

ABSTRACT
Autophagy is a catabolic pathway that helps cells to survive under stressful conditions. Cells also use autophagy to clear microbiological infections, but microbes have learned how to manipulate the autophagic pathway for their own benefit. The experimental evidence obtained in this study suggests that the autophagic flux is blocked at the final steps during the reactivation of Epstein-Barr virus (EBV) from latency. This is indicated by the level of the lipidated form of LC3 that does not increase in the presence of bafilomycin and by the lack of colocalization of autophagosomes with lysosomes, which correlates with reduced Rab7 expression. Since the inhibition of the early phases of autophagy impaired EBV replication and viral particles were observed in autophagic vesicles in the cytoplasm of producing cells, we suggest that EBV exploits the autophagic machinery for its transportation in order to enhance viral production. The autophagic block was not mediated by ZEBRA, an immediate-early EBV lytic gene, whose transfection in Ramos, Akata, and 293 cells promoted a complete autophagic flux. The block occurred only when the complete set of EBV lytic genes was expressed. We suggest that the inhibition of the early autophagic steps or finding strategies to overcome the autophagic block, allowing viral degradation into the lysosomes, can be exploited to manipulate EBV replication.
IMPORTANCE This study shows, for the first time, that autophagy is blocked at the final degradative steps during EBV replication in several cell types. Through this block, EBV hijacks the autophagic vesicles for its intracellular transportation and enhances viral production. A better understanding of virus-host interactions could help in the design of new therapeutic approaches against EBV-associated malignancies.
INTRODUCTION
Herpesviruses are able to switch from latent to lytic phase, either spontaneously or upon stimulation with chemicals acting on different target molecules (1). Epstein-Barr virus (EBV) belongs to the gammaherpesvirus family and infects mainly human B lymphocytes and epithelial cells. EBV is associated with some non-Hodgkin's B cell lymphomas, such as Burkitt's lymphoma, where the majority of the cells harbor a latent infection. Depending on the cell types, several inducers can disrupt latency and activate the EBV lytic program in vitro (2–5). In B95-8, a marmoset B lymphoblastoid cell line, the EBV lytic cycle can be efficiently induced by the protein kinase C (PKC) activator 12-O-tetradecanoylphorbol-13-acetate (TPA) alone or in combination with sodium butyrate (the combination is referred to as T/B), a procedure usually utilized in vitro to produce infectious virus. More recently, several other molecules, such as bortezomib (BZ) and thapsigargin (TG), have been reported to induce the EBV lytic cycle in Burkitt cell lines (6). X-box-binding protein 1 (XPB1 form), activated during the unfolded protein response (UPR), also has been shown to induce the switch from latent to lytic cycle by triggering the immediate-early transactivator proteins ZTA and RTA (7). These proteins can, in turn, activate the entire set of EBV lytic gene promoters, controlling the switch from latency to lytic replication (8). The UPR activation by the endoplasmic reticulum (ER) stressor TG can trigger EBV viral replication in B lymphoblastoid cells (LCL) as well (9). TG is known to activate autophagy, as a prosurvival mechanism, during ER stress (10). However, the impact of autophagy on the reactivation of EBV from latency has not been investigated yet. Autophagy, a cellular process aimed at the degradation and recycling of long-lived proteins and damaged organelles (11, 12), also serves for the degradation and clearance of intracellular pathogens (13). However, microorganisms can subvert this process in a way that turns out to be beneficial for their own survival (14). The autophagic pathway can be induced or blocked at different steps by viruses, depending on the phase of their life cycle and on the host cell types. For example, autophagy is enhanced during Kaposi' sarcoma-associated herpesvirus (KSHV) replication (15), allowing the virus to benefit from the autophagic vesicles, termed autophagosomes, for its transportation into the cell cytoplasm. In particular, the KSHV immediate-early gene ORF50, expressed during KSHV replication, is responsible for autophagy induction. On the other hand, KSHV, during its latent infection, blocks autophagy in dendritic cells to escape from their immune control (16, 17). It also has been reported that the KSHV-encoded protein vFLIP induces an autophagic block in NIH 3T3 cells as well as in the naturally KSHV-infected BCBL1 cells (18). Other herpesviruses, such as herpes simplex virus type 1 (HSV1), also block autophagy to induce neurovirulence (19), and viruses belonging to other families, e.g., hepatitis B virus (20) or hepatitis C virus, activate an incomplete autophagy to enhance their replication (21–23) or block autophagy in cells of the immune system (24). Regarding EBV, it is known that it manipulates autophagy to avoid immune control, as reported for other herpesviruses (25), but whether autophagy is involved in EBV replication has not been investigated yet. In this study, we found that autophagy is activated and plays an important role during the reactivation of EBV from latency in several cell types, such as B95-8, Akata, LCL, and 293/EBV cells, in which the lytic cycle can be induced by ZEBRA transfection. In particular, we found that the early autophagic phases promoted EBV lytic gene expression and viral production and that the autophagic flux was blocked at the late steps. This strategy likely allows the virus to prevent the degradative phases of autophagy that could lead to viral degradation.
The EBV latent-lytic switch can occur in vitro and in vivo, although the cellular events leading to the onset of lytic replication in vivo and how it is regulated remain mostly unknown. Discovering strategies that allow for the manipulation of the transition between the latent and lytic cycle could have translational implications in the treatment of EBV-associated diseases.
MATERIALS AND METHODS
Cells.
B95-8 (marmoset B cell line, EBV infected) (26), Raji (EBV-positive cell line derived from Burkitt's lymphoma harboring a defective viral genome), Akata (EBV-positive cell line derived from a Burkitt's lymphoma) (27), LCL (EBV-positive lymphoblastoid cell line), 293 (a human embryonic epithelial kidney cell line) (28), Ramos (EBV-negative cell line derived from Burkitt's lymphoma) (ATCC), and 2A8 (EBV-negative Akata cell line) were cultured in RPMI 1640–10% fetal calf serum (FCS) (Euroclone), glutamine (2 mM), streptomycin (100 μg/ml), and penicillin (100 U/ml) in 5% CO2 at 37°C. 293/EBV, a cell line in which recombinant EBV-B95-8 genomes were stably transfected into 293 cells (kindly provided by H. J. Delecluse) (29) cultured under the same conditions, also were used.
Cell treatments.
To activate the EBV lytic cycle in B95-8 cells, we used the following chemical drugs: 12-tetradecanoylphorbol 13-acetate (TPA) (20 ng/ml) (P1585; Sigma-Aldrich), sodium butyrate (3 mM) (B5887; Sigma-Aldrich), bortezomib (BZ) (20 nM; Velcade), and thapsigargin (TG) (5 μM) (T9033; Sigma-Aldrich). To induce viral production, Akata cells were treated with goat affinity-purified F(ab′)2 fragment to human IgG (50 μg/ml) (56961; Millipore) (30), and LCL from a B-lymphoblastoid cell line were treated with TG (5 μM) (T9033; Sigma-Aldrich). Finally, the lytic cycle was induced in the 293/EBV cell line by transient transfection with p509 plasmid (kindly provided by H. J. Delecluse), which encodes Zebra protein, for 96 h. To evaluate the impact of the autophagy pathway on EBV replication, cells were pretreated with 3-methyladenine (3-MA) (sc-205596; Santa Cruz Biotechnology) at the indicated concentrations for 30 min or with bafilomycin A1 (BAF) (20 nM) (sc-201550; Santa Cruz Biotechnology) and added to the cell culture for the last 3 h. In order to activate autophagy, B95-8 cells were transfected with pDest-mCherry-EGFP-LC3B plasmid for 48 h and then starved by incubation in Hanks balanced salt solution (HBSS) medium (E6267; Sigma-Aldrich) for 2 h (31). Finally, to inhibit EBV DNA synthesis and late gene expression, B95-8 cells were treated with 1.6 mM phosphonoacetic acid (PAA) (P6909; Sigma-Aldrich) and T/B for 48 h.
Western blot analyses.
Cells (5 × 105) were washed twice with phosphate-buffered saline (PBS) solution and centrifuged at 1,500 rpm for 5 min. The pellet was lysed in a radioimmunoprecipitation assay (RIPA) buffer containing 150 mM NaCl, 1% NP-40, 50 mM Tris-HCl (pH 8), 0.5% deoxycholic acid, 0.1% SDS, protease, and phosphatase inhibitors. The lysates then were subjected to electrophoresis on 4 to 12% NuPage Bis-Tris gels (N00322BOX; Life Technologies) or 3 to 8% gels (EA0375BOX; Life Technologies) to investigate gp350/220 protein expression according to the manufacturer's instructions. To evaluate the lipidated forms of LC3 (LC3II), the cell lysates were denatured in Laemmli buffer for 5 min at 100°C and run on a 15% gel (30% acrylamide-Bis solution; 29:1) (161-015; Bio-Rad) in Tris-glycine-SDS buffer. The gels then were transferred to Protran nitrocellulose membranes (10401196; GE Healthcare) for 2 h in Tris-glycine and overnight (for gp350/220) in Tris-glycine-methanol buffer. The membranes were blocked in PBS-0.1% Tween 20 solution containing 3% bovine serum albumin (BSA), probed with specific antibodies, and developed using ECL blotting substrate (K-12045-D20; Advansta).
Antibodies.
For Western blot analysis, the following primary antibodies were used: mouse monoclonal ZEBRA (1:100) (sc-53904; Santa Cruz Biotechnology), gp350/220 (1:500) (ATCC 72/A1), BMRF1 (EA-D) (1:1,000) (MAB 8186; Millipore), rabbit polyclonal anti-LC3 (1:1,000) (NB100-2220; Novus Biologicals), mouse monoclonal anti-p62 (1:1,000) (610833; BD Transduction Laboratories), rabbit polyclonal anti-ATG5 (1:1,000) (NB110-53818; Novus Biologicals), and rabbit polyclonal anti-Rab7 (sc-10767; Santa Cruz Biotechnology). A monoclonal mouse anti-α-tubulin (1:1,000) (T5168; Sigma-Aldrich) or anti-GAPDH (1:1,000) (sc-137179; Santa Cruz Biotechnology) antibody was used as a marker of equal loading. Goat polyclonal anti-mouse IgG-horseradish peroxidase (HRP) (sc-2005; Santa Cruz Biotechnology) and anti-rabbit IgG-HRP (sc-2004; Santa Cruz Biotechnology) were used as secondary antibodies. All of the primary and secondary antibodies used in this study were diluted in a PBS-0.1% Tween 20 solution containing 3% BSA.
Electron microscopy (EM) analysis.
B95-8 cells were treated with TPA (20 ng/ml) and sodium butyrate (3 mM) for 24 h. Subsequently, cells were fixed in 2% glutaraldehyde in PBS for 24 h at 4°C, postfixed in 1% OsO4 for 2 h, and stained for 1 h in 1% aqueous uranyl-acetate. The samples then were dehydrated in acetone and embedded in Epon-812 (Electron Microscopy Science, Società Italiana Chimici). One-micron-thick sections were cut, stained with 1% methylene blue, and viewed by light microscopy to select representative areas. Ultrathin sections were cut with a Reichert ultramicrotome, counterstained with uranyl-acetate and lead citrate, and examined with a Philips CM10 transmission electron microscope (FEI, Eindhoven, The Netherlands).
Transfection and plasmids.
293 and 293/EBV cell lines were transiently transfected with an empty vector (EV) or with p509 plasmid that contains the BZLF1 gene (pBZLF1) (kindly provided by H. J. Delecluse). Briefly, cells (5 × 105) were seeded on 6-well plates in complete medium without antibiotics for 16 h. The following day, the cells were transfected with the liposome Metafectene (T020; Biontex) using a lipid/DNA ratio of 3 μl to 1 μg. The plasmid DNA (0.5 μg each well) and the liposome were diluted into two solutions of medium without serum and antibiotics; the mixture was incubated for 20 min and then was added to cells for 48 h.
Ramos and Akata cells were transfected by electroporation with a Gene Pulser (Bio-Rad). A total of 3 × 106 log-phase cells were resuspended in 0.3 ml of Opti-MEM medium (31985062; Life Technologies) with 3 μg of EV or p509 plasmid in an electroporation cuvette (4-mm gap width) (FL9554000; M-Medical). Subsequently, it was given as a single pulse (250 V, 960 μF) to the cells, and complete medium was added for 48 h. To control the transfection efficiency, we used a pEGFP-C1 plasmid that expresses green fluorescent protein (GFP) and a fluorescence microscope.
We also performed a transient transfection of pEGFP-LC3 (kindly provided by G. M. Fimia) (32) or pDest-mCherry-EGFP-LC3B (31) in B95-8 cells. Cells (5 × 105) were seeded into 6-well plates and then transfected with 5 μg of plasmid DNA/well with Lipofectamine 2000 according to the manufacturer's instructions. After 48 h, the cells were treated with TPA (20 ng/ml) and sodium butyrate (3 mM) for 24 h. The LC3 dots were visualized by an Apotome Axio Observer Z1 inverted microscope (Zeiss) equipped with an AxioCam MRM Rev.3 at ×40 magnification.
Knockdown of ATG5 and Beclin by siRNA.
The knockdown of ATG5 or Beclin was performed in B95-8 and Akata cells using specific small interfering RNA (sc-41445 and sc-29797, respectively; Santa Cruz Biotechnology). The day before transfection, 3 × 105 cells were seeded in 12-well culture plates in RPMI medium without antibiotics. Subsequently, 75 pmol of small interfering RNA (siRNA) duplex and 7.5 μl of Lipofectamine 2000 transfection reagent (11668027; Life Technologies) were diluted in Opti-MEM medium (31985062; Life Technologies) and added to the cells for 72 h. B95-8 and Akata cells then were treated with TPA (20 ng/ml) and sodium butyrate (3 mM) or with anti-Ig for 24 to 48 h. The transfection efficiency was evaluated by a fluorescein-conjugated control siRNA (sc-36869; Santa Cruz Biotechnology) that also was used as the scrambled control.
The knockdown of Beclin also was performed in 293/EBV cells using a specific small interfering RNA (sc-29797; Santa Cruz Biotechnology). Briefly, cells (5 × 105) were seeded on 6-well plates in complete medium without antibiotics for 16 h. A combination of 150 pmol of Beclin siRNA duplex and 15 μl of Lipofectamine 2000 transfection reagent (11668027; Life Technologies) was diluted in Opti-MEM medium (31985062; Life Technologies) and added to the cells for 72 h.
Virus production.
To investigate whether autophagy inhibition affected EBV production, 293/EBV cells were pretreated with 3-MA (10 mM), a drug that inhibits autophagy at early steps, for 30 min, or a Beclin knockdown was performed for 72 h before ZEBRA transfection. Viral supernatants were harvested, filtered (0.45-μm-pore-size filters), and centrifuged at 29,000 rpm for 2 h. The viral pellets were resuspended in RPMI medium. Functional virus titers were determined via GFP expression. A total of 104 Raji cells were infected into 96-wells plates with a serial dilution of virus stocked 100× and scored for GFP expression after 3 days by UV microscopy. The number of green Raji units per milliliter was calculated as a measurement of the concentration of infectious particles in virus stocks (33). Three different infection experiments were performed.
Immunofluorescence.
The EBV lytic cycle was induced in B95-8 cells as previously described and then left untreated or pretreated with 3-MA or PAA or silenced for ATG5. The cells then were harvested, washed twice in PBS, and seeded on glass slides and air dried. Cells then were permeabilized and fixed in acetone-methanol (1:1) (ZEBRA) or acetone (gp350/220) at −20°C for 10 min and incubated with mouse monoclonal anti-ZEBRA (1:100 in PBS) or anti-gp350/220 (1:100 in PBS) antibody for 1 h. They then were washed three times in PBS and incubated with polyclonal goat anti-mouse Texas red-conjugated antibody (Jackson) (1:50 in PBS) for 30 min or with Alexa Fluor 350 goat anti-mouse (A11045; Life Technologies) (gp350/220). Finally, after several washings, cells were incubated with 1 μg/ml of 4′,6′-diamidino-2-phenylindole (DAPI) to visualize the nuclei, and the coverslips were mounted face down using a PBS-glycerol (1:1) solution. The immunofluorescence was analyzed using an Apotome Axio Observer Z1 inverted microscope (Zeiss) equipped with an AxioCam MRM Rev.3 at ×40 magnification.
LysoTracker red staining.
B95-8 cells transfected with pEGFP-LC3 were treated with TPA (20 ng/ml) and sodium butyrate (3 mM) for 24 h, washed in PBS solution, and incubated with LysoTracker red DND-99 (100 nM) (L7528; Life Technologies), a fluorescent acidotropic probe with high selectivity for acidic organelles, for 30 min at room temperature (10). After several washings with PBS solution, the lysosomes were analyzed using an Apotome Axio Observer Z1 inverted microscope (Zeiss) equipped with an AxioCam MRM Rev.3 at ×40 magnification.
RESULTS
EBV lytic cycle inducers affect the autophagic pathway concomitantly with viral replication.
B95-8 cells were induced into the lytic cycle by several chemicals able to reactivate EBV from latency: tumor promoter phorbol ester (TPA) alone or in combination with sodium butyrate (T/B), bortezomib (BZ), and thapsigargin (TG). As shown in Fig. 1A, TPA, T/B, TG, and, to a lesser extent, BZ were able to induce the expression of the EBV immediate-early lytic protein ZEBRA after 24 h of treatment. We then investigated whether these lytic cycle activators had an impact on autophagy. To this aim, the expression level of the lipidated form of LC3 (LC3II), which is a marker of autophagy, was evaluated by Western blot analysis. As shown in Fig. 1A, all of these drugs, although acting on different cellular targets, induced LC3II accumulation concomitantly with the activation of the complete EBV lytic program in B95-8 cells, including the EBV late lytic gp350/220 protein (data not shown).
FIG 1.

EBV reactivation from latency induces LC3II accumulation. The lytic cycle was activated in B95-8 cells, Akata cells, and LCLs (A and B) by the indicated treatments. ZEBRA and LC3 I/II expression was evaluated after 24 h by Western blot analysis. As control, Ramos and 2A8 EBV-negative cell lines were treated with the same agents used to induce the lytic cycle in B95-8 and Akata cells, respectively. GAPDH was used as a loading control, and a representative experiment out of four is shown. The densitometric analysis of the ZEBRA/GAPDH and LC3II/GAPDH ratios also is reported.
The study then was extended to Akata, a Burkitt lymphoma cell line latently infected with EBV, in which the replicative cycle can be induced by surface Ig cross-linking. We found that, concomitant with the expression of ZEBRA, the appearance of LC3II also was detected in these cells (Fig. 1B), and the same results were obtained in LCL in which the lytic cycle was activated by treatment with TG (Fig. 1B). Finally, as controls, the EBV-negative Burkitt's lymphoma cell lines Ramos and 2A8 were subjected to the same treatments used to induce the EBV lytic cycle in B95-8 and Akata cells, respectively. The results shown in Fig. 1C indicate that all of the treatments were able to induce LC3II accumulation in Ramos cells even in the absence of EBV lytic cycle induction. Unlike Akata cells, the Ig cross-linking reduced the LC3II level in the EBV-negative 2A8 cells (Fig. 1D).
Altogether, these results indicate that EBV lytic cycle inducers affect the autophagic pathway in the EBV-producing cells, and most of them also do this in EBV-negative Burkitt lymphoma cells.
The inhibition of the early autophagic steps reduces EBV lytic gene expression and viral production.
To investigate the role of autophagy on EBV lytic cycle activation, we performed siRNA knockdown of ATG5, a molecule that mediates the lipidation of LC3 and is critical for the formation or elongation of autophagosomes. ATG5 knockdown before T/B exposure or surface Ig cross-linking in B95-8 and Akata cells, respectively, strongly reduced ZEBRA expression (Fig. 2A and C), indicating that autophagy plays a positive role in the EBV replicative cycle. To further demonstrate the importance of the autophagic process in the EBV lytic cycle, we knocked down Beclin (BECN), another essential autophagic gene. As for ATG5, we found a reduction of ZEBRA expression in B95-8 cells knocking down the Beclin gene (Fig. 2B). Mean (± standard deviations [SD]) ZEBRA expression from three different ATG5 or Beclin siRNA experiments is shown (Fig. 2D).
FIG 2.

Autophagy enhances EBV lytic gene expression. ZEBRA expression was evaluated by Western blot analysis in B95-8 cells transfected with ATG5 (A) or Beclin (BECN) (B) siRNA and induced into the lytic cycle with T/B. (C) ZEBRA expression was evaluated by Western blot analysis in Akata cells knocked down with ATG5 before anti-Ig cross-linking. GAPDH was used as a loading control, and a representative experiment out of three is shown. (D) The densitometric analysis of the ZEBRA/GAPDH, ATG5/GAPDH, and Beclin/GAPDH ratios and the mean percentages ± SD of ZEBRA reduction from three different siRNA experiments also is reported.
The involvement of autophagy in the EBV lytic cycle was further demonstrated by immunofluorescence assay (IFA) in B95-8 cells knocked down for ATG5 before T/B exposure. In agreement with the experiments reported above, autophagy inhibition reduced the number of cells expressing the EBV immediate-early lytic protein ZEBRA (Fig. 3A). Mean (± SD) percentages of ZEBRA-positive B95-8 cells with or without ATG5 siRNA were determined (Fig. 3B). Similar results were obtained in Akata cells (data not shown).
FIG 3.

ATG5 knockdown reduces the number of ZEBRA-expressing B95-8 cells. (A) Immunofluorescence analysis showing ZEBRA-positive B95-8 cells left untreated or treated with T/B for 24 h in the presence or absence of ATG5 knockdown. A representative experiment out of three is shown. (B) Mean percentages (±SD) of B95-8 cells expressing ZEBRA from three different ATG5 siRNA experiments also are reported.
Finally, the importance of the first autophagic steps on EBV lytic cycle activation was confirmed by treating B95-8 or Akata cells, before lytic cycle induction, with 3-MA, an inhibitor of class III phosphatidylinositol-3′-kinase (PI3KC3), which is critical for initiating autophagy. Again, a reduction of ZEBRA expression was observed in both cell lines pretreated with 3-MA (Fig. 4A and B).
FIG 4.

Autophagy inhibition reduces EBV lytic gene expression and viral production. ZEBRA expression was evaluated by Western blot analysis in B95-8 (A) and Akata (B) cells pretreated with 3-MA (at the indicated doses) and then induced into the lytic cycle by T/B or anti-Ig treatment, respectively. GAPDH was used as a loading control, and a representative experiment out of three is shown. BMRF1 expression (C) and viral production (D) in terms of Raji cell infection were evaluated in 293/EBV cells after ZEBRA (pBZLF1) or empty vector (EV) transfection, in the presence or absence of 3-MA. GAPDH was used as a loading control, and a representative experiment out of three is shown. (E) 3-MA effect on ZEBRA transfection efficiency was evaluated in 293/EBV cells. BMRF1 expression (F) and viral production (G) in terms of Raji cell infection were evaluated after Beclin silencing in 293/EBV cells that were transfected with ZEBRA (pBZLF1) or with EV.
Whether autophagy inhibition would affect the production of infectious viral particles was evaluated next. 293/EBV cells were induced into the lytic cycle by ZEBRA transfection in the presence or absence of 3-MA. We first confirmed that BMRF1 lytic protein expression was reduced by 3-MA in 293/EBV cells (Fig. 4C) and then collected the cell supernatants, containing GFP-EBV particles, to infect Raji cells. The infection efficiency (or functional titers) was assessed by counting the GFP-positive cells in serial dilutions of the supernatants. 293/EBV supernatants from 3-MA-treated cells showed a reduction of functional titers compared to those of supernatants from untreated 293/EBV (Fig. 4D). To exclude that 3-MA could affect the efficiency of ZEBRA transfection, we tested its expression in untreated or 3-MA-treated 293/EBV cells and found no differences in the presence of 3-MA (Fig. 4E). Similar results, in terms of BMRF1 expression and Raji infection, were obtained by knocking down Beclin before ZEBRA transfection in 293/EBV cells (Fig. 4F and G). These data suggest that autophagy is essential for the production of infectious EBV particles.
An autophagic block occurs in cells undergoing the EBV lytic cycle.
To determine whether the autophagic process, active during EBV replication, would lead to a complete autophagic flux, we examined the expression level of LC3II in the presence or in the absence of bafilomycin (BAF) in B95-8 cells, Akata cells, and LCL. BAF is an inhibitor of the vacuolar proton pump (V-H+-ATPase) that, by preventing the proper acidification of lysosomal compartments, hampers degradation of LC3II. This strategy allows us to distinguish if LC3II accumulation (seen in Fig. 1) was due to its increased formation or to its decreased degradation, with both events normally occurring during a complete autophagic process (34). We found that LC3II levels did not increase in the presence of BAF in B95-8 cells, Akata cells, or LCL treated with T/B, Ig, or TG, respectively (Fig. 5A), indicating that a block of the autophagic flux was occurring in these cells during EBV replication. BAF treatment slightly influenced the EBV lytic gene expression in B95-8 cells, Akata cells, and LCL (Fig. 5A, B, and C), supporting the idea that autophagy was blocked prior to autophagolysosome formation.
FIG 5.

Autophagy is blocked when the lytic cycle is activated in EBV-producing cells. B95-8 cells (A), Akata cells (B), and (C) LCL were induced into the lytic cycle with T/B, anti-Ig, or TG, respectively, and after 24 h were analyzed for LC3I/II and ZEBRA expression in the presence or absence of BAF. GAPDH was used as a loading control, and a representative experiment out of three is shown. The densitometric analysis of the ZEBRA/GAPDH and LC3II/GAPDH ratios also is reported. Ramos cells (D) and Akata cells (E) were transfected with ZEBRA, and after 48 h LC3I/II and ZEBRA expression were evaluated in the presence or in the absence of BAF. (F) Raji cells were treated with T/B for 24 h, and LC3I/II expression was analyzed in the presence or absence of BAF. The activation of the lytic cycle in Raji cells was indicated by ZEBRA and BMRF1 expression. (G) Ramos cells were treated with T/B for 24 h and analyzed for LC3I/II expression in the presence or absence of BAF. (H) B95-8 cells were treated with T/B with or without PAA for 48 h, and LC3I/II expression was analyzed in the presence or absence of BAF. GAPDH was used as a loading control, and a representative experiment out of three is shown. The densitometric analysis of the LC3II/GAPDH ratio also is shown. (I) The effect of PAA on the reduction of gp350/220 expression in B95-8 cells treated with T/B was evaluated by IFA. A representative experiment out of three is shown.
To investigate whether ZEBRA, the first protein expressed upon the activation of the EBV lytic cycle, was responsible for the block of the autophagic flux, we transfected a plasmid encoding BZLF1 into Ramos cells, an EBV-negative Burkitt's lymphoma cell line. Surprisingly, ZEBRA transfection in Ramos cells promoted a complete autophagic flux, indicated by the increase of LC3II in the presence of BAF (Fig. 5D). To further clarify the effect of ZEBRA, we transfected it into Akata cells, a Burkitt's lymphoma cell line harboring the complete EBV genome in a latent state. We found that ZEBRA induced a complete autophagic flux in these cells concomitant with the lack of EBV lytic cycle activation (Fig. 5E). The effect of the EBV early lytic proteins, typically expressed in Raji cells upon T/B treatment, on autophagy was evaluated next. Since a complete autophagic flux was observed in these cells expressing BMRF1 in the absence of EBV late gene expression (Fig. 5F), we suggest that the block of autophagy occurs only when the complete EBV lytic program is activated. In addition, the T/B treatment of EBV-negative Burkitt lymphoma Ramos cells (Fig. 5G), as well as B95-8 cells, treated with phosphonoacetic acid (PAA) to prevent EBV replication (Fig. 5I) led to a complete autophagic flux (Fig. 5H).
Finally, we transfected ZEBRA in 293 and 293/EBV cells to better distinguish between the effect on autophagy mediated by ZEBRA per se (in 293) from the effect mediated by the complete set of EBV lytic genes activated by ZEBRA (in 293/EBV). In agreement with the results obtained in EBV-negative Burkitt's lymphoma cells, we found that ZEBRA transfection in 293 cells induced a complete autophagic flux, indicated by the LC3II increase in combination with BAF (Fig. 6A). In addition, Zebra transfection in these cells promoted the degradation of p62 (Fig. 6B), a protein specifically degraded through autophagy, commonly used to monitor the outcome of the process. On the other hand, when ZEBRA was transfected in 293/EBV cells concomitantly with the EBV lytic cycle activation, it blocked the autophagic flux in terms of LC3II and p62 levels compared to the same cells transfected with empty vector in which a complete autophagic process was induced (Fig. 6A and B).
FIG 6.

ZEBRA transfection in 293 but not in 293/EBV cells leads to an autophagic block. (A) 293 and 293/EBV cells were transfected with ZEBRA (pBZLF1), and after 48 h, LC3I/II and ZEBRA or LC3I/II and BMRF1 were evaluated in the presence or absence of BAF. p62 (B) and Rab7 (C) expression was analyzed in 293 and 293/EBV cells transfected with ZEBRA. GAPDH was used as a loading control, and a representative experiment out of three is shown. The densitometric analysis of the LC3II/GAPDH, p62/GAPDH, and Rab7/GAPDH ratios also is reported.
Since Rab7 plays a fundamental role for complete autophagic flux (35) and is necessary for the fusion between autophagosomes and lysosomes, we next investigated if this molecule would be modulated differently by ZEBRA transfection in 293 and 293/EBV cells. The results shown in Fig. 6C indicate that Rab7 expression was upregulated by ZEBRA transfection in 293 cells, in which complete autophagy was observed. In contrast, in 293/EBV cells, where ZEBRA transfection induced the autophagic block, Rab7 was downregulated concomitantly with the activation of the complete EBV lytic program (Fig. 6A). Rab7 reduction could represent one of the underlying mechanisms leading to the autophagic block observed during EBV replication.
Together, these data indicate that the same treatments that induce a complete autophagic flux in cells in which the EBV lytic program is not complete block autophagy at the final steps in cells undergoing EBV replication. Through this strategy, EBV avoids lysosomal degradation and may use the autophagic vesicles to travel in the cell cytoplasm, redirecting them toward the plasma membrane.
EBV impairs fusion of autophagosomes with lysosomes during replication and travels in the autophagic vesicles.
We analyzed the autophagosome and lysosome localization in B95-8 cells induced into the EBV lytic cycle. We transfected the pDest-mCherry-EGFP-LC3B plasmid into the cells before T/B treatment. The LC3 puncta, indicating autophagosomes arising during autophagy induction, change their color depending on the pH. They appear yellow when both green and red colors are expressed, while, when autophagosomes fuse with lysosomes, the GFP color is shut down and autophagolysosomes become red. As shown in Fig. 7A, B95-8 cells induced into the lytic cycle with T/B contained yellow puncta, indicating that no fusion between autophagosomes and lysosomes was occurring. Conversely, when autophagy was induced by nutrient starvation, red puncta, due to autophagolysosome formation, could be observed in the same cells, indicating a complete autophagic flux (Fig. 7B). To further investigate autophagosome and lysosome localization, B95-8 cells were transfected with GFP-LC3 plasmid, and the lysosomal compartment was stained with LysoTracker red. As shown in Fig. 8, most of the GFP-LC3 puncta (indicating autophagosomes) did not colocalize with lysosomes (stained in red) in the EBV-producing cells identified by a monoclonal antibody against the membrane antigen gp350/220 (blue staining). Together, these results suggest that the fusion between autophagosomes and lysosomes was impaired during EBV replication. Finally, we investigated if the subversion of autophagy would be a strategy utilized by the virus to hijack the autophagic vesicles for its transportation into the cell cytoplasm. To this aim, we performed electron microscopy analysis in B95-8 cells induced into the lytic cycle with T/B and found that viral particles could be seen in double membrane autophagic vesicles at different stages of maturation (36) in the producing cells (Fig. 9A and C to H). The autophagic features were observed in the majority of the cells undergoing viral production, as quantified by analyzing 200 cells (Fig. 9B). Moreover, about 40% of the viral particles were seen in the autophagic vesicles. Knocking down Beclin in B95-8 cells before exposure to T/B treatment strongly reduced the number of vesicles as well as the number of viral particles within them (Fig. 9I and J), confirming that they were autophagosomes.
FIG 7.
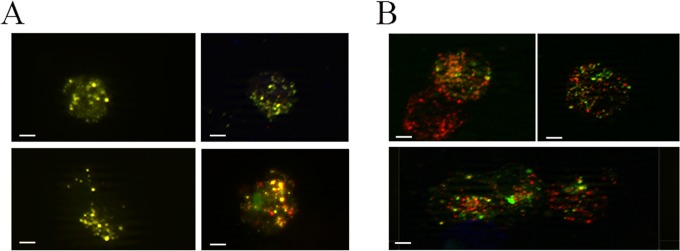
Autophagosomes do not fuse with lysosomes in EBV-producing cells. B95-8 cells transfected with pDest-mCherry-EGFP-LC3B plasmid and treated with T/B for 24 h (A) or starved for 2 h to induce autophagy (B) were analyzed by immunofluorescence. The yellow puncta indicate autophagosomes, while red puncta indicate autophagolysosomes. A representative experiment out of three is shown. Bar, 10 μm.
FIG 8.

Autophagosomes do not colocalize with lysosomes in EBV-producing cells. B95-8 cells transfected with pEGFP-LC3 plasmid and treated with T/B for 24 h were analyzed by immunofluorescence. GFP-LC3 puncta (autophagosomes) and lysosome (stained in red with LysoTracker) localization is shown in EBV-producing cells (stained in blue with EBV anti-gp 350/220 membrane antigen monoclonal antibody and indicated by blue arrowheads). Bar, 10 μm.
FIG 9.

Autophagic vesicles containing viral particles are observed in EBV-producing cells. (A and C to F) B95-8 cells were induced into the lytic cycle with T/B, and electron microscopic analysis shows autophagic vesicles (arrows) transporting viral particles toward the plasma membrane. Empty autophagosomes (Au) and nucleocapsids (arrowheads) also were observed. N, nucleus; PM, plasma membrane. (B) Percentage of B95-8 undergoing replication, autophagy, or both is indicated. B95-8 cells were transfected with scrambled (G) or Beclin (BECN) (H) siRNA before T/B treatment, and the reduction of autophagic vesicles and the viral particles within them by BECN siRNA is shown. (I and J) Reduction of released viruses by BECN siRNA is shown. (A to F) Bar = 0.5 μm. (G to J) Bar = 1 μm.
Based on our results, we propose a model in which the activation of the complete EBV lytic program blocks autophagy, preventing the delivery of the autophagic vesicles that also may contain viral particles, to the lysosomes. Concomitantly, this strategy enables the virus to redirect the autophagic vesicles toward the plasma membrane and to use them for its transportation (Fig. 10).
FIG 10.

Schematic model describing how EBV subverts autophagy during its replication. Adapted from the schematic model reported by Klionsky et al. (34).
DISCUSSION
In this study, we show for the first time that early autophagic phases are involved in the EBV lytic replicative cycle, promoting EBV lytic gene expression and viral production. Conversely, the late autophagic steps do not seem to play an essential role in EBV replication, likely because autophagy is blocked prior to the fusion of the autophagosomes with lysosomes. We propose that through this strategy, EBV appropriates the autophagic vesicles for viral transportation into the cell cytoplasm and avoids viral degradation into the lysosomes. Since ZEBRA is the first EBV protein expressed during lytic cycle activation, we first investigated if it could be responsible for the autophagic block. In agreement with previous results obtained for the KSHV immediate-early gene ORF50 (15), we found that ZEBRA expression per se induced a complete autophagic flux when transfected in Ramos, Akata, or 293 cells, suggesting that the autophagic block did not depend on its expression. The abortive EBV lytic cycle induction in Raji cells by T/B, as well as the concomitant treatment of B95-8 cells with PAA and T/B, also led to complete autophagy. Conversely, a block of autophagy occurred when the complete set of EBV lytic genes was expressed and viral replication was activated in B95-8 cells, Akata cells, LCL, and 293/EBV cells. Of note, 293 and 293/EBV cells were particularly helpful to distinguish the effect on autophagy mediated by ZEBRA per se from the effect mediated by the other EBV lytic genes in the same cell context. It is possible that ZEBRA promotes a complete autophagic flux to degrade transcriptional repressors that hamper EBV lytic replication (37), but further studies will be necessary to address this possibility. Based on our results, we suggest that a possible mechanism underlying the autophagic block observed in cells undergoing EBV replication is the downregulation of Rab7. This protein is essential for the autophagosome maturation (35) and for complete autophagic flux, as demonstrated in a previous study (38). Moreover, Rab7 is involved in lysosome biogenesis (39), and its downregulation could further impair lysosomal degradation by reducing the number of lysosomes. Understanding the relationship between autophagy and viruses could help to find new strategies to control viral infection/replication through autophagy manipulation. From several reports in this field, it has emerged that viruses block autophagy in the immune cells to avoid their degradation (xenophagy) and to interfere with viral antigen presentation (16, 40, 41). The autophagic block mediated by the virus can occur at different levels of the autophagic pathway, from autophagosome formation to lysosome degradation. It seems to be dependent on the virus, on the phases of its life cycle, and on host cell type. Regarding the relationship between autophagy and viral replication, it has been demonstrated that several viruses, such as HCV (42) and HBV, stimulate an incomplete autophagy (43) and that autophagy is involved in KSHV replication (15). However, a KSHV protein, K7, expressed during its lytic phase, is able to block the autophagic process (44). Regarding EBV proteins, it is known that the latent membrane protein 1 (LMP1) regulates autophagy to control its own degradation (45). The results obtained in this study suggest that the autophagy is subverted during the activation of the EBV lytic program, enhancing viral replication and allowing the virus to skip its degradation into the lysosomes.
EBV usually is latent in the majority of the cells that harbor the infection both in vitro and in vivo. Reactivation from latency can be induced in vitro through different stimuli, but how it is regulated in vivo is not completely known. The latent to lytic switch represents a double-edged sword: on the one hand it leads to viral spread and new infections, while on the other hand it is accompanied by the lysis of the productive cells and gives the possibility of treating the infected cells with antiviral agents. In conclusion, we suggest that autophagy manipulation during EBV lytic cycle activation could allow cells to reduce viral production and to benefit from the viral lytic phase activation to successfully utilize antiviral drugs. A better understanding of the mechanisms that regulate the interplay between these two events could help to improve the treatment of EBV-associated diseases.
ACKNOWLEDGMENTS
We thank Sandro Valia and Lucilla Simonelli for technical assistance and T. Johansen and G. M. Fimia for the kind gift of the pDest-mCherry-EGFP-LC3B and pEGFP-LC3 plasmids, respectively. We also thank H. J. Delecluse for providing the 293/EBV cell line and p509 plasmid.
This work was supported by grants from MIUR, Associazione Italiana per la Ricerca SulCancro (AIRC) (no. 10265), Progetto Strategico ISS 9ACF/1, of the Ministero della Salute.
We have no financial or commercial conflicts of interest to declare.
ADDENDUM IN PROOF
Similar results about the positive role of autophagy in the EBV lytic cycle have been reported by C. H. Hung et al. (J. Virol. 88:12133, 2014, doi:10.1128/JVI.02033-14) .
Footnotes
Published ahead of print 20 August 2014
REFERENCES
- 1.Hammerschmidt W, Sugden B. 1988. Identification and characterization of oriLyt, a lytic origin of DNA replication of Epstein-Barr virus. Cell 55:427–433. 10.1016/0092-8674(88)90028-1. [DOI] [PubMed] [Google Scholar]
- 2.Faggioni A, Zompetta C, Grimaldi S, Barile G, Frati L, Lazdins J. 1986. Calcium modulation activates Epstein-Barr virus genome in latently infected cells. Science 232:1554–1556. 10.1126/science.3012779. [DOI] [PubMed] [Google Scholar]
- 3.Gradoville L, Kwa D, El-Guindy A, Miller G. 2002. Protein kinase C-independent activation of the Epstein-Barr virus lytic cycle. J. Virol. 76:5612–5626. 10.1128/JVI.76.11.5612-5626.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lazdins J, Zompetta C, Grimaldi S, Barile G, Venanzoni M, Frati L, Faggioni A. 1987. TPA induction of Epstein-Barr virus early antigens in Raji cells is blocked by selective protein kinase-C inhibitors. Int. J. Cancer 40:846–849. 10.1002/ijc.2910400624. [DOI] [PubMed] [Google Scholar]
- 5.Miller G, El-Guindy A, Countryman J, Ye J, Gradoville L. 2007. Lytic cycle switches of oncogenic human gammaherpesviruses. Adv. Cancer Res. 97:81–109. 10.1016/S0065-230X(06)97004-3. [DOI] [PubMed] [Google Scholar]
- 6.Shirley CM, Chen J, Shamay M, Li H, Zahnow CA, Hayward SD, Ambinder RF. 2011. Bortezomib induction of C/EBPbeta mediates Epstein-Barr virus lytic activation in Burkitt lymphoma. Blood 117:6297–6303. 10.1182/blood-2011-01-332379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun CC, Thorley-Lawson DA. 2007. Plasma cell-specific transcription factor XBP-1s binds to and transactivates the Epstein-Barr virus BZLF1 promoter. J. Virol. 81:13566–13577. 10.1128/JVI.01055-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holley-Guthrie EA, Quinlivan EB, Mar EC, Kenney S. 1990. The Epstein-Barr virus (EBV) BMRF1 promoter for early antigen (EA-D) is regulated by the EBV transactivators, BRLF1 and BZLF1, in a cell-specific manner. J. Virol. 64:3753–3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taylor GM, Raghuwanshi SK, Rowe DT, Wadowsky RM, Rosendorff A. 2011. Endoplasmic reticulum stress causes EBV lytic replication. Blood 118:5528–5539. 10.1182/blood-2011-04-347112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Raciti M, Lotti LV, Valia S, Pulcinelli FM, Di Renzo L. 2012. JNK2 is activated during ER stress and promotes cell survival. Cell Death Dis. 3:e429. 10.1038/cddis.2012.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kroemer G, Marino G, Levine B. 2010. Autophagy and the integrated stress response. Mol. Cell 40:280–293. 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feng Y, He D, Yao Z, Klionsky DJ. 2014. The machinery of macroautophagy. Cell Res. 24:24–41. 10.1038/cr.2013.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deretic V. 2011. Autophagy in immunity and cell-autonomous defense against intracellular microbes. Immunol. Rev. 240:92–104. 10.1111/j.1600-065X.2010.00995.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kirkegaard K, Taylor MP, Jackson WT. 2004. Cellular autophagy: surrender, avoidance and subversion by microorganisms. Nat. Rev. Microbiol. 2:301–314. 10.1038/nrmicro865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wen HJ, Yang Z, Zhou Y, Wood C. 2010. Enhancement of autophagy during lytic replication by the Kaposi's sarcoma-associated herpesvirus replication and transcription activator. J. Virol. 84:7448–7458. 10.1128/JVI.00024-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Santarelli R, Gonnella R, Di Giovenale G, Cuomo L, Capobianchi A, Granato M, Gentile G, Faggioni A, Cirone M. 2014. STAT3 activation by KSHV correlates with IL-10, IL-6 and IL-23 release and an autophagic block in dendritic cells. Sci. Rep. 4:4241. 10.1038/srep04241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blanchet FP, Moris A, Nikolic DS, Lehmann M, Cardinaud S, Stalder R, Garcia E, Dinkins C, Leuba F, Wu L, Schwartz O, Deretic V, Piguet V. 2010. Human immunodeficiency virus-1 inhibition of immunoamphisomes in dendritic cells impairs early innate and adaptive immune responses. Immunity 32:654–669. 10.1016/j.immuni.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee JS, Li Q, Lee JY, Lee SH, Jeong JH, Lee HR, Chang H, Zhou FC, Gao SJ, Liang C, Jung JU. 2009. FLIP-mediated autophagy regulation in cell death control. Nat. Cell Biol. 11:1355–1362. 10.1038/ncb1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Orvedahl A, Levine B. 2008. Autophagy and viral neurovirulence. Cell. Microbiol. 10:1747–1756. 10.1111/j.1462-5822.2008.01175.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sir D, Tian Y, Chen WL, Ann DK, Yen TS, Ou JH. 2010. The early autophagic pathway is activated by hepatitis B virus and required for viral DNA replication. Proc. Natl. Acad. Sci. U. S. A. 107:4383–4388. 10.1073/pnas.0911373107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sir D, Kuo CF, Tian Y, Liu HM, Huang EJ, Jung JU, Machida K, Ou JH. 2012. Replication of hepatitis C virus RNA on autophagosomal membranes. J. Biol. Chem. 287:18036–18043. 10.1074/jbc.M111.320085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dreux M, Gastaminza P, Wieland SF, Chisari FV. 2009. The autophagy machinery is required to initiate hepatitis C virus replication. Proc. Natl. Acad. Sci. U. S. A. 106:14046–14051. 10.1073/pnas.0907344106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dreux M, Chisari FV. 2011. Impact of the autophagy machinery on hepatitis C virus infection. Viruses 3:1342–1357. 10.3390/v3081342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Granato M, Lacconi V, Peddis M, Di Renzo L, Valia S, Rivanera D, Antonelli G, Frati L, Faggioni A, Cirone M. 2014. Hepatitis C virus present in the sera of infected patients interferes with the autophagic process of monocytes impairing their in-vitro differentiation into dendritic cells. Biochim. Biophys. Acta 1843:1348–1355. 10.1016/j.bbamcr.2014.04.003. [DOI] [PubMed] [Google Scholar]
- 25.Cavignac Y, Esclatine A. 2010. Herpesviruses and autophagy: catch me if you can! Viruses 2:314–333. 10.3390/v2010314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miller G, Lipman M. 1973. Release of infectious Epstein-Barr virus by transformed marmoset leukocytes. Proc. Natl. Acad. Sci. U. S. A. 70:190–194. 10.1073/pnas.70.1.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takada K. 1984. Cross-linking of cell surface immunoglobulins induces Epstein-Barr virus in Burkitt lymphoma lines. Int. J. Cancer 33:27–32. 10.1002/ijc.2910330106. [DOI] [PubMed] [Google Scholar]
- 28.Graham FL, Smiley J, Russell WC, Nairn R. 1977. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 36:59–74. 10.1099/0022-1317-36-1-59. [DOI] [PubMed] [Google Scholar]
- 29.Delecluse HJ, Hilsendegen T, Pich D, Zeidler R, Hammerschmidt W. 1998. Propagation and recovery of intact, infectious Epstein-Barr virus from prokaryotic to human cells. Proc. Natl. Acad. Sci. U. S. A. 95:8245–8250. 10.1073/pnas.95.14.8245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takada K, Ono Y. 1989. Synchronous and sequential activation of latently infected Epstein-Barr virus genomes. J. Virol. 63:445–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T. 2007. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 282:24131–24145. 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 32.Fimia GM, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, Nardacci R, Corazzari M, Fuoco C, Ucar A, Schwartz P, Gruss P, Piacentini M, Chowdhury K, Cecconi F. 2007. Ambra1 regulates autophagy and development of the nervous system. Nature 447:1121–1125. 10.1038/nature05925. [DOI] [PubMed] [Google Scholar]
- 33.Granato M, Feederle R, Farina A, Gonnella R, Santarelli R, Hub B, Faggioni A, Delecluse HJ. 2008. Deletion of Epstein-Barr virus BFLF2 leads to impaired viral DNA packaging and primary egress as well as to the production of defective viral particles. J. Virol. 82:4042–4051. 10.1128/JVI.02436-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, Ahn HJ, Ait-Mohamed O, Ait-Si-Ali S, Akematsu T, Akira S, Al-Younes HM, Al-Zeer MA, Albert ML, Albin RL, Alegre-Abarrategui J, Aleo MF, Alirezaei M, Almasan A, Almonte-Becerril M, Amano A, Amaravadi R, Amarnath S, Amer AO, Andrieu-Abadie N, Anantharam V, Ann DK, Anoopkumar-Dukie S, Aoki H, Apostolova N, Arancia G, Aris JP, Asanuma K, Asare NY, Ashida H, Askanas V, Askew DS, Auberger P, Baba M, Backues SK, Baehrecke EH, Bahr BA, Bai XY, Bailly Y, Baiocchi R, Baldini G, Balduini W, Ballabio A, et al. 2012. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8:445–544. 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hyttinen JM, Niittykoski M, Salminen A, Kaarniranta K. 2013. Maturation of autophagosomes and endosomes: a key role for Rab7. Biochim. Biophys. Acta 1833:503–510. 10.1016/j.bbamcr.2012.11.018. [DOI] [PubMed] [Google Scholar]
- 36.Eskelinen EL, Reggiori F, Baba M, Kovacs AL, Seglen PO. 2011. Seeing is believing: the impact of electron microscopy on autophagy research. Autophagy 7:935–956. 10.4161/auto.7.9.15760. [DOI] [PubMed] [Google Scholar]
- 37.Yu X, Wang Z, Mertz JE. 2007. ZEB1 regulates the latent-lytic switch in infection by Epstein-Barr virus. PLoS Pathog. 3:e194. 10.1371/journal.ppat.0030194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ganley IG, Wong PM, Gammoh N, Jiang X. 2011. Distinct autophagosomal-lysosomal fusion mechanism revealed by thapsigargin-induced autophagy arrest. Mol. Cell 42:731–743. 10.1016/j.molcel.2011.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bucci C, Thomsen P, Nicoziani P, McCarthy J, van Deurs B. 2000. Rab7: a key to lysosome biogenesis. Mol. Biol. Cell 11:467–480. 10.1091/mbc.11.2.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Crotzer VL, Blum JS. 2009. Autophagy and its role in MHC-mediated antigen presentation. J. Immunol. 182:3335–3341. 10.4049/jimmunol.0803458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Crotzer VL, Blum JS. 2005. Autophagy and intracellular surveillance: modulating MHC class II antigen presentation with stress. Proc. Natl. Acad. Sci. U. S. A. 102:7779–7780. 10.1073/pnas.0503088102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shrivastava S, Raychoudhuri A, Steele R, Ray R, Ray RB. 2011. Knockdown of autophagy enhances the innate immune response in hepatitis C virus-infected hepatocytes. Hepatology 53:406–414. 10.1002/hep.24073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li J, Liu Y, Wang Z, Liu K, Wang Y, Liu J, Ding H, Yuan Z. 2011. Subversion of cellular autophagy machinery by hepatitis B virus for viral envelopment. J. Virol. 85:6319–6333. 10.1128/JVI.02627-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liang Q, Chang B, Brulois KF, Castro K, Min CK, Rodgers MA, Shi M, Ge J, Feng P, Oh BH, Jung JU. 2013. Kaposi's sarcoma-associated herpesvirus K7 modulates Rubicon-mediated inhibition of autophagosome maturation. J. Virol. 87:12499–12503. 10.1128/JVI.01898-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee DY, Sugden B. 2008. The latent membrane protein 1 oncogene modifies B-cell physiology by regulating autophagy. Oncogene 27:2833–2842. 10.1038/sj.onc.1210946. [DOI] [PubMed] [Google Scholar]