

ABSTRACT
Chikungunya virus (CHIKV) is a mosquito-borne alphavirus that has reemerged to cause profound epidemics of fever, rash, and arthralgia throughout sub-Saharan Africa, Southeast Asia, and the Caribbean. Like other arthritogenic alphaviruses, mechanisms of CHIKV pathogenesis are not well defined. Using the attenuated CHIKV strain 181/25 and virulent strain AF15561, we identified a residue in the E2 viral attachment protein that is a critical determinant of viral replication in cultured cells and pathogenesis in vivo. Viruses containing an arginine at E2 residue 82 displayed enhanced infectivity in mammalian cells but reduced infectivity in mosquito cells and diminished virulence in a mouse model of CHIKV disease. Mice inoculated with virus containing an arginine at this position exhibited reduced swelling at the site of inoculation with a concomitant decrease in the severity of necrosis in joint-associated tissues. Viruses containing a glycine at E2 residue 82 produced higher titers in the spleen and serum at early times postinfection. Using wild-type and glycosaminoglycan (GAG)-deficient Chinese hamster ovary (CHO) cell lines and soluble GAGs, we found that an arginine at residue 82 conferred greater dependence on GAGs for infection of mammalian cells. These data suggest that CHIKV E2 interactions with GAGs diminish dissemination to lymphoid tissue, establishment of viremia, and activation of inflammatory responses early in infection. Collectively, these results suggest a function for GAG utilization in regulating CHIKV tropism and host responses that contribute to arthritis.
IMPORTANCE CHIKV is a reemerging alphavirus of global significance with high potential to spread into new, immunologically naive populations. The severity of CHIKV disease, particularly its propensity for chronic musculoskeletal manifestations, emphasizes the need for identification of genetic determinants that dictate CHIKV virulence in the host. To better understand mechanisms of CHIKV pathogenesis, we probed the function of an amino acid polymorphism in the E2 viral attachment protein using a mouse model of CHIKV musculoskeletal disease. In addition to influencing glycosaminoglycan utilization, we identified roles for this polymorphism in differential infection of mammalian and mosquito cells and targeting of CHIKV to specific tissues within infected mice. These studies demonstrate a correlation between CHIKV tissue tropism and virus-induced pathology modulated by a single polymorphism in E2, which in turn illuminates potential targets for vaccine and antiviral drug development.
INTRODUCTION
Chikungunya virus (CHIKV) is a mosquito-borne alphavirus that has reemerged to cause sudden and severe epidemics throughout sub-Saharan Africa, Southeast Asia, and the Caribbean. CHIKV infection causes a rheumatic disease principally characterized by an abrupt onset of fever, rash, headache, and debilitating polyarthralgia and myalgia (1, 2). Although acute symptoms usually subside within 1 to 2 weeks, chronic joint pain and inflammation occur in many patients for months or years after the initial infection (3–7). The occurrence of chronic arthritis is highly variable, ranging between 15 and 70% of infected individuals depending on the specific study cohort and observed most frequently in the elderly and persons with comorbidities (7).
Since 2004, the geographic distribution of CHIKV has increased significantly, reaching immunologically naive populations in islands of the Indian Ocean, Europe, and the Caribbean (8–13). These epidemics are noteworthy for more severe clinical manifestations, including encephalopathy, hepatitis, meningoencephalitis, and ocular disease and represent the first documented cases of fatal infection (14–16). Currently, there are no licensed CHIKV vaccines or antiviral therapies for infected individuals, and only supportive care for clinical symptoms is available.
CHIKV is an Old World alphavirus and member of the Semliki Forest antigenic complex along with the closely related O'nyong-nyong virus (ONNV), Ross River virus (RRV), and Semliki Forest virus (SFV) (17). The alphavirus genome consists of a single, positive-sense RNA molecule approximately 12 kb in length that contains two open reading frames (18, 19). The first open reading frame encodes four nonstructural proteins (nsP1 to nsP4) that form the replicase complex and mediate synthesis of additional copies of the viral genome and the subgenomic RNA (17–19). The subgenomic, second open reading frame encodes a polyprotein containing three major structural proteins, capsid, pE2, and E1, and the small peptides, 6K and TF (17, 19–22). Following translation of the subgenomic RNA, the capsid protein is autoproteolytically cleaved from the polyprotein (18, 23). The remainder is transported through the endoplasmic reticulum (ER) and Golgi apparatus where the 6K peptide is liberated by cellular proteases and the E1 and pE2 proteins are glycosylated (20, 24). During egress, the cellular protease furin cleaves pE2 to release the E3 peptide and generate the mature heterodimer of E1 fusion and E2 attachment proteins (25–27). E1-E2 heterodimers associate as trimers and stud the host-derived lipid bilayer that surrounds the icosahedral capsid (27). The structural proteins encapsidate the viral genome, forming progeny virions near the plasma membrane, which bud from the host cell to infect neighboring cells and disseminate throughout the host (28).
A live, attenuated CHIKV vaccine was developed by passaging a clinical isolate from Thailand, strain AF15561, extensively in mammalian cell culture to produce a highly attenuated virus, strain 181/25 (29, 30). Strains AF15561 and 181/25 differ at only 10 nucleotide positions and 5 amino acid positions across the genome. A previous study identified two polymorphisms in the E2 attachment protein, T12I and G82R, as important mediators of virus strain 181/25 attenuation in both infant CD1 and immunocompromised mice (31). Accumulation of positively charged residues within E2 as a consequence of cell culture passage has been demonstrated for other alphaviruses to confer binding to negatively charged glycosaminoglycans (GAGs) such as heparan sulfate (32–36). We and others have found that strain 181/25 interacts with GAGs to infect host cells and that this interaction is strengthened through the G82R polymorphism in E2 (37, 38). Given the minimal sequence divergence displayed by strains 181/25 and AF15561, it is not known whether they differ in GAG utilization, nor is it clear whether GAG utilization is the sole mechanism that mediates CHIKV 181/25 attenuation.
Heparan sulfate-binding viruses often exhibit reduced viral dissemination and attenuation for disease in vivo (34, 39–42). However, such viruses can show increased virulence depending on the route of inoculation, as demonstrated for certain strains of eastern equine encephalitis virus (EEEV) (43, 44). A strategy to introduce attenuating heparan sulfate-binding residues within the CHIKV E2 attachment protein has been established as a model for CHIKV vaccine development (45). Wild-type CHIKV strain LR2006 OPY1 (LR) induced significantly less footpad swelling and proinflammatory cytokine production following substitution of E2 Gly82 with arginine and Glu79 with lysine, the latter of which was identified following serial passage in cell culture (45). Introduction of either substitution enhanced sensitivity to blockade of infection by soluble heparin or salt disruption of ionic interactions (45), suggesting that viruses attenuated by virtue of these mutations exhibit increased dependence on GAGs for infection. However, the roles of E2 residue 82 in CHIKV-induced arthritis and viral tropism are not fully understood. Furthermore, mechanisms by which specific CHIKV residues influence viral pathogenesis remain to be elucidated.
In this study, we defined the contribution of sequence polymorphisms displayed by strains 181/25 and AF15561 to CHIKV pathogenesis using a mouse model of CHIKV-induced arthritis. We engineered a panel of CHIKV variants containing these polymorphisms in the genetic background of each parental strain and screened these viruses for differences in infectivity in mammalian and mosquito cells prior to testing in vivo. We found that E2 residue 82 is a determinant of infectivity in mammalian and mosquito cell culture and contributes to CHIKV-induced pathology, including footpad swelling and inflammation and necrosis in the metatarsal muscle ipsilateral to the site of inoculation. In addition, this residue mediates viral dissemination to the spleen and establishment of viremia. Viruses containing an arginine at E2 residue 82 exhibit increased dependence on GAGs for infection of mammalian cells and are more attenuated for musculoskeletal disease. We conclude that CHIKV utilization of GAGs alters viral tropism, tissue inflammation, and tissue injury induced during infection, resulting in attenuated CHIKV disease.
MATERIALS AND METHODS
Cells, viruses, and antibodies.
Baby hamster kidney cells (BHK-21; ATCC CCL-10) were maintained in alpha minimal essential medium (αMEM; Gibco) supplemented to contain 10% fetal bovine serum (FBS; Gibco), 10% tryptose phosphate (TP), 2 mM l-glutamine (Gibco), 100 U/ml of penicillin, 100 μg/ml of streptomycin (Gibco), and 25 ng/ml of amphotericin B (Sigma). Vero cells (ATCC CCL-81) were maintained in αMEM supplemented to contain 5% FBS, l-glutamine, penicillin, streptomycin, and amphotericin B. C6/36 cells were maintained in Leibovitz's L-15 medium (Gibco) supplemented to contain 10% FBS, 10% TP, l-glutamine, penicillin, streptomycin, and amphotericin B. Chinese hamster ovary (CHO) CHO-K1 and CHO-pgsA745 cells (46) were maintained in F-12 nutrient mixture (Gibco) supplemented to contain 10% FBS, l-glutamine, penicillin, streptomycin, and amphotericin B. CHO-K1 and CHO-pgsA745 cell lines were provided by Benhur Lee (University of California, Los Angeles).
The CHIKV 181/25 infectious clone plasmid was generated as described previously (47). The CHIKV AF15561 infectious clone and variant clone plasmids were generated using site-directed mutagenesis of the 181/25 infectious clone plasmid.
CHIKV-specific polyclonal antiserum was obtained from the ATCC (VR-1241AF). CHIKV E2-specific monoclonal antibodies (MAbs) CHK-152 and CHK 48-G8 were provided by Michael Diamond (Washington University).
Site-directed mutagenesis.
Single-amino-acid substitutions (nsP1 I301T, E2 I12T, E2 R82G, 6K P42C, and E1 V404A) were generated by site-directed mutagenesis of the 181/25 infectious clone plasmid using KOD Hot Start DNA polymerase (Novagen). All five amino acid substitutions were introduced into the 181/25 plasmid to generate the AF15561 infectious clone plasmid. Reciprocal, single-amino-acid substitutions (nsP1 T301I, E2 T12I, E2 G82R, 6K C42P, and E1 A404V) were generated similarly in the AF15561 infectious clone plasmid. cDNA from each clone was sequenced to verify that only the desired mutations were introduced.
Generation of virus stocks from infectious clone plasmids.
Infectious clone plasmids were linearized and transcribed in vitro using mMessage mMachine SP6 transcription kits (Ambion). BHK-21 cells were electroporated with viral RNA and incubated at 37°C for 24 h. Supernatants containing progeny virus were collected from electroporated cells and stored at −80°C. For some experiments, supernatants were purified by ultracentrifugation through a 20% sucrose cushion in TNE buffer (50 mM Tris-HCl [pH 7.2], 0.1 M NaCl, and 1 mM EDTA) at ∼115,000 × g in a Beckman 32Ti rotor. Virus pellets were resuspended in virus diluent buffer (VDB) (RPMI medium with HEPES [Gibco] and 1% FBS) and stored at −80°C. Viral titers were determined by plaque assay using Vero cells. All experiments with virus were performed using biosafety level 3 conditions.
CHIKV infectivity assay.
Vero, C6/36, CHO-K1, or CHO-pgsA745 cells seeded onto no. 2 glass coverslips (VWR) in 24-well plates or in 96-well plates (Costar) were adsorbed with CHIKV strains in VDB at a multiplicity of infection (MOI) of 1 (Vero and C6/36) or 10 (CHO-K1 and CHO-pgsA745) PFU/cell at 37°C (Vero, CHO-K1, and CHO-pgsA745) or 30°C (C6/36) for 1 h. The inoculum was removed, complete medium was added, and cells were incubated at 37°C or 30°C for an additional hour. The medium was then supplemented to contain 20 mM ammonium chloride to prevent subsequent rounds of infection. After incubation at 37°C or 30°C for 24 h, cells were fixed with ice-cold 100% methanol, washed with phosphate-buffered saline (PBS), and incubated with PBS containing 5% FBS and 0.1% Triton X-100 (TX) at room temperature for 1 h. The cells were incubated with CHIKV-specific polyclonal antiserum (1:1,500) in PBS with FBS and TX at 4°C overnight. The cells were washed three times with PBS and incubated with Alexa Fluor 488-labeled anti-mouse IgG (1:1,000) in PBS with FBS and TX at room temperature for 2 h. The cells were also incubated with 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen) to stain nuclei. The cells and nuclei were visualized by indirect immunofluorescence using an Axiovert 200 fluorescence microscope (Zeiss). CHIKV-positive cells were enumerated in three fields of view with each field of view containing at least 100 cells for triplicate samples. For some experiments, cells were visualized using an ImageXpress Micro XL imaging system (Molecular Devices) at the Vanderbilt High-Throughput Screening Facility. Total and CHIKV-infected cells were quantified using MetaXpress software (Molecular Devices) in four fields of view containing at least 100 cells per field of view for triplicate samples. The number of CHIKV-positive cells was normalized to the total number of cells per field to determine the percentage of infected cells.
Assessment of CHIKV replication by plaque assay.
Vero or C6/36 cells were adsorbed with CHIKV strains in VDB at an MOI of 0.01 PFU/cell at 37°C (Vero) or 30°C (C6/36) for 1 h. The inoculum was removed, cells were washed with PBS, and complete medium was added. After incubation at 37°C or 30°C for various intervals, 10% of the cell supernatant was collected and replaced with fresh medium. Viral titers in culture supernatants were determined by plaque assay using Vero cells.
Real-time quantitative RT-PCR.
RNA was isolated using a PureLink RNA minikit (Life Technologies). The number of viral genomes/ml for each virus stock was quantified using the qScript XLT one-step reverse transcription-quantitative PCR (RT-qPCR) ToughMix kit (Quanta Biosciences) as described previously (38). CHIKV sequence-specific forward (CHIKVfor [874 5′-AAAGGGCAAGCTTAGCTTCAC-3′]) and reverse (CHIKVrev [961 5′-GCCTGGGCTCATCGTTATTC-3′]) primers were used with an internal fluorogenic probe (CHIKVprobe [899 5′-6-carboxyfluorescein {dFAM}-CGCTGTGATACAGTGGTTTCGTGTG-black hole quencher {BHQ}-3′; Biosearch Technologies). To relate threshold cycle (CT) values to copies of genomic RNA, standard curves were generated from 10-fold dilutions, from 101 to 1010 copies, of in vitro-transcribed genomic 181/25 RNA.
CHIKV binding assay.
Vero cells were adsorbed in suspension with 3 × 1010 genomes of various virus strains in VDB at 4°C for 30 min. The cells were washed with incomplete medium and PBS and fixed in PBS with 1% electron microscopy (EM)-grade paraformaldehyde (Electron Microscopy Sciences). The cells were washed with fluorescence-activated cell sorting (FACS) buffer (PBS with 2% FBS) and incubated with CHIKV E2-specific MAb CHK-152 (1:1,000) in FACS buffer at 4°C for 30 min. The cells were incubated with Alexa Fluor 488-labeled anti-mouse IgG (1:1,000) in FACS buffer at 4°C for 30 min and analyzed using a BD LSRII flow cytometer. Cell staining was quantified using FlowJo software (Tree Star).
Inhibition of CHIKV infection with soluble glycosaminoglycans.
Virus was pretreated with soluble heparin (Sigma) or bovine serum albumin (BSA) (Sigma) at 4°C for 30 min. Vero cells were adsorbed with pretreated virus strains at an MOI of 1 PFU/cell at 37°C for 2 h. The inoculum was removed, and complete medium supplemented to contain 20 mM ammonium chloride was added to prevent subsequent rounds of infection. After incubation at 37°C for 24 h, cells were fixed and incubated with CHIKV-specific polyclonal antiserum and DAPI to detect nuclei. Infection was scored by indirect immunofluorescence. CHIKV-positive cells were enumerated in three fields of view for triplicate samples and normalized to the number of total cells per field.
Heparin-agarose binding assay.
Heparin-conjugated agarose or unconjugated agarose beads were incubated with 5 × 109 genomes of each virus at 4°C for 30 min as described previously (38). The beads were washed with VDB containing 0.02% Tween 20, and beads or input virus were resuspended in sample buffer (50 mM Tris-HCl [pH 6.8], 2% [wt/vol] sodium dodecyl sulfate [SDS], 1% β-mercaptoethanol, 10% [vol/vol] glycerol, 0.04% [wt/vol] bromophenol blue) and boiled for 10 min. Samples were resolved by SDS-polyacrylamide gel electrophoresis (PAGE) in 10% polyacrylamide gels (Bio-Rad) and transferred to an Immun-Blot polyvinylidene difluoride (PVDF) membrane (Bio-Rad). The membranes were incubated with Tris-buffered saline (TBS) containing 5% milk at room temperature for 1 h followed by incubation with CHIKV-specific MAb CHK 48-G8 (1:2,000) in TBS with 0.1% Tween 20 (TBS-T) at 4°C overnight. The membranes were washed three times with TBS-T and incubated with IRDye 750 CW-labeled goat anti-mouse IgG (1:5,000; LI-COR) in TBS-T at room temperature for 2 h. The membranes were washed three times with TBS-T and once with TBS, and CHIKV-specific signal was detected using an Odyssey imaging system (LI-COR).
Infection of mice.
C57BL/6J mice were obtained from The Jackson Laboratory to establish breeding colonies. Mice (20 to 22 days old) were inoculated in the left rear footpad with 103 PFU of virus in PBS containing 1% bovine calf serum (BCS) in a 10-μl volume. Mock-infected animals received diluent alone. Mice were monitored for clinical signs of disease and weighed at 24-h intervals. At various intervals following infection, mice were euthanized by isoflurane overdose, blood was collected by cardiac puncture, and mice were perfused by intracardial injection of PBS. Swelling of the feet of both hind limbs was quantified using calipers to measure the height of the feet. For analysis of viral replication, tissues were collected in PBS containing BCS, weighed, homogenized with a MagNA lyser (Roche), and stored at −80°C. Viral titers in tissue homogenates were determined by plaque assay using BHK-21 cells. For RNA analysis, tissues were collected and homogenized in TRIzol reagent (Life Technologies).
Animal husbandry and experiments were performed in accordance with all University of Colorado School of Medicine Institutional Animal Care and Use Committee guidelines. All mouse studies were performed using biosafety level 3 conditions.
Histological analysis.
At day 7 postinoculation, mice were euthanized and perfused by intracardial injection of 4% paraformaldehyde (PFA) (pH 7.3). Tissues were resected and incubated in PFA at 4°C for at least 72 h. Fixed tissues were embedded in paraffin, and 5-μm sections were stained with hematoxylin and eosin (H&E) to assess histopathologic changes. Tissues were scored by an observer in a blind manner (unaware of the conditions of the experiment) for the presence, distribution, and severity of histologic lesions. For all tissue changes, the following scoring system was used: 0, no lesions; 1, minimal, 0 to 24% of tissue affected; 2, mild, 25 to 49% of tissue affected; 3, moderate, 50 to 75% of tissue affected; 4, marked, >75% of tissue affected.
Analysis of sequence reversion.
RNA was isolated using a PureLink RNA minikit (Life Technologies). cDNA was generated using the SuperScript III first-strand kit (Life Technologies) with random hexamers and used for PCR amplification by KOD polymerase (Novagen) with CHIKV E2 sequence-specific forward (CHIKVE2for [8336 5′-GGGCCGAAGAGTGGAGTCTT-3′]) and reverse (CHIKVE2rev [9089 5′-GACACCCCTGATCGCACATT-3′]) primers. Amplicons were cloned into a TOPO TA vector (Life Technologies) and sequenced over the mutagenized region of the E2 open reading frame.
Statistical analysis.
Mean values for at least duplicate experiments were compared using an unpaired Student's t test, one-way analysis of variance (ANOVA) followed by Bonferroni's posthoc test, or Kruskal-Wallis analysis followed by Dunn's posthoc test (GraphPad Prism). P values of <0.05 were considered to be statistically significant.
RESULTS
E2 residue 82 is a determinant of CHIKV infectivity in mammalian cell culture.
For many viruses, serial passage in mammalian cells enhances viral replicative capacity in cell culture (33, 48–53). To determine whether passage of CHIKV strain AF15561 to generate vaccine strain 181/25 resulted in enhanced replicative capacity, cells were infected with either strain AF15561 or 181/25, and viral titers in culture supernatants were determined by plaque assay over an infectious time course. Relative to strain AF15561, strain 181/25 reached higher titers in several cell lines, including Vero, BHK-21, HeLa, and human brain microvascular endothelial cells (data not shown). These results suggest that strain 181/25 has adapted to mammalian cell culture as a result of cell culture passage, consistent with observations for other passaged viruses.
Virus strains 181/25 and AF15561 differ at five nonsynonymous nucleotides and five synonymous nucleotides across the genome. Polymorphic positions resulting in coding changes in 181/25 are in the nsP1 (T301I), E2 (T12I and G82R), 6K (C42P), and E1 (A404V) proteins (Fig. 1). To define residues that contribute to the replication and infectivity differences observed between strains 181/25 and AF15561, Vero cells were infected at an MOI of 1 PFU/cell with either of the parental strains or viruses containing individual polymorphic residues in the reciprocal genetic background. The percentage of infected cells in each case was quantified after a single round of infection by indirect immunofluorescence (Fig. 2A). As expected, virus strain 181/25 infected a significantly greater percentage of cells than did virus strain AF15561. Substitution of an arginine at E2 residue 82 in the AF15561 background enhanced infectivity to levels even greater than those observed for strain 181/25. Concordantly, substitution of a glycine at E2 residue 82 in the 181/25 background significantly diminished 181/25 infectivity. Interestingly, substituting an isoleucine at E2 residue 12 in the AF15561 background further decreased infectivity relative to AF15561. Introducing any of the other heterologous changes in either background had no effect on infectivity in these cells. To understand how these findings might compare to previous studies with these viruses in mice, the parental and variant viruses were used to infect murine L929 and NIH 3T3 cell lines. As observed in experiments using Vero cells, substitution of an arginine at E2 residue 82 in the AF15561 background enhanced infectivity, whereas substitution of a glycine at E2 residue 82 in the 181/25 background significantly diminished infectivity in these murine cell lines (data not shown). These data suggest that an arginine at residue 82 in the E2 protein confers the enhanced infectivity observed for strain 181/25 in mammalian cells.
FIG 1.
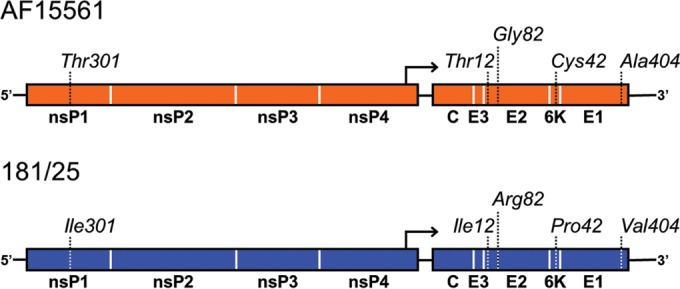
Schematic depiction of polymorphic residues in CHIKV strains AF15561 and 181/25. Distribution of amino acid polymorphisms between strains AF15561 and 181/25 across the ∼12-kb genome. Numbers correspond to the amino acid positions within each protein.
FIG 2.
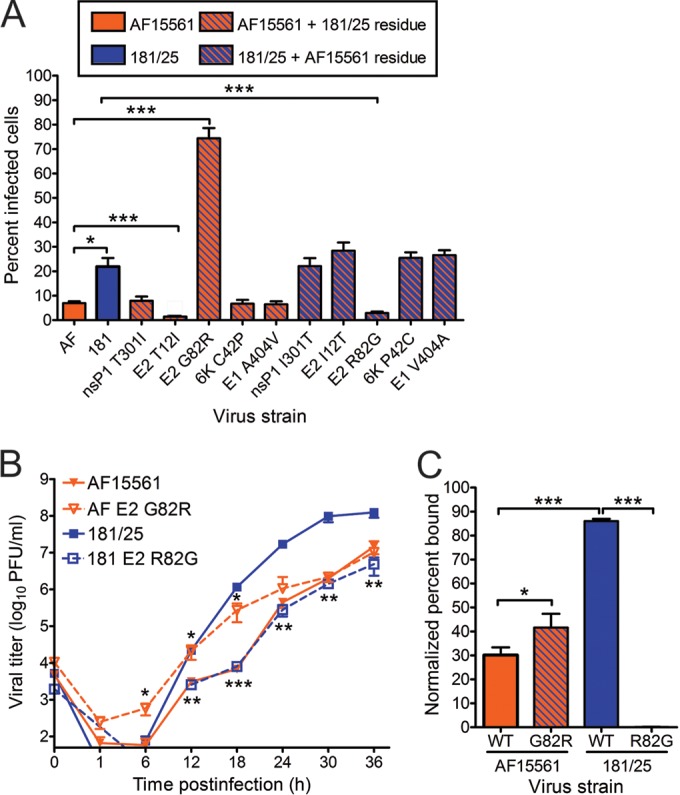
Residue 82 of the E2 attachment protein is a determinant of CHIKV infectivity in mammalian cells. (A) Vero cells were adsorbed with CHIKV strains AF15561 (AF), 181/25 (181), or the variant viruses shown at an MOI of 1 PFU/cell and incubated for 24 h in medium containing 20 mM NH4Cl. The cells were stained with CHIKV-specific antiserum and DAPI to detect nuclei and imaged by fluorescence microscopy. Results are presented as percent infected cells for triplicate experiments. Error bars indicate standard errors of the means. (B) Vero cells were adsorbed with virus strain AF15561, 181/25, AF15561 E2 G82R, or 181/25 E2 R82G at an MOI of 0.01 PFU/cell. At the times shown, viral titers in culture supernatants were determined by plaque assay using Vero cells. Results are presented as the mean viral titers for triplicate samples. Error bars indicate standard deviations. Titers of virus strains AF15561 and AF15561 E2 G82R were significantly different at 6, 12, and 18 h postinfection, and titers of virus strains 181/25 and 181/25 E2 R82G were significantly different at 12, 18, 24, 30, and 36 h postinfection. (C) Vero cells were adsorbed with ∼3 × 1010 genomes of wild-type (WT) virus strain AF15561, 181/25, AF15561 E2 G82R, or 181/25 E2 R82G for 30 min. After 30-min incubation, cells were stained with CHIKV E2-specific MAb, and virus-bound cells were quantified by flow cytometry. Results are presented as percent bound cells for triplicate experiments normalized to cell autofluorescence in the absence of CHIKV MAb. No CHIKV-bound cells were detected for virus strain 181/25 E2 R82G. Error bars indicate standard deviations. Values that are significantly different, as determined by ANOVA, followed by Bonferroni's posthoc test (A and C) and Student's t test (B), are indicated by asterisks as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Because these virus strains differed substantially in infectivity of Vero cells, we quantified the genome/PFU and genome/fluorescent-focus unit (FFU) ratios for each strain (Table 1). In Vero cells, parental strains AF15561 and 181/25 had genome/PFU ratios of 6,598 and 176, respectively. Similarly, strains AF15561 and 181/25 had genome/FFU ratios of 9.497 × 107 and 8.0 × 105, respectively. Viral variants containing residues from strain 181/25 in the AF15561 background had reduced genome/PFU and genome/FFU ratios compared to those of parental strain AF15561. Interestingly, introducing the E2 G82R substitution resulted in the most modest reduction in genome/PFU ratio, but the greatest reduction in genome/FFU ratio. In contrast, introducing the E2 R82G substitution in the 181/25 background resulted in increased genome/PFU and genome/FFU ratios, which were the most dramatic increases of any of the viral variants with AF15561 residues in the 181/25 background. Thus, the E2 82 polymorphism serves as a key determinant of CHIKV infectivity in mammalian cell culture.
TABLE 1.
Infectivity of parental and mutant CHIKV strainsa
| Virus strain | Genome/PFU ratiob | Genome/FFU ratioc (× 104) in: |
|
|---|---|---|---|
| Vero cells | C6/36 cells | ||
| AF15561 | 6,598 | 9,497 | 980 |
| 181/25 | 176 | 80 | 182 |
| AF15561 nsP1 T301I | 1,718 | 2,173 | 250 |
| AF15561 E2 T12I | 1,107 | 7,621 | 344 |
| AF15561 E2 G82R | 5,656 | 760 | 841 |
| AF15561 6K C42P | 3,057 | 4,529 | 436 |
| AF15561 E1 A404V | 4,067 | 6,255 | 586 |
| 181/25 nsP1 I30T | 1,005 | 455 | 381 |
| 181/25 E2 I12T | 99 | 35 | 45 |
| 181/25 E2 R82G | 2,858 | 9,783 | 517 |
| 181/25 6K P42C | 44 | 17 | 16 |
| 181/25 E1 V404A | 121 | 46 | 27 |
Infectivity of virus strains measured in genome equivalents.
Genome copy numbers were determined by real-time quantitative PCRs conducted in triplicate. The number of PFU was determined by plaque assay using Vero cells for three independent replicates for each virus strain.
Fluorescent-focus units (FFU) were determined by indirect immunofluorescence in triplicate for at least two independent experiments.
TABLE 2.
Infectivity of purified CHIKV strainsa
| Virus strain | Genome/PFU ratiob | Genome/FFU ratioc (× 104) in: |
Ratiod | |
|---|---|---|---|---|
| CHO-K1 cells | CHO-pgsA745 cells | |||
| AF15561 | 647 | 1,578 | 11,795 | 7.47 |
| AF15561 E2 G82R | 498 | 867 | 20,494 | 23.65 |
| 181/25 | 92 | 310 | 2,376 | 7.67 |
| 181/25 E2 R82G | 316 | 1,862 | 8,155 | 4.38 |
Infectivity of virus strains measured in genome equivalents.
Genome copy numbers were determined by real-time quantitative PCRs conducted in triplicate. The number of PFU was determined by plaque assay using Vero cells for three independent replicates for each virus strain.
Fluorescent-focus units (FFU) were determined by indirect immunofluorescence in triplicate for at least two independent experiments.
Ratio of the genome/FFU ratio in CHO-pgsA745 cells to the genome/FFU ratio in CHO-K1 cells.
To determine whether E2 residue 82 contributes to the production of infectious virus over multiple rounds of infection, Vero cells were infected at an MOI of 0.01 PFU/cell, and viral titers in culture supernatants were determined by plaque assay at 6-h intervals (Fig. 2B). Compared with titers of AF15561, titers of AF15561 E2 G82R were increased 10-fold by 6 h postinfection, 7-fold by 12 h postinfection, and 40-fold by 18 h postinfection. Compared with titers of virus strain 181/25, titers of 181/25 E2 R82G were reduced 145-fold by 18 h postinfection, 60-fold by 24 h postinfection, and 68-fold by 30 h postinfection. Taken together, these data indicate that E2 residue 82 influences both initial and subsequent rounds of CHIKV infection.
Because this determinant of CHIKV infectivity resides in the viral attachment protein, we tested whether E2 residue 82 influences binding to host cells. For these experiments, Vero cells were adsorbed with each parental strain and the reciprocal E2 82 variant strains, and the percentage of virus-bound cells was quantified using flow cytometry (Fig. 2C). A significantly greater proportion of cells were bound by strain 181/25 than by strain AF15561. In agreement with the infectivity data, introducing the E2 G82R substitution in the AF15561 background significantly enhanced cell attachment. In contrast, introducing the E2 R82G polymorphism in the 181/25 background reduced cell binding to undetectable levels. These data suggest that an arginine at position 82 in the E2 attachment protein is required for the enhanced binding and infectivity observed for strain 181/25 in mammalian cells.
E2 residue 82 contributes to CHIKV infectivity in mosquito cells.
To determine whether an arginine at E2 residue 82 affects infection and replication in invertebrate cells, we tested the parental and reciprocal E2 82 variant strains for infection and replication in mosquito cells (Fig. 3). Aedes albopictus C6/36 cells were infected at an MOI of 1 PFU/cell, and the percentage of infected cells was quantified after a single round of infection by indirect immunofluorescence (Fig. 3A). In sharp contrast to our findings using mammalian cells, strain 181/25 infected significantly fewer C6/36 cells relative to strain AF15561, and substitution of a glycine at E2 residue 82 in the 181/25 background significantly increased 181/25 infectivity in these cells. However, substitution of an arginine at E2 residue 82 in the AF15561 background did not significantly diminish C6/36 infection. Surprisingly, only substitution of isoleucine for threonine at residue 12 in AF15561 E2 was sufficient to reduce infection in these cells, albeit not to the levels observed for strain 181/25. Additionally, AF15561 E2 G82R had a similar genome/FFU ratio compared to that of AF15561, whereas substitutions at the other polymorphic sites with residues from strain 181/25 decreased the genome/FFU ratio. Together, these data indicate that the residue at E2 position 82 provides a fitness advantage for CHIKV infectivity depending on the host cell species.
FIG 3.
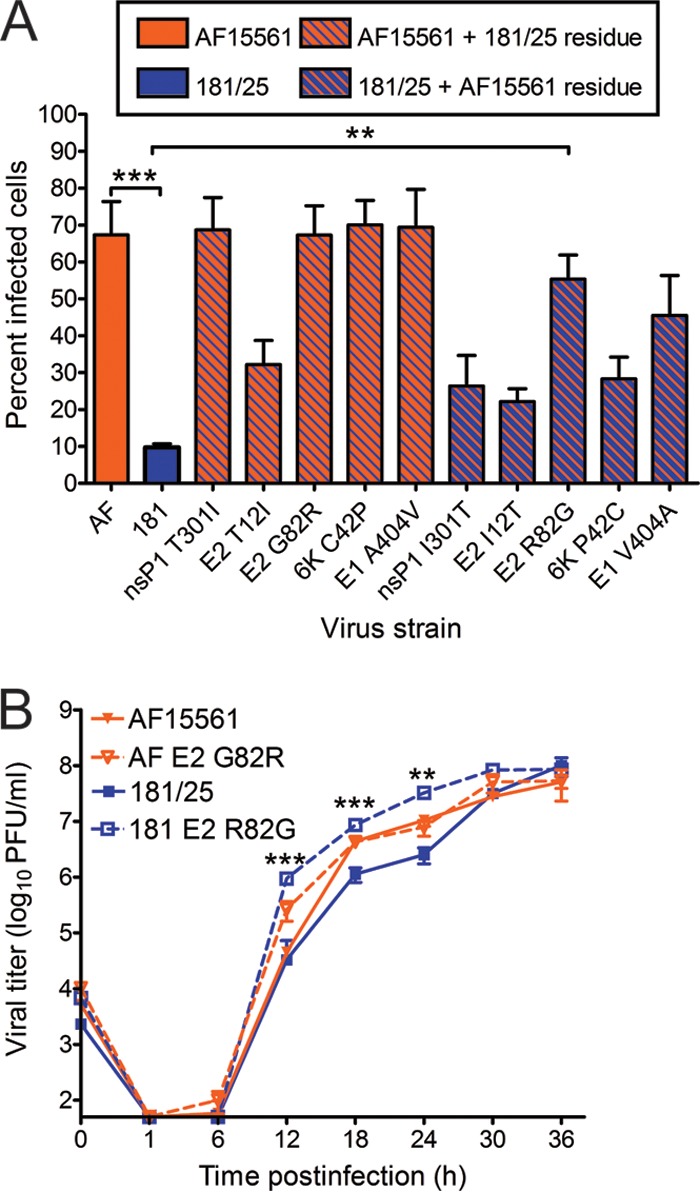
Residue 82 of the E2 attachment protein is a determinant of CHIKV infectivity in mosquito cells. (A) C6/36 mosquito cells were adsorbed with CHIKV strains AF15561 and 181/25 or the variant viruses shown at an MOI of 1 PFU/cell and incubated for 24 h in medium containing 20 mM NH4Cl. The cells were stained with CHIKV-specific antiserum and DAPI to detect nuclei and imaged by fluorescence microscopy. Results are presented as percent infected cells for quadruplicate experiments. Error bars indicate standard errors of the means. (B) C6/36 cells were adsorbed with virus strain AF15561, 181/25, AF15561 G82R, or 181/25 E2 R82G at an MOI of 0.01 PFU/cell. At the times shown, viral titers in culture supernatants were determined by plaque assay using Vero cells. Results are presented as the mean viral titers for triplicate samples. Error bars indicate standard deviations. The titers of virus strains 181/25 and 181/25 E2 R82G were significantly different at 12, 18, and 24 h postinfection. Values that are significantly different, as determined by ANOVA, followed by Bonferroni's posthoc test (A) and Student's t test (B), are indicated by asterisks as follows: **, P < 0.01; ***, P < 0.001.
To assess the role of E2 residue 82 in the production of infectious virus from mosquito cells, C6/36 cells were infected at an MOI of 0.01 PFU/cell, and viral titers in culture supernatants were determined at 6-h intervals (Fig. 3B). Relative to titers of virus strain 181/25, titers of 181/25 E2 R82G were increased 29-fold by 12 h postinfection, 8-fold by 18 h postinfection, and 13-fold by 24 h postinfection. However, relative to titers of virus strain AF15561, there was no decrease in viral titers when an arginine was introduced at E2 82 in the AF15561 strain. These data indicate that E2 Gly82 in the genetic background of strain 181/25 enhances infection of mosquito cells. However, other polymorphisms exhibited by these strains appear to contribute to the enhanced infectivity of AF15561 in mosquito cells in addition to E2 Gly82.
E2 residue 82 influences utilization of glycosaminoglycans.
To understand mechanisms by which E2 residue 82 influences CHIKV attachment to mammalian cells, we next investigated the dependence of virus strains 181/25 and AF15561 on GAGs for infectivity. Wild-type CHO-K1 and GAG-deficient CHO-pgsA745 cells were infected with purified 181/25, AF15561, 181/25 E2 R82G, or AF15561 E2 G82R at an MOI of 10 PFU/cell. The percentage of infected cells for each virus was quantified after a single round of infection by indirect immunofluorescence (Fig. 4A). The CHO-pgsA745 cells were significantly less susceptible to infection by all four viruses relative to infection of CHO-K1 cells (P < 0.0001 for virus strains 181/25 and AF15561, P < 0.001 for AF15561 E2 G82R, and P < 0.05 for 181/25 E2 R82G). However, the CHO-pgsA745 cells were significantly less susceptible to infection by strain 181/25 relative to strain AF15561. Substitution of E2 Gly82 with arginine in the AF15561 background was sufficient to diminish infectivity in these cells to levels observed for strain 181/25. Furthermore, substitution of E2 Arg82 with glycine in the 181/25 background was sufficient to increase infectivity and mitigate GAG dependence to levels observed for strain AF15561. These data confirm that strain 181/25 is more dependent on GAGs for infection than is AF15561 and that E2 residue 82 mediates this dependence.
FIG 4.

An arginine at E2 residue 82 confers greater dependence on glycosaminoglycans. (A) CHO-K1 and CHO-pgsA745 cells were adsorbed with virus strain AF15561, 181/25, AF1561 E2 G82R, or 181/25 E2 R82G at an MOI of 10 PFU/cell and incubated for 24 h. Wild-type (WT) CHIKV or variant strains containing substitutions at E2 residue 82 were tested. The cells were stained with CHIKV-specific antiserum and DAPI to detect nuclei and imaged by fluorescence microscopy. Results are presented as percent infected cells for triplicate experiments normalized to the values for parental CHO-K1 cells. Error bars indicate standard errors of the means. (B) Strains AF15561, 181/25, AF1561 E2 G82R, and 181/25 E2 R82G were treated with BSA at 1,000 μg/ml or heparin at the concentrations shown for 30 min and adsorbed to Vero cells at an MOI of 2.5 PFU/cell. After incubation for 24 h, cells were stained with CHIKV-specific antiserum and DAPI to detect nuclei and imaged by fluorescence microscopy. Results are presented as percent infected cells for triplicate experiments normalized to the values for mock-treated virus. Error bars indicate standard errors of the means. (C) The virus strains shown at 5 × 109 genome copies each were incubated with heparin-conjugated or unconjugated agarose beads for 30 min, resolved by SDS-PAGE, and detected by immunoblotting with CHIKV E2-specific MAb (left). Twenty-five percent of input virus is shown as a control. The percentage of virus bound to beads was quantified by optical densitometry for triplicate experiments (right). Error bars indicate standard deviations. Values that are significantly different, as determined by Student's t test (A and C) and Kruskal-Wallis analysis followed by Dunn's posthoc test (B), are indicated by asterisks as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
We also assessed infectivity by determining genome/PFU ratios using Vero cells and genome/FFU ratios using both CHO-K1 and CHO-pgsA745 cells. We observed a similar trend in the genome/PFU values of purified stocks of the parental and variant viruses used in these experiments. Strain AF15561 had higher genome/PFU ratios than strain 181/25 did in both cell lines, and these differences once again segregated with E2 residue 82. Interestingly, all strains had higher genome/FFU ratios in CHO-pgsA745 cells than in the parental CHO-K1 cells. These data support the hypothesis that viruses containing an arginine at E2 residue 82 are less fit in GAG-deficient cells relative to their glycine-containing counterparts.
On the basis of these and previously published findings (32–36), the GAG dependence mediated by this residue likely occurs at an early step in the infectious life cycle. Therefore, we reasoned that soluble GAGs could act as competitive agonists to block infectivity of GAG-dependent viruses. To test whether competition with soluble GAGs inhibits infection, purified virus was incubated with increasing concentrations of soluble heparin, a highly sulfated GAG, and adsorbed to Vero cells. The percentage of infected cells was quantified after a single round of infection by indirect immunofluorescence (Fig. 4B). Treatment with soluble heparin resulted in a dose-dependent decrease in the percentage of infected cells for all viruses tested. However, this decrease was most substantial for strains 181/25 and AF15561 E2 G82R, for which incubation with the highest concentration of heparin decreased infectivity 15- and 24-fold, respectively. In contrast, incubation of strains AF15561 and 181/25 E2 R82G with the same concentration of heparin resulted in only a 4-fold decrease in infectivity. These findings suggest that CHIKV strains containing an arginine at E2 residue 82 rely on GAGs for efficient cell attachment.
To determine whether the CHIKV strains used in our study interact directly with GAGs, equivalent genome copies of purified parental or variant viruses were incubated with either heparin-conjugated or unconjugated agarose beads. Bound material was resolved by SDS-PAGE and immunoblotted using an E2-specific MAb to detect CHIKV particles (Fig. 4C, left). A significantly greater proportion of strain 181/25 was bound by the heparin-conjugated beads relative to the binding of strain AF15561. Substituting AF15561 E2 Gly82 with arginine increased the proportion of this virus that was bound by the heparin-conjugated beads. Concordantly, substituting strain 181/25 E2 Arg82 with glycine decreased heparin binding. Intensities of the viral protein bands from the particles bound to the heparin-conjugated beads were quantified for three independent experiments and normalized to the intensities of protein bands for input virus (Fig. 4C, right). Approximately 35% and 37% of strains 181/25 and AF15561 E2 G82R, respectively, were captured by the heparin-agarose beads, whereas only 13% and 16% of strains AF15561 and 181/25 E2 R82G, respectively, bound to heparin. These results suggest that CHIKV virions interact directly with GAGs and that these interactions are influenced by E2 residue 82.
CHIKV E2 residue 82 modulates virus-induced pathology.
We next investigated whether E2 residue 82 influences CHIKV pathogenesis using a mouse model of CHIKV disease (54). In this model, 3-week-old mice are inoculated subcutaneously in the left rear footpad. Infected mice develop signs of disease similar to those observed in humans infected with CHIKV, including swelling of the feet and ankles, arthritis, myositis, and tenosynovitis. Mice were inoculated with 103 PFU of strain 181/25, AF15561, 181/25 E2 R82G, or AF15561 E2 G82R, and virulence was assessed by weight gain and swelling of feet (Fig. 5). Mice infected with virus strain AF15561 gained less weight than mice infected with virus strain 181/25, indicating that strain AF15561 is more virulent in these animals (Fig. 5A). Mice inoculated with AF15561 E2 G82R gained weight in parallel with mock-infected mice, suggesting that an arginine at E2 residue 82 in the AF15561 background significantly reduces CHIKV virulence. However, mice inoculated with 181/25 E2 R82G did not exhibit impaired weight gain, suggesting that substitution of a glycine at E2 residue 82 in strain 181/25 is not sufficient to recapitulate the virulent phenotype.
FIG 5.

CHIKV E2 residue 82 modulates virus-induced pathology. C57BL/6J mice (20 to 22 days old) were inoculated with 103 PFU of CHIKV strain AF15561, 181/25, AF1561 E2 G82R, or 181/25 E2 R82G in the left rear footpad. (A) Mice were weighed at 24-h intervals postinoculation. Results are presented as the mean percent starting weight (weight on day 0). Error bars indicate standard deviations. The number of mice at different time points follow: day 0 to day 1 (D0-D1), n = 17; D2-D3, n = 15; D4-D5, n = 8; D6-D7, n = 3. (B) Swelling of the left and right hind feet was quantified using calipers at the times shown (in days). Error bars indicate standard deviations. The number of mice at different time points follow: D1, n = 2; D3, n = 7; D5, n = 5; D7, n = 3. Values that are significantly different as determined by ANOVA followed by Bonferroni's posthoc test are indicated by asterisks as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To determine the effect of E2 residue 82 on CHIKV-induced arthritis, we assessed swelling of the left and right feet at 1, 3, 5, and 7 days postinoculation (Fig. 5B). At 3, 5, and 7 days postinoculation, mice infected with virus strain AF15561 exhibited significant swelling of the left feet. No swelling was observed in the uninoculated feet of any of the animals. Inoculation with AF15561 E2 G82R resulted in reduced swelling in the left hind limb at 3 and 5 days postinoculation relative to AF15561, whereas inoculation with 181/25 E2 R82G only modestly increased swelling at 5 days postinoculation relative to 181/25. Together, these data suggest that a glycine at E2 residue 82 is necessary but not sufficient for full virulence of strain AF15561, as assessed by weight gain and foot swelling.
To determine whether E2 residue 82 influences the magnitude of pathological injury, left hind limbs of infected mice were processed for histology and assigned a pathology score based on histologic changes (Fig. 6). Tissue damage appeared more severe for mice infected with virus strain AF15561 than for mice infected with virus strain 181/25 at day 7 postinoculation (Fig. 6A and B). In particular, there was slightly more inflammation and necrosis in the metatarsal muscles of AF15561-infected mice compared to those of 181/25-infected mice. However, the levels of myositis and tendonitis induced by these strains were comparable. Substitution of an arginine at E2 82 in the AF15561 strain led to reduced tissue damage, inflammation, and necrosis compared to mice inoculated with the parental AF15561 strain (Fig. 6B). More dramatically, substitution of a glycine at this residue in the 181/25 strain led to consistently more inflammation and necrosis of the metatarsal muscle as well as more severe tendonitis in the hind limb relative to the hind limbs of 181/25-infected mice. These data suggest that E2 residue 82 modulates CHIKV-induced disease and that a glycine at this residue is sufficient to mediate tissue injury in CHIKV-infected mice.
FIG 6.

An arginine at E2 residue 82 diminishes CHIKV-induced arthritis. C57BL/6J mice (20 to 22 days old) were inoculated with PBS or 103 PFU of virus strain AF15561, 181/25, AF1561 E2 G82R, or 181/25 E2 R82G in the left rear footpad. At day 7 postinoculation, mice were euthanized and perfused with 4% PFA. Consecutive 5-μm sections of the left hind limb were stained with H&E. (A) Representative sections of three mice per group are shown for mock-infected mice or mice inoculated with the parental strains and the reciprocal E2 82 variant strains. Bars, 50 μm. (B) H&E-stained sections were scored for histological evidence of inflammation and necrosis in the metatarsal muscle and tendonitis. Results are expressed as pathology score of tissues from individual animals. Horizontal black lines indicate mean pathology score. Scores were assigned based on the following scale: 0, no lesions; 1, minimal, 0 to 24% of tissue affected; 2, mild, 25 to 49% of tissue affected; 3, moderate, 50 to 75% of tissue affected; 4, marked, >75% of tissue affected.
CHIKV titers in the spleen and serum are influenced by E2 residue 82.
To determine whether differences in virus-induced pathology are a consequence of differences in viral replication, mice were inoculated subcutaneously in the left rear footpad with 103 PFU of virus strain 181/25, AF15561, 181/25 E2 R82G, or AF15561 E2 G82R. Tissues were harvested at 1, 3, and 5 days postinoculation, and viral titers were determined by plaque assay (Fig. 7). At 1 day postinoculation, all viruses produced equivalent titers in the left and right hind limbs (Fig. 7A). However, virus strain AF15561 produced higher titers in the spleen and serum relative to virus strain 181/25. The titers of strain AF15561 E2 G82R were reduced in the spleen and serum compared to those of strain AF15561 at 1 day postinoculation, and strain 181/25 E2 R82G produced higher titers in the spleen and serum compared to those of strain 181/25, although this difference was not statistically significant. These data suggest that a glycine at E2 residue 82 contributes to higher viral titers in the spleen and serum at early times postinoculation.
FIG 7.
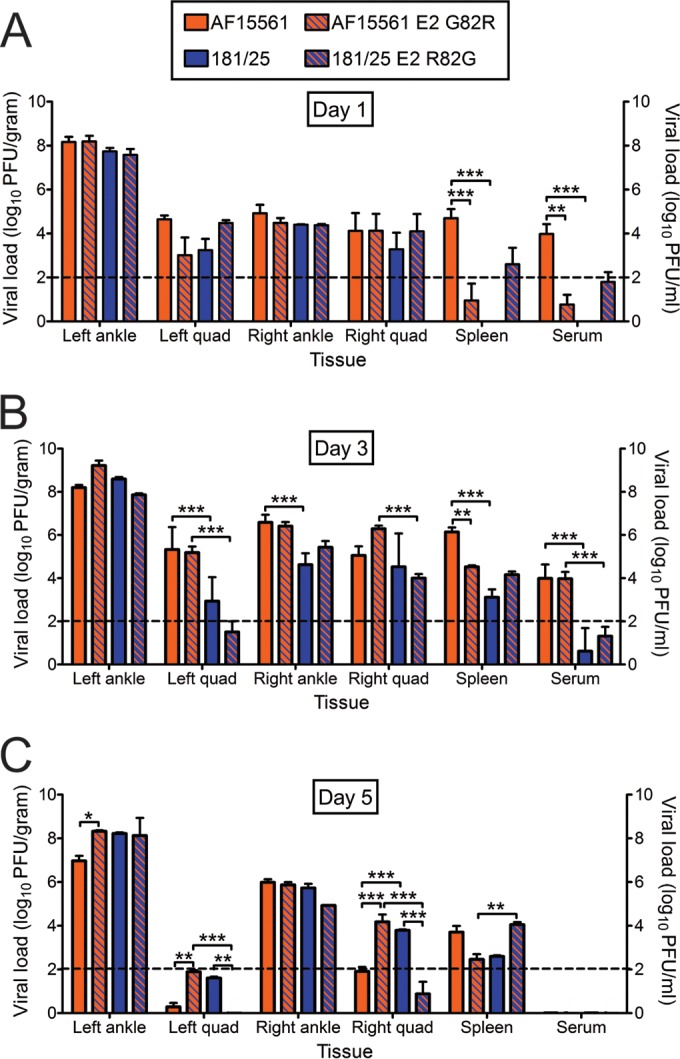
Viral loads following inoculation of parental strains and the reciprocal E2 82 variant strains. C57BL/6J mice (20 to 22 days old) were inoculated with PBS or 103 PFU of virus strain AF15561, 181/25, AF1561 E2 G82R, or 181/25 E2 R82G in the left rear footpad. At days 1, 3, and 5 postinoculation, mice were euthanized, their ankles, quadriceps (Quad), and spleens were excised, and serum was collected. Viral titers in tissue and serum homogenates were determined by plaque assay using BHK-21 cells. Results are expressed as the mean PFU/gram (tissue) or PFU/ml (serum). Error bars indicate standard errors of the means. Dashed lines indicate the limit of detection. The number of mice at different time points follow: D1, n = 5; D3, n = 7; D5, n = 5. Values that are significantly different as determined by ANOVA followed by Bonferroni's posthoc test are indicated by asterisks and bars as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
By 3 days postinoculation, all viruses produced comparable titers in the left ankle, but strain AF15561 replicated to significantly higher titers in the left quadriceps and right ankle relative to strain 181/25 (Fig. 7B). However, this difference in replication did not segregate with E2 residue 82, as mice inoculated with AF15561 E2 G82R displayed higher viral titers in the left quadriceps and right ankle relative to 181/25 E2 R82G. Similarly, higher titers of AF15561 E2 G82R were detected in the right quadriceps and serum relative to 181/25 E2 R82G. In the spleen, we observed a trend similar to the day 1 time point with AF15561 E2 G82R producing lower titers in that organ relative to AF15561, and 181/25 E2 R82G producing slightly higher titers relative to 181/25.
By 5 days postinoculation, no virus was detected in the serum. In the left and right quadriceps, titers of virus strain AF15561 E2 G82R were higher relative to those of strain AF15561. In addition, titers of virus strain 181/25 were higher in these tissues relative to those of 181/25 E2 R82G. These data suggest that an arginine at E2 residue 82 correlates with higher viral titers in the quadriceps muscles at later times postinoculation. In contrast, viruses containing a glycine at E2 82 produced higher titers in the spleen at earlier times postinoculation. Thus, residue 82 in the E2 glycoprotein contributes to either viral dissemination to or replication within the hind limbs and spleen and influences establishment of viremia.
A glycine at E2 residue 82 is selected in the spleens of CHIKV-infected mice.
Since high mutation rates are associated with replication of positive-sense RNA viruses, we determined the sequences of viral isolates from CHIKV-infected mice to assess the stability of the engineered mutations. RNA was isolated from the spleens of CHIKV-infected mice at 1 day postinoculation, cDNA was generated, and sequences of the E2 open reading frame from multiple clones were determined. Of the clones derived from mice inoculated with strain AF15561 E2 G82R, 21 of 23 clones (91%) encoded a glycine at E2 residue 82. In contrast, only 1 of 7 (14%) of the clones derived from mice inoculated with strain AF15561 encoded an arginine at this position. Viral RNA could not be recovered from the spleens of animals infected with virus strain 181/25 at this early time point, which precluded sequence analysis. To confirm that these mutations were not present in the viral inocula, RNA was isolated from virus stocks, cDNA was generated, and E2 sequences were determined. Of the 20 clones sequenced from each virus stock, all encoded the engineered residues across the E2 open reading frame. These results suggest that a glycine at E2 82 is preferentially selected early in CHIKV infection in the spleen, but a residual population of isolates with an arginine at this position is maintained.
DISCUSSION
CHIKV causes both an acute and chronic disease characterized by debilitating joint pain and inflammation (1, 2). However, the viral and host determinants responsible for CHIKV disease have not been fully defined. Additionally, mechanisms of pathogenesis for arthritogenic alphaviruses like CHIKV are not completely understood. CHIKV strain 181/25, which was isolated following serial passage in mammalian cell culture, differs from its closest known parental strain, AF15561, at 5 synonymous and 5 nonsynonymous nucleotide positions. One nonsynonymous polymorphism, G82R in the E2 attachment protein, influences interactions with GAGs (38) and attenuates virulence in some mouse models (31, 45). In this study, we demonstrate a role for E2 residue 82 in the differential infection of mammalian and mosquito cells and in CHIKV-induced musculoskeletal disease. Viruses containing a glycine at E2 82 replicated to higher titers in lymphoid tissues and established higher levels of viremia. Moreover, these viruses induced greater pathological injury, including inflammation and necrosis, in joint-associated tissues compared with viruses containing an arginine at this position. Data presented here expand our knowledge of how CHIKV E2 residue 82 influences virus-cell interactions and provide new information about the function of this residue in vivo.
Our results along with previously published data (31, 38, 45) indicate that an arginine at E2 residue 82 enhances utilization of GAGs by CHIKV to infect cultured cells and attenuates the virus in mice. However, it is not clear how an increase in GAG dependence leads to attenuation of CHIKV. For other alphaviruses, such as Sindbis virus and Venezuelan equine encephalitis virus, higher-affinity interactions with GAGs prevent viral spread to sites of secondary replication or facilitate viral clearance from the bloodstream (34, 55, 56). We found that CHIKV strains containing an arginine at E2 82 produce lower titers at sites of secondary replication, including the spleen, and reduced viremia in immunocompetent mice. These findings are consistent with those obtained by Gardner et al. using a similar mouse model (45). Thus, it appears that GAG-dependent strains of CHIKV disseminate less efficiently, are cleared more rapidly, or perhaps both.
E2 residue 82 may also contribute to differences in tissue tropism by both GAG-dependent and GAG-independent mechanisms. Differential distribution and levels of GAGs throughout the body may influence targeting of GAG-dependent viruses to specific tissues or alter the capacity of these viruses to interact with specific cell types. Virus strains 181/25 and AF15561 differ in GAG dependence but not to the extent observed for strain 181/25 and other clinical isolates (37, 38, 45). Strain 181/25 infectivity in CHO-pgsA745 cells, which lack all GAGs, was decreased less than 4-fold relative to strain AF15561. In addition, the 50% inhibitory concentration (IC50) of heparin inhibition for strains 181/25 and AF15561 were comparable (9.4 and 10.8 μg/ml, respectively). These data suggest that additional mechanisms underlie the attenuation of strain 181/25. Since E2 82 is solvent exposed and lies within a putative receptor-binding domain of E2 (27), it is possible that this residue participates in interactions with cellular receptors other than GAGs. Substituting AF15561 E2 Gly82 with an arginine enhances infectivity in Vero cells relative to strain AF15561, likely by promoting interactions with GAGs or other cell surface moieties to mediate attachment. Enhanced viral attachment to the cell surface would facilitate more rapid internalization of the virus and consequent infection. However, infectivity of AF15561 E2 G82R was also significantly greater than that observed for the 181/25 strain in Vero cells, although both strains bound heparin-conjugated agarose beads to a similar extent. Furthermore, substituting AF15561 E2 Gly82 with an arginine only modestly enhanced binding to Vero cells. Thus, these findings suggest that an arginine at this residue in the AF15561 background provides a replication advantage in mammalian cell culture in addition to GAG engagement and attachment to host cells.
As early as 1 day postinoculation of strain AF15561 E2 G82R, nucleotide sequences of most isolates detected in the spleens of infected mice encode a glycine at E2 residue 82. Reversion was also observed by Gorchakov et al. (31) in which 100% isolates in blood encode a glycine at E2 82 by 3 days postinoculation. In contrast, following inoculation with the E2 T12I variant, only 22% of isolates encoded a threonine at E2 residue 12 (31). E2 82 may mediate viral tropism specifically in lymphoid tissues early in infection, and the presence of an arginine at this residue may limit the capacity of the virus to replicate in lymphoid cells. Consistent with this idea, lower levels of viral RNA were detected in the popliteal lymph nodes of mice inoculated with viruses containing an arginine at E2 82 (data not shown). The rapid selection of a glycine at this residue may explain the higher titers of virus strains 181/25 and AF15561 E2 G82R in the spleens and other tissues of infected mice at later times postinoculation and might support a role for this site early in infection. Although viral isolates from the spleens of AF15561-infected mice encoded a glycine at E2 82, the population was not homogeneous, as one clone encoded an arginine at this residue. These data suggest that selection at this position is not absolute in vivo.
Residues in E2 regulate virion stability by influencing the folding of the protein and by mediating interactions with the E1 and capsid proteins on the virion surface (57–61). Therefore, changes in E2, such as the nonconservative G82R substitution, may alter the stability of the CHIKV virion as demonstrated for other alphaviruses (57–61). When the E2 Arg82 residue was modeled into the crystal structure of the CHIKV E1/E2 heterodimer (Protein Data Bank [PDB] accession no. 3N42 [27]), this residue closely apposes Glu79, which likely results in the formation of a salt bridge between the cationic guanidinium group of arginine and the anionic carboxylate group of glutamate (data not shown). The formation of this salt bridge may stabilize the E2 protein and, in turn, promote CHIKV infectivity in cell culture and at sites of initial replication but limit dissemination to sites of secondary replication.
Beyond viral attachment and tissue tropism, E2 82 may contribute to host responses to CHIKV early in infection. Although there were significant differences in swelling and pathology in the left hind limb, titers produced at this site by strains that vary solely at E2 82 were comparable. These data support the hypothesis that differences in pathological injury produced by CHIKV strains with an E2 residue polymorphism result from differences in immune and inflammatory responses. Such responses could be tissue specific and dependent upon replication efficiency in the discrete cell subsets that are targeted within those tissues. Concordant with this hypothesis, relative to the virulent CHIKV LR strain, LR E2 Arg82 induces lower levels of proinflammatory cytokines and chemokines (45). Our data suggest that E2 residue 82 also modulates the induction of necrotic pathways that contribute to differences in swelling elicited by these viruses during infection. Despite similar levels of myositis and tendonitis induced by these strains, viruses containing E2 Arg82 induced less necrosis in the hind limb metatarsal muscle than viruses containing E2 Gly82. In mice deficient for alpha/beta interferon (IFN-α/β) receptors or the signal transducer and activator of transcription factor 1 (STAT1) signal transducer, similar levels of inflammatory infiltrates were observed in the hind limbs following infection with CHIKV strains LR and 181/25, despite reduced swelling in mice infected with strain 181/25 (37). These data suggest a correlation between induction of necrotic pathways and events that contribute to swelling in the infected host, which may occur independently of immune cell infiltration.
Our study demonstrates that a glycine at E2 82 is required for virulence in the AF15561 background but not sufficient to confer virulence to the attenuated 181/25 strain. Substituting 181/25 E2 Arg82 with glycine failed to recapitulate the virulent phenotype, as assessed by weight gain and footpad swelling. However, introducing a glycine at E2 82 was sufficient to induce histopathological injury in the hind limbs of infected mice to levels induced by the parental AF15561 strain. Therefore, the effects of this residue are dependent on the genetic background in which the polymorphism is present. Accordingly, substitution of AF15561 E2 Thr12 with isoleucine from strain 181/25, which infects mammalian cells more efficiently, decreased infectivity to levels less than those observed for AF15561. This substitution was demonstrated previously to contribute to 181/25 attenuation (31) but was not identified in our study in which we used infectivity in mammalian cells as an in vitro correlate of virulence to screen viral variants. Therefore, additional polymorphisms displayed by strains AF15561 and 181/25 likely contribute to attenuation of 181/25 in this mouse model of CHIKV disease.
An arginine at E2 residue 82 does not mediate enhancement of infectivity in mosquito cells as in mammalian cells. These data suggest a GAG-independent function for this residue in invertebrate cells. Several mosquito species express the enzymes capable of synthesizing GAGs (62), but a thorough characterization of GAGs expressed by C6/36 cells has not been reported. Additionally, GAGs are present on the surfaces of midgut and salivary gland cells of certain mosquito species (62, 63), but it is not clear whether these molecules contribute to CHIKV infection in the invertebrate host. In contrast to mammalian cells, substituting 181/25 E2 Arg82 with glycine enhances infection of C6/36 cells. This enhancement might result from disrupting interactions with negatively charged GAGs or promoting different or enhanced interactions with other mosquito cell factors either at the cell surface or during later steps in infection. The former would indicate that interactions with GAGs on the surfaces of C6/36 cells impede CHIKV infection. We think this is not the case, as substituting AF15561 E2 Gly82 with arginine does not substantially diminish infection of these cells. Additional cell surface entry mediators may or may not be the same for mammalian and mosquito hosts. The importance of a glycine at this residue for viral fitness is evidenced by its high conservation among CHIKV isolates (38), suggesting that it confers an advantage for replication in mosquitoes.
Understanding mechanisms of virus entry and the interactions that promote dissemination and pathogenesis is critical for development of pathogen-specific therapeutic and prophylactic intervention strategies. Our study suggests that E2 residue 82 is a determinant of infection in both mammalian and mosquito cells and defines a role for E2 residue 82 in GAG engagement and CHIKV virulence in vivo. We demonstrate that viruses containing E2 Arg82 display enhanced infectivity in mammalian cells, while viruses containing E2 Gly82 display enhanced infectivity in mosquito cells. The enhancement provided by an arginine at E2 82 was only partly attributable to increased binding, suggesting that this residue promotes interactions with other cell surface molecules or influences steps in the virus life cycle following attachment. In infected mice, viruses encoding an arginine at E2 residue 82 exhibit defects in replication in lymphoid tissues, establishment of viremia, and production of pathological injury in joint-associated muscle. A glycine at E2 82 is under strong selective pressure in the spleens of CHIKV-infected mice, but this residue was not static, as an arginine was also selected at low frequency. Our findings support new functions for E2 residue 82 in host-specific and GAG-independent processes and in the development of joint swelling through necrosis-mediated events. Future studies to understand mechanisms by which E2 residue 82 influences CHIKV tropism and host responses during infection will reveal both viral and host targets to restrict infection and diminish disease.
ACKNOWLEDGMENTS
We thank Jennifer Konopka-Anstadt, Bernardo Mainou, and Clint Smith for critical reviews of the manuscript. We are grateful to members of the Dermody and Morrison laboratories for useful discussions and to John Williams for helpful suggestions during these studies. We thank Halil Aydin and Jonathan Cook from the laboratory of Jeffrey Lee for assistance with structural analysis. A subset of infectivity assays was conducted at the Vanderbilt High-Throughput Screening Facility. The flow cytometry experiments were performed in the Vanderbilt Cytometry Shared Resource.
This work was supported by Public Health Service awards T32 HL07751 (A.W.A.), F32 AI096833 (L.A.S.), and U54 AI057157 (T.S.D.). Additional support was provided by the Elizabeth B. Lamb Center for Pediatric Research.
Footnotes
Published ahead of print 20 August 2014
REFERENCES
- 1.Burt FJ, Rolph MS, Rulli NE, Mahalingam S, Heise MT. 2012. Chikungunya: a re-emerging virus. Lancet 379:662–671. 10.1016/S0140-6736(11)60281-X. [DOI] [PubMed] [Google Scholar]
- 2.Suhrbier A, Jaffar-Bandjee MC, Gasque P. 2012. Arthritogenic alphaviruses–an overview. Nat. Rev. Rheumatol. 8:420–429. 10.1038/nrrheum.2012.64. [DOI] [PubMed] [Google Scholar]
- 3.Borgherini G, Poubeau P, Jossaume A, Gouix A, Cotte L, Michault A, Arvin-Berod C, Paganin F. 2008. Persistent arthralgia associated with Chikungunya virus: a study of 88 adult patients on Reunion Island. Clin. Infect. Dis. 47:469–475. 10.1086/590003. [DOI] [PubMed] [Google Scholar]
- 4.Sissoko D, Malvy D, Ezzedine K, Renault P, Moscetti F, Ledrans M, Pierre V. 2009. Post-epidemic Chikungunya disease on Reunion Island: course of rheumatic manifestations and associated factors over a 15-month period. PLoS Negl. Trop. Dis. 3:e389. 10.1371/journal.pntd.0000389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoarau JJ, Jaffar Bandjee MC, Krejbich Trotot P, Das T, Li-Pat-Yuen G, Dassa B, Denizot M, Guichard E, Ribera A, Henni T, Tallet F, Moiton MP, Gauzere BA, Bruniquet S, Jaffar Bandjee Z, Morbidelli P, Martigny G, Jolivet M, Gay F, Grandadam M, Tolou H, Vieillard V, Debre P, Autran B, Gasque P. 2010. Persistent chronic inflammation and infection by Chikungunya arthritogenic alphavirus in spite of a robust host immune response. J. Immunol. 184:5914–5927. 10.4049/jimmunol.0900255. [DOI] [PubMed] [Google Scholar]
- 6.Lynch N, Ellis Pegler R. 2010. Persistent arthritis following Chikungunya virus infection. N. Z. Med. J. 123:79–81. [PubMed] [Google Scholar]
- 7.Schilte C, Staikowsky F, Couderc T, Madec Y, Carpentier F, Kassab S, Albert ML, Lecuit M, Michault A. 2013. Chikungunya virus-associated long-term arthralgia: a 36-month prospective longitudinal study. PLoS Negl. Trop. Dis. 7:e2137. 10.1371/journal.pntd.0002137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Simon F, Parola P, Grandadam M, Fourcade S, Oliver M, Brouqui P, Hance P, Kraemer P, Ali Mohamed A, de Lamballerie X, Charrel R, Tolou H. 2007. Chikungunya infection: an emerging rheumatism among travelers returned from Indian Ocean islands. Report of 47 cases. Medicine (Baltimore) 86:123–137. [DOI] [PubMed] [Google Scholar]
- 9.Rezza G, Nicoletti L, Angelini R, Romi R, Finarelli AC, Panning M, Cordioli P, Fortuna C, Boros S, Magurano F, Silvi G, Angelini P, Dottori M, Ciufolini MG, Majori GC, Cassone A, CHIKV Study Group 2007. Infection with chikungunya virus in Italy: an outbreak in a temperate region. Lancet 370:1840–1846. 10.1016/S0140-6736(07)61779-6. [DOI] [PubMed] [Google Scholar]
- 10.Kee AC, Yang S, Tambyah P. 2010. Atypical Chikungunya virus infections in immunocompromised patients. Emerg. Infect. Dis. 16:1038–1040. 10.3201/eid1606.091115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grandadam M, Caro V, Plumet S, Thiberge JM, Souares Y, Failloux AB, Tolou HJ, Budelot M, Cosserat D, Leparc-Goffart I, Despres P. 2011. Chikungunya virus, southeastern France. Emerg. Infect. Dis. 17:910–913. 10.3201/eid1705.101873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leparc-Goffart I, Nougairede A, Cassadou S, Prat C, de Lamballerie X. 2014. Chikungunya in the Americas. Lancet 383:514. 10.1016/S0140-6736(14)60185-9. [DOI] [PubMed] [Google Scholar]
- 13.Van Bortel W, Dorleans F, Rosine J, Blateau A, Rousset D, Matheus S, Leparc-Goffart I, Flusin O, Prat C, Cesaire R, Najioullah F, Ardillon V, Balleydier E, Carvalho L, Lemaitre A, Noel H, Servas V, Six C, Zurbaran M, Leon L, Guinard A, van den Kerkhof J, Henry M, Fanoy E, Braks M, Reimerink J, Swaan C, Georges R, Brooks L, Freedman J, Sudre B, Zeller H. 2014. Chikungunya outbreak in the Caribbean region, December 2013 to March 2014, and the significance for Europe. Euro Surveill. 19(13):pii=20759 http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=20759. [DOI] [PubMed] [Google Scholar]
- 14.Borgherini G, Poubeau P, Staikowsky F, Lory M, Le Moullec N, Becquart JP, Wengling C, Michault A, Paganin F. 2007. Outbreak of Chikungunya on Reunion Island: early clinical and laboratory features in 157 adult patients. Clin. Infect. Dis. 44:1401–1407. 10.1086/517537. [DOI] [PubMed] [Google Scholar]
- 15.Economopoulou A, Dominguez M, Helynck B, Sissoko D, Wichmann O, Quenel P, Germonneau P, Quatresous I. 2009. Atypical Chikungunya virus infections: clinical manifestations, mortality and risk factors for severe disease during the 2005–2006 outbreak on Reunion. Epidemiol. Infect. 137:534–541. 10.1017/S0950268808001167. [DOI] [PubMed] [Google Scholar]
- 16.Das T, Jaffar-Bandjee MC, Hoarau JJ, Krejbich Trotot P, Denizot M, Lee-Pat-Yuen G, Sahoo R, Guiraud P, Ramful D, Robin S, Alessandri JL, Gauzere BA, Gasque P. 2010. Chikungunya fever: CNS infection and pathologies of a re-emerging arbovirus. Prog. Neurobiol. 91:121–129. 10.1016/j.pneurobio.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 17.Griffin DE. 2001. Alphaviruses, p 917–962 In Knipe DM, Howley PM. (ed), Fields virology, 4th ed. Lippincott-Raven Press, Philadelphia, PA. [Google Scholar]
- 18.Khan AH, Morita K, Parquet MDC, Hasebe F, Mathenge EG, Igarashi A. 2002. Complete nucleotide sequence of chikungunya virus and evidence for an internal polyadenylation site. J. Gen. Virol. 83:3075–3084. [DOI] [PubMed] [Google Scholar]
- 19.Solignat M, Gay B, Higgs S, Briant L, Devaux C. 2009. Replication cycle of Chikungunya: a re-emerging arbovirus. Virology 393:183–197. 10.1016/j.virol.2009.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Simizu B, Yamamoto K, Hashimoto K, Ogata T. 1984. Structural proteins of Chikungunya virus. J. Virol. 51:254–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Firth AE, Chung BY, Fleeton MN, Atkins JF. 2008. Discovery of frameshifting in Alphavirus 6K resolves a 20-year enigma. Virol. J. 5:108. 10.1186/1743-422X-5-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Snyder JE, Kulcsar KA, Schultz KL, Riley CP, Neary JT, Marr S, Jose J, Griffin DE, Kuhn RJ. 2013. Functional characterization of the alphavirus TF protein. J. Virol. 87:8511–8523. 10.1128/JVI.00449-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Strauss JH, Strauss EG. 1994. The alphaviruses: gene expression, replication, and evolution. Microbiol. Rev. 58:491–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Konishi E, Hotta S. 1980. Studies on structural proteins of Chikungunya Virus. I. Separation of three species of proteins and their preliminary characterization. Microbiol. Immunol. 24:419–428. [DOI] [PubMed] [Google Scholar]
- 25.de Curtis I, Simons K. 1988. Dissection of Semliki Forest virus glycoprotein delivery from the trans-Golgi network to the cell surface in permeabilized BHK cells. Proc. Natl. Acad. Sci. U. S. A. 85:8052–8056. 10.1073/pnas.85.21.8052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ozden S, Lucas-Hourani M, Ceccaldi PE, Basak A, Valentine M, Benjannet S, Hamelin J, Jacob Y, Mamchaoui K, Mouly V, Despres P, Gessain A, Butler-Browne G, Chretien M, Tangy F, Vidalain PO, Seidah NG. 2008. Inhibition of Chikungunya virus infection in cultured human muscle cells by furin inhibitors: impairment of the maturation of the E2 surface glycoprotein. J. Biol. Chem. 283:21899–21908. 10.1074/jbc.M802444200. [DOI] [PubMed] [Google Scholar]
- 27.Voss JE, Vaney MC, Duquerroy S, Vonrhein C, Girard-Blanc C, Crublet E, Thompson A, Bricogne G, Rey FA. 2010. Glycoprotein organization of Chikungunya virus particles revealed by X-ray crystallography. Nature 468:709–712. 10.1038/nature09555. [DOI] [PubMed] [Google Scholar]
- 28.Jose J, Snyder JE, Kuhn RJ. 2009. A structural and functional perspective of alphavirus replication and assembly. Future Microbiol. 4:837–856. 10.2217/fmb.09.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Levitt NH, Ramsburg HH, Hasty SE, Repik PM, Cole FE, Jr, Lupton HW. 1986. Development of an attenuated strain of Chikungunya virus for use in vaccine production. Vaccine 4:157–162. [DOI] [PubMed] [Google Scholar]
- 30.Edelman R, Tacket CO, Wasserman SS, Bodison SA, Perry JG, Mangiafico JA. 2000. Phase II safety and immunogenicity study of live Chikungunya virus vaccine TSI-GSD-218. Am. J. Trop. Med. Hyg. 62:681–685. [DOI] [PubMed] [Google Scholar]
- 31.Gorchakov R, Wang E, Leal G, Forrester NL, Plante K, Rossi SL, Partidos CD, Adams AP, Seymour RL, Weger J, Borland EM, Sherman MB, Powers AM, Osorio JE, Weaver SC. 2012. Attenuation of Chikungunya virus vaccine strain 181/clone 25 is determined by two amino acid substitutions in the E2 envelope glycoprotein. J. Virol. 86:6084–6096. 10.1128/JVI.06449-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Byrnes AP, Griffin DE. 1998. Binding of Sindbis virus to cell surface heparan sulfate. J. Virol. 72:7349–7356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klimstra WB, Ryman KD, Johnston RE. 1998. Adaptation of Sindbis virus to BHK cells selects for use of heparan sulfate as an attachment receptor. J. Virol. 72:7357–7366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bernard KA, Klimstra WB, Johnston RE. 2000. Mutations in the E2 glycoprotein of Venezuelan equine encephalitis virus confer heparan sulfate interaction, low morbidity, and rapid clearance from blood of mice. Virology 276:93–103. 10.1006/viro.2000.0546. [DOI] [PubMed] [Google Scholar]
- 35.Heil ML, Albee A, Strauss JH, Kuhn RJ. 2001. An amino acid substitution in the coding region of the E2 glycoprotein adapts Ross River virus to utilize heparan sulfate as an attachment moiety. J. Virol. 75:6303–6309. 10.1128/JVI.75.14.6303-6309.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smit JM, Waarts BL, Kimata K, Klimstra WB, Bittman R, Wilschut J. 2002. Adaptation of alphaviruses to heparan sulfate: interaction of Sindbis and Semliki Forest viruses with liposomes containing lipid-conjugated heparin. J. Virol. 76:10128–10137. 10.1128/JVI.76.20.10128-10137.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gardner CL, Burke CW, Higgs ST, Klimstra WB, Ryman KD. 2012. Interferon-alpha/beta deficiency greatly exacerbates arthritogenic disease in mice infected with wild-type Chikungunya virus but not with the cell culture-adapted live-attenuated 181/25 vaccine candidate. Virology 425:103–112. 10.1016/j.virol.2011.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Silva LA, Khomandiak S, Ashbrook AW, Weller R, Heise MT, Morrison TE, Dermody TS. 2014. A single-amino-acid polymorphism in Chikungunya virus E2 glycoprotein influences glycosaminoglycan utilization. J. Virol. 88:2385–2397. 10.1128/JVI.03116-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ryman KD, Klimstra WB, Johnston RE. 2004. Attenuation of Sindbis virus variants incorporating uncleaved PE2 glycoprotein is correlated with attachment to cell-surface heparan sulfate. Virology 322:1–12. 10.1016/j.virol.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 40.Bear JS, Byrnes AP, Griffin DE. 2006. Heparin-binding and patterns of virulence for two recombinant strains of Sindbis virus. Virology 347:183–190. 10.1016/j.virol.2005.11.034. [DOI] [PubMed] [Google Scholar]
- 41.Ryman KD, Gardner CL, Burke CW, Meier KC, Thompson JM, Klimstra WB. 2007. Heparan sulfate binding can contribute to the neurovirulence of neuroadapted and nonneuroadapted Sindbis viruses. J. Virol. 81:3563–3573. 10.1128/JVI.02494-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee E, Wright PJ, Davidson A, Lobigs M. 2006. Virulence attenuation of dengue virus due to augmented glycosaminoglycan-binding affinity and restriction in extraneural dissemination. J. Gen. Virol. 87:2791–2801. 10.1099/vir.0.82164-0. [DOI] [PubMed] [Google Scholar]
- 43.Gardner CL, Ebel GD, Ryman KD, Klimstra WB. 2011. Heparan sulfate binding by natural eastern equine encephalitis viruses promotes neurovirulence. Proc. Natl. Acad. Sci. U. S. A. 108:16026–16031. 10.1073/pnas.1110617108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gardner CL, Choi-Nurvitadhi J, Sun C, Bayer A, Hritz J, Ryman KD, Klimstra WB. 2013. Natural variation in the heparan sulfate binding domain of the eastern equine encephalitis virus E2 glycoprotein alters interactions with cell surfaces and virulence in mice. J. Virol. 87:8582–8590. 10.1128/JVI.00937-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gardner CL, Hritz J, Sun C, Vanlandingham DL, Song TY, Ghedin E, Higgs S, Klimstra WB, Ryman KD. 2014. Deliberate attenuation of Chikungunya virus by adaptation to heparan sulfate-dependent infectivity: a model for rational arboviral vaccine design. PLoS Negl. Trop. Dis. 8:e2719. 10.1371/journal.pntd.0002719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Esko JD, Stewart TE, Taylor WH. 1985. Animal cell mutants defective in glycosaminoglycan biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 82:3197–3201. 10.1073/pnas.82.10.3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mainou B, Zamora PF, Ashbrook AW, Dorset DC, Kim KS, Dermody TS. 2013. Reovirus cell entry requires functional microtubules. mBio 4(4):e00405–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weaver SC, Brault AC, Kang W, Holland JJ. 1999. Genetic and fitness changes accompanying adaptation of an arbovirus to vertebrate and invertebrate cells. J. Virol. 73:4316–4326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krieger N, Lohmann V, Bartenschlager R. 2001. Enhancement of hepatitis C virus RNA replication by cell culture-adaptive mutations. J. Virol. 75:4614–4624. 10.1128/JVI.75.10.4614-4624.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Blaney JE, Jr, Manipon GG, Firestone CY, Johnson DH, Hanson CT, Murphy BR, Whitehead SS. 2003. Mutations which enhance the replication of dengue virus type 4 and an antigenic chimeric dengue virus type 2/4 vaccine candidate in Vero cells. Vaccine 21:4317–4327. [DOI] [PubMed] [Google Scholar]
- 51.Greene IP, Wang E, Deardorff ER, Milleron R, Domingo E, Weaver SC. 2005. Effect of alternating passage on adaptation of Sindbis virus to vertebrate and invertebrate cells. J. Virol. 79:14253–14260. 10.1128/JVI.79.22.14253-14260.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Coffey LL, Vignuzzi M. 2011. Host alternation of Chikungunya virus increases fitness while restricting population diversity and adaptability to novel selective pressures. J. Virol. 85:1025–1035. 10.1128/JVI.01918-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Volchkova VA, Dolnik O, Martinez MJ, Reynard O, Volchkov VE. 2011. Genomic RNA editing and its impact on Ebola virus adaptation during serial passages in cell culture and infection of guinea pigs. J. Infect. Dis. 204(Suppl 3):S941–S946. 10.1093/infdis/jir321. [DOI] [PubMed] [Google Scholar]
- 54.Morrison TE, Oko L, Montgomery SA, Whitmore AC, Lotstein AR, Gunn BM, Elmore SA, Heise MT. 2011. A mouse model of Chikungunya virus-induced musculoskeletal inflammatory disease: evidence of arthritis, tenosynovitis, myositis, and persistence. Am. J. Pathol. 178:32–40. 10.1016/j.ajpath.2010.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Klimstra WB, Ryman KD, Bernard KA, Nguyen KB, Biron CA, Johnston RE. 1999. Infection of neonatal mice with Sindbis virus results in a systemic inflammatory response syndrome. J. Virol. 73:10387–10398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Byrnes AP, Griffin DE. 2000. Large-plaque mutants of Sindbis virus show reduced binding to heparan sulfate, heightened viremia, and slower clearance from the circulation. J. Virol. 74:644–651. 10.1128/JVI.74.2.644-651.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gidwitz S, Polo JM, Davis NL, Johnston RE. 1988. Differences in virion stability among Sindbis virus pathogenesis mutants. Virus Res. 10:225–239. 10.1016/0168-1702(88)90018-4. [DOI] [PubMed] [Google Scholar]
- 58.Snyder AJ, Sokoloski KJ, Mukhopadhyay S. 2012. Mutating conserved cysteines in the alphavirus E2 glycoprotein causes virus-specific assembly defects. J. Virol. 86:3100–3111. 10.1128/JVI.06615-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fields W, Kielian M. 2013. A key interaction between the alphavirus envelope proteins responsible for initial dimer dissociation during fusion. J. Virol. 87:3774–3781. 10.1128/JVI.03310-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cutler DF, Melancon P, Garoff H. 1986. Mutants of the membrane-binding region of Semliki Forest virus E2 protein. II. Topology and membrane binding. J. Cell Biol. 102:902–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lopez S, Yao JS, Kuhn RJ, Strauss EG, Strauss JH. 1994. Nucleocapsid-glycoprotein interactions required for assembly of alphaviruses. J. Virol. 68:1316–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sinnis P, Coppi A, Toida T, Toyoda H, Kinoshita-Toyoda A, Xie J, Kemp MM, Linhardt RJ. 2007. Mosquito heparan sulfate and its potential role in malaria infection and transmission. J. Biol. Chem. 282:25376–25384. 10.1074/jbc.M704698200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dinglasan RR, Alaganan A, Ghosh AK, Saito A, van Kuppevelt TH, Jacobs-Lorena M. 2007. Plasmodium falciparum ookinetes require mosquito midgut chondroitin sulfate proteoglycans for cell invasion. Proc. Natl. Acad. Sci. U. S. A. 104:15882–15887. 10.1073/pnas.0706340104. [DOI] [PMC free article] [PubMed] [Google Scholar]