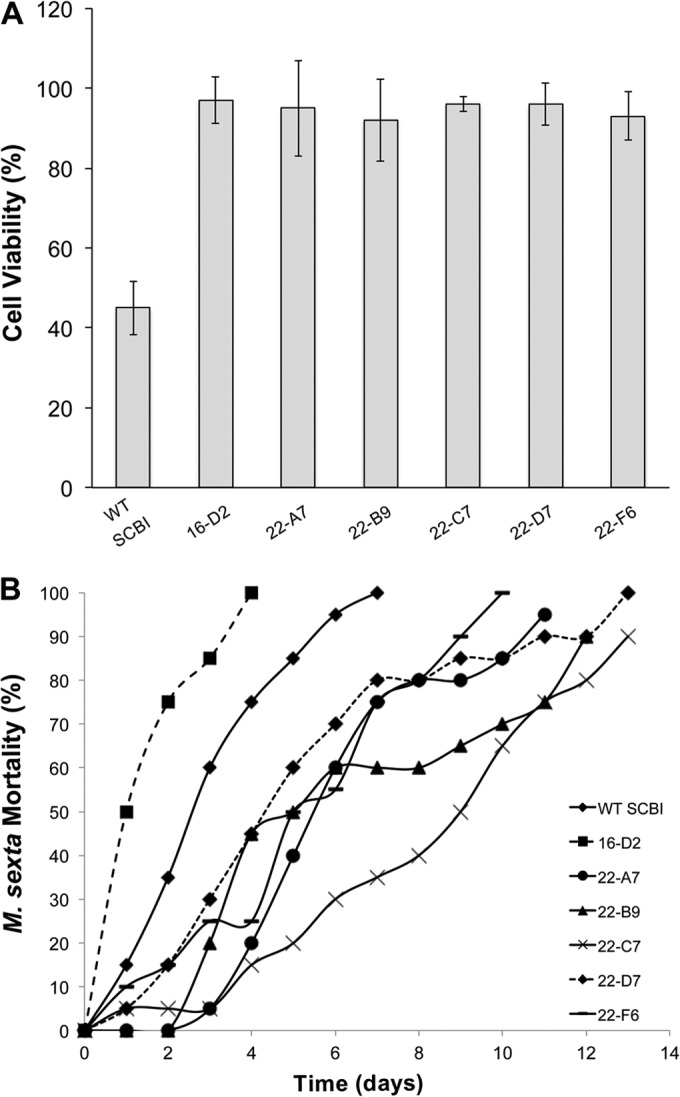
All six protease mutants were no longer cytotoxic to BGMK cells and displayed different levels of virulence toward M. sexta. (A) BGMK cells were exposed to bacterial supernatant (2 mg/ml total protein) for 24 h at 37°C, and cell viability was tested using a MTT assay. Untreated cells were used as a negative control and represented 100% viability. Values are means ± standard deviations (error bars) from at least two independent experiments. WT, wild type. (B) Third-instar M. sexta larvae were injected with 2 × 103 CFU and held individually at 37°C for 14 days. Mortality was measured as the percentage of the larva population killed over time.
All six protease mutants were altered in insect virulence.
The ability of the protease mutants to kill third-instar M. sexta larva was assessed over a 14-day period following an intrahemocoelic injection and incubation at 37°C (Fig. 2B). Mutant 16-D2 killed insect larva at a significantly higher rate than wild-type Serratia sp. SCBI did (an LT50 value of 1.48 days versus 2.96 days, respectively, P < 0.05). The other protease mutants killed at a significantly lower rate (P < 0.05) than wild-type Serratia sp. SCBI did. Mutants 22-A7, 22-B9, 22-C7, 22-D7, and 22-F6 had LT50 values of 5.98, 6.59, 8.35, 5.29, and 5.22 days, respectively. Successful genetic complementation of mutants 16-D2, 22-C7, 22-D7, and 22-F6 restored wild-type rates of killing (Table 2).
Mutants 22-B9 and 22-C7 showed decreased rates of hemolytic activity.
The hemolytic capabilities of the protease mutants were determined using a quantitative sheep blood assay. Mutants 16-D2, 22-A7, 22-D7, and 22-F6 all had wild-type rates of hemolytic activity, lysing between 90 and 100% of SRBCs within the 4-h incubation period (Fig. 3). Mutants 22-B9 and 22-C7 demonstrated reduced rates of hemolytic activity (P < 0.05), lysing only 60% and 35% of SRBCs, respectively.
FIG 3.
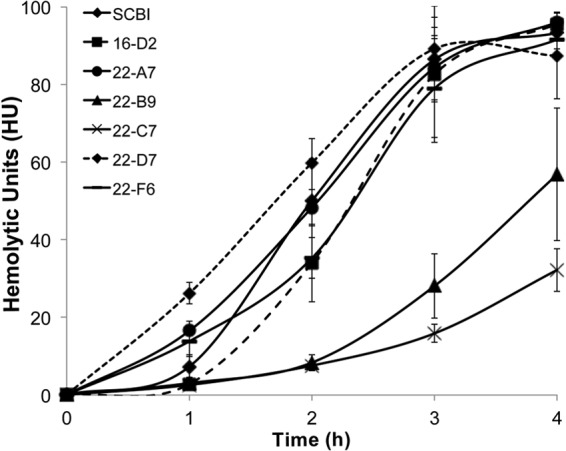
Comparison of the rates of hemolysis by wild-type Serratia sp. SCBI and mutants. Hemolytic activity was determined against SRBCs over a 4-h incubation period at 28°C. Values are means ± standard deviations (error bars) from at least two independent experiments.
Mutant 16-D2 showed enhanced swarming motility, but mutant 22-C7 exhibited reduced rates of swarming motility.
The swarming motility of the protease mutants was determined using 0.65% agar PP3 plates and measuring the diameter of the swarm ring. The optimal swarming motility of wild-type Serratia sp. SCBI is 28°C (4), and therefore, the ability of each protease mutant to swarm was initially determined for all six protease mutants at this temperature. Following 42 h of incubation at 28°C, mutant 22-C7 demonstrated reduced swarming motility (P < 0.05) compared to wild-type Serratia sp. SCBI (Fig. 4C). Wild-type Serratia sp. SCBI swarmed across the entire plate by 42 h (88 mm), but mutant 22-C7 had an average swarm ring diameter of 31.7 ± 9.6 mm. Mutants 22-A7, 22-B9, 22-D7, and 22-F6 demonstrated wild-type swarming motility. In contrast, mutant 16-D2 had an increased rate of swarming motility (P < 0.05) and swarmed across the entire plate in less than 18 h. Due to its high rate of motility, the swarming assay was repeated at 22°C with all six mutants and wild-type Serratia sp. SCBI to assess how fast mutant 16-D2 swarmed at a suboptimal temperature. Following 18 h at 22°C, wild-type Serratia sp. SCBI and mutants 22-A7, 22-B9, 22-C7, 22-D7, and 22-F6 had not begun swarming and appeared as a normal colony with a diameter of between 8 and 12 mm. During that same incubation period, mutant 16-D2 had completely swarmed across the entire plate (88 mm) (Fig. 4A).
FIG 4.
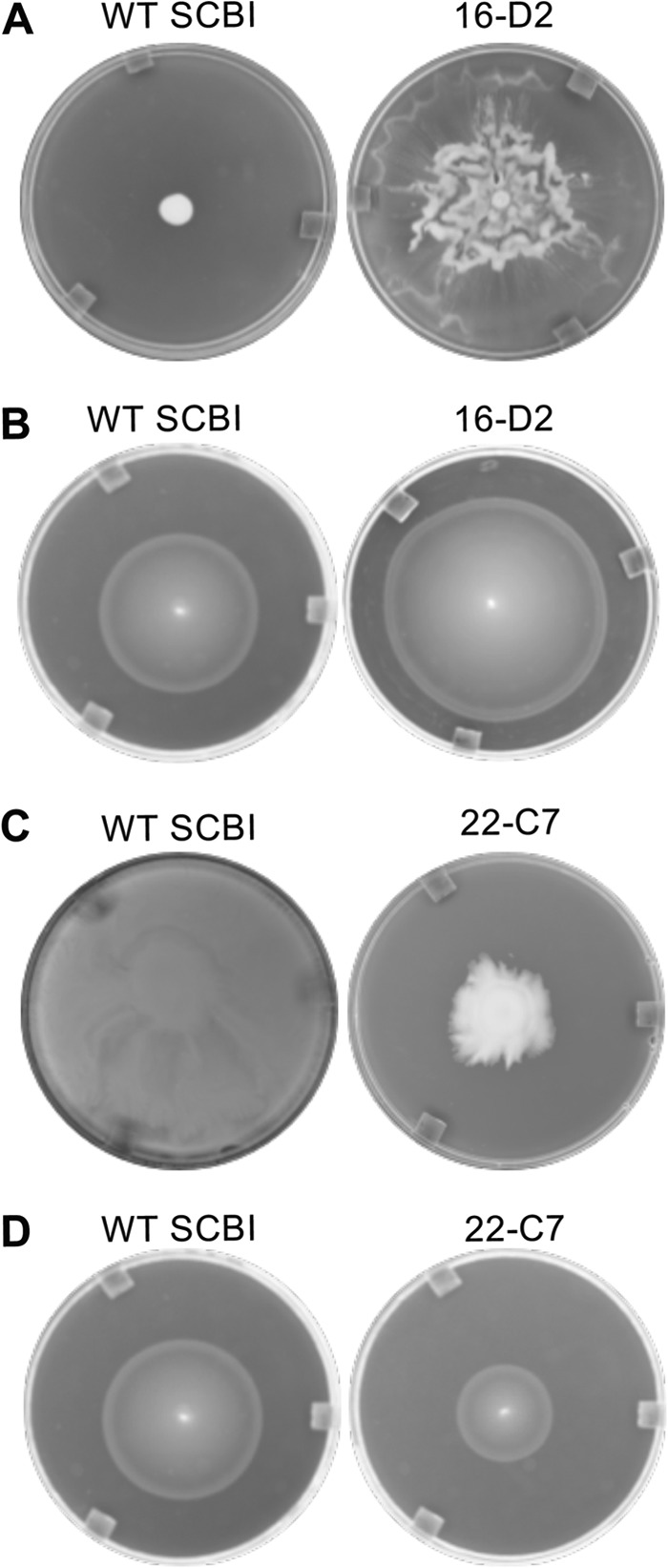
Swarming motility by wild-type Serratia sp. SCBI and mutants. Mutant 16-D2 displayed increased rates of motility, but mutant 22-C7 had reduced rates of motility. (A) Photographs of wild-type (WT) Serratia sp. SCBI and mutant 16-D2 following 18 h at 22°C on 0.65% agar PP3 swarm plates. (B) Photographs of wild-type Serratia sp. SCBI and mutant 16-D2 following 8 h at 37°C in 0.2% agar swim plates. (C) Photographs of wild-type Serratia sp. SCBI and mutant 22-C7 following 42 h at 28°C on 0.65% agar PP3 swarm plates. (D) Photographs of wild-type Serratia sp. SCBI and mutant 22-C7 following 8 h at 37°C in 0.2% agar swim plates.
The rate of swimming motility was determined using 0.2% agar plates and measuring the diameter of the swim ring. Following 8 h of incubation at 37°C, wild-type Serratia sp. SCBI had an average swim ring diameter of 55.2 ± 3.2 mm (Fig. 4B). Mutant 16-D2, in contrast, had an average swim ring diameter of 69.7 ± 4.2 mm (P < 0.05). Mutant 22-C7 had a lower rate of swimming motility compared to the parental wild-type strain (P < 0.05) with an average swim ring diameter of 27.5 ± 1.2 mm (Fig. 4D). All other protease mutants demonstrated wild-type swimming motility.
RNA-seq transcriptomic analysis on protease mutants.
Since mutants 16-D2, 22-C7, and 22-F6 had defects within either regulatory genes or the transporter gene, we were interested in their effect on global gene expression and performed transcriptome analysis on these mutants. RNA was extracted from these mutants and wild-type Serratia sp. SCBI during stationary-phase growth and used for RNA-seq analysis.
A comparison of genes expressed by mutant 16-D2 to those in the parental wild type generated a total of 244 genes that had significant changes in their levels (1.5-fold change; P < 0.05) in their expression. Of these genes, 142 were significantly upregulated (see Table S3 in the supplemental material). Eight genes involved in motility showed increased expression. A PhoPQ-activated pathogenicity-related protein was upregulated 9.82-fold. A type VI secretion protein, type VI secretory pathway peptidase PulO, and type IV secretion protein IcmF were upregulated 9.26-, 5.77-, and 5.62-fold, respectively. An extracellular serine protease and serralysin (PrtA4) were upregulated 3.45- and 2.32-fold, respectively. An ABC-type bacteriocin/antibiotic exporter was upregulated 4-fold, and a microcin H47 secretion protein was upregulated 3.88-fold. A colonic acid capsular biosynthesis protein was upregulated 1.54-fold. Also of note, the PIN domain protein inactivated in mutant 22-F6 was upregulated 1.75-fold in mutant 16-D2. A total of 31% of all upregulated genes were hypothetical genes, and most of these had no assigned COG category. A total of 102 genes were significantly downregulated in mutant 16-D2 compared to wild-type Serratia sp. SCBI (see Table S4 in the supplemental material). Overall, many of these genes were involved in transcriptional regulation and metabolism. In addition, at least three genes involved in cytochrome c synthesis were downregulated. A total of 12 hypothetical genes were significantly downregulated.
A total of 258 genes had significantly altered expression (P < 0.05) in mutant 22-C7 compared to wild-type Serratia sp. SCBI. Of these 258 genes, 144 had increased expression (see Table S5 in the supplemental material). Fourteen genes involved in transcription and translation were significantly upregulated. Eight genes in signal transduction had increased expression. Three genes involved in plasmid maintenance and stabilization were significantly upregulated. A total of 45 hypothetical genes were upregulated. A total of 114 genes were significantly downregulated in mutant 22-C7 compared to wild-type Serratia sp. SCBI (see Table S6 in the supplemental material). A phospholipase D gene had a 10.18-fold decrease in expression. A flagellar biosynthesis protein and FlgN family protein, also involved in flagellar production, had a 9.41-fold and 2.93-fold decrease in expression, respectively. Three genes involved in iron transport encoding a siderophore-interacting protein, ABC-type Fe3+ transport periplasmic protein, and frataxin (an iron transport protein) showed a 4.64-, 3.32-, and 2.19-fold decrease in expression, respectively. Three genes involved in fimbrial production were downregulated. In addition, a collagen-binding surface adhesion gene, spaP, was downregulated 2.12-fold. The PrtA homologues were all downregulated; however, their P values were not below 0.05, and therefore not statistically significant.
A total of 280 genes had significantly altered expression (P < 0.05) in mutant 22-F6 compared to wild-type Serratia sp. SCBI. Of these 280 genes, 162 had increased expression (see Table S7 in the supplemental material). A total of 10 genes involved in motility were upregulated. A significant number of genes in the COG (clusters of orthologous groups of proteins) categories replication/repair/recombination, transcription, and translation were upregulated. This included the PIN domain protein inactivated in mutant 22-F6 and its operon partner, the CopG family transcriptional regulator (see Fig. S1G in the supplemental material). The PIN transcript was a truncated mRNA that ended at the start of the transposon insertion. Two chitinase genes were upregulated 3.11-fold and 2.63-fold. An esterase/lipase was upregulated 2.48-fold. Forty-three hypothetical genes were upregulated, comprising 25.9% of all genes that had increased expression, and three of the top five most significantly upregulated genes were hypothetical genes. A total of 118 genes had significantly decreased expression in mutant 22-F6 (see Table S8 in the supplemental material). Twelve genes involved in nucleotide metabolism and transport were significantly downregulated. An intracellular protease/amidase was downregulated 18.3-fold, and an ATP-dependent protease was downregulated 2.61-fold. A peptidase M60 viral enhancin protein was downregulated 4-fold. At least 14 genes involved in transcriptional or translational regulated had significantly decreased expression. Seven genes associated with the outer membrane were downregulated, including two permeases, an outer membrane protein, an EnvZ/OmpR regulon moderator, tolA, ompX, and an outer membrane lipoprotein.
Quantitative RT-PCR was performed on four genes with significant changes in expression in the RNA-seq analysis (the hemolysin-coregulated protein ImpD, the heme exporter protein CcmC, a bleomycin resistance protein, and flagellar protein FliZ). Expression data from the normalized RNA-seq data compared to qRT-PCR data demonstrated the same trends, thus confirming that data from both platforms were of high quality (see Table S9 in the supplemental material).
For the three mutants, the number of common genes that were up- or downregulated was determined (Fig. 5). There were 12 genes upregulated by all three mutants and 19 downregulated. No obvious patterns were found among these genes. Most of the upregulated genes encoded hypothetical proteins and two DNA regulatory proteins (SCBI_0214 and SCBI_3467). In general, each of the three mutants shared a similar number of up- or downregulated genes. Mutants 22-C7 and 22-F6 shared the most upregulated genes, including several genes encoding transcriptional regulators (SCBI_0963, SCBI_3139, 3610, SCBI_0196, SCBI_2463, SCBI_3253, SCBI_0339, SCBI_1671, SCBI_1757, SCBI_0983, and SCBI_3467). However, the majority of the up- and downregulated genes were specific to the individual mutant.
FIG 5.

Analysis of the common up- and downregulated genes found among the three SCBI mutants.
EDTA has a strong inhibitory effect on protease activity.
The results of the transposon mutant library screen failed to produce a mutant with a disruption in a protease gene, so we performed further analysis on wild-type Serratia sp. SCBI to determine what types of proteases are being secreted and what environmental conditions had the greatest influence on the expression of predicted protease genes. First, an azocasein assay was carried out using several protease inhibitors in order to determine whether the protease activity was based on a metalloprotease or serine protease. PMSF is an irreversible serine protease inhibitor, while EDTA inhibits metalloproteases by chelating the divalent cations necessary for their activity. To determine whether the presence of these inhibitors would have an inhibitory effect on Serratia sp. strain SCBI proteolytic activity, 1 mM PMSF and 50 mM EDTA were incorporated into a liquid azocasein assay. Without any inhibitors, protease activity was measured at 11.21 U (data not shown). The addition of PMSF did not significantly affect protease activity, lowering activity slightly to 9.56 U. However, EDTA lowered proteolytic activity significantly, dropping levels to 5.66 U, a 49.5% decrease (P < 0.05). A combination of PMSF and EDTA added to Serratia sp. SCBI supernatant resulted in the lowest proteolytic activity of only 3.65 U and represents only 32.6% of the activity seen without any protease inhibitors (P < 0.05). The differences seen between the addition of EDTA alone and the addition of a combination of PMSF and EDTA were not significant. These results indicate that the majority of Serratia sp. SCBI proteolytic activity was due to a combination of metalloprotease and serine protease activities, but the larger portion of its activity was driven by metalloprotease.
Temperature influences the expression of proteases.
Analysis of the Serratia sp. SCBI genome indicates that this bacterium contains the genes for at least two extracellular serine proteases, a zinc metalloprotease (yfgC), a secreted metalloprotease, and four alkaline metalloproteases (see Table S2 in the supplemental material). The two serine protease genes (SCBI_4441 and SCBI 4442) are located next to each other in the genome and were termed serine protease 1 and serine protease 2 for the location at which they fall in the genome. The serine protease 1 gene is 3,093 bp, and the serine protease 2 gene is 3,024 bp. The zinc metalloprotease yfgC (SCBI_3465) and the secreted metalloprotease gene (SCBI_3545) are 1,470 bp and 1,026 bp, respectively. The four alkaline metalloproteases are homologues of the metalloprotease prtA (also known as serralysin). These four prtA homologues were designated prtA1 (SCBI_0187), prtA2 (SCBI_1952), prtA3 (SCBI_2172), and prtA4 (SCBI_3084), named for the order in which they appear in the sequenced genome. COG analysis of all four prtA homologues classifies these proteases as RTX toxins due to multiple calcium-binding repeats and peptidase M10 serralysin C termini. prtA1 is a 1,515-bp gene that shares high identity with prtA in S. marcescens Db11. prtA2 is a 1,419-bp gene homologous to the prtA gene found in Photorhabdus species. prtA3 is a 1,617-bp gene homologous to the plant-degrading proteases found in Dickeya and Erwinia species. prtA4 is a 1,383-bp gene homologous to proteases found in other Serratia species and in Dickeya species.
Temperature is an important influence on protease activity in Serratia sp. SCBI (4). To assess the optimal temperature at which these eight protease genes were expressed, qRT-PCR was done utilizing RNA extracted from wild-type Serratia sp. SCBI cells grown to stationary phase (18 h of incubation) at 22, 28, or 37°C. Previous growth curve analysis of Serratia sp. SCBI showed that optimal growth is seen at 28°C and 37°C, yet the rate of growth at 22°C is not significantly lower (4). Optical density readings were determined prior to RNA extraction to ensure that the population density of each culture was the same in order to achieve an accurate comparison of gene expression. For all four prtA homologues, the highest mRNA levels were at 28°C and the lowest mRNA levels were at 22°C (Fig. 6A and B). The prtA2 gene showed the most significant reduction in mRNA levels at 37°C compared to 28°C. mRNA levels of the two serine proteases, yfgC, and the secreted metalloprotease were highest at 22°C, but the metalloprotease showed only slightly reduced mRNA levels at 28°C (Fig. 6C and D). The mRNA levels of all four of these genes were significantly reduced at 37°C.
FIG 6.
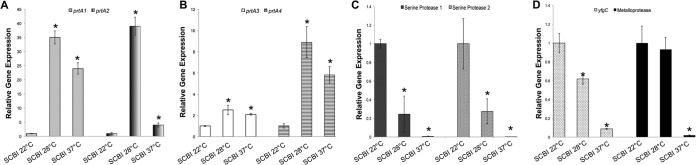
Temperature influences the expression of protease genes in Serratia sp. SCBI. qRT-PCR was performed on RNA extracted from Serratia sp. SCBI cultures grown in LB medium for 18 h at the test temperature (22, 28, or 37°C). mRNA levels were normalized to the rplU housekeeping gene and compared to the calibrator (SCBI at 22°C). Data are presented as the relative changes in gene expression between the values obtained with the test conditions and the calibrator. (A to D) Levels of expression of prtA1 and prtA2 (A), prtA3 and prtA4 (B), serine protease 1 and serine protease 2 (C), and yfgC and the secreted metalloprotease (D). Values are means ± standard deviations (error bars) from at least two independent experiments. Relative gene expression values that are significantly different (P < 0.05) from the value for the calibrator (SCBI at 22°C) are indicated by an asterisk.
PrtA homologues were upregulated following insect death, while the serine proteases and other metalloproteases showed higher mRNA levels during active infection of M. sexta.
To assess the importance of protease activity during insect pathogenesis, qRT-PCR was performed utilizing RNA extracted from exponential-growth-phase cultures, from cells during hemolysis of SRBCs, and from cells collected from either live or dead M. sexta larvae following intrahemocoelic injection. All experiments from which RNA was obtained were performed at the optimal temperature for virulence (37°C). The prtA homologues were all significantly upregulated in Serratia sp. SCBI-killed M. sexta larvae compared to exponential-phase cultures (Fig. 7A and B). prtA1 and prtA2 mRNA levels increased over 200-fold compared to mRNA levels found in exponential-phase culture. Exposure of the cultures to SRBCs did not significantly increase prtA1 and prtA2 mRNA levels (Fig. 7A). A 7-fold and 10-fold increase in prtA3 and prtA4 mRNA levels, respectively, was observed for Serratia sp. SCBI obtained from dead M. sexta larvae compared to cells obtained from exponential-growth cultures (Fig. 7B). Among the prtA homologues, only the prtA4 mRNA level showed an increase during SRBC hemolysis. In contrast to the prtA homologues, the serine proteases, yfgC, and the secreted metalloprotease genes showed the highest mRNA levels in cells from active M. sexta infections (Fig. 7C and D). Serine protease 2, yfgC, and the metalloprotease genes also showed an increase in mRNA levels during hemolysis of SRBCs compared to growth in LB medium.
FIG 7.
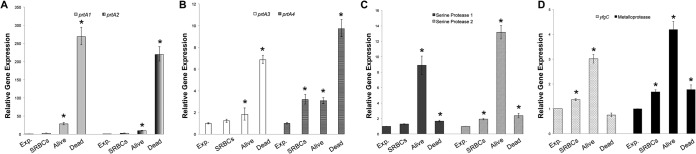
Expression of protease genes in wild-type Serratia sp. SCBI under different environmental conditions. qRT-PCR was performed on RNA extracted from Serratia sp. SCBI during exponential-phase growth (Exp.) in LB medium, during hemolysis of SRBCs, during active infection of M. sexta, and following death of M. sexta. mRNA levels were normalized to the mRNA level of rplU housekeeping gene and compared to the calibrator (exponential-phase cells). Data are presented as the relative changes in gene expression between the values obtained with the test conditions and the calibrator. (A to D) Expression levels of prtA1 and prtA2 (A), prtA3 and prtA4 (B), serine protease 1 and serine protease 2 (C), and yfgC and the secreted metalloprotease (D). Values are means ± standard deviations (error bars) from at least two independent experiments. Relative gene expression values that are significantly different (P < 0.05) from the value for the calibrator (exponential-phase cells) are indicated by an asterisk.
Expression of protease genes was downregulated in all six transposon mutants.
The mRNA levels for the known protease genes were determined for the miniHimar RB1 mutants by the use of qRT-PCR. RNA was extracted from cultures grown to stationary phase at the optimal temperature for expression (28°C or 22°C). With mutants 22-A7, 22-B9, 22-C7, 22-D7, and 22-F6, prtA1 and prtA2 mRNA levels were significantly downregulated, showing up to a 95% decrease (Fig. 8A), while prtA3 and prtA4 mRNA levels were moderately downregulated (Fig. 8B). The serine protease mRNA levels were also significantly downregulated in these mutants to the point where very little mRNA was measured (Fig. 8C). For these mutants, the levels of yfgC and the secreted metalloprotease mRNA showed 60 to 80% reduction from the parental wild-type level (Fig. 8D). mRNA levels of yfgC and the secreted metalloprotease were largely not affected in mutant 16-D2; however, there was a reduction in mRNA of between 60 and 80% in mutants 22-A7, 22-B9, 22-C7, 22-D7, and 22-F6.
FIG 8.
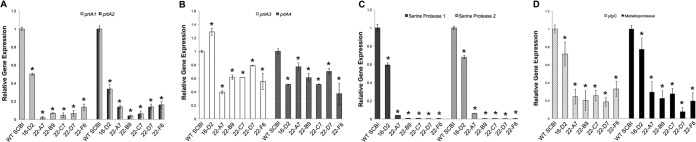
Expression of protease genes in wild-type Serratia sp. SCBI and mutants. qRT-PCR was performed on RNA extracted from wild-type Serratia sp. SCBI and mutant cultures grown in LB medium for 18 h at 22°C or 28°C. mRNA levels were normalized to the rplU housekeeping gene and compared to the calibrator (wild-type [WT] SCBI). Data are presented as the relative changes in gene expression between the values obtained from the protease mutants and the calibrator. (A to D) Expression levels of prtA1 and prtA2 at 28°C (A), prtA3 and prtA4 at 28°C (B), serine protease 1 and serine protease 2 at 22°C (C), and the secreted metalloprotease (exponential-phase cells) and yfgC at 22°C (D). Values are means ± standard deviations (error bars) from at least two independent experiments. Relative gene expression values that are significantly different (P < 0.05) from the value for the calibrator (WT SCBI) are indicated by an asterisk.
DISCUSSION
Proteases are known virulence factors in bacterial pathogens, such as Serratia marcescens (18), Photorhabdus luminescens (38), and Shigella flexneri (39). Protease activity plays an important role in suppression of the host innate immune response and degradation of host tissue. Results from our study suggest that proteases secreted by Serratia sp. SCBI, in particular the PrtA proteases, play a role similar to that of the proteases found in S. marcescens, P. luminescens, and S. flexneri. The identification of six Serratia sp. SCBI miniHimar RB1 mutants with altered proteolytic activity provided insight into how protease genes within this organism are regulated and how they are secreted. All six protease mutants were no longer toxic toward the BGMK cell line, providing clear evidence that protease activity is essential for cytotoxicity. However, insect viability assays using M. sexta as a model organism gave different results, eliminating any possibility of linking proteolytic activity levels directly to insect pathogenesis. Nevertheless, identifying the genes inactivated in mutants 16-D2, 22-A7, 22-B9, 22-C7, 22-D7, and 22-F6 uncovered several regulatory mechanisms that, in addition to regulating proteolytic activity, appear to play important roles in motility and virulence.
The location of the transposon insertion in mutant 16-D2 was mapped to a predicted alkaline protease secretion protein, aprE. aprE falls in the middle of a three-gene operon that is predicted to transcribe an ABC transport system that is highly homologous to the lipBCD system of S. marcescens (16). The inactivation of aprE likely resulted in loss of expression of the third gene in the operon, the outer membrane protein tolC. With two out of the three genes no longer being transcribed, this ABC transport system would no longer be able to secrete proteases, providing a clear-cut explanation as to why there was a complete loss of detectable protease activity in mutant 16-D2. However, upon further analysis, mutant 16-D2 also demonstrated unexpected changes in motility and virulence. Mutant 16-D2 also demonstrated a hypervirulent phenotype, killing M. sexta larvae in half the time it takes for wild-type Serratia sp. SCBI to kill M. sexta.
To investigate the possible mechanisms behind the hypervirulent phenotype of mutant 16-D2, RNA-seq was utilized to compare levels of gene expression between this mutant and wild-type Serratia sp. SCBI during stationary-phase growth. Genes involved in adhesion, proteolytic activity, and direct competition with other microbes may contribute to the hypervirulent phenotype of mutant 16-D2. Genes involved in fimbria and P-pilus production were upregulated, which are known virulence factors for a number of pathogens. The gene encoding the PhoPQ-activated protein PqaA was expressed at nearly 10-fold-higher levels in the mutant 16-D2 relative to wild-type Serratia sp. SCBI. PhoPQ-regulated genes are responsible for Salmonella virulence against mice, and mutation of pqaA in Salmonella enterica serovar Typhimurium affects virulence and sensitivity to the antimicrobial peptide melittin (40). Proteins involved in a type VI secretion pathway were upregulated at least 5-fold in mutant 16-D2. In S. marcescens Db10, a type VI secretion system (T6SS) that is nonessential for virulence but efficiently kills other bacterial cells exists. Its activity is required for survival when cultured with Enterobacter cloacae (41, 42). Other genes upregulated in mutant 16-D2, which may be playing a vital role in warding off competitors and thereby indirectly contribute to virulence, were an ABC-type bacteriocin/antibiotic exporter and the microcin H47. Overall, transcriptomic analysis uncovered possible genetic mechanisms to explain the hypervirulence of mutant 16-D2. However, it does not explain how loss of a protease exporter gene results in such drastic global effects. One hypothesis is that a buildup of proteases within the cell may be exerting inadvertent effects on intracellular proteins, including regulatory proteins, which may influence expression of virulence factors.
The location of the transposon insertion in mutant 22-A7 was mapped to a predicted BglB (also known as BglG) family antiterminator. In E. coli, BglG regulates the expression of the β-glucoside utilization (bgl) operon bglGFBH (43). The Bgl system in E. coli plays a vital role in the phosphotransferase system. There are four Bgl family proteins in Serratia sp. SCBI, but none reside in the beginning of an operon containing other genes involved in β-glucoside utilization, as is found in E. coli. The BglB family antiterminator inactivated in mutant 22-A7 appears to be the second gene in a two-gene operon, the first gene also being a BglB protein. How a BglB family antiterminator could influence proteolytic activity is unknown, as there is no precedence in the literature linking this type of antiterminator to protease expression. It is possible that this BglB is regulating protease mRNA transcripts; however, further work, including obtaining successful complementation of this gene in mutant 22-A7, is necessary to uncover the mechanism connecting BglB to proteolytic activity.
The location of the transposon insertion in mutant 22-B9 was mapped to a predicted inosine/xanthosine triphosphatase. During oxidative stress, chemical mutagenesis and oxidative deamination result in damage to chromosomal DNA and the free nucleotide pool (44–46). ITP and XTP are produced from the deamination of adenine and guanine, respectively, and their incorporation into DNA produces mutations. In order to control the concentration of these damaging nucleotides, inosine and xanthosine triphosphatases target ITP and XTP for degradation. With the loss of an inosine/xanthosine triphosphatase, as is seen in mutant 22-B9, the mutation rate would likely increase. Therefore, a high number of mutations may have affected genes or regulation of genes responsible for the reduction in protease activity, cytotoxicity, virulence, and hemolytic activity in mutant 22-B9.
The location of the transposon insertion in mutant 22-C7 was mapped to a predicted tRNA uridine 5-carboxymethyl-aminomethyl modification enzyme, GidA. The function of GidA has been investigated in several Gram-negative bacteria. GidA is required for the addition of carboxymethylaminomethyl groups to the C-5 carbon of uridine at position 34 of tRNAs that read codons ending with A or G in mixed-codon families, thereby preventing misincorporation and frameshift mutations (47, 48). The inactivation of GidA in Pseudomonas, Streptococcus, Aeromonas, and Salmonella has global effects due to loss of translational fidelity, negatively influencing such phenotypes as virulence, antibiotic production, hemolytic activity, proteolytic activity, cytotoxicity, and swarming motility (49–52).
Mutant 22-C7 displayed attenuation of proteolytic activity, cytotoxicity, hemolysis, motility, and virulence, suggesting that GidA plays a role in Serratia sp. SCBI similar to that in other Gram-negative pathogens. At 1,890 bp, gidA is the same size as gidA in Aeromonas hydrophila. Microarray analysis of an A. hydrophila gidA mutant showed significant changes in the expression of 278 genes, and phenotypic analyses of this mutant uncovered deficiencies in the invasion of intestinal epithelial cells, changes in bacterial motility, reduced intracellular survival in macrophages, and a reduction in cytotoxicity (51). Upregulated genes in the A. hydrophila gidA mutant included transcriptional regulators, heat shock proteins, and proteins involved in iron transport, whereas downregulated genes included genes important in invasion, flagellar genes, type 3 secretion system pathway genes, and motility and chemotaxis genes. Inactivation of gidA in Serratia sp. SCBI also appears to have an effect on gene expression levels. qRT-PCR results showed that prtA1, prtA2, serine proteases 1 and 2, yfgC, and the secreted metalloprotease were all significantly downregulated. RNA-seq analysis showed 258 genes with altered expression in mutant 22-C7. The types of genes showing alterations in gene expression were similar to those in S. enterica. A high number of genes involved in transcriptional and translational regulation were upregulated in mutant 22-C7. In terms of downregulated genes, several genes involved in flagellar production had significantly decreased expression as well as the surface adhesion, spaP, which is involved in type 3 secretion systems, similar to the S. enterica gidA mutant. In contrast to the S. enterica gidA mutant, Serratia sp. SCBI mutant 22-C7 had a decrease in iron transport genes. Also of note in mutant 22-C7 was a decrease in expression of a phospholipase D gene and three genes involved in fimbrial production, all of which likely contributed to the decrease in virulence of this mutant.
The location of the transposon insertion in mutant 22-D7 was mapped to methyl-accepting chemotaxis protein (MCP) II. MCPs are transmembrane sensor proteins in bacteria that function in the detection of molecules in that environment. Recently, it has been shown that in addition to motility, MCPs also play a role in regulating virulence in such pathogens as Pseudomonas aeruginosa (53, 54). The MCP inactivated in mutant 22-D7 falls into the category of an orphan MCP, as its two-component system partners are not immediately apparent. With a severe reduction in proteolytic activity, cytotoxicity, and virulence, as well as significant downregulation of expression in a number of protease genes, it is possible that this MCP interacts with a pathway involved in the regulation of either a second messenger, such as cAMP, or that its corresponding response regulator is a DNA-binding protein the directly controls transcription of protease and virulence-related genes. Further work is needed to characterize this orphan MCP and its partners in order to understand its role in proteolytic activity and virulence.
The location of the transposon insertion in mutant 22-F6 was mapped to a 381-bp nucleic acid-binding protein, which contains a PIN domain. PIN domain proteins are found in bacteria, archaea, and eukaryotes. The function of PIN domain proteins in eukaryotes, including humans, is to eliminate mRNA transcripts containing premature stop codons, a process otherwise known as nonsense-mediated mRNA decay (55–57). In prokaryotes, PIN domain proteins often function as the toxin in toxin-antitoxin (TA) systems. The largest class of type II toxin-antitoxin systems in prokaryotes is the VapBC TA system, in which VapC is the toxin and VapB serves as the matching antitoxin. VapBC TAs in such organisms as Sinorhizobium meliloti, Mycobacterium smegmatis, and Neisseria gonorrhoeae appear be involved in metabolic processes during host interactions and under stressful conditions by targeting an array of mRNA transcripts for degradation (58–60). The VapBC TAs in Salmonella enterica and Shigella flexneri are induced by amino acid starvation and chloramphenicol, resulting in a bacteriostatic condition (61). The PIN domain protein inactivated in mutant 22-F6 shows high sequence similarity with VapC toxins from Sinorhizobium meliloti, Salmonella enterica, and Shigella flexneri. Although further work is needed to confirm that this PIN domain protein and the upstream CopG DNA-binding protein constitute a bona fide TA locus, they do appear to be transcribed polycistronically and are the same size as other VapBC TAs, at 126 and 73 amino acids, respectively. Phenotypic and qRT-PCR results suggest that one function of this PIN domain protein in Serratia sp. SCBI is to regulate proteolytic activity. In addition, RNA-seq analysis of mutant 22-F6 showed significant changes in the expression of a high number of genes involved in transcriptional or translational regulation, including increased expression of the PIN domain protein hit by the transposon in this mutant and its operon partner, CopG. Given the nature of regulation of VapBC TAs, the mutation of the PIN domain protein likely results in a decreased ability to bind to CopG and prevent transcription of these two genes. Therefore, without negative autoregulation, there is increased expression of both genes. Further genetic analysis of both the PIN domain protein and CopG should shed light on the function of this predicted TA system.
Though the results of the transposon mutant screen were successful in uncovering mechanisms important in protease activity, there was a distinct lack of data on the individual protease genes in Serratia sp. SCBI. Therefore, the genome of Serratia sp. SCBI was analyzed to identify predicted protease genes and to perform expression studies of these genes to gain further understanding of their possible role in various environmental conditions, including pathogenesis. The high number of prtA homologues found within Serratia sp. SCBI indicated that these types of proteases are important to this organism. There is high sequence similarity between prtA1 and prtA2 to the prtA genes found in S. marcescens and P. luminescens, respectively, giving an indication of the function of each protease. In S. marcescens, PrtA is linked to destruction of mammalian immune system components (15, 25) and cytotoxicity (27). Similarly, PrtA from P. luminescens is important in cytotoxicity and shows its highest expression following death of the insect host (62, 63). Given the high mRNA levels seen for prtA1 and prtA2 following the death of Serratia sp. SCBI-killed M. sexta larvae and loss of cytotoxicity in all six attenuated mutants, these two proteases could be largely responsible for destruction of host tissue and acquisition of cell-bound nutrients. The results of the azocasein assay, which showed that the majority of protease activity by Serratia sp. SCBI is metalloprotease based, further supports the possibility that prtA1 and prtA2 are the genes encoding the two most important enzymes involved in proteolytic activity. In addition, prtA3 and prtA4 also showed elevated mRNA levels following the death of Serratia sp. SCBI-killed M. sexta larva, they but did not show as great of an increase as the prtA1 and prtA2 genes did. The expression data for the remaining protease genes analyzed in this study, two serine proteases and two metalloproteases, indicates that these enzymes may be more important in active infection. Without deletion of these genes individually, it is impossible to determine their true function, and unfortunately, screening of the Serratia sp. SCBI miniHimar RB1 transposon library failed to provide any hits in a protease gene. Increasing the number of mutants in the library could have provided a mutant with an inactivated protease gene, as only 2,100 mutants were tested, and the genome of Serratia sp. SCBI contains approximately 4,849 genes. However, there is a high redundancy of protease genes in the Serratia sp. SCBI genome, and inactivation of one protease gene is unlikely to affect the phenotype to any significant extent. Attempts at site-directed mutagenesis of prtA1 and prtA2 by lambda Red recombination were unsuccessful. Further analysis of these genes, and a successful site-directed mutagenesis system for Serratia sp. SCBI, is needed to understand the individual function of prtA1, prtA2, and the other protease genes.
Overall, this study has uncovered the importance of protease activity in the cytotoxic effects seen on mammalian cells by Serratia sp. SCBI. This study has also laid the foundation for further work on characterizing the molecular pathways governing protease activity in Serratia sp. SCBI. The redundancy of protease genes within the Serratia sp. SCBI genome underscores the importance of proteolytic activity in the lifestyle of this insect pathogen. It is likely that Serratia sp. SCBI secretes proteases throughout the infective process in the insect host, yet the data collected more strongly supports the role of proteases in the breakdown of the insect cadaver and bioconversion of host tissue. Results from the transposon library provided evidence that Serratia sp. SCBI proteolytic activity drives the destruction of mammalian cells. The major secretion pathway for extracellular proteases appears to be through an ABC transport system, though other secretory pathways may exist. Regulation of Serratia sp. SCBI protease genes appears to be controlled by several different mechanisms that have no precedence in the literature. A methyl-accepting chemotaxis protein, BglB family antiterminator, a PIN domain protein, and the global regulator GidA are all putatively involved in the regulation of proteases at either the transcriptional or translational level. RNA-seq analysis highlighted a number of genes that are likely contributing to the hypervirulence of mutant 16-D2 and include the PhoPQ operon, adhesion genes, type VI secretion genes, and PrtA4. Transcriptome analysis showed that the regulatory role of GidA in Serratia sp. SCBI appears to be similar to its role in other Gram-negative pathogens. The PIN domain protein inactivated in mutant 22-F6 is likely part of a toxin-antitoxin system, and its loss resulted in significant changes in transcription and translation, which is not surprising given the nature of toxin-antitoxin systems in bacteria. Further investigations into the six genes inactivated by transposon insertion should shed light on the specific mechanisms that not only control protease activity but other important physiological characteristics as well.
Supplementary Material
ACKNOWLEDGMENTS
This investigation was supported in part by Hatch grant NH00496, USDA NIFA grant 2009-35302-05257, and the College of Life Science and Agriculture, University of New Hampshire—Durham.
We thank Cintia Felix, Julie Morrison, and Vaughn S. Cooper for their contributions to the production of the Serratia sp. SCBI mutant library. We also thank W. Kelley Thomas, Krystalynne Morris, and Feseha Abebe-Akele for their assistance with RNA-seq and access to the Serratia sp. strain SCBI genome database and fosmid library.
Footnotes
Published ahead of print 2 September 2014
This article is scientific contribution number 2559 from the New Hampshire Agricultural Experiment Station.
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.01908-14.
REFERENCES
- 1.Grimont F, Grimont PAD. 2006. The genus Serratia, p 219–244 In Dworkin M, Falkow S, Rosenberg E, Schleifer K-H, Stackebrandt E. (ed), The prokaryotes, 3rd ed. A handbook on the biology of bacteria: proteobacteria: gamma subclass, vol 6 Springer, Berlin, Germany. [Google Scholar]
- 2.Petersen LM, Tisa LS. 2013. Friend or foe? A review of the mechanisms that drive Serratia towards diverse lifestyles. Can. J. Microbiol. 59:627–640. 10.1139/cjm-2013-0343. [DOI] [PubMed] [Google Scholar]
- 3.Abebe E, Jumba M, Bonner K, Gray V, Morris K, Thomas WK. 2010. An entomopathogenic Caenorhabditis briggsae. J. Exp. Biol. 213:3223–3229. 10.1242/jeb.043109. [DOI] [PubMed] [Google Scholar]
- 4.Petersen LM, Tisa LS. 2012. Influence of temperature on the physiology and virulence of the insect pathogen Serratia sp. strain SCBI. Appl. Environ. Microbiol. 78:8840–8844. 10.1128/AEM.02580-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Monreal J, Reese ET. 1969. The chitinase of Serratia marcescens. Can. J. Microbiol. 15:689–696. 10.1139/m69-122. [DOI] [PubMed] [Google Scholar]
- 6.Braun V, Schmitz G. 1980. Excretion of a protease by Serratia marcescens. Arch. Microbiol. 124:55–61. 10.1007/BF00407028. [DOI] [PubMed] [Google Scholar]
- 7.Givskov M, Olsen L, Molin S. 1988. Cloning and expression in Escherichia coli of the gene for extracellular phospholipase A1 from Serratia liquefaciens. J. Bacteriol. 170:5855–5862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen YC, Shipley GL, Ball TK, Benedik MJ. 1992. Regulatory mutants and transcriptional control of the Serratia marcescens extracellular nuclease gene. Mol. Microbiol. 6:643–651. 10.1111/j.1365-2958.1992.tb01512.x. [DOI] [PubMed] [Google Scholar]
- 9.Kaska M, Lysenko O, Chaloupka J. 1976. Exocellular proteases of Serratia marcescens and their toxicity to larvae of Galleria mellonella. J. Invertebr. Pathol. 21:465–473. [DOI] [PubMed] [Google Scholar]
- 10.Braun V, Giinther H, Neub B, Tautz C. 1985. Hemolytic activity of Serratia marcescens. Arch. Microbiol. 141:371–376. 10.1007/BF00428852. [DOI] [PubMed] [Google Scholar]
- 11.Akatsuka H, Kawai E, Omori K, Komatsubara S, Shibatani T, Tosa T. 1994. The lipA gene of Serratia marcescens which encodes an extracellular lipase having no N-terminal signal peptide. J. Bacteriol. 176:1949–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Flyg C, Xanthopoulos KG. 1983. Insect pathogenic properties of Serratia marcescens. Passive and active resistance to insect immunity studied with protease-deficient and phage-resistant mutants. J. Gen. Microbiol. 129:453–464. [DOI] [PubMed] [Google Scholar]
- 13.Molla A, Matsumura Y, Yamamoto T, Okamura R, Maeda H. 1987. Pathogenic capacity of proteases from Serratia marcescens and Pseudomonas aeruginosa and their suppression by chicken egg white ovomacroglobulin. Infect. Immun. 55:2509–2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matsumoto K, Maeda H, Takata K, Kamata R, Okamura R. 1984. Purification and characterization of four proteases from a clinical isolate of Serratia marcescens kums 3958. J. Bacteriol. 157:225–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Molla A, Matsumoto K, Oyamada I, Katsuki T, Maeda H. 1986. Degradation of protease inhibitors, immunoglobulins, and other serum proteins by Serratia protease and its toxicity to fibroblasts in culture. Infect. Immun. 53:522–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Akatsuka H, Kawai E, Omori K, Shibatani T. 1995. The three genes lipB, lipC, and lipD involved in the extracellular secretion of the Serratia marcescens lipase which lacks an N-terminal signal peptide. J. Bacteriol. 177:6381–6389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Akatsuka H, Binet R, Kawai E, Wandersman C, Omori K. 1997. Lipase secretion by bacterial hybrid ATP-binding cassette exporters: molecular recognition of the LipBCD, PrtDEF, and HasDEF exporters. J. Bacteriol. 179:4754–4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kamata R, Matsumoto K, Okamura R, Yamamoto T, Maeda H. 1985. The serratial 56K protease as a major pathogenic factor in serratial keratitis. Ophthalmology 92:1452–1459. 10.1016/S0161-6420(85)33855-1. [DOI] [PubMed] [Google Scholar]
- 19.Matsumoto K, Yamamoto T, Kamata R, Maeda H. 1984. Pathogenesis of serratial infection: activation of the Hageman factor-prekallikrein cascade by serratial protease. Biochemistry 96:739–749. [DOI] [PubMed] [Google Scholar]
- 20.Kamata R, Yamamoto T, Matsumoto K, Maeda H. 1985. A serratial protease causes vascular permeability reaction by activation of the Hageman factor-dependent pathway in guinea pigs. Infect. Immun. 48:747–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Molla A, Yamamoto T, Akaike T, Miyoshi S, Maeda H. 1989. Activation of Hageman factor and prekallikrein and generation of kinin by various microbial proteinases. J. Biol. Chem. 264:10589–10594. [PubMed] [Google Scholar]
- 22.Molla A, Akaike T, Maeda H. 1989. Inactivation of various proteinase inhibitors and the complement system in human plasma by the 56-kilodalton proteinase from Serratia marcescens. Infect. Immun. 57:1868–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oda T, Kojima Y, Akaike T, Ijiri S, Molla A, Maeda H. 1990. Inactivation of chemotactic activity of C5a by the serratial 56-kilodalton protease. Infect. Immun. 58:1269–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maeda H, Molla A, Oda T, Katsuki T. 1987. Internalization of serratial protease into cells as an enzyme-inhibitor complex with alpha 2-macroglobulin and regeneration of protease activity and cytotoxicity. J. Biol. Chem. 262:10946–10950. [PubMed] [Google Scholar]
- 25.Molla A, Kagimoto T, Maeda H. 1988. Cleavage of immunoglobulin G (IgG) and IgA around the hinge region by proteases from Serratia marcescens. Infect. Immun. 56:916–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kida Y, Inoue H, Shimizu T, Kuwano K. 2007. Serratia marcescens serralysin induces inflammatory responses through protease-activated receptor 2. Infect. Immun. 75:164–174. 10.1128/IAI.01239-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marty KB, Williams CL, Guynn LJ, Benedik MJ, Blanke SR. 2002. Characterization of a cytotoxic factor in culture filtrates of Serratia marcescens. Infect. Immun. 70:1121–1128. 10.1128/IAI.70.3.1121-1128.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Demidyuk IV, Kalashnikov AE, Gromova TY, Gasanov EV, Safina DR, Zabolotskaya MV, Rudenskaya GN, Kostrov SV. 2006. Cloning, sequencing, expression, and characterization of protealysin, a novel neutral proteinase from Serratia proteamaculans representing a new group of thermolysin-like proteases with short N-terminal region of precursor. Protein Expr. Purif. 47:551–561. 10.1016/j.pep.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 29.Tsaplina OA, Efremova TN, Kever LV, Komissarchik YY, Demidyuk IV, Kostrov SV, Khaitlina SY. 2009. Probing for actinase activity of protealysin. Biochemistry 74:648–654. 10.1134/S0006297909060091. [DOI] [PubMed] [Google Scholar]
- 30.Tsaplina O, Efremova T, Demidyuk I, Khaitlina S. 2012. Filamentous actin is a substrate for protealysin, a metalloprotease of invasive Serratia proteamaculans. FEBS J. 279:264–274. 10.1111/j.1742-4658.2011.08420.x. [DOI] [PubMed] [Google Scholar]
- 31.Bozhokina E, Tsaplina OA, Efremova TN, Kever LV, Demidyuk IV, Kostrov SV, Adam T, Kommissarchik YY, Khaitlina SY. 2011. Bacterial invasion of eukaryotic cells can be mediated by actin-hydrolysing metalloproteases grimelysin and protealysin. Cell Biol. Int. 35:111–118. 10.1042/CBI20100314. [DOI] [PubMed] [Google Scholar]
- 32.Riedel K, Ohnesorg T, Krogfelt KA, Hansen TS, Omori K, Givskov M, Eberl L. 2001. N-Acyl-l-homoserine lactone-mediated regulation of the Lip secretion system in Serratia liquefaciens MG1. J. Bacteriol. 183:1805–1809. 10.1128/JB.183.5.1805-1809.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shanks RMQ, Stella NA, Arena KE, Fender JE. 2013. Mutation of crp mediates Serratia marcescens serralysin and global secreted protein production. Res. Microbiol. 164:38–45. 10.1016/j.resmic.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Givskov M, Molin S. 1993. Secretion of Serratia liquefaciens phospholipase from Escherichia coli. Mol. Microbiol. 8:229–242. 10.1111/j.1365-2958.1993.tb01567.x. [DOI] [PubMed] [Google Scholar]
- 35.Bouhenni R, Gehrke A, Saffarini D. 2005. Identification of genes involved in cytochrome c biogenesis in Shewanella oneidensis, using a modified mariner transposon. Appl. Environ. Microbiol. 71:4935–4937. 10.1128/AEM.71.8.4935-4937.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Michaels B, Tisa LS. 2011. Swarming motility by Photorhabdus temperata is influenced by environmental conditions and uses the same flagella as that used in swimming motility. Can. J. Microbiol. 57:196–203. 10.1139/W10-119. [DOI] [PubMed] [Google Scholar]
- 37.Guzman L, Belin D, Carson M, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 177:4121–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Felföldi G, Marokházi J, Képiró M, Venekei I. 2009. Identification of natural target proteins indicates functions of a serralysin-type metalloprotease, PrtA, in anti-immune mechanisms. Appl. Environ. Microbiol. 75:3120–3126. 10.1128/AEM.02271-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Egile C, D'Hauteville H, Parsot C, Sansonetti PJ. 1997. SopA, the outer membrane protease responsible for polar localization of IcsA in Shigella flexneri. Mol. Microbiol. 23:1063–1073. 10.1046/j.1365-2958.1997.2871652.x. [DOI] [PubMed] [Google Scholar]
- 40.Baker SJ, Daniels C, Morona R. 1997. PhoP/Q regulated genes in Salmonella typhi: identification of melittin sensitive mutants. Microb. Pathog. 22:165–179. 10.1006/mpat.1996.0099. [DOI] [PubMed] [Google Scholar]
- 41.Murdoch SL, Trunk K, English G, Fritsch MJ, Pourkarimi E, Coulthurst SJ. 2011. The opportunistic pathogen Serratia marcescens utilizes type VI secretion to target bacterial competitors. J. Bacteriol. 193:6057–6069. 10.1128/JB.05671-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fritsch MJ, Trunk K, Diniz JA, Guo M, Trost M, Coulthurst SJ. 2013. Proteomic identification of novel secreted antibacterial toxins of the Serratia marcescens type VI secretion system. Mol. Cell. Proteomics 12:2735–2749. 10.1074/mcp.M113.030502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schnetz K, Toloczyki C, Rak B. 1987. Beta-glucoside (bgl) operon of Escherichia coli K-12: nucleotide sequence, genetic organization, and possible evolutionary relationship to regulatory components of two Bacillus subtilis genes. J. Bacteriol. 169:2579–2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lindahl T. 1979. DNA glycolases, endonucleases for apurinic/apyrimidinic sites, and base excision-repair. Prog. Nucleic Acid Res. Mol. Biol. 22:135–192. 10.1016/S0079-6603(08)60800-4. [DOI] [PubMed] [Google Scholar]
- 45.Lu AL, Li X, Gu Y, Wright PM, Chang DY. 2001. Repair of oxidative DNA damage: mechanisms and functions. Cell Biochem. Biophys. 35:141–170. 10.1385/CBB:35:2:141. [DOI] [PubMed] [Google Scholar]
- 46.Petit C, Sancar A. 1999. Nucleotide excision repair: from E. coli to man. Biochimie 81:15–25. 10.1016/S0300-9084(99)80034-0. [DOI] [PubMed] [Google Scholar]
- 47.Bregeon D, Colot V, Radman M, Taddei F. 2001. Translational misreading: a tRNA modification counteracts a +2 ribosomal frameshift. Genes Dev. 15:2295–2306. 10.1101/gad.207701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yim L, Moukadiri I, Bjork GR, Armengod ME. 2006. Further insights into the tRNA modification process controlled by proteins MnmE and GidA of Escherichia coli. Nucleic Acids Res. 34:5892–5905. 10.1093/nar/gkl752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kinscherf TG, Willis DK. 2002. Global regulation by gidA in Pseudomonas syringae. J. Bacteriol. 184:2281–2286. 10.1128/JB.184.8.2281-2286.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cho KH, Caparon MG. 2008. tRNA modification by GidA/MnmE is necessary for Streptococcus pyogenes virulence: a new strategy to make live attenuated strains. Infect. Immun. 76:3176–3186. 10.1128/IAI.01721-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shippy DC, Eakley NM, Bochsler PN, Chopra AK, Fadl AA. 2011. Biological and virulence characteristics of Salmonella enterica serovar Typhimurium following deletion of glucose-inhibited division (gidA) gene. Microb. Pathog. 50:303–313. 10.1016/j.micpath.2011.02.004. [DOI] [PubMed] [Google Scholar]
- 52.Sha J, Kozlova EV, Fadl AA, Olano JP, Peterson JW, Chopra AK, Houston CW. 2004. Molecular characterization of a glucose-inhibited division gene, gidA, that regulates cytotoxic enterotoxin of Aeromonas hydrophila. Infect. Immun. 72:1084–1095. 10.1128/IAI.72.2.1084-1095.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fulcher NB, Holliday PM, Klem E, Cann MJ, Wolfgang MC. 2010. The Pseudomonas aeruginosa Chp chemosensory system regulates intracellular cAMP levels by modulating adenylate cyclase activity. Mol. Microbiol. 76:889–904. 10.1111/j.1365-2958.2010.07135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McLaughlin HP, Caly DL, McCarthy Y, Ryan RP, Dow JM. 2012. An orphan chemotaxis sensor regulates virulence and antibiotic tolerance in the human pathogen Pseudomonas aeruginosa. PLoS One 7:e42205. 10.1371/journal.pone.0042205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Clissold PM, Pinting CP. 2000. PIN domains in nonsense-mediated mRNA decay and RNAI. Curr. Biol. 10:R888–R890. 10.1016/S0960-9822(00)00858-7. [DOI] [PubMed] [Google Scholar]
- 56.Glavan F, Behm-Ansmant I, Izaurraide E, Conti E. 2006. Structures of the PIN domains of SMG6 and SMG5 reveal a nuclease within the mRNA surveillance complex. EMBO J. 25:5117–5125. 10.1038/sj.emboj.7601377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huntzinger E, Kashima I, Fauser M, Sauliere J, Izaurralde E. 2008. SMG6 is the catalytic endonuclease that cleaves mRNAs containing nonsense codons in metazoan. RNA 14:2609–2617. 10.1261/rna.1386208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bodagai M, Ferenczi S, Bashtovyy D, Miclea P, Papp P, Dusha I. 2006. The ntrPR operon of Sinorhizobium meliloti is organized and functions as toxin-antitoxin module. Mol. Plant-Microbe Interact. 19:811–822. 10.1094/MPMI-19-0811. [DOI] [PubMed] [Google Scholar]
- 59.Mattison K, Wilbur JS, So M, Brennan RG. 2006. Structure of FitAB from Neisseria gonorrhoeae bound to DNA reveals a tetramer of toxin-antitoxin heterodimers containing pin domains and ribbon-helix-helix motifs. J. Biol. Chem. 281:37942–37951. 10.1074/jbc.M605198200. [DOI] [PubMed] [Google Scholar]
- 60.Frampton R, Aggio RBM, Villas-Boas SG, Arcus VL, Cook GM. 2012. Toxin-antitoxin systems of Mycobacterium smegmatis are essential for cell survival. J. Biol. Chem. 287:5340–5356. 10.1074/jbc.M111.286856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Winther KS, Gerdes K. 2009. Ectopic production of VapCs from Enterobacteria inhibits translation and trans-activates YoeB mRNA interferase. Mol. Microbiol. 72:918–930. 10.1111/j.1365-2958.2009.06694.x. [DOI] [PubMed] [Google Scholar]
- 62.Silva CP, Waterfield NR, Daborn PJ, Dean P, Chilver T, Au CPY, Sharma S, Potter U, Reynolds SE, Ffrench-Constant RH. 2002. Bacterial infection of a model insect: Photorhabdus luminescens and Manduca sexta. Cell. Microbiol. 4:329–339. 10.1046/j.1462-5822.2002.00194.x. [DOI] [PubMed] [Google Scholar]
- 63.Bowen DJ, Rocheleau TA, Grutzmacher CK, Meslet L, Valens M, Marble D, Dowling A, Ffrench-Constant RH, Blight MA. 2003. Genetic and biochemical characterization of PrtA, an RTX-like metalloprotease from Photorhabdus. Microbiology 149:1581–1591. 10.1099/mic.0.26171-0. [DOI] [PubMed] [Google Scholar]
- 64.Simon R, Priefer U, Puhler A. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in Gram-negative bacteria. Biotechnology 1:784–791. 10.1038/nbt1183-784. [DOI] [Google Scholar]
- 65.Kolter R, Helinski DR. 1978. Construction of plasmid R6K derivatives in vitro: characterization of the R6K replication origin. Plasmid 1:571–580. 10.1016/0147-619X(78)90014-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.