

Abstract
RecQ family helicases function as safeguards of the genome. Unlike Escherichia coli, the Gram-positive Bacillus subtilis bacterium possesses two RecQ-like homologues, RecQ[Bs] and RecS, which are required for the repair of DNA double-strand breaks. RecQ[Bs] also binds to the forked DNA to ensure a smooth progression of the cell cycle. Here we present the first biochemical analysis of recombinant RecQ[Bs]. RecQ[Bs] binds weakly to single-stranded DNA (ssDNA) and blunt-ended double-stranded DNA (dsDNA) but strongly to forked dsDNA. The protein exhibits a DNA-stimulated ATPase activity and ATP- and Mg2+-dependent DNA helicase activity with a 3′→5′ polarity. Molecular modeling shows that RecQ[Bs] shares high sequence and structure similarity with E. coli RecQ. Surprisingly, RecQ[Bs] resembles the truncated Saccharomyces cerevisiae Sgs1 and human RecQ helicases more than RecQ[Ec] with regard to its enzymatic activities. Specifically, RecQ[Bs] unwinds forked dsDNA and DNA duplexes with a 3′-overhang but is inactive on blunt-ended dsDNA and 5′-overhung duplexes. Interestingly, RecQ[Bs] unwinds blunt-ended DNA with structural features, including nicks, gaps, 5′-flaps, Kappa joints, synthetic replication forks, and Holliday junctions. We discuss these findings in the context of RecQ[Bs]'s possible functions in preserving genomic stability.
INTRODUCTION
DNA helicases use the chemical energy derived from the binding and hydrolysis of ATP to catalyze the unwinding of double-stranded DNA (dsDNA). They are found in all living organisms and perform essential functions during DNA metabolism, including replication, transcription, repair, and recombination. The RecQ family helicases, which were named after the Escherichia coli RecQ (RecQ[Ec]) helicase (1, 2), belong to the superfamily (SF) 2 of DNA helicases (3). RecQ family members have been found in diverse organisms, including bacteria, yeasts, fungi, flies, frogs, and humans. Prokaryotes and unicellular eukaryotes usually possess a single RecQ homologue, while multicellular organisms express multiple ones (4–6). In E. coli, RecQ[Ec] is the solitary RecQ helicase. In humans, five family members, RECQ1, BLM, WRN, RECQ4, and RECQ5, have been identified. Mutations in BLM, WRN, and RECQ4 are linked to the human hereditary disorders Bloom, Werner, and Rothmund-Thomson syndromes, respectively. The affected individuals are characterized by a rapid onset of cancer, accelerated aging, growth abnormalities, and other defects (7). At the cellular level, the RecQ enzymes maintain genomic integrity through various mechanisms, such as dsDNA break repair (8).
In E. coli, RecBCD is a major component of the dsDNA break (DSB) repair machinery. The RecQ[Ec] enzyme is active on both partially and fully duplex DNA substrates (2). In the absence of RecBCD in recBC sbcB sbcC mutant cells, RecQ[Ec] initiates homologous recombination for DSB repair via the RecF pathway, which is the major homologous recombination pathway for single-stranded DNA (ssDNA) gap repair (9, 10). In this pathway, RecQ[Ec] unwinds a DNA duplex or a duplex with a 3′-overhang to generate a 3′-terminated ssDNA, which is used for homologous pairing by the RecA protein (11, 12). The RecQ[Ec] enzyme also participates in the hybrid pathways, in which the parts of E. coli RecBCD and RecF recombination machines interexchange (13). In these pathways, RecQ[Ec] can initiate homologous recombination on the substrates that are not suitable for RecBCD (14, 15) because of its substrate specificity, which is wider than that of RecBCD (11). Furthermore, RecQ[Ec] can not only promote recombination but also disrupt joint molecule intermediates in vitro (10, 11). Thus, RecQ[Ec] contributes to recombination fidelity (16) and to recombinational repair without crossing over. Finally, RecQ[Ec] interacts functionally with topoisomerase III and therefore promotes the catenation and decatenation of dsDNA (17, 18).
The E. coli paradigm may not be valid for all other bacteria. At least for the DSB repair, different pathways are operative in Bacillus subtilis (19). B. subtilis is an aerobic, endospore-forming, rod-shaped, Gram-positive bacterium commonly found in soil and water sources and in association with plants. Unlike E. coli, firmicutes may possess two RecQ-like homologues, RecQ[Bs] and RecS. These two homologous proteins might have some overlapping activities, and both are needed for the full resistance of growing cells to DNA-damaging agents (19–21). In B. subtilis, the two-ended DSBs are repaired mainly via homologous recombination and with very low efficiency by nonhomologous end joining (22). Homologous recombination initiates with 5′→3′ end processing by the RecJ exonuclease in collaboration with RecQ[Bs] or RecS, collectively termed RecQ[Bs](S)-RecJ, or by the AddAB nuclease-helicase enzyme complex (the counterpart of E. coli RecBCD) (23, 24). The resulting 3′-ssDNA tails are bound by the ssDNA-binding [SSB] protein (SSB[Bs]) (homologous to E. coli SSB, or SSB[Ec]) (24, 25). RecN facilitates tethering of DNA ends and promotes the relocalization to a discrete repair center (25–29). In this model, when processing of DNA ends, AddAB and RecQ[Bs](S)-RecJ are two complementary pathways with similar efficiency, while the RecQ[Bs](S)-RecJ repair avenue is operative even in the presence of AddAB (19). Furthermore, the cellular localization is different in RecQ[Ec] and in RecQ[Bs]. In E. coli, RecQ[Ec] colocalizes with the replisome in the absence of DSBs and disappears when replication rounds are completed (30). However, in B. subtilis, RecQ[Bs] localizes throughout the nucleoids in the presence or absence of DSBs, indicating that RecQ[Bs] is constitutively associated with chromosomal DNA (19). Finally, RecQ[Bs] and two other SF2 family repair helicases, PriA[Bs] and RecG[Bs], localize at the replication factory in living B. subtilis cells during active duplication of the chromosome through interaction with SSB[Bs]. These helicases bind specifically to the forked DNA molecules. Upon inactivation of the replication fork provoked, the retained helicases can act on the fork to attempt its rescue, thereby ensuring a smooth progression of the cell cycle and maintaining the stability of the genome (31).
RecQ[Bs], YocI, a 591-residue protein, is a putative member of the RecQ helicase family. However, its biochemical activities have not yet been characterized. It remains to be determined if RecQ[Bs] is a bona fide DNA helicase and whether its enzymatic activities correlate with the observed in vivo functions in DNA metabolism, in particular, recombination and repair (22, 31). In this study, we first carried out structural modeling studies on RecQ[Bs], based upon known crystal structures of the RecQ[Ec] fragments (32, 33). As expected, RecQ[Bs] and RecQ[Ec] have similar secondary structural elements and three-dimensional (3D) structures. We then expressed and purified RecQ[Bs] protein in E. coli. The results demonstrate that the recombinant protein binds preferentially to the forked dsDNA substrates and possesses a DNA-dependent ATPase activity. Furthermore, the protein is an ATP- and Mg2+-dependent DNA helicase with a 3′→5′ polarity that is inactive on the blunt-ended dsDNA molecules but active on the forked dsDNA, DNA duplexes with 3′-overhang, and several blunt-ended DNA duplexes with added structural features, including nicks, gaps, 5′-flaps, Kappa joints, synthetic replication forks, and Holliday junctions (HJ), which are commonly found as the intermediates in the DNA replication, repair, and recombination. Remarkably, our comprehensive biochemical characterization shows that RecQ[Bs] resembles the truncated Saccharomyces cerevisiae Sgs1 and some of human RecQ (hRecQ) helicases more than RecQ[Ec] with regard to its enzymatic activities, in spite of high sequence and structure similarity between RecQ[Bs] and RecQ[Ec]. The implications of these results are discussed in terms of the characterized cellular functions for RecQ[Bs] (22, 31).
MATERIALS AND METHODS
Materials.
ATP, dATP, CTP, dCTP, GTP, dGTP, UTP, dTTP, 5′-adenylylimidodiphosphate (AMPPNP), EDTA, dithiothreitol (DTT), and SSB[Ec] protein were purchased from Sigma Co. (St. Louis, MO). [γ-32P]ATP (3,000 Ci/mmol) was purchased from Amersham (Piscataway, NJ). T4 polynucleotide kinase (PNK) and bovine serum albumin (BSA) were purchased from New England BioLabs (Beverly, MA). Chelex resin and Bio-Gel HTP hydroxyapatite were purchased from Bio-Rad (Hercules, CA). Puratronic CaCl2, MgCl2, MnCl2, NiCl2, and ZnCl2 were from Alfa Aesar (Ward Hill, MA). Thrombin was purchased from Sigma (St. Louis, MO). HiTrap benzamidine FF agarose and Ni2+-charged HiTrap chelating columns were purchased from GE Healthcare (Piscataway, NJ).
Synthetic peptides.
N-terminal Alexa Fluor dye-labeled peptides SSB[Bs]-C10 (Ile-Asp-Ile-Ser-Asp-Asp-Asp-Leu-Pro-Phe) and YwpH-C10 (Asp-Pro-Lys-Pro-Glu-Lys-Ala-Ala-Asp) were synthesized and purified by Biomart (Dalian, China).
Nucleic acid substrates and labeling.
The polyacrylamide gel electrophoresis (PAGE)-purified single-stranded oligodeoxynucleotides (Table 1) were purchased from either Genset or MWG-Biotech (High Point, NC). The ϕX174 viral circle ssDNA was obtained from New England BioLabs (Beverly, MA). The DNA concentrations were determined from A260 measurements using absorption coefficients calculated from the nucleotide sequence for the oligomeric substrates (34). A single-stranded oligonucleotide was labeled at its 5′-end using [γ-32P]ATP and T4 PNK. For the kinase reaction, 50 pmol of oligonucleotide was incubated with an equal amount of radioactive ATP (3,000 Ci/mmol) and 1 U of PNK in 50 μl of the kinase buffer (70 mM Tris-HCl [pH 7.6], 10 mM MgCl2, 5 mM DTT) at 37°C for 1 h. The reaction was terminated by heating to 70°C for 15 min, and the labeled oligonucleotide was then purified using a G25 spin column to remove nonincorporated [γ-32P]ATP. The purity of the labeled oligonucleotide was evaluated using 18% denaturing PAGE.
TABLE 1.
Oligonucleotides used in this study
| Name | No. of bases (nt) | Sequence (5′→3′) or description |
|---|---|---|
| S1 | 44 | GCACTGGCCGTCGTTTTACGGTCGTGACTGGGAAAACCCTGGCG |
| S2 | 45 | TTTTTTTTTTTTTTTTTTTTTTCCAAGTAAAACGACGGCCAGTGC |
| S3 | 5,386 | ϕX174 circle ssDNA |
| S4 | 20 | TCAGCACCAGCACGCTCCCA |
| S5 | 40 | TCAGCACCAGCACGCTCCCATTTTTTTTTTTTTTTTTTTT |
| S6 | 35 | AAAAAAAAAAAAAAATCAGCACCAGCACGCTCCCA |
| S7 | 70 | TGCAGTAGCGCCAATATGAGAAGAGCCATACCGCTGATTCTGCGTTTGCTGATGAACTAAGTCAACCTCA |
| S8 | 19 | TCATATTGGCGCTACTGCA |
| S9 | 19 | TGAGGTTGACTTAGTTCAT |
| S10 | 19 | GTGTGGAAAATCTCTAGCA |
| S11 | 19 | TGCTAGAGATTTTCCACAC |
| S12 | 40 | ACGTGGGCAAAGGTTCGTCAATGGACTGACAGCTGCATGG |
| S13 | 19 | GACGAACCTTTGCCCACGT |
| S14 | 21 | CCATGCAGCTGTCAGTCCATT |
| S15 | 19 | CCATGCAGCTGTCAGTCCA |
| S16 | 16 | CCATGCAGCTGTCAGT |
| S17 | 19 | GTAAAACGACGGCCAGTGC |
| S18 | 20 | AGTAAAACGACGGCCAGTGC |
| S19 | 24 | TCCAAGTAAAACGACGGCCAGTGC |
| S20 | 29 | TTTTTTCCAAGTAAAACGACGGCCAGTGC |
| S21 | 46 | TTTTTTTTTTTTTTTTTTTTTTTCCAAGTAAAACGACGGCCAGTGC |
| S22 | 20 | GCACTGGCCGTCGTTTTACG |
| S23 | 24 | GCACTGGCCGTCGTTTTACGGTCG |
| S24 | 29 | GCACTGGCCGTCGTTTTACGGTCGTGACT |
| S25 | 44 | GCACTGGCCGTCGTTTTACGGTCGTGACTGGGAAAACCCTGGCG |
| S26 | 25 | CGCCAGGGTTTTCCCAGTCACGACC |
| S27 | 44 | CGCCAGGGTTTTCCCAGTCACGACCAACCCCTTTTTTTTTTCAA |
| S28 | 26 | TTGGAAAAAAAAAAAAAAAAAAAAAA |
| S29 | 40 | CCGTGATCACCAATGCAGATTGACGAACCTTTGCCCACGT |
| S30 | 40 | ACGTGGGCAAAGGTTCGTCAATGGACTGACAGCTGCATGG |
| S31 | 40 | CCATGCAGCTGTCAGTCCATTGTCATGCTAGGCCTACTGC |
| S32 | 40 | GCAGTAGGCCTAGCATGACAATCTGCATTGGTGATCACGG |
| S33 | 60 | TGCAGTAGCGCCAATATGAGAAGAGCCATACCGCTGATTCTGCGTTTGCTGATGAACTAA |
| S34 | 19 | GCACTGGCCGTCGTTTTAC |
The labeled oligonucleotide was incubated with one, two, or three unlabeled ssDNA oligonucleotides in order to construct various dsDNA substrates with different structural features (Table 2). Typically, 50 pmol of labeled ssDNA oligonucleotide was incubated with 75 to 100 pmol of complementary nonlabeled ssDNA oligonucleotides in 100 μl of hybridization buffer (200 mM Tris-HCl [pH 8.0], 250 mM NaCl, and 10 mM EDTA). The mixture was incubated at 95°C for 10 min and then allowed to slowly cool to room temperature. These annealed dsDNA substrates were further purified using Bio-Gel HTP hydroxyapatite to remove the excess ssDNA substrates. The efficiency of hybridization was verified by migration on a native 20% native PAGE.
TABLE 2.
Structures of the DNA substrates in this study
Construction of the plasmids encoding the RecQ[Bs] and SSB[Bs] proteins.
A full coding region of the RecQ[Bs] cDNA was generated by PCR-mediated amplification using B. subtilis strain QST 713 chromosome DNA as a template. A 1.8-kb PCR product whose sequence was shown to be identical to that of the published RecQ[Bs] gene (GenBank accession number AF027868) was cloned into pGEM-T Easy Vector (Promega). The Easy Vector system was then used to construct the expression plasmid pET15b-RecQ[Bs] by PCR with primers 5′-CATATGTTACATAGAGCCCAATCCCTTCTGGCTCAT, which contains an NdeI site (underlined) at the 5′ end, and 5′-ATACAGGCTTATGCAAGGATGACAGACTAACTCGAG, which contains an XhoI site (underlined) at the 3′ end. The pET15b-RecQ[Bs] expression cassette encoded an N-terminal hexa-histidine affinity tag and the residues 1 to 591 of RecQ[Bs]. This system produces a soluble fusion protein. pET15b-SSB[Bs] expression plasmid was constructed by PCR using primers 5′-GGAATTCCATATGCTTAACCGAGTTGTATTAGTCGGAAG (NdeI site underlined) and 5′-CCGTTACTCGAGGTCGACGAATGGAAGATCATCATCCGAGATGT (XhoI site underlined).
Expression and purification of the recombinant proteins.
The plasmids encoding RecQ[Bs] protein were transformed into the E. coli strain BL21(DE3) (Novagen), and protein overexpression was induced in the log phase (A600 = 0.6) by 0.2 mM isopropyl β-d-thiogalactopyranoside (IPTG) for 12 to 14 h at 15°C. The harvested cells were suspended in 30 ml of suspension buffer (20 mM Tris-HCl, pH 7.9, 5 mM imidazole, 500 mM NaCl) and were lysed using a French press, followed by sonication. The lysate was centrifuged (70,000 × g for 30 min at 4°C), and the supernatant was applied to a column charged with histidine binding resin (Novagen). The column was extensively washed with wash buffer (20 mM Tris-HCl [pH 7.9], 500 mM NaCl, and 5 mM imidazole). The proteins bound to the column were eluted stepwise using the wash buffer containing 50, 65, 80, 200, or 500 mM imidazole. The RecQ[Bs]-containing fractions, identified by both DNA-dependent ATP hydrolysis and helicase activity assays, were pooled. The RecQ[Bs] protein was further purified by fast protein liquid chromatography (FPLC) size exclusion chromatography (Superdex 200; Amersham Biosciences). The active fractions were pooled, dialyzed against storage buffer (20 mM Tris-HCl [pH 7.9], 500 mM NaCl, 2 mM DTT, 10% glycerol), and stored at −80°C. Some of the histidine-tagged fusion protein was digested with thrombin to remove the His tag (1 mg fusion protein/1 U thrombin) for 1 h at 25°C. The digested mixture was applied to a 1-ml HiTrap benzamidine FF agarose column and subsequently applied to a 5-ml Ni2+-charged HiTrap chelating column. The thrombin-cleaved RecQ[Bs] was eluted at 30 mM imidazole and was then loaded onto a 320-ml Sephacryl 100 gel filtration column equilibrated with a buffer containing 20 mM Tris-HCl (pH 7.4) and 50 mM NaCl for further purification. The histidine-tagged SSB[Bs] protein was purified using a similar procedure. The protein purities were judged by Coomassie blue staining and electrospray mass spectrometry, and protein concentrations were determined spectrophotometrically using a Bio-Rad protein Assay.
Determination of oligomerization state of RecQ[Bs] by size exclusion chromatography.
Size exclusion chromatography was performed at 18°C using an FPLC system (ÄKTA Purifier; Amersham Bioscience) on a Superdex 200 (analytical-grade) column equilibrated with elution buffer (50 mM Tris-HCl [pH 7.5], 400 mM NaCl, 0.1 mM EDTA). Fractions of 0.5 ml were collected at a flow rate of 0.4 ml/min, and absorbance was measured at 280 and 260 nm. The partition coefficient Kav was calculated using the formula Kav = (Ve − V0)/(Vt − V0), where Ve is the elution volume of the sample, V0 is the excluded volume of the column, and Vt is the total volume of the column. The excluded volume, V0 (7.52 ml), and the total volume, Vt (23.5 ml), were measured by calibration with dextran blue and thymidine. The calibration graph of log Rs (Stokes radius) versus Kav was constructed using a high- and low-molecular-mass calibration kit from Sigma: cytochrome c (molecular mass = 12.4 kDa; Rs = 12 Å), carbonic anhydrase (molecular mass = 29 kDa; Rs = 22.5 Å), albumin (molecular mass = 67 kDa; Rs = 35.5 Å), phosphorylase b (molecular mass = 97.4 kDa; Rs = 38.75 Å), and thyroglobulin (molecular mass = 669 kDa; Rs = 85 Å). Assuming similar shape factors, the plot calibration of log molecular mass versus Kav allowed the determination in first approximation of the molecular mass of the RecQ[Bs] protein.
Determination of oligomerization state of RecQ[Bs] by dynamic light scattering.
Dynamic light scattering measurements were performed using a DynaPro NanoStar instrument (Wyatt Technology, USA) equipped with a thermostated cell holder using filtered (0.1-μm filters) solutions in disposable cuvettes (UVette; Eppendorf). The protein concentration was at 1.5 μM in a buffer composed of 50 mM Tris-HCl (pH 8.0), 200 mM NaCl, and 1 mM DTT (total volume, 50 μl). The scattered light was collected at an angle of 90°. The recording times were typically between 3 and 5 min (20 to 30 cycles of an average of 10 s each). The analysis was performed with Dynamics 7.0 software using regularization methods (Wyatt Technology). The molecular mass (M) was calculated from the hydrodynamic radius using the empirical equation M = (1.68 × HR)2.34, where M and HR are molecular mass (in kDa) and hydrodynamic radius (in nm), respectively.
ATPase assay.
The ATPase activity of RecQ[Bs] was determined by measuring the release of the inorganic phosphate (Pi) produced from [γ-32P]ATP hydrolysis (35). The reaction was carried out using an ATPase reaction buffer (50 mM Tris-HCl [pH 8.0], 35 mM NaCl, 2 mM MgCl2, 0.1 mg/ml BSA, and 1 mM DTT) at 37°C in a volume of 40 μl. The reactions were initiated by the addition of an appropriate concentration of the RecQ[Bs] protein into 500 μl of reaction mixture containing the indicated amounts of [γ-32P]ATP and an oligonucleotide. The reaction was stopped by transferring 40 μl of aliquot from the reaction mixture every 30 s into a hydrochloric solution of ammonium molybdate. The liberated radioactive 32Pi was extracted with a solution of 2-butanol–cyclohexane–acetone–ammonium molybdate (250:250:50:0.1) saturated with water. An aliquot of the organic phase was removed, and the radioactivity was quantified using a liquid scintillation counter. The DNA or RNA used as a cofactor for the ATPase activity of RecQ[Bs] was as follows: ssDNA (oligonucleotide S33 [Table 1]), 60-nucleotide (-nt) ssDNA; linear dsDNA, 7-kb PN6 linear dsDNA; circular dsDNA, 5.5-kb GC70-plasmid DNA; denatured circular dsDNA, 5.5-kb GC70-plasmid DNA; denatured RNA, 678-nt RNA.
Helicase assays.
The helicase assay measures the unwinding of a 32P-labeled DNA fragment from a DNA duplex. Helicase assays were performed in 20 μl of a reaction mixture containing helicase buffer (50 mM Tris-HCl [pH 8.0], 1 mM DTT, 35 mM NaCl, 2 mM MgCl2, 2 mM ATP, 100 μg/ml BSA) and an indicated 32P-labeled dsDNA substrate (0.45 nM). The buffer and substrate concentrations were used in all unwinding assays, except when stated otherwise. The reaction was initiated by adding the indicated amount of the RecQ[Bs] protein into the mixture. The reaction mixture was assembled on ice, equilibrated briefly at room temperature, and incubated for 30 min at 37°C. The reaction was terminated by the addition of 5 μl of the quench solution (150 mM EDTA, 2% SDS, 30% glycerol, and 0.1% bromphenol blue). Reaction products were resolved using 6 or 12% native PAGE. The gel was dried, and the bands were visualized and quantified by PhosphorImager (Molecular Dynamics, Amersham, Piscataway, NJ). The intensity of each band in the PhosphorImager was determined using the volume integration function of the PhosphorImager. The concentration of each labeled fragment i in lane j was calculated using the following equation: Fragmentij = (intensityij/totalj) × DNA0, where intensityij is the integrated intensity of band i in lane j, totalj is the sum of all integrated bands in lane j, and DNA0 is the initial DNA concentration.
DNA-binding measurements by electrophoretic mobility shift assays (EMSAs).
The binding reactions (20-μl volumes) were conducted in a binding buffer (50 mM Tris-HCl [pH 7.5], 1 mM DTT, 50 mM NaCl, and 10% [vol/vol] glycerol). 5′-32P-labeled DNA substrate (0.45 nM) and the protein (at the concentrations indicated in the figure legends) were incubated in the binding buffer at 25°C for 20 min. After adding 4 μl of the native loading dye (0.25% bromophenol blue in 30% glycerol), the products were loaded on a 15% native PAGE (acrylamide/bis-acrylamide, 37.5:1 [wt/wt]). Electrophoresis was carried out in Tris-borate EDTA (TBE) buffer (40 mM Tris-acetate [pH 8.0], 1 mM EDTA) at 100 V. The gel was dried and visualized by autoradiography.
DNA-binding and protein-binding assays by fluorescence polarization.
The binding of RecQ[Bs] to different DNA substrates was analyzed by fluorescence polarization using a Beacon fluorescence polarization spectrophotometer (PanVera) as described previously (36, 37). The 5′-fluorescein-labeled ss- and dsDNA were used. Various amounts of the protein were added to a 150-μl aliquot of binding buffer [25 mM Tris-HCl (pH 8.0), 30 mM NaCl, 3 mM Mg(OAc)2, and 1 mM DTT] containing 2 nM the fluorescein-labeled ssDNA or dsDNA. Each sample was allowed to equilibrate in the solution for 5 min before the fluorescence polarization was measured. A second reading was taken after 10 min, in order to ensure that the mixture had reached equilibrium. Generally speaking, the binding equilibrium can be obtained in about 5 min, as less than 5% change in fluorescence polarization was observed after an additional 25-min incubation. The equilibrium dissociation constant of the protein-DNA complex was determined by plotting polarization as a function of the protein concentration and fitting the data to the Michaelis-Menten or Hill equation using the program KaleidaGraph (Synergy Software).
The binding affinities of RecQ[Bs] to the C-terminal peptides of two SSB[Bs] proteins, SSB[Bs] and YwpH (38), were determined as for the ss- and dsDNA substrates described above, and 5 nM Alexa Fluor dye-labeled peptides and various amounts of RecQ[Bs] were used.
Quantification of the protein-bound Zn2+ ion.
The protein-bound Zn2+ ion was measured using 4-(2-pyridylazo)resorcinol (PAR), a reporter dye that absorbs light at 490 nm when bound to Zn2+. To precisely quantify the Zn2+ content, all buffers were treated with Chelex-100 resins (Bio-Rad). The protein was dialyzed against the EDTA-free Chelex-treated buffer, passed over a 10-cm column of Chelex-100, and reconcentrated. To facilitate the Zn2+ release, the protein (about 1 nmol in 40 μl) was first denatured with Chelex-treated 7 M guanidine-HCl and then transferred to a 1-ml cuvette. PAR was added to the cuvette to a final concentration of 100 μM, and the volume was adjusted to 1 ml with PAR buffer (20 mM Tris-HCl [pH 8.0], 150 mM NaCl). The absorbance was recorded from 300 to 600 nm on Uvikon spectrophotometer 941 (Kontron) at 25°C. The quantity of Zn2+ was determined using the absorbance coefficient for the Zn(PAR)2 complex (ε500 = 6.6 × 104 M−1 cm−1). As a control, 20 nmol of the pure ZnCl2 was quantified under identical conditions.
Molecular modeling.
A template search was performed by comparing the sequence of RecQ[Bs] against the PDBSTR database (containing protein sequences for which three-dimensional structures are available in the Protein Data Bank) using BLAST (39). Two crystal structures were selected as the templates for N- and C-terminal parts of RecQ[Bs]. The first structure selected as a template corresponds to the N-terminal catalytic core of RecQ[Ec] (Ala-2 to Val-516) excluding the helicase/RNase D C-terminal (HRDC) domain (PDB code 1OYW) (32). The sequence identity and similarity between this fragment of RecQ[Ec] and the corresponding fragment of RecQ[Bs] (Met-1 to Glu-506) are 39% and 62%, respectively. The second template structure selected corresponds to the HRDC domain of RecQ[Ec] (Asn-530 to Asp-606) (PDB code 1WUD) (33). The sequence identity and similarity between this fragment of RecQ[Ec] and the corresponding fragment of RecQ[Bs] (Asn-516 to Thr-590) are 41% and 65%, respectively.
The sequence alignments were achieved using program ClustalW (40) (see Fig. S1 in the supplemental material). The relatively high sequence identities (above the well-established limit of 30%) made it possible to perform molecular modeling on the two parts of RecQ[Bs]. Two structural models were built using Nest (41). The model structures were further refined by energy minimization employing conjugate gradient optimization from the Charmm27 force field (42). Finally, the structural models were validated using Procheck (for stereochemistry) and the multievaluation tool Eval123D (43).
RESULTS
RecQ[Bs] has an RecQ[Ec]-like fold as suggested by molecular modeling.
Comparative sequence analysis between RecQ[Ec] and the reading frame from the B. subtilis genome has identified RecQ[Bs] as a homologue (19–21). The sequence alignment between two proteins over the whole sequence shows that two proteins have 40% identity and 58% similarity (see Fig. S1 in the supplemental material). This degree of sequence homology allowed us to perform molecular modeling on RecQ[Bs] using the known three-dimensional crystal structures of RecQ[Ec]. Two crystal structures of the RecQ[Ec] fragments, the N-terminal catalytic core domain fragment (32) and the C-terminal HRDC domain-containing fragment (33, 44), were used as the templates for the modeling studies.
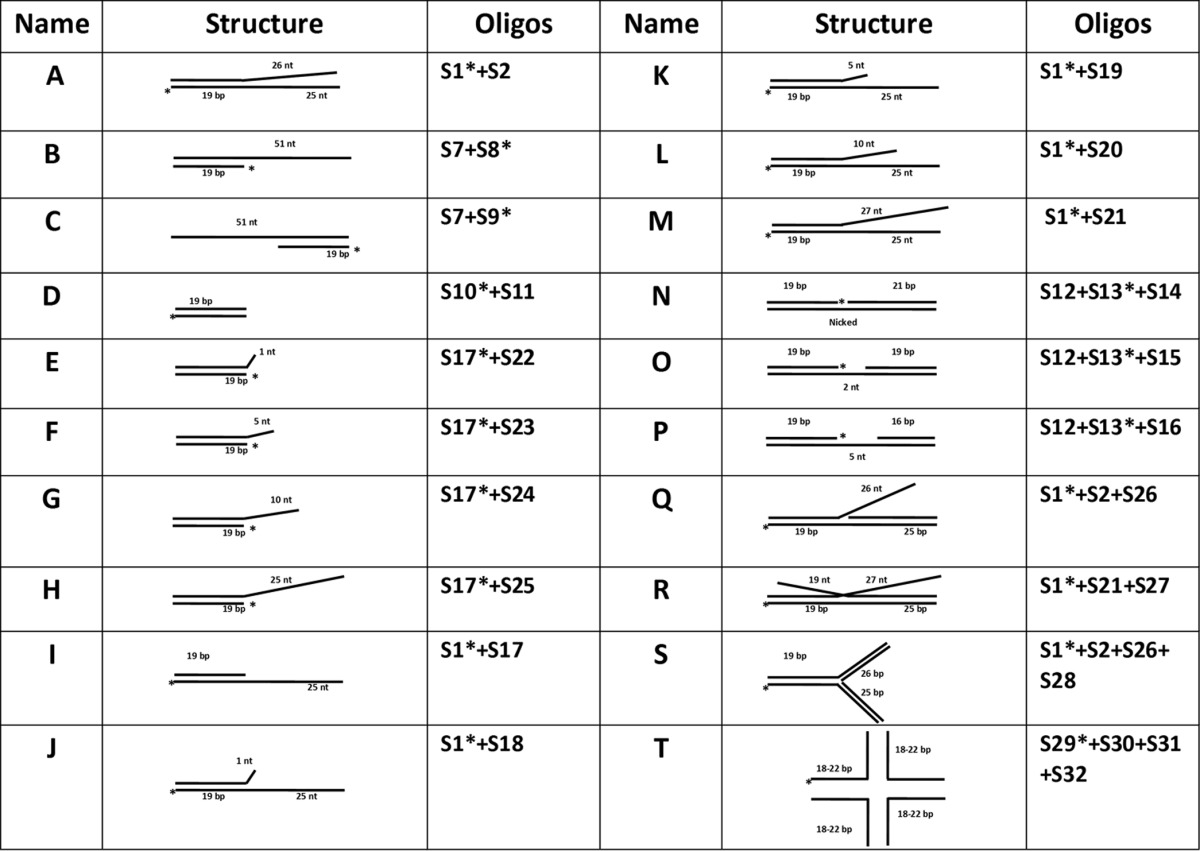
The first modeling was performed on the 506-residue N-terminal fragment (from Met-1 to Glu-506) of RecQ[Bs] using the structure of the catalytic core domain of RecQ[Ec], which included the helicase and RecQ conserved C-terminal (RQC) domains (32). The modeling studies showed that the N terminus of RecQ[Bs] also contained a helicase domain and an RecQ-specific RQC domain (Fig. 1A and B). In particular, the RQC domain consisted of a conserved zinc-binding subdomain (ZBD) (Fig. 1C) (32) and a less conserved winged helix (WH) subdomain (Fig. 1A) (32, 45). Furthermore, the results show that the structures of the N-terminal regions of the two proteins are very similar. For the ZBD subdomain, our structural model predicted that four cysteine residues, Cys-374, Cys-391, Cys-394, and Cys-397, of RecQ[Bs] were responsible for the zinc binding (Fig. 1C). The existence of the ZBD domain was further confirmed by a PAR assay, which showed that RecQ[Bs] indeed contained one equivalent Zn2+ ion per protein molecule (Table 3). However, the direct involvement of the four modeled cysteines remains to be demonstrated by the mutagenesis studies.
FIG 1.
Three-dimensional structure models of RecQ[Bs]. (A) Ribbon representation of the RecQ[Bs] helicase model. The first and second lobes of the helicase domain are colored in red and blue, respectively, the highly conserved ZBD subdomain is represented in yellow, and the WH subdomain of the RQC domain is in green. (B) Tube representation of the RecQ[Bs] helicase model (orange) superimposed on the crystal structure of the RecQ[Ec] helicase catalytic core, used as a template (PDB number 1OYW, in blue). The ZBD subdomains are in cartoon representation. The HRDC domain is not included in the structures represented. (C) Detail of the ZBD subdomain in the model of RecQ[Bs]. The four conserved cysteine residues coordinate a zinc ion, but their direct involvement remains to be demonstrated experimentally.
TABLE 3.
Summary of ATPase activity, DNA-binding properties, and Zn2+ contenta of the RecQ[Bs] helicase
| DNA typeb | ATPase activity |
DNA-binding affinity (Kd [nM]) | |
|---|---|---|---|
| Km (μM) | kcat (s−1) | ||
| ssDNA (−) | NDc | ND | |
| ssDNA (+) | 502.0 ± 17.8 | 6.9 ± 0.2 | ND |
| dsDNA | ND | ||
| Forked DNA | 38.0 ± 14.2 | ||
The ratio of Zn2+ to RecQ protein was 0.98 ± 0.12.
− and + indicate absence and presence, respectively.
ND, not determined (the value could not be determined accurately).
Next, we performed a modeling study on the C-terminal fragment of RecQ[Bs] using the crystal structure of the E. coli HRDC domain as a template (PDB code 1WUD) (33). The result showed that the RecQ[Bs] C-terminal fragment from Asn-516 to Thr-590 also possessed an HRDC domain (see Fig. S1 in the supplemental material). The HRDC domain of RecQ[Ec] has been implicated in conferring DNA substrate specificity (33, 46). In BLM, the HRDC domain is essential for dissolution of a double Holliday junction (47). Taken together, these results indicate that RecQ[Bs] has an RecQ[Ec]-like fold (Fig. 1).
RecQ[Bs] is a monomeric protein in the presence and absence of DNA and AMPPNP.
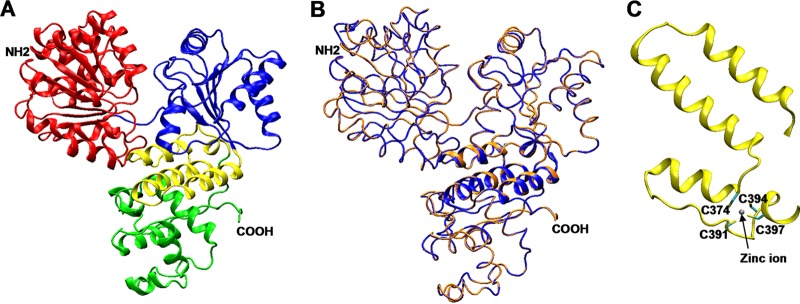
The RecQ[Bs] protein was expressed in E. coli and purified as an N-terminal His6-tagged fusion protein using Ni2+-NTA agarose, followed by size exclusion chromatography to near homogeneity (see Fig. S2 in the supplemental material). The protein used for the biochemical characterization in this study contained an N-terminal His6 tag followed by 591 resides of the full-length RecQ[Bs] sequence. The recombinant protein had a molecular mass of 68 kDa and migrated at ∼60 kDa on an 8% SDS-PAGE (see Fig. S2 in the supplemental material). Gel filtration chromatography was used to evaluate the oligomerization state of the purified protein in solution. The results showed that at the concentrations of 1 and 15 mg/ml (Fig. 2A), the recombinant RecQ[Bs] eluted as a globular protein with an apparent molecular mass of 69 ± 2 kDa (Fig. 2), suggesting that the protein exists as a monomer in solution.
FIG 2.
Monomeric state of the recombinant RecQ[Bs] protein. (A) Gel filtration analysis of the purified RecQ[Bs]. Elution profiles of the purified RecQ[Bs] from a Superdex 200 column at two concentrations (1 and 15 mg/ml). Only the region showing the absorption profile of RecQ[Bs] is shown. The molecular mass calibration was done as described in Materials and Methods. (B) Dynamic light scattering analysis of the recombinant RecQ[Bs] in the presence of 2 mM AMPPNP and/or 3 μM forked duplex DNA (oligonucleotides S29/S32). The measurement was performed as described in Materials and Methods. The determined radius and the calculated molecular mass of protein alone and in complex with different ligands are shown in the table.
To further determine the oligomeric state of RecQ[Bs] in the presence and in the absence of AMPPNP, a nonhydrolyzable analogue of ATP, and/or DNA, we performed dynamic light scattering measurement and found that the protein remains in a monomeric state when it is in a complex with 2 mM AMPPNP and/or 3 μM forked duplex DNA in solution (Fig. 2B).
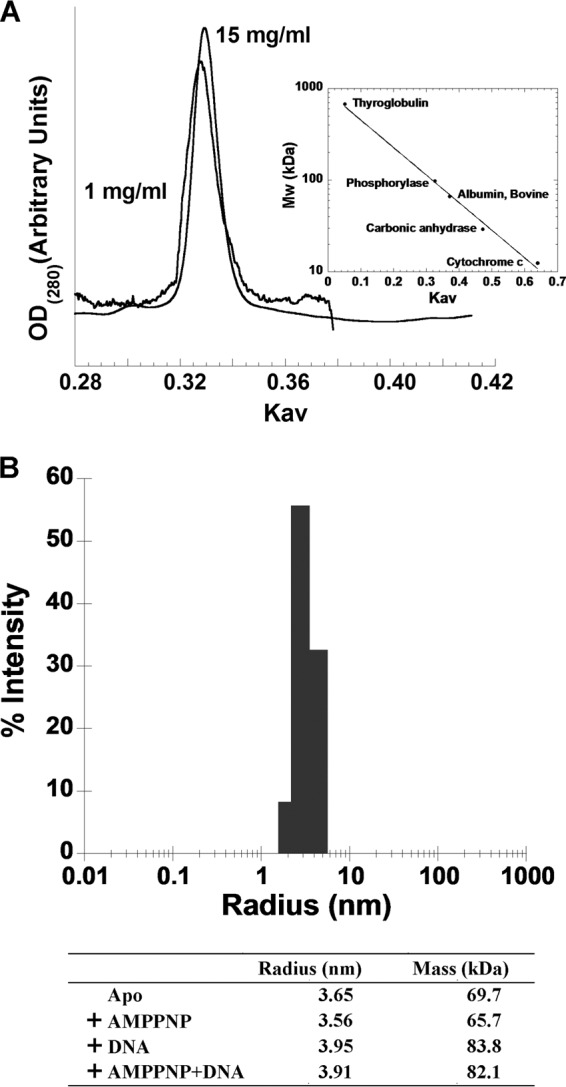
RecQ[Bs] binds both ssDNA and blunt-ended dsDNA weakly but forked dsDNA strongly.
We first analyzed the ability of RecQ[Bs] to bind ssDNA and dsDNA substrates by EMSAs (Fig. 3A). In the absence of ATP, the protein bound to either ssDNA or blunt-ended dsDNA. However, the apparent binding affinities of the protein for these substrates were rather low, judged from the extent of the DNA-protein complex formation (Fig. 3A, left panel for ssDNA; data not shown for blunt-ended dsDNA). Interestingly, RecQ[Bs] formed a more stable protein-DNA complex with a forked DNA duplex (Fig. 3A, right panel). The DNA-binding affinities of the protein were determined quantitatively under the equilibrium conditions using the fluorescein-labeled DNA substrates (Fig. 3B and Table 3), as described for other RecQ proteins (36, 37). The results show that the forked DNA duplex reaches 100% binding at a protein concentration of ∼250 nM, while ssDNA and blunt-ended dsDNA fail to reach maximum binding, even at a considerably higher protein concentration (Fig. 3B). The binding affinity (Kd) of the protein for the forked dsDNA is 38 ± 12.4 nM. These results show that RecQ[Bs] has a preference for the forked DNA duplex over blunt-ended dsDNA and ssDNA.
FIG 3.
RecQ[Bs] binds weakly to ssDNA and blunt-ended dsDNA but strongly to forked dsDNA. (A) DNA-binding affinities of RecQ[Bs] were measured by EMSAs. The assays were performed as a function of RecQ[Bs] concentration with either a 44-nt ssDNA (oligonucleotide S1 [Table 1]) or a 19-bp forked dsDNA substrate (DNA substrate A; 0.50 nM [Table 2]). Shown are representative 6% native PAGEs. The DNA substrates are schematically depicted on the left or right. Asterisks (*) indicate the position of the 32P label. (B) Anisotropy-based DNA binding isotherms of RecQ[Bs]. The same DNA molecules (S1 in Table 1 and A and D in Table 2) used in the gel shift assays were labeled with fluorescein at the 3′-end of the DNA substrates. Fluorescence anisotropy values of the different DNA substrates were determined as a function of the protein concentration. Data represent the means from at least three experiments with standard deviations indicated by error bars. The solid lines represent the best fits of the data to the Michaelis-Menten equation. The apparent Kd values determined (36, 37) are summarized in Table 3.
RecQ[Bs] possesses ATPase activity that is stimulated by either ssDNA or dsDNA.
The ATPase activity of the RecQ[Bs] protein was analyzed in the presence or absence of a DNA or RNA cofactor (see Fig. S3A in the supplemental material). No activity was detected in the presence of an RNA molecule or in the absence of a DNA cofactor. The ATPase activity of RecQ[Ec] was more strongly stimulated by ssDNA than by dsDNA (2). However, the ATPase activity of RecQ[Bs] was stimulated at similar levels by ssDNA and dsDNA.
We next characterized the effects of pH, divalent cations, BSA, DTT, and ATP on the ATPase activity of RecQ[Bs]. The solution pH had a noticeable effect, with an optimal effect at pH 7.5 to 8.2 (see Fig. S3B in the supplemental material). Divalent metal ions also affected the ATPase activity of the enzyme. Mg2+ ion appeared to be required for the observed ATPase activity of the enzyme, as omitting the cation or removing the cation by EDTA completely abolished the activity (see Fig. S3C in the supplemental material). Other cations, Mn2+, Ca2+, and in particular Zn2+, could not replace Mg2+ (see Fig. S3C in the supplemental material). BSA was able to enhance the ATPase of the enzyme, while DTT was not required for the reaction, suggesting that the protein does not contains any solvent-exposed cysteine residues (see Fig. S3C in the supplemental material). As expected, replacing ATP with nonhydrolyzable AMPPNP nearly abolished the observed ATPase activity (see Fig. S3C in the supplemental material). Altogether, these results show that RecQ[Bs] is an Mg2+- and DNA-dependent ATPase.
The steady-state kinetic parameters Km and kcat for the RecQ[Bs]-catalyzed ATP hydrolysis were measured in the presence and in the absence of ssDNA (see Fig. S3D in the supplemental material). To determine the Km values, ATP concentrations of 20 μM to 3.0 mM were used, and the ATPase activity was hyperbolic with respect to the ATP concentration (see Fig. S3D in the supplemental material). A Km value of 502.0 ± 17.8 μM for ATP was measured in the presence of ssDNA (Table 3). However, the Km value could not be determined accurately in the absence of ssDNA, as the ATP hydrolysis reaction was rather slow under the identical conditions. A kcat value of 6.9 s−1 was determined for ATPase in the presence of ssDNA (Table 3). To estimate the minimum number of nucleotide-binding sites involved in ATP hydrolysis, the ATP hydrolysis data were fitted to the Hill equation. The Hill coefficient of 1.1 ± 0.1 was determined from the slope of the plot (see Fig. S3D, inset, in the supplemental material), indicating that there was a single ATP-binding site in one enzyme molecule. Thus, the RecQ[Bs] enzyme functions as a monomer during ATP hydrolysis in the presence of an ssDNA molecule. This result is consistent with the gel filtration and dynamic light scattering studies described previously.
RecQ[Bs] is an ATP-dependent DNA helicase.
To characterize the helicase activity of the putative RecQ[Bs] helicase, a substrate composed of 5′-32P-labeled 20-nt oligonucleotide annealed to ϕX174 circle ssDNA was used (Fig. 4A). This partial DNA duplex has neither a 5′- nor a 3′-tail. Thus, similar DNA duplexes containing either a 5′- or a 3′-tail were also included in the helicase assay (Fig. 4A). The RecQ[Bs] protein unwound all three dsDNA substrates with almost the same efficiency at a protein concentration of 0.5 μM (Fig. 4A).
FIG 4.
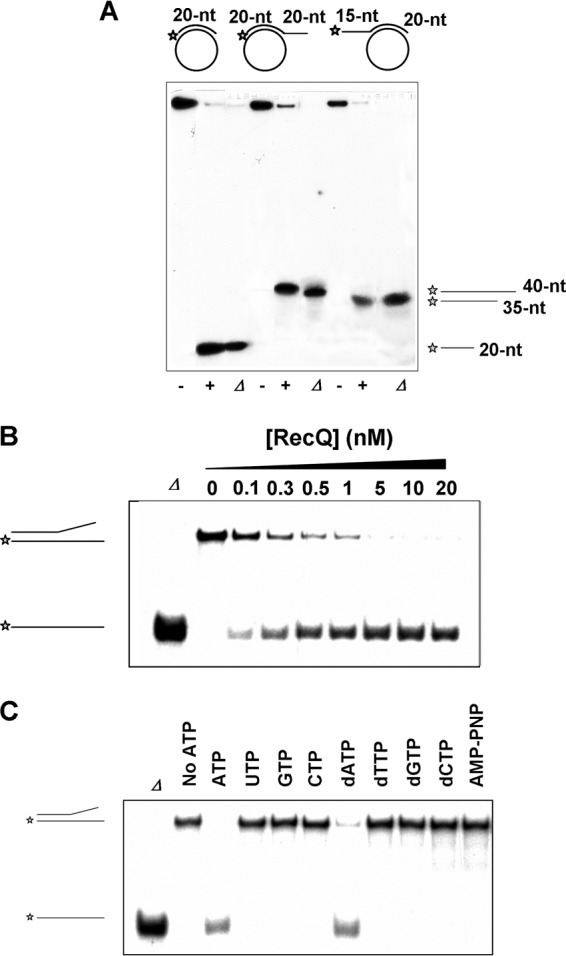
RecQ[Bs] is an ATP-dependent DNA helicase. (A) RecQ[Bs] unwinds dsDNA substrates formed by annealing three different 5′-32P-labeled oligonucleotides with ϕX174 circular ssDNA. The 20-nt (S4), 40-nt (S5), and 35-nt (S6) oligonucleotides (Table 1) were used to form three different substrates. The panel shows the structures of the substrates (0.45 nM) and autoradiograms of the 12% native PAGE. The reactions were carried out in the presence (+) and absence (−) of the enzyme (0.5 μM) at 37°C for 30 min. Lanes labeled with Δ represent the boiled substrates. The reaction products are depicted on the right. (B) RecQ[Bs] unwinds a forked dsDNA. A radiolabeled 19-bp forked DNA molecule (substrate A in Table 2) (0.45 nM) was incubated with increasing amounts (top of the panel) of enzyme in the helicase buffer at 37°C for 30 min and separated by native PAGE. Lane Δ represents the boiled substrate. (C) RecQ[Bs] unwinds the forked DNA (substrate A in Table 2) in the presence of ATP. This activity is dependent on ATP (or dATP) hydrolysis, as it is not seen in the absence of ATP (or dATP) or in the presence of AMPPNP or any other triphosphate nucleotide. Helicase assays were conducted as described for panel B, in the presence of same amount of ATP or any other triphosphate nucleotide and RecQ[Bs] (80 nM).
To determine the optimal enzyme concentration required for unwinding dsDNA, the protein concentrations of 2.5 to 80 nM and a 19-bp forked dsDNA substrate were used (Fig. 4B), as most RecQ family helicases display a strong preference for the forked DNA duplexes and also as the RecQ[Bs] bound strongly to the forked DNA (Fig. 3B). The unwinding percentage of the dsDNA substrate increased with increasing enzyme concentration, and 90% of the dsDNA substrate was unwound at the enzyme concentration of 5 nM. The helicase activity of the enzyme was found to be sensitive to free magnesium concentration, which was inhibitory to the helicase activity of RecQ[Bs], and the enzyme displayed optimal unwinding at a ratio of Mg2+/ATP of 0.8 to 1.2 (data not shown).
To determine the nucleotide preference of the RecQ[Bs] helicase, four nucleotides and four deoxynucleotides were used to evaluate the enzyme-mediated unwinding of the same 19-bp forked dsDNA substrate (Fig. 4C). Under identical experimental conditions, only ATP and dATP promoted the helicase reaction, with ATP being preferred (100% unwinding) over dATP (82% unwinding). When AMPPNP was substituted for ATP, no helicase activity was observed for the enzyme. All these results indicate that RecQ[Bs] is an ATP-dependent DNA helicase.
RecQ[Bs] has a 3′→5′ polarity with a low unwinding activity that can be enhanced by an ssDNA-binding protein.
The RecQ family helicases characterized so far exhibit a 3′→5′ polarity. The polarity of unwinding by a helicase is defined by the strand to which the enzyme binds and the direction in which it moves along that strand. To determine the polarity of the RecQ[Bs] helicase action, the partial dsDNA substrates with either 3′- or 5′-single-stranded tails were used. The RecQ[Bs] helicase unwound only the 3′-overhung dsDNA substrate (Fig. 5), indicating that RecQ[Bs] is a unidirectional helicase with a 3′→5′ polarity.
FIG 5.
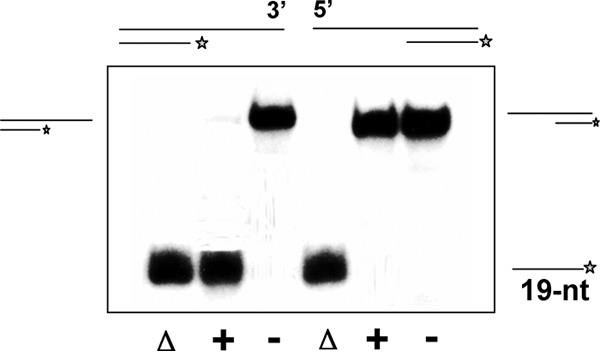
RecQ[Bs] is a 3′→5′ DNA helicase. The enzyme unwinds a substrate with a 3′- but not a 5′-single-stranded region. Radiolabeled oligonucleotide S8 or S9 was annealed with unlabeled oligonucleotide S7 (Table 1) to form 3′- or 5′-overhung 19-bp dsDNA molecules, substrate B or C (Table 2). The DNA substrate (0.45 nM) was incubated in the presence (+) or absence (−) of the enzyme (0.1 μM) at 37°C for 30 min, and the products were analyzed by electrophoresis using a 12% native PAGE and autoradiography. Substrates and reaction products are depicted on the left or right. Lanes labeled with Δ represent the heat-denatured substrates.
Next, we characterized the effect of the length of either 5′- or 3′-ssDNA tail on the unwinding activity of RecQ[Bs]. To evaluate the effect of the 3′-ssDNA tail length, four DNA duplexes (19 bp) with 3′-ssDNA tails of 1, 5, 10, and 25 nt were constructed (see Fig. S4A in the supplemental material). Under identical conditions, the enzyme was able to unwind about 50% of the substrates with 10- and 25-nt 3′-ssDNA tails and 30% of the substrate with 5-nt 3′-ssDNA tail. However, the enzyme was inactive on the substrate with only 1-nt 3′-ssDNA tail. These results suggest that the enzyme requires a minimal number of 3′-ssDNA nucleotides to bind in order to initiate unwinding, and this number is at least 2 but less than 5.
As RecQ[Bs] is a 3′→5′ DNA helicase, it could bind the 3′-ssDNA tail and move along that strand to unwind the dsDNA substrate. If that is the case, as long as the minimal number of 3′-ssDNA nucleotides is available for the enzyme binding, the length of the noncomplementary 5′-ssDNA tail on the other strand of the duplex should have no noticeable effect on the unwinding of the forked DNA substrate. Indeed, when four forked substrates, all containing a 25-nt 3′-ssDNA tail but differing in the number of nucleotides (0, 1, 5, or 10) in the noncomplementary 5′-ssDNA tail were used, similar unwinding efficiencies (62 to 69%) were observed under identical conditions (see Fig. S4B in the supplemental material).
To evaluate RecQ[Bs]-mediated helicase activity toward different-length duplex DNAs, two partial DNA duplex substrates were formed by annealing either 20 nt or 70 nt to a circular ϕX174 ssDNA. Figure 6 shows a time course of the RecQ[Bs]-mediated dsDNA unwinding. The short (20-bp) partial DNA duplex was unwound within 5 min, and the unwound product increased with time (Fig. 6A). In contrast, under the same conditions, no dsDNA unwinding could be detected for the longer (70-bp) substrate even after 50 min (Fig. 6B). Thus, RecQ[Bs] alone can unwind only short dsDNA substrates in vitro.
FIG 6.

RecQ[Bs] has a low unwinding activity toward long duplex DNA, which can be enhanced by an ssDNA-binding protein. A 32P-labeled oligonucleotide (20 or 70 nt; S4 or S7 in Table 1) was annealed to ϕX174 ssDNA (0.45 nM) to create a dsDNA substrate that was used for the helicase activity assays of RecQ[Bs] (0.5 μM) at 37°C; the reaction was stopped at the indicated time, and the products were analyzed by electrophoresis using a 12% native PAGE. (A, B) Representative gels for the assays carried out with the dsDNA substrate produced from either the 20-nt (S4) (A) or the 70-nt (S7) (B) oligonucleotide. Substrates and reaction products are depicted on the left; Δ, heat-denatured substrate. (C) Binding affinity of SSB[Bs] to RecQ[Bs]. The binding isotherm of N-terminal Alexa Fluor dye-labeled SSB[Bs]-C10 peptide to RecQ[Bs] was determined as a function of the RecQ[Bs] concentration as described in Materials and Methods. (D) Enhancement of unwinding activity of RecQ[Bs] by SSB[Bs]. The preformed 70-bp partial duplex DNA (0.45 nM, ϕX174/S7) was incubated with various amounts of RecQ[Bs] or SSB[Bs] alone for 30 min to determine the dsDNA unwinding. The results of these helicase assays are shown as a function of the protein (RecQ[Bs] or SSB[Bs]) concentration. Then, the experiments were repeated with a fixed amount of RecQ[Bs] (0.1 μM) and various amounts of SSB[Bs] or SSB[Bs]ΔC10 for 30 min. The results of the unwinding assays are presented as a function of the protein (SSB[Bs] or SSB[Bs]ΔC10) concentration. The results are the averages from at least three replicate experiments for each combination. Error bars in these experiments indicate standard deviations (n = 3).
It has been shown that SSB[Ec] stimulates the RecQ[Ec]-promoted unwinding (48, 49). SSB is composed of two domains: an N-terminal oligonucleotide/oligosaccharide-binding (OB) domain and a C-terminal domain of about 60 residues that end with a peptide characterized by a consensus signature, D-D-D-I/L-P-F, usually implicated in the interaction with interactome. The structural OB domain of SSB functions as the ssDNA-binding motif. Two paralogous genes, ssb and ywpH, coding for SSB were found in B. subtilis. The amino acid sequences of SSB[Bs] and YwpH show 80% similarity and 63% identity, respectively. Additionally, SSB[Bs] and YwpH share 75% and 56% sequence similarities with SSB[Ec], respectively. Moreover, YwpH appears to be lacking 59 residues of the C terminus of SSB[Bs] (see Fig. S5 in the supplemental material). Previously, SSB[Ec] has been shown to bind to RecQ[Ec] through its C-terminal domain, in particular, the last 9 residues (48, 49). Therefore, we first determined whether the C-terminal peptides of the SSB[Bs] and YwpH proteins could bind to RecQ[Bs]. Two peptides, SSB[Bs]-C10 and YwpH-C10, which correspond to the last 10 amino acids of the C terminus of SSB[Bs] and YwpH, respectively, were N-terminally labeled with Alexa Fluor dye. Their binding isotherms to RecQ[Bs] were determined. The binding curve with the SSB[Bs]-C10 peptide was hyperbolic, with a derived Kd value of 9.3 ± 1.4 μM (Fig. 6C). However, the binding affinity of the YwpH-C10 peptide to RecQ[Bs] was too weak to measure. Next, the effect of SSB[Bs] on the RecQ[Bs]-mediated unwinding of the 70-bp duplex was examined, and a strong enhancement was observed (Fig. 6D). This enhancement effect by SSB[Bs] was specific, because first, SSB[Bs] alone could not denature the 70-bp DNA duplex, and second, the SSB[Bs]-mediated enhancement increased as a function of the SSB[Bs] concentration; and furthermore, when the last 10 residues of the SSB[Bs] C terminus were removed, its stimulation effect was significantly decreased (Fig. 6D). Taken together, the results indicate that RecQ[Bs] exhibits a 3′→5′ polarity with a low in vitro unwinding activity toward longer duplex DNA that can be enhanced by a direct interaction with the C terminus of SSB.
RecQ[Bs] is inactive on the blunt-ended dsDNA but is active on the forked and gapped DNA duplexes.
The results for 3′-overhung DNA duplex substrates suggest that RecQ[Bs] would be inactive on the blunt-ended dsDNA molecules (see Fig. S4A in the supplemental material). This is somewhat surprising, as the RecQ[Ec] helicase has been shown to efficiently unwind such substrates at a high enzyme concentration (2, 11, 48). We therefore examined the activity of the enzyme on the blunt-ended dsDNA. First, a 50-bp blunt-ended dsDNA substrate was employed, and no unwinding activity was observed for RecQ[Bs] (data not shown). Next, a 19-bp blunt-ended dsDNA was used instead, and the same result was observed. RecQ[Bs] was inactive on such a substrate even at a concentration of 1 μM (Fig. 7, left panel). Varying the concentrations of ATP and Mg2+ had no noticeable effect on such a substrate either (data not shown). Finally, we characterized the helicase activity of RecQ[Bs] on the variants of the blunt-ended dsDNA, namely, nicked and gapped dsDNA duplexes. Under conditions identical to those shown for the blunt-ended dsDNA, the enzyme had a moderate activity on the gapped dsDNA substrates with gap sizes of 0 (nicked dsDNA), 2, and 5, which had 40-bp, 38-bp, and 35-bp dsDNA regions, respectively (Fig. 7, right panel). Although the length of the dsDNA region of these gapped dsDNAs was considerably longer than that of the 19-bp blunt-ended dsDNA, the unwinding efficiencies were determined to be 18, 61, and 82% for the substrates with gap sizes of 0, 2, and 5, respectively (Fig. 7).
FIG 7.
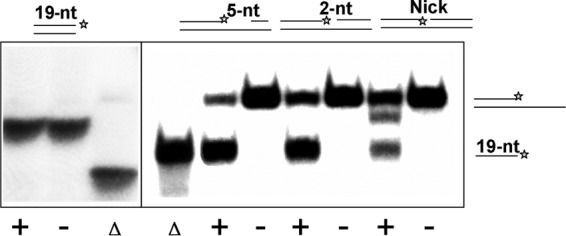
RecQ[Bs] is inactive on the blunt-ended dsDNA but is active on the nicked and gapped dsDNA. Radiolabeled dsDNA substrates (0.45 nM; substrate D in Table 2) were incubated in the presence (+) or absence (−) of RecQ[Bs] (0.1 μM) for 30 min at 37°C and separated by native PAGE. The substrates are depicted on the top of the panels. The position of the 5′-32P label is marked by an asterisk. Δ, heat-denatured substrate. RecQ[Bs] has no effect on the blunt-ended dsDNA (substrate D in Table 2) but can unwind nicked (substrate N in Table 2) or gapped (substrates O and P in Table 2) dsDNA substrates. For nicked dsDNA, the reaction intermediate, 3′-overhung dsDNA, can be observed.
Interestingly, we observed an additional band immediately below the substrate band in the reaction with the nicked substrate, which corresponds to the reaction intermediate product, 3′-overhung 19-bp dsDNA, formed by the substrates S13* and S12 (Tables 1 and 2 and Fig. 7). Compared to the gapped dsDNA (substrates O and P in Table 2), the nicked DNA, similar to the blunt-ended dsDNA, binds RecQ[Bs] weakly. Because of its poor binding affinity and unwinding efficiency, the unwinding intermediate can be observed.
RecQ[Bs] targets the DNA replication, repair, and recombination intermediates.
After showing that the RecQ[Bs] helicase preferentially unwound forked DNA duplexes with noncomplementary 3′-ssDNA tail and was active on gapped dsDNA, we then tested whether the enzyme could unwind other blunt-ended dsDNA with added structure features, such as 5′-flap DNA, Kappa joint molecules, synthetic replication fork, and Holliday junction molecules. These substrates resemble the DNA replication, repair, and recombination intermediates.
5′-Flap DNA molecules mimic the intermediates for the Okazaki fragment processing during lagging-strand DNA replication and for the DNA repair pathways such as base excision repair. These substrates are similar to the forked DNA in having a junction but lack one of the preexisting ssDNA tracts flanking the duplex region. Therefore, this type of substrate can be envisioned as a replication fork in which the synthesis of the lagging strand has been blocked or has not yet reached the point of the leading strand. To determine whether RecQ[Bs] can unwind one of such substrates or not, a 19-bp DNA duplex with its downstream 25-nt ssDNA region hybridized to a 25-nt ssDNA oligonucleotide was constructed. As shown in Fig. 8A, the enzyme unwound such substrate, releasing the labeled partial duplex with 5′-ssDNA overhang.
FIG 8.
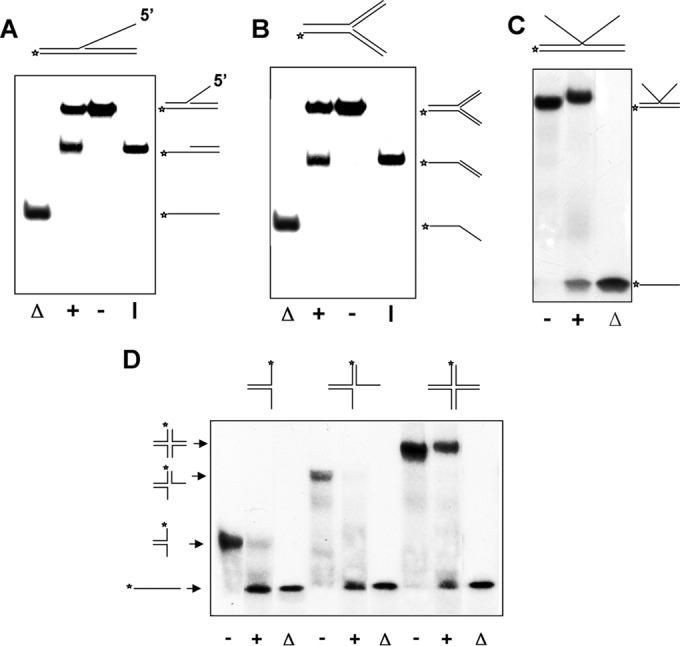
RecQ[Bs] targets DNA replication, repair, and recombination intermediates. The helicase assays were carried out with the indicated 5′-32P-labeled DNA substrates (0.45 nM). All reaction mixtures containing RecQ[Bs] (0.15 μM) were incubated for 30 min at 37°C, and the products were analyzed by electrophoresis using a 12% native PAGE. The substrate and intermediate and final products are depicted on either the right or the left. The position of the 5′-32P label is marked by an asterisk. (A) Representative gel showing unwinding of a 5′-flap DNA molecule (substrate Q in Table 2) in the presence (+) or absence (−) of the enzyme. Lane Δ, heat-denatured substrate; lane I, control intermediate. (B) Representative gel showing unwinding of a synthetic fork dsDNA molecule (substrate S in Table 2) in the presence (+) or absence (−) of the enzyme. Lane Δ, heat-denatured substrate; lane I, control intermediate. (C) Representative gel showing unwinding of a Kappa joint DNA molecule (substrate R in Table 2) in the presence (+) or absence (−) of the enzyme. Lane Δ, heat-denatured substrate. The slightly slower migration of the DNA substrate band represents the substrate bound to the protein. (D) Representative gel showing unwinding of the two-, three-, and four-way junction DNA substrates. The substrates were formed by annealing 5′-32P-labeled oligonucleotide S29 with oligonucleotide S32, or with oligonucleotides S32 and S30, or with oligonucleotides S30, S31, and S32 (Table 1) to form two-, three-, and four-way (substrate T in Table 2) junction DNA molecules, respectively. The unwinding reactions were carried out in the presence (+) or absence (−) of the enzyme at 37°C for 30 min. Lanes labeled with Δ represent the boiled substrates. The reaction substrates and products are depicted on the left and top of the gel.
A synthetic forked-DNA molecule is a three-way junction DNA molecule in which both 5′- and 3′-tails of a forked duplex are double stranded. The synthetic forked DNA resembles a stalled replication fork with double-stranded leading and lagging strands. As shown in Fig. 8B, RecQ[Bs] unwound one of such substrates at an enzyme concentration of 0.15 μM. Interestingly, only the DNA duplex product with a 5′-labeled overhang was observed (Fig. 8B). This result is consistent with the 3′→5′ polarity of the helicase.
A Kappa joint molecule can be formed by a linear dsDNA molecule invaded by a homologous ssDNA, and it resembles the letter K. Hence, it is generally referred to as a Kappa intermediate. The Kappa intermediates are often formed in the DNA recombination and repair processes. As shown in Fig. 8C, RecQ[Bs] unwound a Kappa substrate at an enzyme concentration of 0.15 μM.
Several RecQ helicases, including BLM, WRN, RecQ[Ec], and Sgs1, are known to disrupt synthetic immobile four-way DNA junctions, which resemble a Holliday junction (HJ or X structure) intermediate normally formed during homologous recombination (11, 50–52). We expected that RecQ[Bs] would catalyze the same reaction. Substrates of two-, three-, and four-stranded synthetic DNA junctions were prepared by annealing two, three, or four partially complementary oligonucleotides, each 40 nt in length (Tables 1 and 2). Indeed, as shown in Fig. 8D, the enzyme disrupted the synthetic Holliday junction substrate formed by four partially complementary oligonucleotides, each 40 nt in length (Tables 1 and 2). The substrate contained a short region (4 bp) of homology at the central core, which closely mimics the structure of a natural Holliday junction present at the crossover point (53). Interestingly, under our experimental conditions, we did not observe two- and three-stranded intermediates for three- and four-stranded junction DNA substrates. This behavior is reminiscent of that of the full-length Sgs1 (54) but different from that of the truncated Sgs1 (50), WRN (52), or BLM (51). Taken together, all these results show that RecQ[Bs] can target the DNA replication, repair, and recombination intermediates.
DISCUSSION
In this report, we describe the first biochemical characterization of the recombinant RecQ[Bs] protein. Our results reveal that RecQ[Bs] is indeed an RecQ homologue and shares similar structure with RecQ[Ec]. However, our analysis indicates that RecQ[Bs] resembles the truncated Sgs1 and some of the human RecQ homologues more than RecQ[Ec] with regard to its enzymatic properties. These results suggest functional similarities and differences between RecQ[Bs] and other RecQ members of the family.
Comparative sequence analysis has identified RecQ[Bs] as an RecQ[Ec] homologue. These two proteins have a high sequence similarity (58%) and therefore likely share a similar three-dimensional structure. This postulation has been confirmed by the molecular modeling. The gel filtration results indicate that RecQ[Bs] is monomeric at least up to a concentration of 15 mg/ml in solution in the absence of a DNA cofactor (Fig. 2A). Furthermore, the Hill analysis shows that it is monomeric as an ATPase during ATP hydrolysis in the presence of an ssDNA cofactor (see Fig. S3D in the supplemental material). Dynamic light scattering study shows that RecQ[Bs] remains monomeric in the presence of AMPPNP and/or DNA (Fig. 2B), indicating that RecQ[Bs] is a monomeric helicase, similar to RecQ[Ec] (55).
RecQ[Bs] displays very low ATPase activity in the presence of an RNA molecule or in the absence of a DNA cofactor. Unlike RecQ[Ec], whose ATPase activity is stimulated by ssDNA but not by dsDNA (2), the ATPase activity of RecQ[Bs] is stimulated by both forms of DNA (see Fig. S3A in the supplemental material), with an optimal pH of 7.5 to 8.2 (see Fig. S3B in the supplemental material). In the presence of ssDNA, the apparent kcat for RecQ[Bs] ATPase is 6.9 s−1 at 37°C (pH 8.0). The kcat values for the ssDNA-dependent ATP hydrolysis for RecQ[Ec], Sgs1, hRECQ1, hBLM, hWRN, and hRECQ5 are 36.1 (37°C, pH 8.0), 260 (30°C, pH 7.5), 0.5 to 2.1 (37°C, pH 7.5), 2.4 to 19.4 (37°C, pH 7.4), 1.0 to 2.5 (24°C, pH 7.4), and 15.6 s−1 (37°C, pH 8.0), respectively (36, 54, 56–59). Thus, RecQ[Bs] is about 5-fold less active than RecQ[Ec] but is about 2- to 4-fold more active than human members of the family, except hRECQ5. These results suggest that RecQ[Bs] enzymatically resembles some of the hRecQ helicases more than RecQ[Ec], although RecQ[Bs] shares high sequence homogeneity and 3D structure similarity with RecQ[Ec]. This conclusion is somewhat unexpected but is not impossible. The full-length Sgs1 is shown to resemble RecQ[Ec] more than any of the hRecQ helicases with respect to its enzymatic activities (54).
RecQ[Ec] and Sgs1 helicases can initiate duplex DNA unwinding from a blunt-ended terminus (2, 11, 54). However, truncated Sgs1 and the hRecQ helicases RECQ1, BLM, WRN, RECQ4, and RECQ5β cannot initiate unwinding from the blunt ends; instead, they are capable of unwinding the forked structures (4, 50). Similar to what is seen with hRecQ helicases, the unwinding of a blunt-ended dsDNA by RecQ[Bs] was undetectable under similar conditions (Fig. 7), in spite of varying free Mg2+ and enzyme concentrations. RecQ[Bs] efficiently unwinds forked DNA with a 3′→5′ polarity, the property shared by all known RecQ family helicases. These results are consistent with the observed DNA-binding affinities of the protein.
Similar to hRecQ helicases and truncated Sgs1 (4), the unwinding efficiency catalyzed by RecQ[Bs] decreases sharply with increasing length of the DNA duplex (Fig. 6). This may be related to the processivity of the unwinding reaction. However, the unwinding assay that we used in this study detects only complete unwinding, not the partially unwound fragments. It is therefore plausible that the longer unwound DNA strands reanneal behind the translocating RecQ[Bs] and thereby reduce the observed unwinding efficiency. Alternatively, RecQ[Bs] might really exhibit a low processivity and fall off the DNA before completing the unwinding of the annealed fragment. We found that the presence of SSB[Ec] or SSB[Bs] significantly increased the extent of unwinding of the 70-bp partial duplex by RecQ[Bs] (Fig. 6C). Presumably, SSB[Bs] interacts with RecQ[Bs] through its C terminus, to stimulate the RecQ[Bs]-mediated unwinding, thereby increasing the unwinding activity toward the longer duplex DNA. It is noteworthy that the above observation may or may not be related to the processivity. Definitive determination of the processivity should be performed under single-turnover conditions with stopped-flow apparatus in future experiments.
One striking feature of RecQ[Bs]-mediated DNA unwinding is that RecQ[Bs] unwinds well blunt-ended DNA duplexes with certain structural features, including nicks, gaps, 5′-flap resembling Okazaki fragment maturation, Kappa joint, synthetic replication fork, and Holliday junction, although it is inactive on blunt-ended dsDNA. This suggested that RecQ[Bs] is more implicated in resolving the DNA intermediates formed during DNA replication, repair, and recombination. DNA damage causes perturbations in DNA replication, arrests cell cycle progression, and triggers a transcriptional response that increases the chances of survival. The detection and subsequent repair of DSBs is critical for the survival of all organisms. In B. subtilis, the site-specific or randomly induced DSBs are repaired mainly by homologous recombination (22). Here we have shown that RecQ[Bs] is indeed an RecQ helicase that could be involved in homologous recombination. Consistent with this proposal, RecQ[Bs] has been shown to localize throughout the nucleoids in the presence or absence of DSBs, indicating that RecQ[Bs] is constitutively associated with chromosomal DNA (19). However, mutations in addA5 addB72 (60) or null mutations in the recS (20), recJ, or the recQ[Bs] gene (19) render cells only moderately sensitive to the killing action of DNA-damaging agents. Thus, Sanchez and coworkers have proposed that (i) RecQ[Bs] and RecS likely have some overlapping activities, (ii) AddAB helicase/nuclease and RecQ[Bs](S)-RecJ helicase-nuclease provide two complementary pathways for processing DNA ends with relatively similar efficiency, and (iii) the RecQ[Bs](S)-RecJ repair pathway is operative even in the presence of AddAB (19). Therefore, different avenues are operative in B. subtilis and E. coli during DSB repair (19). Consequently, in B. subtilis, homologous recombination could initiate with the 5′→3′ end processing by the RecJ exonuclease in collaboration with RecQ[Bs] or RecS or by the AddAB nuclease-helicase complex (23, 24). The resulting 3′-single-stranded tails are then bound by SSB[Bs], with RecN binding to the 3′-OH ends (24, 25). Further biochemical characterization of RecQ[Bs] in greater detail is obviously needed to provide the biochemical basis of the functional roles of RecQ[Bs] and to understand biochemical similarities and differences between RecQ[Bs] and other RecQ helicases, in particular RecQ[Ec], Sgs1, and hRecQ enzymes.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Science Foundation of China (no. 31370798, 11304252, and 31301632) and Projects 985 and 211 from the Ministry of Education of China (to X.G.X.), the American Cancer Society (RSG-03-238-01), the American Heart Association (0335253N), and the fund from Kunming University of Science and Technology (to J.-S.H.).
Footnotes
Published ahead of print 22 September 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.06367-11.
REFERENCES
- 1.Nakayama H, Nakayama K, Nakayama R, Irino N, Nakayama Y, Hanawalt PC. 1984. Isolation and genetic characterization of a thymineless death-resistant mutant of Escherichia coli K12: identification of a new mutation (recQ1) that blocks the RecF recombination pathway. Mol. Gen. Genet. 195:474–480. 10.1007/BF00341449. [DOI] [PubMed] [Google Scholar]
- 2.Umezu K, Nakayama K, Nakayama H. 1990. Escherichia coli RecQ protein is a DNA helicase. Proc. Natl. Acad. Sci. U. S. A. 87:5363–5367. 10.1073/pnas.87.14.5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gorbalenya AE, Koonin EV. 1993. Helicases: amino acid sequence comparisons and structure-function relationships. Curr. Opin. Struct. Biol. 3:419–429. 10.1016/S0959-440X(05)80116-2. [DOI] [Google Scholar]
- 4.Bachrati CZ, Hickson ID. 2003. RecQ helicases: suppressors of tumorigenesis and premature aging. Biochem. J. 374:577–606. 10.1042/BJ20030491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Killoran MP, Keck JL. 2006. Sit down, relax and unwind: structural insights into RecQ helicase mechanisms. Nucleic Acids Res. 34:4098–4105. 10.1093/nar/gkl538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brosh RM, Jr, Bohr VA. 2007. Human premature aging, DNA repair and RecQ helicases. Nucleic Acids Res. 35:7527–7544. 10.1093/nar/gkm1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ouyang KJ, Woo LL, Ellis NA. 2008. Homologous recombination and maintenance of genome integrity: cancer and aging through the prism of human RecQ helicases. Mech. Ageing Dev. 129:425–440. 10.1016/j.mad.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 8.Mankouri HW, Hickson ID. 2007. The RecQ helicase-topoisomerase III-Rmi1 complex: a DNA structure-specific ‘dissolvasome'? Trends Biochem. Sci. 32:538–546. 10.1016/j.tibs.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 9.Horii Z, Clark AJ. 1973. Genetic analysis of the recF pathway to genetic recombination in Escherichia coli K12: isolation and characterization of mutants. J. Mol. Biol. 80:327–344. 10.1016/0022-2836(73)90176-9. [DOI] [PubMed] [Google Scholar]
- 10.Kowalczykowski SC. 2000. Initiation of genetic recombination and recombination-dependent replication. Trends Biochem. Sci. 25:156–165. 10.1016/S0968-0004(00)01569-3. [DOI] [PubMed] [Google Scholar]
- 11.Harmon FG, Kowalczykowski SC. 1998. RecQ helicase, in concert with RecA and SSB proteins, initiates and disrupts DNA recombination. Genes Dev. 12:1134–1144. 10.1101/gad.12.8.1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Handa N, Morimatsu K, Lovett ST, Kowalczykowski SC. 2009. Reconstitution of initial steps of dsDNA break repair by the RecF pathway of E. coli. Genes Dev. 23:1234–1245. 10.1101/gad.1780709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amundsen SK, Smith GR. 2003. Interchangeable parts of the Escherichia coli recombination machinery. Cell 112:741–744. 10.1016/S0092-8674(03)00197-1. [DOI] [PubMed] [Google Scholar]
- 14.Kuzminov A. 1999. Recombinational repair of DNA damage in Escherichia coli and bacteriophage lambda. Microbiol. Mol. Biol. Rev. 63:751–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ryder L, Whitby MC, Lloyd RG. 1994. Mutation of recF, recJ, recO, recQ, or recR improves Hfr recombination in resolvase-deficient ruv recG strains of Escherichia coli. J. Bacteriol. 176:1570–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hanada K, Ukita T, Kohno Y, Saito K, Kato J, Ikeda H. 1997. RecQ DNA helicase is a suppressor of illegitimate recombination in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 94:3860–3865. 10.1073/pnas.94.8.3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harmon FG, Brockman JP, Kowalczykowski SC. 2003. RecQ helicase stimulates both DNA catenation and changes in DNA topology by topoisomerase III. J. Biol. Chem. 278:42668–42678. 10.1074/jbc.M302994200. [DOI] [PubMed] [Google Scholar]
- 18.Suski C, Marians KJ. 2008. Resolution of converging replication forks by RecQ and topoisomerase III. Mol. Cell 30:779–789. 10.1016/j.molcel.2008.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sanchez H, Kidane D, Castillo Cozar M, Graumann PL, Alonso JC. 2006. Recruitment of Bacillus subtilis RecN to DNA double-strand breaks in the absence of DNA end processing. J. Bacteriol. 188:353–360. 10.1128/JB.188.2.353-360.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fernandez S, Sorokin A, Alonso JC. 1998. Genetic recombination in Bacillus subtilis 168: effects of recU and recS mutations on DNA repair and homologous recombination. J. Bacteriol. 180:3405–3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fernandez S, Ayora S, Alonso JC. 2000. Bacillus subtilis homologous recombination: genes and products. Res. Microbiol. 151:481–486. 10.1016/S0923-2508(00)00165-0. [DOI] [PubMed] [Google Scholar]
- 22.Sanchez H, Carrasco B, Ayora S, Alonso JC. 2007. Dynamics of DNA double-strand break repair in Bacillus subtilis. Caister Academic Press, Norfolk, United Kingdom. 10.1111/j.1365-2958.2007.05835.x. [DOI] [Google Scholar]
- 23.Michel B, Grompone G, Flores MJ, Bidnenko V. 2004. Multiple pathways process stalled replication forks. Proc. Natl. Acad. Sci. U. S. A. 101:12783–12788. 10.1073/pnas.0401586101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sanchez H, Alonso JC. 2005. Bacillus subtilis RecN binds and protects 3′-single-stranded DNA extensions in the presence of ATP. Nucleic Acids Res. 33:2343–2350. 10.1093/nar/gki533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sanchez H, Cardenas PP, Yoshimura SH, Takeyasu K, Alonso JC. 2008. Dynamic structures of Bacillus subtilis RecN-DNA complexes. Nucleic Acids Res. 36:110–120. 10.1093/nar/gkm759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kidane D, Sanchez H, Alonso JC, Graumann PL. 2004. Visualization of DNA double-strand break repair in live bacteria reveals dynamic recruitment of Bacillus subtilis RecF, RecO and RecN proteins to distinct sites on the nucleoids. Mol. Microbiol. 52:1627–1639. 10.1111/j.1365-2958.2004.04102.x. [DOI] [PubMed] [Google Scholar]
- 27.Sanchez H, Kidane D, Reed P, Curtis FA, Cozar MC, Graumann PL, Sharples GJ, Alonso JC. 2005. The RuvAB branch migration translocase and RecU Holliday junction resolvase are required for double-stranded DNA break repair in Bacillus subtilis. Genetics 171:873–883. 10.1534/genetics.105.045906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carrasco B, Manfredi C, Ayora S, Alonso JC. 2008. Bacillus subtilis SsbA and dATP regulate RecA nucleation onto single-stranded DNA. DNA Repair 7:990–996. 10.1016/j.dnarep.2008.03.019. [DOI] [PubMed] [Google Scholar]
- 29.Manfredi C, Carrasco B, Ayora S, Alonso JC. 2008. Bacillus subtilis RecO nucleates RecA onto SsbA-coated single-stranded DNA. J. Biol. Chem. 283:24837–24847. 10.1074/jbc.M802002200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sherratt DJ, Soballe B, Barre FX, Filipe S, Lau I, Massey T, Yates J. 2004. Recombination and chromosome segregation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 359:61–69. 10.1098/rstb.2003.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lecointe F, Serena C, Velten M, Costes A, McGovern S, Meile JC, Errington J, Ehrlich SD, Noirot P, Polard P. 2007. Anticipating chromosomal replication fork arrest: SSB targets repair DNA helicases to active forks. EMBO J. 26:4239–4251. 10.1038/sj.emboj.7601848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bernstein DA, Zittel MC, Keck JL. 2003. High-resolution structure of the E. coli RecQ helicase catalytic core. EMBO J. 22:4910–4921. 10.1093/emboj/cdg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bernstein DA, Keck JL. 2005. Conferring substrate specificity to DNA helicases: role of the RecQ HRDC domain. Structure 13:1173–1182. 10.1016/j.str.2005.04.018. [DOI] [PubMed] [Google Scholar]
- 34.Cantor CR, Warshaw MM, Shapiro H. 1970. Oligonucleotide interactions. 3. Circular dichroism studies of the conformation of deoxyoligonucleotides. Biopolymers 9:1059–1077. 10.1002/bip.1970.360090909. [DOI] [PubMed] [Google Scholar]
- 35.Guo RB, Rigolet P, Zargarian L, Fermandjian S, Xi XG. 2005. Structural and functional characterizations reveal the importance of a zinc binding domain in Bloom's syndrome helicase. Nucleic Acids Res. 33:3109–3124. 10.1093/nar/gki619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ren H, Dou SX, Zhang XD, Wang PY, Kanagaraj R, Liu JL, Janscak P, Hu JS, Xi XG. 2008. The zinc-binding motif of human RECQ5beta suppresses the intrinsic strand-annealing activity of its DExH helicase domain and is essential for the helicase activity of the enzyme. Biochem. J. 412:425–433. 10.1042/BJ20071150. [DOI] [PubMed] [Google Scholar]
- 37.Dou SX, Wang PY, Xu HQ, Xi XG. 2004. The DNA binding properties of the Escherichia coli RecQ helicase. J. Biol. Chem. 279:6354–6363. 10.1074/jbc.M311272200. [DOI] [PubMed] [Google Scholar]
- 38.Linderoth J, Jerkeman M, Cavallin-Stahl E, Kvaloy S, Torlakovic E. 2003. Immunohistochemical expression of CD23 and CD40 may identify prognostically favorable subgroups of diffuse large B-cell lymphoma: a Nordic Lymphoma Group Study. Clin. Cancer Res. 9:722–728. [PubMed] [Google Scholar]
- 39.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403–410. 10.1006/jmbi.1990.9999. [DOI] [PubMed] [Google Scholar]
- 40.Thompson JD, Higgins DG, Gibson TJ. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673–4680. 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Petrey D, Xiang Z, Tang CL, Xie L, Gimpelev M, Mitros T, Soto CS, Goldsmith-Fischman S, Kernytsky A, Schlessinger A, Koh IY, Alexov E, Honig B. 2003. Using multiple structure alignments, fast model building, and energetic analysis in fold recognition and homology modeling. Proteins 53(Suppl 6):430–435. 10.1002/prot.10550. [DOI] [PubMed] [Google Scholar]
- 42.MacKerell J, AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher I, Roux WEB, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, Karplus M. 1998. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102:3586–3616. 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 43.Gracy J, Chiche L, Sallantin J. 1993. Improved alignment of weakly homologous protein sequences using structural information. Protein Eng. 6:821–829. 10.1093/protein/6.8.821. [DOI] [PubMed] [Google Scholar]
- 44.Morozov V, Mushegian AR, Koonin EV, Bork P. 1997. A putative nucleic acid-binding domain in Bloom's and Werner's syndrome helicases. Trends Biochem. Sci. 22:417–418. 10.1016/S0968-0004(97)01128-6. [DOI] [PubMed] [Google Scholar]
- 45.Hu JS, Feng H, Zeng W, Lin GX, Xi XG. 2005. Solution structure of a multifunctional DNA- and protein-binding motif of human Werner syndrome protein. Proc. Natl. Acad. Sci. U. S. A. 102:18379–18384. 10.1073/pnas.0509380102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu Z, Macias MJ, Bottomley MJ, Stier G, Linge JP, Nilges M, Bork P, Sattler M. 1999. The three-dimensional structure of the HRDC domain and implications for the Werner and Bloom syndrome proteins. Structure 7:1557–1566. 10.1016/S0969-2126(00)88346-X. [DOI] [PubMed] [Google Scholar]
- 47.Wu L, Chan KL, Ralf C, Bernstein DA, Garcia PL, Bohr VA, Vindigni A, Janscak P, Keck JL, Hickson ID. 2005. The HRDC domain of BLM is required for the dissolution of double Holliday junctions. EMBO J. 24:2679–2687. 10.1038/sj.emboj.7600740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Harmon FG, Kowalczykowski SC. 2001. Biochemical characterization of the DNA helicase activity of the Escherichia coli RecQ helicase. J. Biol. Chem. 276:232–243. 10.1074/jbc.M006555200. [DOI] [PubMed] [Google Scholar]
- 49.Shereda RD, Bernstein DA, Keck JL. 2007. A central role for SSB in Escherichia coli RecQ DNA helicase function. J. Biol. Chem. 282:19247–19258. 10.1074/jbc.M608011200. [DOI] [PubMed] [Google Scholar]
- 50.Bennett RJ, Keck JL, Wang JC. 1999. Binding specificity determines polarity of DNA unwinding by the Sgs1 protein of S. cerevisiae. J. Mol. Biol. 289:235–248. 10.1006/jmbi.1999.2739. [DOI] [PubMed] [Google Scholar]
- 51.Karow JK, Constantinou A, Li JL, West SC, Hickson ID. 2000. The Bloom's syndrome gene product promotes branch migration of holliday junctions. Proc. Natl. Acad. Sci. U. S. A. 97:6504–6508. 10.1073/pnas.100448097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Constantinou A, Tarsounas M, Karow JK, Brosh RM, Bohr VA, Hickson ID, West SC. 2000. Werner's syndrome protein (WRN) migrates Holliday junctions and co-localizes with RPA upon replication arrest. EMBO Rep. 1:80–84. 10.1093/embo-reports/kvd004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bennett RJ, West SC. 1996. Resolution of Holliday junctions in genetic recombination: RuvC protein nicks DNA at the point of strand exchange. Proc. Natl. Acad. Sci. U. S. A. 93:12217–12222. 10.1073/pnas.93.22.12217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cejka P, Kowalczykowski SC. 2010. The full-length Saccharomyces cerevisiae Sgs1 protein is a vigorous DNA helicase that preferentially unwinds holliday junctions. J. Biol. Chem. 285:8290–8301. 10.1074/jbc.M109.083196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang XD, Dou SX, Xie P, Hu JS, Wang PY, Xi XG. 2006. Escherichia coli RecQ is a rapid, efficient, and monomeric helicase. J. Biol. Chem. 281:12655–12663. 10.1074/jbc.M513089200. [DOI] [PubMed] [Google Scholar]
- 56.Liu JL, Rigolet P, Dou SX, Wang PY, Xi XG. 2004. The zinc finger motif of Escherichia coli RecQ is implicated in both DNA binding and protein folding. J. Biol. Chem. 279:42794–42802. 10.1074/jbc.M405008200. [DOI] [PubMed] [Google Scholar]
- 57.Brosh RM, Jr, Orren DK, Nehlin JO, Ravn PH, Kenny MK, Machwe A, Bohr VA. 1999. Functional and physical interaction between WRN helicase and human replication protein A. J. Biol. Chem. 274:18341–18350. 10.1074/jbc.274.26.18341. [DOI] [PubMed] [Google Scholar]
- 58.Brosh RM, Jr, Li JL, Kenny MK, Karow JK, Cooper MP, Kureekattil RP, Hickson ID, Bohr VA. 2000. Replication protein A physically interacts with the Bloom's syndrome protein and stimulates its helicase activity. J. Biol. Chem. 275:23500–23508. 10.1074/jbc.M001557200. [DOI] [PubMed] [Google Scholar]
- 59.Cui S, Arosio D, Doherty KM, Brosh RM, Jr, Falaschi A, Vindigni A. 2004. Analysis of the unwinding activity of the dimeric RECQ1 helicase in the presence of human replication protein A. Nucleic Acids Res. 32:2158–2170. 10.1093/nar/gkh540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Alonso JC, Stiege AC, Luder G. 1993. Genetic recombination in Bacillus subtilis 168: effect of recN, recF, recH and addAB mutations on DNA repair and recombination. Mol. Gen. Genet. 239:129–136. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.