

Abstract
SlrP is an E3 ubiquitin ligase that can be translocated into eukaryotic host cells by the two type III secretion systems that are expressed by Salmonella enterica serovar Typhimurium and are encoded in Salmonella pathogenicity islands 1 (SPI1) and 2 (SPI2). Expression of slrP and translocation of its product were examined using lac, 3×FLAG, and cyaA′ translational fusions. Although slrP was expressed in different media, optimal expression was found under conditions that imitate the intravacuolar environment and promote synthesis of the SPI2-encoded type III secretion system. Translocation into mammalian cells took place through the SPI1- or the SPI2-encoded type III secretion system, depending on specific host cell type and timing. A search for genetic factors involved in controlling the expression of slrP unveiled LeuO, Lon, and the two-component system PhoQ/PhoP as novel regulators of slrP. Our experiments suggest that LeuO and Lon act through HilD under SPI1-inducing conditions, whereas PhoP directly interacts with the slrP promoter to activate transcription under SPI2 inducing conditions.
INTRODUCTION
Type III secretion systems (T3SSs) are employed by Gram-negative bacterial pathogens and symbionts to translocate proteins, known as effectors, directly into the cytosol of eukaryotic host cells. These effectors can manipulate certain cellular signal transduction pathways to create a bacterial survival or replicative niche (1).
The animal pathogen Salmonella enterica serovar Typhimurium encodes two distinct T3SSs, T3SS1 and T3SS2, in Salmonella pathogenicity islands 1 (SPI1) and 2 (SPI2), respectively (2–4). At least 38 effectors are secreted through these systems: 7 through T3SS1, 22 through T3SS2, and 9 through both systems (5). Although some effectors are encoded in SPI1 or SPI2, most of them, including those secreted by both systems, are encoded outside these islands. Effectors secreted by T3SS1 upon contact with the host cell are involved in remodeling of the host cell cytoskeleton to permit Salmonella invasion of nonphagocytic cells through a trigger mechanism and are responsible for symptoms associated to enteric salmonellosis (6). In contrast, expression of T3SS2 is optimal inside the Salmonella-containing vacuole in response to signals such as low pH and low Mg2+ concentration. This system is essential for intracellular survival in macrophages (7).
Finely tuned regulation of both systems is necessary to ensure adaptation to different environmental conditions. HilA is the central regulator in the SPI1 regulatory scheme, activating genes encoding all the components necessary for a functional T3SS1. Expression of hilA is controlled by the combined action of three AraC-like transcriptional activators: HilC, HilD, and RtsA (8). The two-component regulatory system SsrA/SsrB is the central regulator of T3SS2, with PhoQ/PhoP and EnvZ/OmpR also playing an important role (9). Interestingly, although PhoP has a positive effect on SPI2 expression and a general negative effect on SPI1 expression, it also activates some SPI1 genes (10). The transition from T3SS1 to T3SS2 expression, observed in certain infection models, is driven by processes of differential regulation and cross talk between regulatory proteins. There are, however, examples of expression of T3SS1 effectors at late stages of infection (11, 12) and circumstances of coexpression and cooperation between both T3SSs, depending on the host cell type or the entry mode (13).
Effectors should be synthesized in coordination with their cognate secretion system. This is straightforward for those that are encoded in SPI1 or SPI2. The regulation, however, may be more complex for others, in particular for effectors, such as SlrP, that have been shown to be secreted by both T3SSs.
SlrP (for Salmonella leucine-rich repeat protein) was identified as a host-specific virulence factor (14). Analysis of the amino acid sequence of this protein together with the sequence of the Salmonella effectors SspH1, SspH2, and other related proteins from Shigella and Yersinia, identified four domains (15). The N-terminal domains A and B are involved in T3SS secretion and translocation into RAW264.7 macrophages, which is dependent on T3SS1 at 1 h postinfection (p.i.) and on T3SS2 at 6 h p.i. (16). The third domain contains several leucine-rich repeats that could function as a protein-binding motif (17). The C-terminal domain is similar to the C-terminal domain of the IpaH family in Shigella flexneri. This domain is responsible for the E3 ubiquitin ligase activity that has been shown for SspH1, SspH2, SlrP, and members of the IpaH family. They constitute a new class of ubiquitin ligases known as NEL (for novel E3 ligase) (18–21). We showed previously that SlrP binds and ubiquitinates human thioredoxin-1 (22). The localization of SlrP in host epithelial cells is mainly cytosolic, but it is also partially located in the endoplasmic reticulum, where it can interact with the chaperone ERdj3 (23). SlrP could promote cell death by interfering with the functions of these target proteins.
The regulation of slrP expression has been studied specifically in the context of its coordination with the expression of T3SS1 (24, 25). In this context, slrP is induced by overexpression of HilC, HilD, and RtsA independently of the central SPI1 regulator HilA, with RtsA being the best inducer. However, since SlrP is also a potential substrate for T3SS2, we were interested in carrying out a comparative analysis of the environmental and genetic factors that control slrP expression under SPI1- and SPI2-inducing conditions and of the relative importance of both T3SSs in the translocation of SlrP during infection of different host cell types.
In this study, we show that slrP is expressed under both SPI1- and SPI2-inducing conditions, but the highest expression was observed under SPI2-inducing conditions. Translocation can occur through both T3SSs or specifically through T3SS1 or T3SS2, depending on the host cell type and infection timing. We show that, under SPI1-inducing conditions, LeuO and Lon are indirect regulators of the slrP expression that act through HilD. We also show that the main activator of this gene under SPI2-inducing conditions is the two-component system PhoQ/PhoP and that PhoP can directly bind to the promoter region of slrP.
MATERIALS AND METHODS
Bacterial strains, bacteriophages, and strain construction.
Escherichia coli and S. enterica serovar Typhimurium strains used in this study are described in Table 1. Salmonella strains were derived from the mouse-virulent strain ATCC 14028. Transductional crosses using phage P22 HT 105/1 int201 (26) were used for strain construction (27). To obtain phage-free isolates, transductants were purified by streaking on green plates. Green plates were prepared as described previously (28), except that methyl blue (Sigma) was substituted for aniline blue. Phage sensitivity was tested by cross-streaking with the clear-plaque mutant P22 H5.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Relevant characteristic(s) | Source or reference(s) |
|---|---|---|
| E. coli strains | ||
| DH5α | supE44 ΔlacU169 (ϕ80 lacZΔM15) hsdR17 recA1 endA1 gyrA96 thi-1 relA1 | 51 |
| XL1-Blue | recA1 endA1 gyrA96 thi-1 hsdR17 supE44 relA1 Δlac-pro/F′ proAB lacIq lacZΔM15 Tn10 (Tetr) | 52 |
| M15 | lac ara gal mtl | Qiagen |
| S. enterica serovar Typhimurium strainsa | ||
| 14028 | Wild type | ATCC |
| 55130 | 14028 phoQ24 (PhoP constitutive) | E.A. Groisman |
| SV4699 | 14028 phoP7953::Tn10 Tcr | 53, 54 |
| SV5193 | 14028 slrP::3×FLAG Kmr | 22 |
| SV5373 | 14028 ΔhilA | J. López-Garrido |
| SV5452 | 14028 ΔssrB::Cmr | 55 |
| SV5487 | 14028 Δlon | J. López-Garrido |
| SV6016 | 14028 slrP(1–130)::mini-Tn5cyaA′ | 33 |
| SV6017 | 14028 ΔSPI2::Cmr | 33 |
| SV6048 | 14028 ΔleuO::Cmr | E. Espinosa |
| SV6055 | 14028 ΔSPI1::Kmr | 33 |
| SV6084 | 14028 slrP::lacZ Kmr | This study |
| SV6402 | 14028 ΔhilD::Cmr | J. López-Garrido |
| SV7859 | 14028 slrP::lacZ T-POP-leuO Kmr Tcr | This study |
| SV7960 | 14028 slrP::lacZ T-POP-hilD Kmr Tcr | This study |
| Plasmids | ||
| p3138 | lacZ template vector | 29 |
| pIC552 | Parent for lacZ transcriptional fusions, Apr | 56 |
| pIZ1949 | pQE30-phoP | 35 |
| pIZ2028 | pIC552-slrP(−460/+21) | This study |
| pIZ2029 | pIC552-slrP(−139/+21) | This study |
| pIZ2033 | pIZ2028-(G-35C) | This study |
| pIZ2035 | pIZ2029-(G-35C) | This study |
| pIZ2040 | pIZ2028-(T-40C) | This study |
| pIZ2041 | pIZ2029-(T-40C) | This study |
| pKD46 | bla PBAD gam bet exo pSC101 oriTS | 57 |
| pREP4 | lacI Kmr | Qiagen |
Derivatives of these strains were used as indicated in the text.
Bacterial culture.
The standard culture medium for S. enterica and E. coli was Luria-Bertani (LB) broth. For SPI1-inducing conditions, S. enterica strains were grown overnight at 37°C in LB–0.3 M NaCl medium under static conditions. For SPI2-inducing conditions, bacteria from cultures in LB were washed and diluted 1:100 with low-phosphate, low-magnesium minimal medium (LPM) at pH 5.8 and incubated overnight at 37°C with shaking. LPM contained 80 mM 2-(N-morpholino)ethanesulfonic acid (pH 5.8), 5 mM KCl, 7.5 mM (NH4)2SO4, 0.5 mM K2SO4, 0.1% Casamino Acids, 38 mM glycerol, 337.5 μM K2HPO4-KH2PO4 (pH 7.4), and 8 μM MgCl2. For some experiments, the concentration of NaCl, the concentration of MgCl2, or the pH of the medium was modified as indicated in Results. Solid media contained 1.5% agar. Antibiotics were used at the following final concentrations in rich medium: kanamycin (Km), 50 μg ml−1; chloramphenicol (Cm), 20 μg ml−1; ampicillin (Ap), 100 μg ml−1; and tetracycline (Tc), 20 μg ml−1. In minimal medium, antibiotics were used at the following concentrations: Km, 125 μg ml−1; Cm, 5 μg ml−1; Ap, 50 μg ml−1; and Tc, 10 μg ml−1. Plates for monitoring β-galactosidase activity contained 5-bromo-4-chloro-indolyl-β-d-galactopyranoside (X-Gal; final concentration, 40 μg ml−1).
DNA amplification with the PCR.
Amplification reactions were carried out in a T100 thermal cycler (Bio-Rad). The final volume of reaction mixtures was 50 μl, and the final concentration of MgCl2 was 1.5 mM. Reagents were used at the following concentrations: deoxynucleoside triphosphates (dNTPs), 300 μM; primers, 0.3 μM; and Taq polymerase (Kapa HiFi DNA polymerase; Kapa Biosystems), 1 unit per reaction. The thermal program included the following steps: (i) initial denaturation, 3 min at 95°C; (ii) 30 cycles of denaturation (98°C, 20 s), annealing (55°C, 15 s), and extension (72°C, 30 s per kb); and (iii) final incubation at 72°C for 7 min, to complete extension. To generate point mutations in the slrP promoter cloned in pIC552 the thermal program included the following steps: (i) initial denaturation, 30 s at 95°C and (ii) 12 cycles of denaturation (95°C, 30 s), annealing (55°C, 1 min), and extension (68°C, 5 min). Primers are listed in Table 2. PCR constructs were sequenced with an automated DNA sequencer (Stab Vida, Oeiras, Portugal).
TABLE 2.
Oligonucleotides used in this study
| Purpose and oligonucleotide | Sequence (5′–3′) |
|---|---|
| Generation of slrP::lacZ | |
| slrPRedLacZdir | GTTCAGATCAGGTAGGGAAAATATGTTTAATATTACTAATCCCGTCGTTTTACAACGTCG |
| slrPRedRev | TAAACAGGGCTCTCTCCCTCTTCTGATAAACTGCGTTCAGCGTGTAGGCTGGAGCTGCTTC |
| ST-PCR | |
| tpop1 | GCCTTCTTATTCGGCCTTGAATTGATCATATGCGG |
| stACGCC | GGCCACGCGTCGACTAGTACNNNNNNNNNNACGCC |
| stGATAT | GGCCACGCGTCGACTAGTACNNNNNNNNNNGATAT |
| tpop2 | CTTTTTCCGTGATGGTAACC |
| st1 | GGCCACGCGTCGACTAGTAC |
| Construction of pIZ2028 | |
| slrP(−460)5′ | CATGAGATCTCGATCGCCAGCGAGTCATCG |
| slrP(+21)3′ | GATCCTCGAGATTTTCCCTACCTGATCTG |
| Construction of pIZ2029 | |
| slrP(−139)5′ | CATGAGATCTCTACTTAAAAAAGAGGTGATG |
| slrP(+21)3′ | As above |
| Construction of pIZ2033/2035 | |
| slrPG35C5′ | GTGACCTCTTATTTAAACTTGAGAATATCCTTATC |
| slrPG35C3′ | GATAAGGATATTCTCAAGTTTAAATAAGAGGTCAC |
| Construction of pIZ2040/2041 | |
| slrPT40C5′ | CGACTGTGACCTCTTATCTAAAGTTGAGAATATCC |
| slrPT40C3′ | GGATATTCTCAACTTTAGATAAGAGGTCACAGTCG |
| slrP promoter | |
| promslrPdir | CGATCGCCAGCGAGTCATCG |
| promslrPrev | ATTTTCCCTACCTGATCTG |
| slyB promoter | |
| promslyBdir | AGACTTGCCTGTTGCGCAAC |
| promslyBrev | AAACGCTATTTCAGCATCCC |
| phoN promoter | |
| promphoNdir | AATGCGTGTCAGTCAGGCAC |
| promphoNrev | TTAGCTACGATCAGTGGTAG |
Plasmids.
Plasmids used in this study are listed in Table 1. Plasmids expressing transcriptional lacZ fusions were derivatives of pIC552. To construct these plasmids, DNA from strain 14028 was used as a template for PCR amplification with the primers listed in Table 2. The amplified fragments were digested with BglII and XhoI and ligated with BglII/XhoI-digested pIC552. To generate point mutations in the slrP promoter, pIZ2028 and pIZ2029 were used as templates for PCR amplification using primer pairs slrPG35C5′/slrPG35C3′ and slrPT40C5′/slrPT40C3′. Products were digested with 1 μl of DpnI (10 U μl−1) for 1 h at 37°C and used to transform E. coli DH5α. All constructs were confirmed by DNA sequencing.
Construction of a chromosomal lacZ fusion.
A previously described one-step procedure was used to generate a translational slrP::lacZ fusion (29). Plasmid p3138 was used as the template for amplification of lacZ and a Kmr gene using the primers slrPRedLacZdir and slrPRedRev. The PCR product was used to transform the wild-type strain ATCC 14028 carrying the Red recombinase expression plasmid pKD46.
β-Galactosidase assays.
Levels of β-galactosidase were assayed as described previously (30), using the CHCl3-SDS permeabilization procedure. Bacteria were grown overnight in LB medium at 37°C with shaking. Cells were then diluted 1:50 in LB medium or washed and diluted 1:50 in LPM and incubated at 37°C with shaking, for LB and LPM, or without shaking, only for LB. Cultures were incubated for 1.5 to 2 h to reach exponential phase or 8 h to reach stationary phase.
Mammalian cell culture.
HeLa cells (human epithelial; ECAC no. 93021013), RAW264.7 cells (murine macrophages; ECACC no. 91062702), and NRK-49F cells (normal rat kidney fibroblasts; ATCC CRL1570) were cultured in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal calf serum. Two millimolar l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin were included in the culture media. Cells were maintained in a 5% CO2 humidified atmosphere at 37°C.
Bacterial infection of cultured cells.
Mammalian cells were plated in 24-well plates at 1.5 × 105 cells per well and incubated 24 h at 37°C with 5% CO2 in media without antibiotics. For infections under SPI1-inducing (invasive) conditions, bacteria grown overnight in LB–0.3 M NaCl in a tightly closed tube without shaking were added at a multiplicity of infection of 150. For infections of RAW264.7 cells under noninvasive conditions, bacteria were grown in LB for 24 h at 37°C with shaking. Bacteria were centrifuged onto the cell monolayer at 200 × g for 5 min and then incubated at 37°C with 5% CO2. The cell culture was washed twice with phosphate-buffered saline (PBS) at 1 h p.i., overlaid with DMEM containing 100 μg ml−1 gentamicin, and incubated for another hour. The culture was then washed twice with PBS, covered with DMEM with gentamicin 16 μg ml−1, and incubated for 2 to 22 h.
Protein translocation assays.
Following the infections described above, the translocation of the SlrP-CyaA′ fusion into the eukaryotic cells was monitored by measuring the levels of cyclic AMP (cAMP). The infected cells were lysed, and the level of cAMP in the lysates was determined using a colorimetric direct cAMP enzyme immunoassay kit (Arbor Assays) according to the manufacturer's instructions.
Mutagenesis with T-POP.
Strain 14028 of S. enterica serovar Typhimurium was mutagenized with T-POP, a derivative of transposon Tn10dTc that lacks terminators for tetA and tetR, allowing Tc-dependent overexpression of genes adjacent to the point of insertion from internal T-POP promoters (31). Pools of 5,000 colonies, each carrying an independent T-POP insertion, were then prepared and lysed with phage P22 HT. The lysates were used to transduce strain SV6084 (14028 slrP::lacZ), selecting Tc-resistant transductants on LB and LPM plates supplemented with X-Gal, Tc, and Km.
Molecular characterization of T-POP insertions.
A semirandom, two-step PCR protocol (32) was used to amplify genomic regions adjacent to the T-POP insertions. The reactions were carried out as previously described (33) using primers listed in Table 2. The final products were sequenced using the primers tpop2 and st1. Sequence analysis was performed with molecular biology algorithms from the National Center for Biotechnology Information at www.ncbi.nlm.nih.gov.
Western blotting and antibodies.
Salmonella strains were cultured as explained above for β-galactosidase assays. The bacteria were then pelleted by centrifugation and resuspended in SDS-PAGE sample buffer. Proteins were separated by SDS-PAGE (mini-Protean TGX precast gels, 12% or 4 to 15%) and electrophoretically transferred to nitrocellulose filters for Western blot analysis using anti-FLAG M2 monoclonal antibodies (1:5,000; Sigma), anti-DnaK (8E2/2) monoclonal antibodies (1:5,000; Assay Designs), and anti-GroEL polyclonal antibodies (1:20,000; Sigma). Goat anti-mouse horseradish peroxidase (HRP)-conjugated antibodies (1:5,000; Bio-Rad) and goat anti-rabbit HRP-conjugated antibodies (1:10,000; GE Healthcare) were used as secondary antibodies.
Protein purification and phosphorylation.
His6-PhoP protein was produced and purified as previously described (34) with some modifications (35). For slot blot assays, S. enterica His6-PhoP was phosphorylated with acetyl phosphate as previously described (36) with modifications. Briefly, His6-PhoP was incubated in 20 μl of phosphorylation buffer (50 mM Tris-HCl [pH 7.5], 50 mM KCl, 10 mM MgCl2) containing 10 mM acetyl phosphate (Sigma-Aldrich) for 1 h at 37°C.
Slot blot for DNA-protein interaction.
DNA fragments used for the PhoP binding assay were amplified by PCR using Salmonella 14028 as a template. The reverse primers, listed in Table 2, were labeled with FAM fluorochrome. PCR amplification rendered fragments of 281, 355, and 481 bp for the phoN, slyB, and slrP promoters, respectively. The binding assay was carried out as previously described (36) with modifications. Briefly, a solution of 10 nM FAM-labeled DNA and 0, 0.125, 0.25, 0.5, 1, and 2 μM phosphorylated His6-PhoP was prepared in binding buffer (50 mM Tris-HCl [pH 8.0], 50 mM KCl) in a total volume of 20 μl and incubated for 30 min at room temperature. Nitrocellulose filters were blocked with 0.5% (wt/vol) skim milk powder and 50 μg herring sperm DNA per ml in PBS and washed with PBS. Binding reaction mixtures were diluted 1:10 with PBS and blotted onto blocked nitrocellulose filters using a PR 600 slot blot manifold (Hoefer Scientific Instruments) connected to a portable vacuum/pressure pump (Millipore). Wells were then washed five times with PBS, and membranes were air dried. Images were acquired using a Fujifilm FLA-5100 system, and quantification was performed using Image J software.
Statistical analyses.
Student's t test was used to analyze differences in enzymatic activities. P values of 0.05 or less were considered significant.
RESULTS
Expression of slrP in different media.
Because SlrP is a potential substrate for T3SS1 and T3SS2, it is expected to be synthesized under a wide range of environmental conditions. To study the critical factors that may influence the expression of slrP, we used a lacZ fusion that was created in the bacterial chromosome under the control of the native transcriptional and translational signals. First, we compared the activity of the fusion under previously described SPI1- and SPI2-inducing conditions. As seen in Fig. 1A, although slrP was expressed in both cases, expression was increased more than 2-fold under SPI2-inducing conditions.
FIG 1.
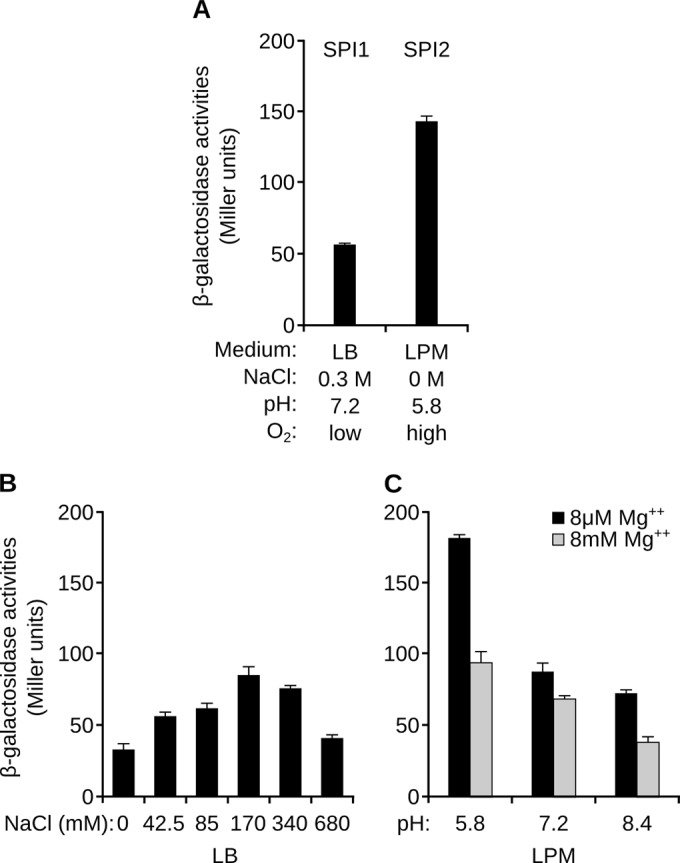
Expression of slrP in different culture media. β-Galactosidase activities were measured from overnight cultures of an S. enterica strain carrying a chromosomal slrP::lacZ translational fusion. (A) Bacteria were incubated overnight at 37°C in LB with 0.3 M NaCl without shaking (SPI1) or in LPM at pH 5.8 with shaking (SPI2). (B) Different concentrations of NaCl were used to test the role of osmolarity in sseK1 expression under SPI1-inducing conditions. (C) Activities were measured after growth in LPM with different pH and Mg2+ concentrations, as indicated. Means and standard deviations from three independent β-galactosidase measurements are shown.
One of the factors that induce expression of T3SS1 is high osmolarity (37). The role of this factor in the expression of slrP was tested in LB with different concentrations of NaCl. Optimal expression in this medium was obtained at 170 mM NaCl (Fig. 1B), whereas slight repression was observed with the extreme concentrations used (0 and 680 mM).
Two relevant factors that are present in the LPM used for induction of SPI2 are low Mg2+ concentration and low pH. As seen in Fig. 1C, both factors are important to achieve an optimal expression of slrP: both the increase of pH from 5.8 to 7.2 and the increase of Mg2+ concentration from 8 μM to 8 mM significantly reduced the level of β-galactosidase activity associated with the slrP::lacZ fusion.
Kinetics of translocation of SlrP into different cell types.
The results presented above support the idea that the expression of slrP, although overlapping the expression of T3SS1 and T3SS2, is maximal under SPI2-inducing conditions. To investigate the role of these systems in the secretion of SlrP under a variety of physiological conditions, we carried out bacterial infections of different mammalian cell types and measured the translocation of the effector into the host cytosol over time. For these experiments we took advantage of a chromosomal translational slrP::cyaA′ fusion that was previously generated in our laboratory as a derivative of strain ATCC 14028 of S. enterica serovar Typhimurium (33). The translocation of SlrP fused to the catalytic domain of CyaA from Bordetella pertussis was detected as an increase in the concentration of cAMP (38). Three mammalian cell lines previously validated as appropriate models for in vitro Salmonella infections were used: (i) human epithelial HeLa cells, (ii) murine macrophage-like RAW264.7, and (iii) NRK-49F rat fibroblasts. Infections of nonphagocytic cells (HeLa and NRK cells) were carried out with invasive bacteria, i.e., cells were grown under SPI1-inducing conditions. The same conditions were used to infect RAW cells for 1 or 2 h. However, noninvasive bacteria were used for longer infections of RAW cells to prevent rapid cell death induced by invasive bacteria in macrophages (39). Infections were also carried out using derivatives of S. enterica serovar Typhimurium lacking T3SS1, T3SS2, or both. As seen in Fig. 2 the kinetics of translocation was different for every cell type. (i) Translocation into epithelial cells was observed from 1 h to 24 h p.i. and was always dependent on T3SS1 (Fig. 2A). (ii) The situation for macrophages was more complex. SlrP was injected through T3SS1 at 1 h and 2 h p.i. when invasive bacteria were used; however, at 8, 16, and 24 h p.i. (when noninvasive bacteria were used), SlrP was the substrate of T3SS2 (Fig. 2B). (iii) Interestingly, the results for NRK suggest that translocation of SlrP into fibroblasts is specifically T3SS1 dependent from 1 to 8 h p.i. but can occur by T3SS1 and T3SS2 at later times p.i., since both systems needed to be inactivated simultaneously to prevent translocation (Fig. 2C).
FIG 2.

Kinetics of translocation of SlrP into different cell types. Human epithelial HeLa cells (A), RAW264.7 murine macrophage-like cells (B), and NRK-49F normal rat kidney fibroblasts (C) were infected with derivatives of S. enterica serovar Typhimurium 14028 (wild-type [wt], ΔSPI1, ΔSPI2, and ΔSPI1 ΔSPI2 strains) carrying a chromosomal SlrP-CyaA′ fusion expressed under the native slrP promoter. To detect translocation, the level of cAMP was measured 1, 2, 4, 8, 16, and 24 h p.i. Bacteria were grown under SPI1-inducing conditions for most infections. Noninvasive bacteria were used specifically for infections of RAW264.7 cells for 4, 8, 16, and 24 h. Means and standard deviations from triplicate experiments are represented.
Regulation of slrP expression by PhoP, Lon, LeuO, and HilD.
Next, we decided to look for genetic factors controlling the expression of slrP using two complementary approaches. First we tested the effect of mutations in genes encoding regulators known to control the expression of T3SS1 and T3SS2: phoP, phoQ, ssrB, hilD, and hilA. The level of expression was assessed in liquid bacterial cultures grown under SPI1 (Fig. 3A)- or SPI2 (Fig. 3B)-inducing conditions using the translational slrP::lacZ fusion mentioned above. The results suggest that the PhoQ/PhoP system is a major regulator of slrP expression: there is a 10-fold decrease in expression in a phoP-null mutant under SPI2-inducing conditions and a 4-fold increase in a phoQ24 mutant (a mutant with constitutive activation of the system) under SPI1-inducing conditions. Some effects were also observed for the hilD and ssrB mutations under SPI1- and SPI2-inducing conditions, respectively. These results were confirmed at the protein level using an SlrP-3×FLAG fusion that was detected by Western blotting using an anti-FLAG antibody (Fig. 3, bottom). Interestingly, the effect of the hilD mutation was more dramatic at this level, suggesting that an indirect posttranslational regulatory mechanism may also operate.
FIG 3.
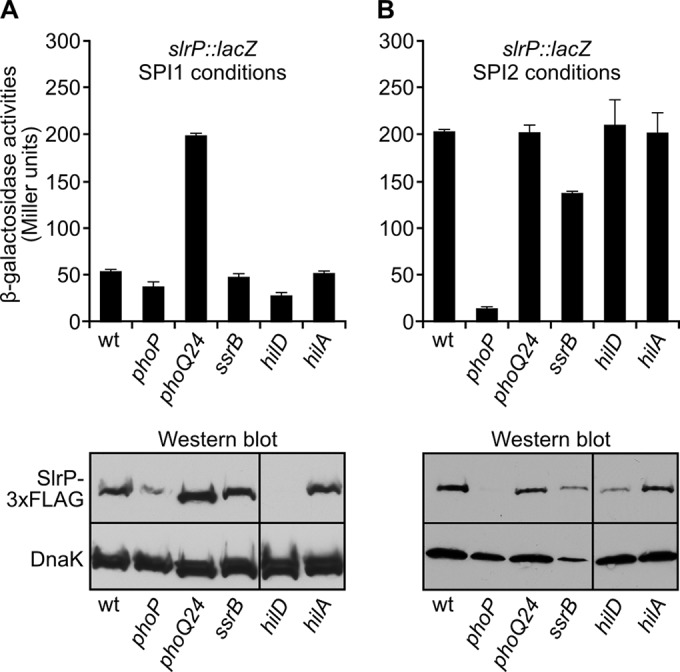
Regulation of slrP by the PhoQ/PhoP system. Expression of slrP was studied in different genetic backgrounds, as indicated, under SPI1 (A)- or SPI2 (B)-inducing conditions. Means and standard deviations from three independent β-galactosidase measurements are shown for strains carrying a slrP::lacZ fusion (top). Expression at the protein level was assessed by immunoblotting using strains expressing SlrP-3×FLAG (bottom). A monoclonal anti-FLAG antibody was used to detect the fusion protein, and a polyclonal anti-DnaK antibody was used as a loading control.
The existence of additional regulators was investigated using a second approach based on a T-POP screen (31). Bacteria carrying the slrP::lacZ fusion were mutagenized with the defective transposon and plated on media containing Tc, to select for the presence of T-POP, and X-Gal, a chromogenic indicator of slrP::lacZ expression. The screens were carried out in LB with 0.3 M NaCl and in LPM, and colonies exhibiting some color shift, darker or lighter blue, were analyzed. About 40,000 colonies were screened in each medium, and a total of 48 colonies were initially selected. Finally, only 4 T-POP insertions caused differential expression of slrP in β-galactosidase assays in a reproducible way after reconstruction experiments. All of them showed their effects under SPI1-inducing conditions (LB–0.3 M NaCl). The insertions were mapped by sequencing DNA adjacent to the transposon.
Two insertions increased expression of slrP in a Tc-independent manner, suggesting that a gene encoding a negative regulator was disrupted by the transposon in these cases. These insertions were located in lon, the gene encoding the protease Lon. Confirmation that the disruption of lon was responsible for the observed effect was obtained by deleting the lon gene in the strain carrying the slrP::lacZ fusion and measuring β-galactosidase activity (Fig. 4A).
FIG 4.
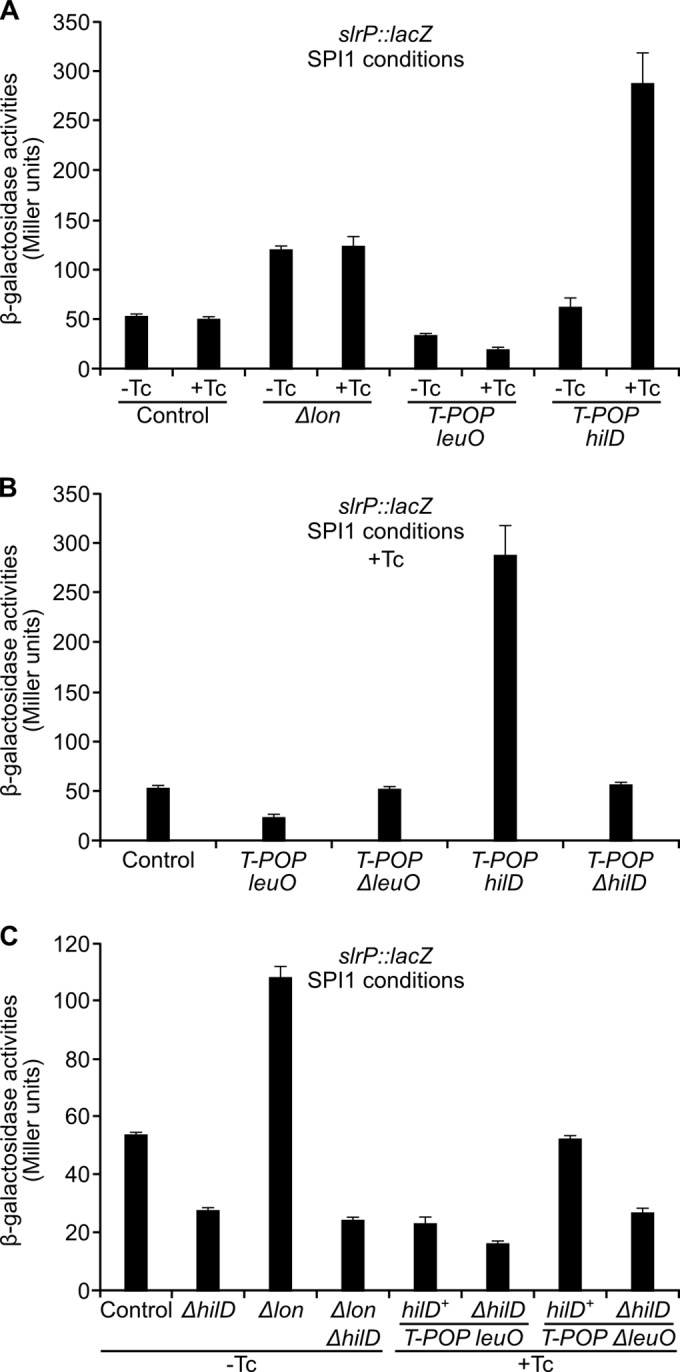
Regulation of slrP by lon, leuO, and hilD. (A) β-Galactosidase activity of slrP::lacZ in different genetic backgrounds: Δlon, T-POP leuO, and T-POP hilD. Tetracycline was added to the indicated cultures in order to express leuO or hilD from an internal T-POP promoter. (B) β-Galactosidase activity of slrP::lacZ in the presence and in the absence of LeuO or HilD. (C) Epistasis analysis of slrP expression. Effect of HilD expression on the β-galactosidase activity of slrP::lacZ in the presence and in the absence of Lon and LeuO.
The remaining two insertions had opposite effects on slrP expression, but both were dependent on Tc (Fig. 4A). One was adjacent to leuO and caused a decrease in the expression of slrP. The effect of the transposon was suppressed by a deletion of leuO, suggesting that expression of leuO from an outward Tc-dependent T-POP promoter was responsible for the observed effect. The other insertion was located near hilD. This insertion increased expression of slrP in the presence of Tc, and the increase was suppressed by deleting hilD (Fig. 4B).
It was shown previously that Lon protease degrades HilD (40) and that the transcriptional regulator LeuO directly activates transcription of hilE (41), whose product inhibits HilD activity (42). As a consequence, both Lon and LeuO modulate the expression of SPI1 through HilD. To investigate whether slrP regulation follows the same pattern, we carried out epistasis analysis. Results shown in Fig. 4C support the model that Lon and LeuO regulate slrP in a manner that is mostly HilD dependent.
The role of Lon, LeuO, and HilD in the regulation of slrP and the relationships between the three regulators were further studied at the protein level using strains expressing SlrP-3×FLAG (Fig. 5). The results of this analysis support the conclusions obtained with the lac fusion.
FIG 5.
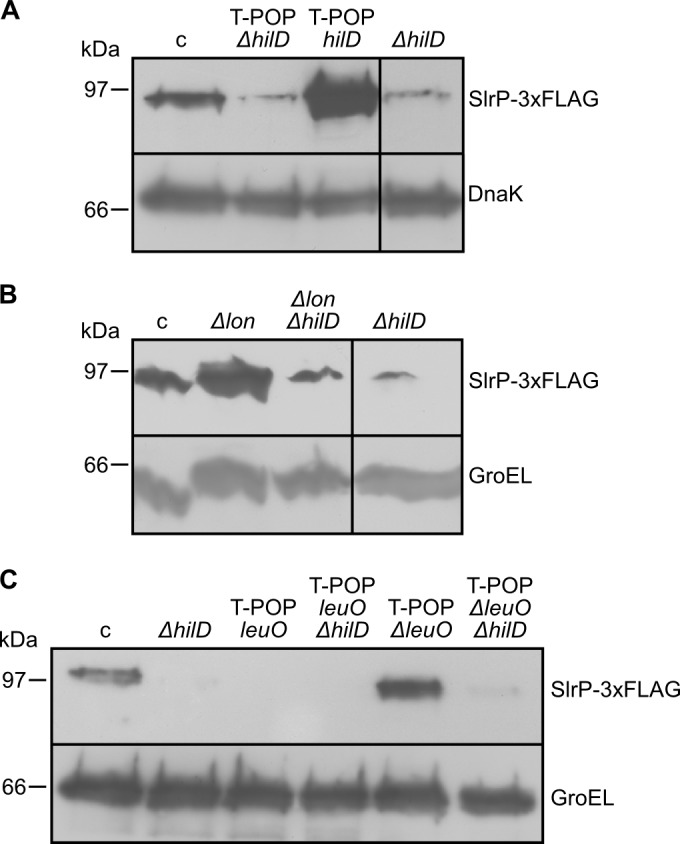
Regulation of slrP by lon, leuO, and hilD at the protein level. The level of SlrP was studied by immunoblotting using strains expressing a 3×FLAG fusion. Extracts from stationary-phase cultures of these strains were resolved by SDS-PAGE (12%), and a monoclonal anti-FLAG antibody was used for Western blotting (upper bands). Polyclonal anti-GroEL or anti-DnaK antibodies were used as a loading control (lower bands). Molecular mass markers are indicated on the left. c, wild-type background. (A) Effects of overexpression and deletion of hilD on slrP expression. (B) Effect of Lon on slrP expression in the presence and in the absence of HilD. (C) Effect of LeuO on slrP expression in the presence and in the absence of HilD.
Direct regulation of slrP expression by PhoP.
The data above suggest that the PhoQ/PhoP two-component system has a major role in the regulation of the expression of slrP. To study if PhoP was a direct activator of slrP transcription, first we analyzed the promoter region of this gene. The transcriptional start site (TSS) of slrP was determined in a recent global analysis carried out on strain S. enterica serovar Typhimurium strain SL1344 using RNA sequencing techniques (43). The +1 nucleotide was found to be a T located 21 nucleotides upstream of the translational start codon. Putative −10 and −35 σ70 promoter consensus motifs were identified by visual inspection, together with a putative PhoP-binding site (Fig. 6A). A similar motif is found in the promoters of a subset of PhoP-activated genes, including steA, phoP, mgtA, pmrD, slyB, and orgB (Fig. 6B).
FIG 6.
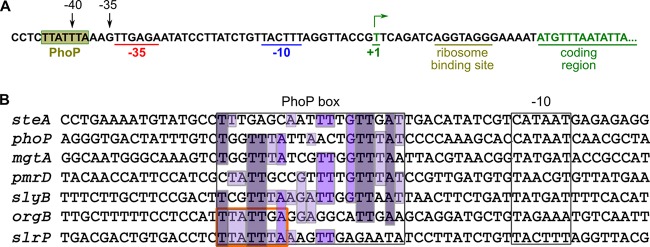
Analysis of the promoter region of slrP. (A) The sequence surrounding the transcriptional start site (+1) is shown. The consensus sequences for σ70-dependent transcription, putative PhoP-binding site, ribosomal-binding site, and start of the coding region are indicated. (B) Alignment of the promoter region of slrP and six PhoP-activated genes with a similar architecture. Putative −10 and PhoP-binding boxes are indicated.
The role of this region in driving transcription and PhoP-regulation of slrP was tested using the promoter-probe vector pIC552, which permits the construction of transcriptional lac fusions. DNA fragments containing the promoter and 5′ untranslated regions of slrP, from −460 to +21 and from −139 to +21, were cloned into this plasmid and introduced into the wild type and a phoP derivative of S. enterica serovar Typhimurium. The level of expression of the lac fusions indicated that these fragments contained the motifs necessary for transcription and PhoP regulation of slrP (Fig. 7A). To analyze the relevance of the predicted PhoP box, we constructed two point mutations: a transition inside the putative PhoP motif, at position −40, and a transversion outside this motif, at position −35 (Fig. 6A). Whereas the second mutation had no effect, the first resulted in a dramatic reduction of expression and PhoP-dependent regulation (Fig. 7A), supporting a role for this site in mediating PhoP regulation. Finally, we investigated the possibility of a direct interaction of PhoP with the promoter of slrP using a slot blot technique. Phosphorylated His6-PhoP was used for these assays together with PCR-amplified fragments derived from the slrP promoter and the promoters of slyB and phoN, which were used as positive and negative controls, respectively (35, 44). As seen in Fig. 7B, PhoP was able to bind to the promoters of slrP and slyB but not to the phoN promoter. Interestingly, the mutation at position −40, which abrogated PhoP regulation, also prevented PhoP binding.
FIG 7.
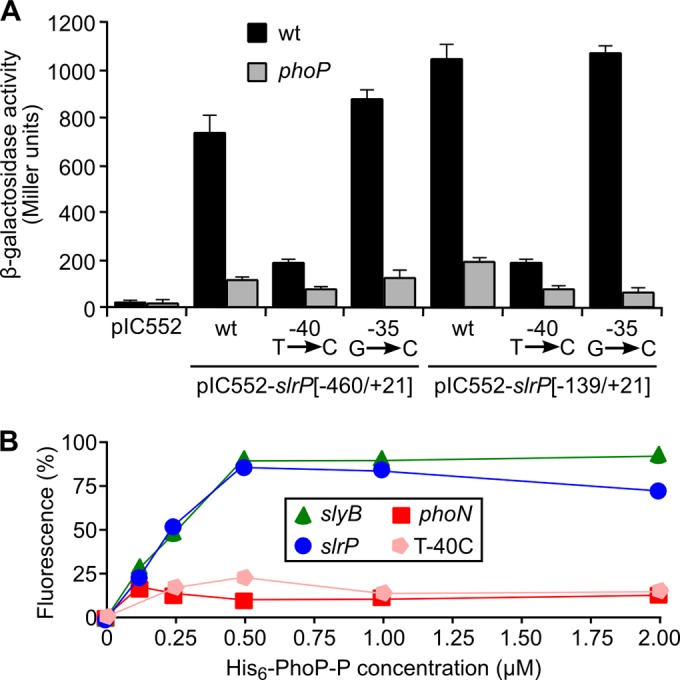
PhoP binds to the promoter of slrP. (A) Two fragments of DNA containing the promoter region of slrP, −460/+21 and −139/+21, were inserted into plasmid pIC552 to generate lacZ transcriptional fusions. These plasmids and derivatives carrying the indicated point mutations were introduced into S. enterica serovar Typhimurium strain 14028 (wild type) or a phoP-null mutant, and β-galactosidase activities were measured in cultures grown to stationary phase in liquid LPM. Means and standard deviations from two independent β-galactosidase measurements are shown. (B) Purified His6-PhoP was phosphorylated in vitro using acetyl phosphate. DNA fragments containing the promoter region of slrP, the same region with a point mutation (T-40C), and the promoter regions of slyB and phoN, as positive and negative controls, respectively, were PCR amplified using fluorochrome-labeled primers and incubated with the indicated concentrations of phosphorylated His6-PhoP (His6-PhoP-P). Slot blotting was used to quantify binding. Results from a representative experiment of three independent experiments are shown.
DISCUSSION
Definition of the environmental and genetic factors that control the expression of genes encoding T3SS effectors as well as of the conditions and timing for translocation into host cells is essential to understand the function of these virulence factors. This is particularly relevant in the case of an effector like SlrP that is encoded outside SPI1 and SPI2 and is secreted though two distinct and differentially regulated T3SSs. Previous work showed that this effector is capable of being secreted through T3SS1 and T3SS2 into macrophage-like cells (16). However, these experiments were carried out expressing slrP from a constitutive promoter in a plasmid. In addition, some reports suggested a low level of expression of slrP in SPI2-inducing media (16, 45). These data may cast some doubts on the physiological significance of T3SS2-dependent secretion of SlrP.
Here, we used a combination of translational lac and 3×FLAG fusions to study expression of slrP under different conditions and translational cyaA′ fusions to study the kinetics of translocation of SlrP into different host cell types. All the fusions were constructed in the native locus so that they conserved the native promoter and ribosome-binding site. A first conclusion of our experiments is that, although slrP is expressed at significant levels in SPI1- and SPI2-inducing media (Fig. 1 and 3), expression is optimal in the latter, a medium that partially mimics the intravacuolar environment. The discrepancy of these results with previous data mentioned above may be due to the different kinds of fusions used, luc transcriptional versus lac translational fusions, and more likely to the different media used to establish SPI2-inducing conditions. Whereas in previous studies either low Mg2+ concentration (16) or acidic pH (45) was used as the inducing signal, here we used a medium that combines both signals. Indeed, we show that these signals are equally important for induction of slrP (Fig. 1).
The physiological relevance of the induction of slrP expression under different conditions was further confirmed by our translocation experiments using a chromosomal slrP::cyaA′ fusion (Fig. 2). This fusion has the advantage of being expressed at physiological levels and, therefore, is optimal for obtaining insights about the kinetics of translocation into host cells. The main conclusions that can be drawn from these experiments are the following. (i) Both T3SS1 and T3SS2 are functional in SlrP translocation, but different patterns are observed depending on the host cells. (ii) Translocation into epithelial HeLa cells is strictly dependent on T3SS1. This may be explained, at least partially, by the fact that the T3SS1-dependent trigger mechanism is essentially the only way for Salmonella to invade nonphagocytic epithelial cells. Therefore, T3SS2-dependent translocation from the vacuole cannot be easily tested in these cells, since a T3SS1 mutant is unable to invade. (iii) Translocation of SlrP into NRK-49K fibroblasts is T3SS1 dependent from 1 to 4 h p.i. but occurs through both T3SSs at later times. These results suggest that in these cells, T3SS2 became functional between 4 and 8 h p.i., whereas T3SS1 was active throughout the experiment. We were able to study T3SS2 translocation in these cells because Salmonella invades fibroblasts by multiple routes, including T3SS1-independent routes (46). Hence, fibroblasts could be more suitable than epithelial cells as a model to study T3SS effector translocation into nonphagocytic cells. (iv) In macrophages, translocation was T3SS1 dependent from 1 to 2 h p.i. and T3SS2 dependent from 8 to 24 h p.i. The differences with fibroblasts could be ascribed to cell type differences and to the diverse means of entry used by Salmonella in each case. Translocation of SlrP into these cells was not detected when noninvasive bacteria were used at 1 h, 2 h (data not shown), or 4 h (Fig. 2B), suggesting that, in noninvasive phagocytized bacteria, T3SS1 is not induced at all and T3SS2 is induced between 4 and 8 h p.i. One factor that should be taken into account is the role of T3SS2 in the proliferation of S. enterica serovar Typhimurium inside RAW264.7 macrophages (47). However, the lack of intracellular proliferation of the SPI2 mutant does not completely explain the absence of secretion of SlrP, since the number of intracellular bacteria in this case would be still enough to detect translocation if T3SS1 was active.
In this study, we also investigated the genetic factors involved in the regulation of the expression of slrP under SPI1- and SPI2-inducing conditions. A genetic screen identified LeuO and Lon as negative regulators and HilD as a positive regulator under SPI1-inducing conditions (Fig. 4 and 5). Epistasis analysis suggests that LeuO and Lon act through HilD, a previously described slrP regulator (24). This is consistent with HilD being a substrate for the Lon protease (40) and with previous findings indicating that LeuO mediates SPI1 downregulation mainly through activation of the transcription of hilE, a gene encoding a HilD inhibitor (41). Interestingly, LeuO also seems to repress SPI1 by some minor HilD-independent mechanisms. The partial additive effect on the expression of the slrP::lacZ fusion that was observed upon combination of deletion of hilD and expression of leuO from a T-POP promoter suggests that similar mechanisms could operate in the regulation of slrP expression.
Our results also indicate that the PhoQ/PhoP two-component regulatory system is the main activator of slrP expression under SPI2-inducing conditions (Fig. 3). Under these conditions, a null mutation in phoP generated a 14-fold decrease in slrP expression. In addition, the activating phoQ24 mutation increased expression under SPI1-inducing conditions to the levels observed in the wild-type strain under SPI2 conditions. The PhoQ/PhoP system controls expression of SPI2 and some T3SS2-related effectors encoded outside the island through the SsrA/SsrB system. However, an ssrB-null mutation had a minor effect on slrP expression, a result that was consistent with previous reports (16, 45) and suggested that PhoP could act directly on the slrP promoter. Support for this hypothesis was obtained from in vitro slot blot-based experiments showing binding of PhoP to DNA fragments containing the promoter region of slrP (Fig. 7). In addition, binding and PhoP regulation were abolished by a point mutation in a putative PhoP box found in this region (Fig. 6 and 7). The distal half of this motif is very similar to the consensus sequence previously defined for a subset of PhoP-regulated genes that contain a promoter pattern known as architecture I (48), whereas the other half of the motif is not conserved in slrP. This observation may explain why this promoter was not detected in previous global searches for PhoP-regulated genes based on sequence similarities (49). We previously found a similar situation for the promoter of steA (35), a gene encoding another Salmonella T3SS effector, although for steA, the conserved part of the PhoP box was the proximal half (Fig. 6). Both effectors are substrates of T3SS1 and T3SS2, and in both cases there is a direct regulation by PhoP and a minor regulatory role for SsrB. This is in contrast with genes encoding other Salmonella effectors that are specifically secreted through T3SS2 and whose regulation is strictly dependent on the SsrA/SsrB two-component system (45, 50).
The dually positive regulation that we describe here for slrP by LeuO, Lon, and HilD under SPI1-inducing conditions and by PhoP under SPI2-inducing conditions partially explains the ability of SlrP to be secreted by both T3SSs under different circumstances. Both systems are interconnected in several ways (9, 13), and this or a similar regulatory model could operate for other effectors that are substrates of T3SS1 and T3SS2.
ACKNOWLEDGMENTS
This work was supported by grants SAF2010-15015 and SAF2013–46229-R from the Spanish Ministry of Economy and Competitiveness and the European Regional Development Fund and by grant P08-CVI-03487 from the Consejería de Economía, Innovación y Ciencia, Junta de Andalucía, Spain.
We are grateful to Javier López-Garrido and Elena Espinosa for strain constructions, to Joaquín Bernal-Bayard and Bárbara Calderón for help in some experiments, and to Josep Casadesús for critical reading of the manuscript.
Footnotes
Published ahead of print 2 September 2014
REFERENCES
- 1.Galán JE, Lara-Tejero M, Marlovits TC, Wagner S. 2014. Bacterial type III secretion systems: specialized nanomachines for protein delivery into target cells. Annu. Rev. Microbiol. 10.1146/annurev-micro-092412-155725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Galán JE, Curtiss R., III 1989. Cloning and molecular characterization of genes whose products allow Salmonella typhimurium to penetrate tissue culture cells. Proc. Natl. Acad. Sci. U. S. A. 86:6383–6387. 10.1073/pnas.86.16.6383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ochman H, Soncini FC, Solomon F, Groisman EA. 1996. Identification of a pathogenicity island required for Salmonella survival in host cells. Proc. Natl. Acad. Sci. U. S. A. 93:7800–7804. 10.1073/pnas.93.15.7800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shea JE, Hensel M, Gleeson C, Holden DW. 1996. Identification of a virulence locus encoding a second type III secretion system in Salmonella typhimurium. Proc. Natl. Acad. Sci. U. S. A. 93:2593–2597. 10.1073/pnas.93.6.2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ramos-Morales F. 2012. Impact of Salmonella enterica type III secretion system effectors on the eukaryotic host cell. ISRN Cell Biol. 2012:787934. 10.5402/2012/787934. [DOI] [Google Scholar]
- 6.Zhang S, Santos RL, Tsolis RM, Stender S, Hardt WD, Baumler AJ, Adams LG. 2002. The Salmonella enterica serotype Typhimurium effector proteins SipA, SopA, SopB, SopD, and SopE2 act in concert to induce diarrhea in calves. Infect. Immun. 70:3843–3855. 10.1128/IAI.70.7.3843-3855.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hensel M, Shea JE, Waterman SR, Mundy R, Nikolaus T, Banks G, Vazquez-Torres A, Gleeson C, Fang FC, Holden DW. 1998. Genes encoding putative effector proteins of the type III secretion system of Salmonella pathogenicity island 2 are required for bacterial virulence and proliferation in macrophages. Mol. Microbiol. 30:163–174. 10.1046/j.1365-2958.1998.01047.x. [DOI] [PubMed] [Google Scholar]
- 8.Ellermeier JR, Slauch JM. 2007. Adaptation to the host environment: regulation of the SPI1 type III secretion system in Salmonella enterica serovar Typhimurium. Curr. Opin. Microbiol. 10:24–29. 10.1016/j.mib.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 9.Fass E, Groisman EA. 2009. Control of Salmonella pathogenicity island-2 gene expression. Curr. Opin. Microbiol. 12:199–204. 10.1016/j.mib.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aguirre A, Cabeza ML, Spinelli SV, McClelland M, García Vescovi E, Soncini FC. 2006. PhoP-induced genes within Salmonella pathogenicity island 1. J. Bacteriol. 188:6889–6898. 10.1128/JB.00804-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Giacomodonato MN, Uzzau S, Bacciu D, Caccuri R, Sarnacki SH, Rubino S, Cerquetti MC. 2007. SipA, SopA, SopB, SopD and SopE2 effector proteins of Salmonella enterica serovar Typhimurium are synthesized at late stages of infection in mice. Microbiology 153:1221–1228. 10.1099/mic.0.2006/002758-0. [DOI] [PubMed] [Google Scholar]
- 12.Giacomodonato MN, Noto Llana M, Aya Castañeda MDR, Buzzola FR, Sarnacki SH, Cerquetti MC. 2014. AvrA effector protein of Salmonella enterica serovar Enteritidis is expressed and translocated in mesenteric lymph nodes at late stages of infection in mice. Microbiology 160:1191–1199. 10.1099/mic.0.077115-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moest TP, Meresse S. 2013. Salmonella T3SSs: successful mission of the secret(ion) agents. Curr. Opin. Microbiol. 16:38–44. 10.1016/j.mib.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 14.Tsolis RM, Townsend SM, Miao EA, Miller SI, Ficht TA, Adams LG, Baumler AJ. 1999. Identification of a putative Salmonella enterica serotype Typhimurium host range factor with homology to IpaH and YopM by signature-tagged mutagenesis. Infect. Immun. 67:6385–6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miao EA, Scherer CA, Tsolis RM, Kingsley RA, Adams LG, Baumler AJ, Miller SI. 1999. Salmonella typhimurium leucine-rich repeat proteins are targeted to the SPI1 and SPI2 type III secretion systems. Mol. Microbiol. 34:850–864. 10.1046/j.1365-2958.1999.01651.x. [DOI] [PubMed] [Google Scholar]
- 16.Miao EA, Miller SI. 2000. A conserved amino acid sequence directing intracellular type III secretion by Salmonella typhimurium. Proc. Natl. Acad. Sci. U. S. A. 97:7539–7544. 10.1073/pnas.97.13.7539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kobe B, Kajava AV. 2001. The leucine-rich repeat as a protein recognition motif. Curr. Opin. Struct. Biol. 11:725–732. 10.1016/S0959-440X(01)00266-4. [DOI] [PubMed] [Google Scholar]
- 18.Quezada CM, Hicks SW, Galan JE, Stebbins CE. 2009. A family of Salmonella virulence factors functions as a distinct class of autoregulated E3 ubiquitin ligases. Proc. Natl. Acad. Sci. U. S. A. 106:4864–4869. 10.1073/pnas.0811058106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rohde JR, Breitkreutz A, Chenal A, Sansonetti PJ, Parsot C. 2007. Type III secretion effectors of the IpaH family are E3 ubiquitin ligases. Cell Host Microbe 1:77–83. 10.1016/j.chom.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 20.Singer AU, Rohde JR, Lam R, Skarina T, Kagan O, Dileo R, Chirgadze NY, Cuff ME, Joachimiak A, Tyers M, Sansonetti PJ, Parsot C, Savchenko A. 2008. Structure of the Shigella T3SS effector IpaH defines a new class of E3 ubiquitin ligases. Nat. Struct. Mol. Biol. 15:1293–1301. 10.1038/nsmb.1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu Y, Li H, Hu L, Wang J, Zhou Y, Pang Z, Liu L, Shao F. 2008. Structure of a Shigella effector reveals a new class of ubiquitin ligases. Nat. Struct. Mol. Biol. 15:1302–1308. 10.1038/nsmb.1517. [DOI] [PubMed] [Google Scholar]
- 22.Bernal-Bayard J, Ramos-Morales F. 2009. Salmonella type III secretion effector SlrP is an E3 ubiquitin ligase for mammalian thioredoxin. J. Biol. Chem. 284:27587–27595. 10.1074/jbc.M109.010363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bernal-Bayard J, Cardenal-Muñoz E, Ramos-Morales F. 2010. The Salmonella type III secretion effector, Salmonella leucine-rich repeat protein (SlrP), targets the human chaperone ERdj3. J. Biol. Chem. 285:16360–16368. 10.1074/jbc.M110.100669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ellermeier CD, Slauch JM. 2003. RtsA and RtsB coordinately regulate expression of the invasion and flagellar genes in Salmonella enterica serovar Typhimurium. J. Bacteriol. 185:5096–5108. 10.1128/JB.185.17.5096-5108.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ellermeier CD, Slauch JM. 2004. RtsA coordinately regulates DsbA and the Salmonella pathogenicity island 1 type III secretion system. J. Bacteriol. 186:68–79. 10.1128/JB.186.1.68-79.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schmieger H. 1972. Phage P22-mutants with increased or decreased transduction abilities. Mol. Gen. Genet. 119:75–88. 10.1007/BF00270447. [DOI] [PubMed] [Google Scholar]
- 27.Maloy SR. 1990. Experimental techniques in bacterial genetics. Jones & Bartlett, Boston, MA. [Google Scholar]
- 28.Chan RK, Botstein D, Watanabe T, Ogata Y. 1972. Specialized transduction of tetracycline resistance by phage P22 in Salmonella typhimurium. II. Properties of a high-frequency-transducing lysate. Virology 50:883–898. [DOI] [PubMed] [Google Scholar]
- 29.Gerlach RG, Holzer SU, Jackel D, Hensel M. 2007. Rapid engineering of bacterial reporter gene fusions by using Red recombination. Appl. Environ. Microbiol. 73:4234–4242. 10.1128/AEM.00509-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 31.Rappleye CA, Roth JR. 1997. A Tn10 derivative (T-POP) for isolation of insertions with conditional (tetracycline-dependent) phenotypes. J. Bacteriol. 179:5827–5834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chun KT, Edenberg HJ, Kelley MR, Goebl MG. 1997. Rapid amplification of uncharacterized transposon-tagged DNA sequences from genomic DNA. Yeast 13:233–240. . [DOI] [PubMed] [Google Scholar]
- 33.Baisón-Olmo F, Cardenal-Muñoz E, Ramos-Morales F. 2012. PipB2 is a substrate of the Salmonella pathogenicity island 1-encoded type III secretion system. Biochem. Biophys. Res. Commun. 423:240–246. 10.1016/j.bbrc.2012.05.095. [DOI] [PubMed] [Google Scholar]
- 34.Gal-Mor O, Elhadad D, Deng W, Rahav G, Finlay BB. 2011. The Salmonella enterica PhoP directly activates the horizontally acquired SPI-2 gene sseL and is functionally different from a S. bongori ortholog. PLoS One 6:e20024. 10.1371/journal.pone.0020024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cardenal-Muñoz E, Ramos-Morales F. 2013. DsbA and MgrB regulate steA expression through the two-component system PhoQ/PhoP in Salmonella enterica. J. Bacteriol. 195:2368–2378. 10.1128/JB.00110-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tang YT, Gao R, Havranek JJ, Groisman EA, Stock AM, Marshall GR. 2012. Inhibition of bacterial virulence: drug-like molecules targeting the Salmonella enterica PhoP response regulator. Chem. Biol. Drug Des. 79:1007–1017. 10.1111/j.1747-0285.2012.01362.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bajaj V, Lucas RL, Hwang C, Lee CA. 1996. Co-ordinate regulation of Salmonella typhimurium invasion genes by environmental and regulatory factors is mediated by control of hilA expression. Mol. Microbiol. 22:703–714. 10.1046/j.1365-2958.1996.d01-1718.x. [DOI] [PubMed] [Google Scholar]
- 38.Sory MP, Cornelis GR. 1994. Translocation of a hybrid YopE-adenylate cyclase from Yersinia enterocolitica into HeLa cells. Mol. Microbiol. 14:583–594. 10.1111/j.1365-2958.1994.tb02191.x. [DOI] [PubMed] [Google Scholar]
- 39.Fink SL, Cookson BT. 2007. Pyroptosis and host cell death responses during Salmonella infection. Cell Microbiol. 9:2562–2570. 10.1111/j.1462-5822.2007.01036.x. [DOI] [PubMed] [Google Scholar]
- 40.Takaya A, Kubota Y, Isogai E, Yamamoto T. 2005. Degradation of the HilC and HilD regulator proteins by ATP-dependent Lon protease leads to downregulation of Salmonella pathogenicity island 1 gene expression. Mol. Microbiol. 55:839–852. 10.1111/j.1365-2958.2004.04425.x. [DOI] [PubMed] [Google Scholar]
- 41.Espinosa E, Casadesús J. 2014. Regulation of Salmonella enterica pathogenicity island 1 (SPI-1) by the LysR-type regulator LeuO. Mol. Microbiol. 91:1057–1069. 10.1111/mmi.12500. [DOI] [PubMed] [Google Scholar]
- 42.Baxter MA, Fahlen TF, Wilson RL, Jones BD. 2003. HilE interacts with HilD and negatively regulates hilA transcription and expression of the Salmonella enterica serovar Typhimurium invasive phenotype. Infect. Immun. 71:1295–1305. 10.1128/IAI.71.3.1295-1305.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kroger C, Dillon SC, Cameron AD, Papenfort K, Sivasankaran SK, Hokamp K, Chao Y, Sittka A, Hebrard M, Handler K, Colgan A, Leekitcharoenphon P, Langridge GC, Lohan AJ, Loftus B, Lucchini S, Ussery DW, Dorman CJ, Thomson NR, Vogel J, Hinton JC. 2012. The transcriptional landscape and small RNAs of Salmonella enterica serovar Typhimurium. Proc. Natl. Acad. Sci. U. S. A. 109:E1277–E1286. 10.1073/pnas.1201061109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lejona S, Aguirre A, Cabeza ML, García Vescovi E, Soncini FC. 2003. Molecular characterization of the Mg2+-responsive PhoP-PhoQ regulon in Salmonella enterica. J. Bacteriol. 185:6287–6294. 10.1128/JB.185.21.6287-6294.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu X, Hensel M. 2010. Systematic analysis of the SsrAB virulon of Salmonella enterica. Infect. Immun. 78:49–58. 10.1128/IAI.00931-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aiastui A, Pucciarelli MG, Garcia-del Portillo F. 2010. Salmonella enterica serovar typhimurium invades fibroblasts by multiple routes differing from the entry into epithelial cells. Infect. Immun. 78:2700–2713. 10.1128/IAI.01389-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cirillo DM, Valdivia RH, Monack DM, Falkow S. 1998. Macrophage-dependent induction of the Salmonella pathogenicity island 2 type III secretion system and its role in intracellular survival. Mol. Microbiol. 30:175–188. 10.1046/j.1365-2958.1998.01048.x. [DOI] [PubMed] [Google Scholar]
- 48.Zwir I, Latifi T, Pérez JC, Huang H, Groisman EA. 2012. The promoter architectural landscape of the Salmonella PhoP regulon. Mol. Microbiol. 84:463–485. 10.1111/j.1365-2958.2012.08036.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harari O, Park SY, Huang H, Groisman EA, Zwir I. 2010. Defining the plasticity of transcription factor binding sites by Deconstructing DNA consensus sequences: the PhoP-binding sites among gamma/enterobacteria. PLoS Comput. Biol. 6:e1000862. 10.1371/journal.pcbi.1000862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cordero-Alba M, Bernal-Bayard J, Ramos-Morales F. 2012. SrfJ, a Salmonella type III secretion system effector regulated by PhoP, RcsB, and IolR. J. Bacteriol. 194:4226–4236. 10.1128/JB.00173-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hanahan D. 1983. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 166:557–580. 10.1016/S0022-2836(83)80284-8. [DOI] [PubMed] [Google Scholar]
- 52.Bullock WO, Fernandez JM, Short JM. 1987. XL1-BLUE: a high efficiency plasmid transforming RecA Escherichia coli strain with beta-galactosidase selection. Biotechniques 5:376–379. [Google Scholar]
- 53.Groisman EA, Chiao E, Lipps CJ, Heffron F. 1989. Salmonella typhimurium phoP virulence gene is a transcriptional regulator. Proc. Natl. Acad. Sci. U. S. A. 86:7077–7081. 10.1073/pnas.86.18.7077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Segura I, Casadesús J, Ramos-Morales F. 2004. Use of mixed infections to study cell invasion and intracellular proliferation of Salmonella enterica in eukaryotic cell cultures. J. Microbiol. Methods 56:83–91. 10.1016/j.mimet.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 55.García-Calderón CB, Casadesús J, Ramos-Morales F. 2007. Rcs and PhoPQ regulatory overlap in the control of Salmonella enterica virulence. J. Bacteriol. 189:6635–6644. 10.1128/JB.00640-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Macián F, Pérez-Roger I, Armengod ME. 1994. An improved vector system for constructing transcriptional lacZ fusions: analysis of regulation of the dnaA, dnaN, recF and gyrB genes of Escherichia coli. Gene 145:17–24. 10.1016/0378-1119(94)90317-4. [DOI] [PubMed] [Google Scholar]
- 57.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645. 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]