

ABSTRACT
Ebola virus (EBOV) belongs to the group of nonsegmented negative-sense RNA viruses. The seven EBOV genes are separated by variable gene borders, including short (4- or 5-nucleotide) intergenic regions (IRs), a single long (144-nucleotide) IR, and gene overlaps, where the neighboring gene end and start signals share five conserved nucleotides. The unique structure of the gene overlaps and the presence of a single long IR are conserved among all filoviruses. Here, we sought to determine the impact of the EBOV gene borders during viral transcription. We show that readthrough mRNA synthesis occurs in EBOV-infected cells irrespective of the structure of the gene border, indicating that the gene overlaps do not promote recognition of the gene end signal. However, two consecutive gene end signals at the VP24 gene might improve termination at the VP24-L gene border, ensuring efficient L gene expression. We further demonstrate that the long IR is not essential for but regulates transcription reinitiation in a length-dependent but sequence-independent manner. Mutational analysis of bicistronic minigenomes and recombinant EBOVs showed no direct correlation between IR length and reinitiation rates but demonstrated that specific IR lengths not found naturally in filoviruses profoundly inhibit downstream gene expression. Intriguingly, although truncation of the 144-nucleotide-long IR to 5 nucleotides did not substantially affect EBOV transcription, it led to a significant reduction of viral growth.
IMPORTANCE Our current understanding of EBOV transcription regulation is limited due to the requirement for high-containment conditions to study this highly pathogenic virus. EBOV is thought to share many mechanistic features with well-analyzed prototype nonsegmented negative-sense RNA viruses. A single polymerase entry site at the 3′ end of the genome determines that transcription of the genes is mainly controlled by gene order and cis-acting signals found at the gene borders. Here, we examined the regulatory role of the structurally unique EBOV gene borders during viral transcription. Our data suggest that transcriptional regulation in EBOV is highly complex and differs from that in prototype viruses and further the understanding of this most fundamental process in the filovirus replication cycle. Moreover, our results with recombinant EBOVs suggest a novel role of the long IR found in all filovirus genomes during the viral replication cycle.
INTRODUCTION
Ebola virus (EBOV) belongs to the filovirus family and is one of the most pathogenic viruses known, causing a severe hemorrhagic fever in humans, with case fatality rates up to 90% (1). Currently, there are no licensed vaccines or specific therapeutics available to prevent or treat EBOV disease. Filoviruses have been classified as tier 1 select agents by the CDC and as category A priority pathogens by NIAID, and work with these pathogens must be performed under the highest biosafety level (BSL) conditions, BSL-4. Due to this requirement, fundamental processes in the viral replication cycle are still not completely understood, including the regulation of filoviral transcription. Although EBOV follows the basic transcription mechanism described for nonsegmented negative-sense (NNS) RNA viruses (2), it has evolved some distinct features to transcribe its genome, including an unconventional protein requirement for transcription, cotranscriptional editing of the mRNA encoding the glycoprotein (GP), and the structure of cis-acting regulatory elements on the genome (reviewed in reference 3). The last was further examined in the work presented here. The 19-kb NNS RNA genome of EBOV is tightly wrapped by the nucleoprotein (NP) and contains seven genes in the order 3′ NP-VP35-VP40-GP-VP30-VP24-L 5′ (Fig. 1A). Transcription of the encapsidated genome is mediated by the RNA-dependent RNA polymerase L, the polymerase cofactor VP35, and the transcription factor VP30 (4, 5). According to the stop-start model describing NNS RNA virus transcription, the viral polymerase complex binds to a single transcription promoter at the 3′ end of the genome and moves toward the 5′ end, sequentially synthesizing mostly monocistronic mRNAs (reviewed in reference 2). Primarily at the gene borders, it occasionally dissociates from the template and then has to rebind to the promoter at the 3′ end of the genome (6). This results in a gradient of transcript abundance in EBOV-infected cells, with high mRNA levels transcribed from the promoter-proximal NP gene and low expression levels from the promoter-distal L gene (7).
FIG 1.

Northern blot analysis of EBOV mRNAs. (A) Scheme of the EBOV genome showing the diverse structures of the gene borders. Gene borders examined in panels C to F are circled. ORFs are shown as gray boxes and UTRs flanking the ORFs as white boxes. GS signals are illustrated as white triangles, GE signals as black bars, and the editing site of the GP gene as a white bar. IRs are depicted as gray lines, and their lengths are indicated underneath. Gene borders with overlapping transcription signals that lack an IR are indicated by “overlap.” le, leader; tr, trailer. The consensus sequences of the gene borders are shown above the genome. The conserved pentamer shared by overlapping GS and GE signals is highlighted in boldface. Nucleotide variations are indicated. N represents A, C, or G; asterisk, missing nucleotide. (B) Scheme of mRNA species transcribed from gene borders NP-VP35, VP35-VP40, and VP30-VP24. Shown are all mRNAs detected by Northern hybridization in panels C to F. Numbers are assigned as shown in panels C to F. (C to F) Northern hybridization of EBOV mRNAs transcribed from gene borders with different structures. Huh-T7 cells were infected with EBOV at an MOI of 3 or left uninfected. Poly(A)+ mRNA was isolated at 24 h p.i. and analyzed by Northern hybridization using gene-specific riboprobes. Probe specificities are shown underneath each blot. The numbers on the right indicate expected mRNAs based on their predicted sizes excluding poly(A) tails. The poly(A) tails of filoviral transcripts are assumed to be about 100 to 300 nt in length, as described for other NNS RNA viruses (2). (C) NP-VP35 gene border. 1, NP mRNA (2,971 nt); 2, VP35 mRNA (1,376 nt); 3, NP-VP35 rth-mRNA (4,352 nt); 4, VP35-VP40 rth-mRNA (2,863 nt). (D) VP35-VP40 gene border. 2, VP35 mRNA (1,376 nt); 5, VP40 mRNA (1,505 nt); 3, NP-VP35 rth-mRNA (4,352 nt); 4, VP35-VP40 rth-mRNA (2,863 nt); 6, VP40-GP-editstop rth-mRNA (2,535 nt), indicating an rth-mRNA that is terminated at the editing signal of the GP gene; 7, VP40-GP rth-mRNA (3,916 nt). (E) VP30-VP24 gene border. 8, VP30 mRNA (1,453 nt); 9, VP24 mRNA (1,612 nt); 10, VP30-VP24 rth-mRNA (3,209 nt); 11, GP-VP30 rth-mRNA (3,841 nt). (F) Longer exposure time of panel E showing that a VP24-L rth-mRNA band (8,398 nt) was not detected.
The gene order is not the only transcriptional-regulatory mechanism used by NNS RNA viruses. Polymerase activity is additionally modulated by cis-acting elements encountered at the gene borders, which encompass the region from the gene end (GE) signal of the upstream gene to the gene start (GS) signal of the downstream gene (Fig. 1A). The GE and GS signals are usually separated by an intergenic region (IR) that is not transcribed in the monocistronic mRNAs and is believed to be scanned by the viral polymerase during transcription (8). The EBOV GS and GE signals are highly conserved (Fig. 1A). Analogous to other NNS RNA viruses, the GS signals are likely involved in directing transcription initiation and capping of the nascent mRNA, while the GE signals mediate transcript termination (reviewed in references 2 and 3). The GE signals of all NNS RNA viruses contain a uridine stretch that serves as a template for polymerase stuttering, leading to the addition of a poly(A) tail at the 3′ end of the transcripts (9, 10). Curiously, the EBOV VP24 gene contains two consecutive GE signals (11) (Fig. 2A). In an EBOV minigenome system, both GEs were needed for efficient expression of the downstream gene (12). However, the functional implication of this unique arrangement for EBOV remains unclear.
FIG 2.
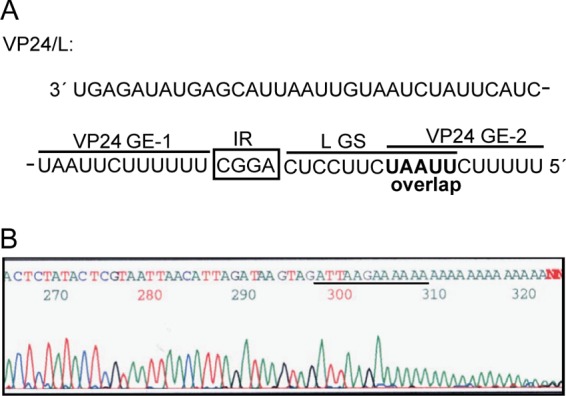
The EBOV polymerase primarily terminates transcription of the VP24 gene at the first GE signal. (A) Sequence of the VP24-L gene border, including the last 33 nt of the VP24 5′ UTR. The first GE signal of the VP24 gene (VP24 GE-1) is separated by a 4-nt-long IR from the GS signal of the L gene (L GS), whereas the second GE signal (VP24 GE-2) directly overlaps the L GS. The sequence is shown in negative-sense orientation. (B) 3′ RACE to determine the 3′ end of VP24 mRNA. Shown is a representative sequencing result from 1 out of 13 clones. The sequence of VP24 GE-1 is underlined. The sequence is shown in positive-sense orientation.
Based on the structures of their IRs, the NNS RNA viruses fall into two groups. The first group comprises viruses with short IRs with conserved lengths (2 or 3 nucleotides [nt]) and sequences and includes vesicular stomatitis virus Indiana (VSVIndiana), Sendai virus (SeV), and measles virus. The genomes of the second group of viruses contain IRs that vary in both size and sequence. This group includes VSV New Jersey, rabies virus (RABV), Newcastle disease virus (NDV), simian virus 5 (SV5) and SV41, and respiratory syncytial virus (RSV) (reviewed in references 2 and 13). Filoviruses belong to the second group. Their genomes contain several short IRs ranging from 4 to 9 nt and a single long IR that varies in length from 97 nt (Marburg virus) to 144 nt (EBOV) (Fig. 1A). The filoviral IRs lack obvious sequence homology. While the location of the long IR between the VP30 and VP24 genes is maintained in all ebolavirus genomes, it is located one gene border upstream in marburgvirus genomes, separating the GP and VP30 genes (14). To our knowledge, among the NNS RNA viruses, equally long IRs (over 100 nt) have so far been described only for the metapneumoviruses, and their function remains to be determined (15). It is intriguing why such a long untranscribed region is retained within all filovirus genomes. Previous studies with bicistronic EBOV reporter minigenomes suggested that the long IR is important for the expression of the neighboring genes, but its regulatory function during viral RNA synthesis has not yet been examined (12).
Three of the seven EBOV genes are not separated by IRs, but the GE of the upstream gene directly overlaps the GS of the downstream gene, sharing a highly conserved pentameric sequence motif (11) (Fig. 1A). While overlapping genes are known for other NNS RNA viruses, directly overlapping transcription signals are unique to filoviruses (16–20). Separating the overlapping VP35 GE and VP40 GS signals in an EBOV bicistronic minigenome (Bici) led to a decrease in downstream reporter activity and an increase in upstream reporter activity, indicating that the overlaps are of functional importance for EBOV gene expression (12). As viral mRNA products synthesized from overlapping EBOV genes have not yet been analyzed, it is not known if this unique arrangement of the GE and GS signals influences the behavior of the transcribing viral polymerase. Passing over the GS signal might cause the polymerase to slow down or pause and thus could improve recognition of the overlapping GE signal, leading to the inhibition of readthrough transcription. Readthrough mRNA (rth-mRNA) synthesis caused by inefficient transcriptional termination at a GE signal has been frequently observed for NNS RNA virus genes separated by IRs (21–28). Failure to recognize a GE signal results in transcription of bicistronic or even polycistronic mRNAs. The bicistronic transcripts comprise two open reading frames (ORFs), the second of which is not accessible for translation (29). As readthrough transcription bypasses one gene border and thereby one transcriptional attenuation step, it leads not only to reduced expression of the second ORF, but also to increased mRNA synthesis of all subsequent genes. Modulation of termination efficiency can therefore be used to regulate viral gene expression (reviewed in references 2 and 30).
In the present study, we analyzed the regulatory effects of EBOV cis-acting elements, including directly overlapping transcription signals and the exceptionally long IR, on mRNA transcription. We examined whether readthrough transcription takes place at the highly variable EBOV gene borders and whether the unique arrangement of the GE signals within overlapping gene borders interferes with polymerase readthrough. Our data reveal that readthrough mRNA synthesis occurs during EBOV infection and seems to take place largely independently of gene border structure, with the exception of the VP24-L gene border. There, two consecutive GE signals of the VP24 gene might inhibit transcriptional readthrough. Furthermore, we studied the effect of the unusually long IR separating the VP30 and VP24 genes on transcription. We present data suggesting that the long IR is not only involved in the regulation of transcription reinitiation, but is also essential for efficient viral replication. This might explain why it is preserved across all filoviral genomes.
MATERIALS AND METHODS
Cell lines and viruses.
VeroE6 cells (American Type Culture Collection), Vero cells, and the human hepatoma cell lines Huh7 and Huh-T7, constitutively expressing the T7 RNA polymerase (kindly provided by V. Gaussmüller, University of Lübeck, Lübeck, Germany), were grown in Dulbecco's modified Eagle medium (DMEM) containing penicillin (50 U/ml) and streptomycin (50 μg/ml) (P/S) and supplemented with 10% fetal bovine serum (FBS). To maintain expression of the T7 RNA polymerase, the culture medium for Huh-T7 cells was additionally supplemented with 1 mg/ml of Geneticin (31). BSR-T7/5 cells constitutively expressing the T7 RNA polymerase (kindly provided by K. K. Conzelmann, Max von Pettenkofer Institute and Gene Center, Munich, Germany) were cultured as described previously (32).
Ebola virus, species Zaire ebolavirus, Mayinga variant (GenBank number AF086833), was used for the infections in this study and to generate Bicis and recombinant EBOVs (recEBOVs) (11). EBOV and recEBOVs were propagated in VeroE6 cells. Virus titers were determined by 50% tissue culture infectious dose (TCID50) assay for EBOV and by focus-forming-unit (FFU) assay for recEBOV. All work with infectious virus was performed under BSL-4 conditions at the Institute of Virology, Philipps University of Marburg, Marburg, Germany, and at the Integrated Research Facility in the Rocky Mountain Laboratories, Division of Intramural Research, NIAID, NIH, Hamilton, MT, following standard operating procedures approved by the Institutional Biosafety Committees.
3′ Rapid amplification of cDNA ends (RACE) of EBOV VP24 mRNA.
Total cellular RNA was isolated from EBOV-infected Huh-T7 cells at 40 h postinfection (p.i.) using the RNeasy kit (Qiagen) following the manufacturer's recommendations. VP24 mRNA was reverse transcribed and subsequently amplified using the One Step RT-PCR kit (Qiagen) with an oligo(dT)-specific and a VP24 gene-specific primer. The 518-bp PCR product was further amplified by nested PCR with Taq polymerase using an internal VP24 gene-specific primer and the oligo(dT) primer, generating a 483-bp DNA fragment. The PCR fragment was cloned into the pCRII-Topo vector (Stratagene). Vector-specific primers were used for sequencing of the inserts.
RNA isolation and Northern blot analysis.
Total RNA of transfected or infected cells was isolated using the RNeasy kit (Qiagen) following the manufacturer's protocol for purification of total RNA from animal cells. Polyadenylated RNA was purified using oligo(dT) cellulose as described previously (33) or using the Micro Poly(A) Purist kit (Ambion) as recommended by the manufacturer. The purified mRNA was analyzed by Northern hybridization performed with digoxigenin-labeled negative-sense riboprobes directed against the chloramphenicol acetyltransferase (CAT) gene as described previously (33). Transcripts of EBOV-infected cells were detected with digoxigenin-labeled, gene-specific riboprobes. The probes were produced by in vitro transcription with SP6 polymerase using the DIG RNA labeling kit (Roche) from gene-specific PCR fragments containing the SP6 promoter sequence attached to the 5′ end of the reverse primer. All data presented for a given experiment were taken from the same blot and exposure, while lanes not relevant to this study were excised. For RNA quantification, purified mRNA was quantified, and equal RNA amounts were loaded onto the gel prior to Northern hybridization. mRNA amounts in scanned blots were quantified with Quantity One software (Bio-Rad Laboratories).
qRT-PCR analysis.
Approximately 6 × 105 Huh7 cells grown in 6-well plates were infected with the respective recEBOV at a multiplicity of infection (MOI) of 0.3 FFU/cell. Total RNA was isolated from the infected cells at the indicated time points by using TRIzol reagent (Invitrogen) according to the manufacturer's recommendations. Polyadenylated mRNA species were concentrated with the Micro Poly(A) Purist kit (Ambion). Five nanograms of each mRNA sample was analyzed in duplicate by quantitative reverse transcription-PCR (qRT-PCR) with VP30 and VP24 gene-specific primers using the QuantiFast SYBR green RT-PCR kit (Qiagen) and a CFX96 Real-Time PCR cycler (Bio-Rad Laboratories). Since the amplification efficiencies of the VP24 and VP30 genes were determined to be comparable (data not shown), VP30 was used as a viral reference gene. The expression rate of VP24 mRNA was calculated in relation to those of corresponding samples infected with recEBOV wild-type (wt) (recEBOV-wt-IR 144nt) using the ΔΔCT method.
Construction of Bicis.
Bici NP/VP35-wt IR 5nt, containing the NP-VP35 gene border (nt 2728 to 3128), is identical to minigenome E-bici-1,2 described previously (5). Bici VP30/VP24-wt IR 144nt was cloned using the same strategy. Briefly, the VP30-VP24 gene border (nt 9425 to 9982) was amplified by reverse transcription-PCR with primers flanked by BglII and BamHI restriction sites. The PCR fragment was cloned into construct 3E-5E-ΔCAT (5) containing the first 300 nt of the CAT gene and the full-length CAT gene separated by a BglII restriction site that was used for insertion of the PCR fragment. Mutant Bicis containing truncations of the long IR were generated by PCR with phosphorylated primers flanking the region to be deleted. Following the PCR, samples were digested with DpnI to remove template DNA, purified by gel extraction, and religated. Bici VP30/VP24 IR 0nt was created by QuikChange site-directed mutagenesis (Stratagene) of Bici VP30/VP24 IR 5nt. To clone Bici VP30/VP24 IR 10ntmut1 and Bici VP30/VP24 IR 10ntmut2, the IR was replaced by QuikChange mutagenesis using Bici VP30/VP24 IR 10nt as a template. To create Bici VP30/VP24 IR 293nt, the authentic 144-nt-long IR was amplified by PCR with primers containing KpnI sites and inserted into a KpnI site introduced into the central region of the IR by replacement of 2 nucleotides (A9793 to G and A9794 to G) using QuikChange mutagenesis, bringing its total length to more than double the size of the authentic IR. Positive clones were verified by sequencing.
Transfection of cells.
BSR-T7/5 and Huh-T7 cells grown in 6-well plates to 60 to 70% confluence were transfected using FuGene 6 (Roche Molecular Applied Science) or TransIt-LT1 (Mirus), following the manufacturer's recommendations. For testing the transcription of minigenomes in EBOV-infected cells, Huh-T7 cells were transfected with 2 μg plasmid DNA of each Bici or the monocistronic minigenome 3E-5E (4), along with 1 μg of pCAGGS-T7, expressing the T7 RNA polymerase (kindly provided by T. Takimoto, St. Jude Children's Research Hospital, Memphis, TN, and Y. Kawaoka, University of Wisconsin, Madison, WI). To analyze the transcriptional activities of Bicis in minigenome assays, BSR-T7/5 or Huh-T7 cells were transfected with 1.0 μg pT/LEBO, 0.5 μg pT/NPEBO, 0.5 μg pT/VP35EBO, and 0.1 μg pT/VP30EBO, along with 1.5 μg of the respective Bici constructs (4). When Huh-T7 cells were used, 0.5 μg of pCAGGS-T7 was added. For analysis of CAT reporter activity, 0.3 μg of pGL3-Control plasmid (Promega) expressing firefly luciferase was added as the transfection control. All transfection mixtures were adjusted to the same amount of DNA using the empty vector pT/empty. As negative controls, plasmid pT/LEBO was replaced by the appropriate amount of pT/empty (lanes shown as −L in the relevant figures).
Infection of transfected Huh-T7 cells and passaging of Bici-containing supernatants.
To analyze minigenome activity in EBOV-infected cells, Huh-T7 cells were transfected with minigenome plasmid DNA, along with the pCAGGS-T7 plasmid as described above. At 12 h posttransfection, the transfected cells were infected with EBOV at an MOI of 5 TCID50 units/cell and subjected to Northern blot analysis at 48 h or 72 h p.i.
For infection with Bici-containing supernatants, the supernatants of Bici-transfected and infected cells were harvested at 48 h or 72 h p.i. and cleared of cell debris by low-speed centrifugation. Approximately 2 × 105 Huh-T7 cells were infected with 250 μl supernatant mixed with 250 μl DMEM. Following virus adsorption, 2.5 ml of DMEM plus P/S containing 2.5% FBS was added. Cells were harvested at 48 h p.i. and subjected to Northern blot analysis.
Quantitative CAT assay.
Approximately 6 × 105 to 8 × 105 BSR-T7/5 cells grown in 6-well plates were transfected as described above. At 2 days posttransfection, the cells were washed twice with phosphate-buffered saline (PBS) and lysed with 150 μl reporter lysis buffer (luciferase assay system; Promega). Luciferase activity (representing the transfection efficiency) was measured using the luciferase assay system (Promega) according to the manufacturer's protocol. Quantitative CAT assays were performed with normalized amounts of cell lysates following the protocol of the Fast CAT Green (deoxy) Chloramphenicol Acetyltransferase Assay Kit (Molecular Probes). Fluorescent samples were detected in a GelDoc 2000 system and analyzed using Quantity One software (Bio-Rad Laboratories). Relative CAT activity is represented as the measured amount of acetylated chloramphenicol out of total chloramphenicol detected with local background subtraction.
Construction and rescue of recombinant EBOV.
The plasmid p15AK-EBOV HRSC carries the EBOV antigenome flanked by the hammerhead ribozyme at the 5′ end (34) and the supercut version of the hepatitis delta virus antigenomic ribozyme at the 3′ end (35) under the control of a T7 RNA polymerase promoter. In this construct, the EBOV genome contains two silent mutations in the NP gene (A1258 to G and C2206 to U) and one in the GP gene (C7732 to U) that have been introduced to remove BsmBI restriction sites for cloning purposes. To generate EBOV full-length clones with a truncated VP30-VP24 IR, a shuttle plasmid was used containing the ApaI/SacII fragment of the EBOV genome spanning nt 4727 to 11860 of the viral genome. Within this shuttle plasmid, the IR was truncated to 10 nt or 5 nt, respectively, by fusion PCR (36) and cloned back into p15AK-EBOV HRSC using ApaI and SacI restriction sites. The constructs were sequenced prior to use in rescue experiments. recEBOVs were rescued in VeroE6 cells as previously described (7, 37). Briefly, VeroE6 cells seeded in 6-well plates at a confluence of 50% were transfected using TransIt-LT1 (Mirus) according to the manufacturer's protocol with plasmids encoding recEBOV (0.25 μg), along with helper plasmids expressing the T7 RNA polymerase (pCAGGS-T7; 0.25 μg) and the EBOV polymerase complex (0.125 μg pCAGGS-NP, 0.125 μg pCAGGS-VP35, 0.075 μg pCAGGS-VP30, 1 μg pCAGGS-L). Twenty-four hours posttransfection, the medium was exchanged, and 7 days later, the supernatant was transferred to fresh VeroE6 cells (passage 1 [p1]). Supernatants were harvested when cytopathic effect (CPE) was observed and used to grow virus stocks. To confirm viral sequences, RNA was extracted from the supernatants of infected cells using the QIAamp Viral RNA minikit (Qiagen) and subjected to sequencing. The stability of the introduced mutations was confirmed by sequencing this region in the recombinant viruses throughout the course of the experiments (see Fig. 7B). Sequencing also revealed that the viruses started to develop additional mutations at different locations within the viral genome with increasing passage numbers.
FIG 7.

Gene expression, replication kinetics, and focus morphology of recombinant EBOV with a truncated IR at the VP30-VP24 gene border. (A) Scheme of the EBOV genome. The highlighted IR was truncated from 144 nt in the wild-type virus (recEBOV-wt-IR 144nt) to 5 nt (recEBOV-IR 5nt) or 10 nt (recEBOV-IR 10nt). VP30 mRNA and VP24 mRNA are indicated as large arrows. The binding sites of primer pairs used for qRT-PCR, as well as the sizes of PCR fragments, are shown underneath. (B) Flowchart showing the experimental procedure and sequenced virus passages. (C) Relative expression levels of VP24 mRNA determined by qRT-PCR analysis. Huh7 cells were infected with mutant virus or recombinant wild-type virus at an MOI of 0.3 FFU/cell. Cells were harvested at 24 h and 48 h p.i., polyadenylated mRNA was isolated, and the amounts of VP30 and VP24 transcripts were determined by qRT-PCR. Shown are the mean relative expression levels of VP24 mRNA (+SD). (D) Replication kinetics of recombinant EBOVs. Huh7 cells were infected with recEBOV-wt-IR 144nt, recEBOV-IR 5nt, or recEBOV-IR 10nt at an MOI of 0.3 FFU/cell. Supernatants were collected at the indicated time points, and viral titers were determined by focus-forming-unit assay. Shown are the mean values (±SD) of 3 experiments. The asterisks indicate statistically significant differences (P < 0.001) between each mutant virus and the wild type as determined by ANOVA (including Bonferroni correction for multiple testing). The values of recEBOV-IR 5nt were not significantly different from those of recEBOV-IR 10nt, except for the data point 1 day p.i. (P = 0.0248). (E) Morphology of foci formed in Vero cells infected with recombinant EBOVs at 4 days p.i. The foci were visualized by immunofluorescent staining. (Top) Representative overview of large- and small-focus phenotypes observed for the different EBOVs. (Bottom) Higher magnification showing immunofluorescent foci (left) and corresponding bright-field images (right).
Growth kinetics and focus-forming-unit assay of recombinant EBOV.
Approximately 3 × 105 Huh7 cells in 6-well plates were infected with recEBOVs at an MOI of 0.3 FFU/cell. Following adsorption of the virus, the cell monolayer was washed once, and 3 ml of DMEM plus P/S containing 3% FBS was added onto the cells. Supernatant (500 μl) was collected immediately after this washing step (day 0). Supernatant samples were collected each day for 6 days, and the harvested supernatant was replaced with an equal volume of DMEM plus P/S supplemented with 3% FBS. Virus titers (FFU/ml) were determined by counting the infected cell foci in Vero cells using an indirect immunofluorescent-antibody assay as described previously (38). Briefly, 90% confluent Vero cells grown in 96-well plates were infected with serial 10-fold-diluted virus supernatants. Following virus adsorption, the cells were overlaid with modified Eagle medium containing 5% FBS and 1.2% carboxymethyl cellulose sodium salt (Wako Pure Chemical, Osaka, Japan) and incubated for 4 days at 37°C. The cells were fixed with 10% formalin for 30 min, thoroughly washed with PBS, and incubated in 10% formalin overnight to inactivate the EBOV. Foci were visualized by immunofluorescence staining with a VP40-specific rabbit polyclonal antibody and detected with an anti-rabbit fluorescein isothiocyanate (FITC) antibody (Sigma).
Statistical analysis.
For quantitative analyses of CAT activity or mRNA amounts, a one-sample t test or one-way analysis of variance (ANOVA) was performed using GraphPad Prism 5 software.
RESULTS
Readthrough transcription occurs independently of gene border structure in EBOV-infected cells.
As EBOV genes either overlap or are separated by long (144-nt) or short (4- or 5-nt) IRs, we examined whether the activity of the EBOV polymerase is influenced by the diverse structures of the gene borders. Therefore, we analyzed the expression of genes separated by a short IR (NP-VP35), a long IR (VP30-VP24), or a gene overlap (VP35-VP40) during EBOV infection (Fig. 1A). We examined polyadenylated mRNAs transcribed in EBOV-infected cells by Northern hybridization, using pairs of RNA probes targeting the upstream and the downstream genes of each gene border (Fig. 1B). In addition to the monocistronic mRNAs, we detected transcripts that hybridized with both gene-specific probes and matched the estimated sizes of readthrough mRNAs (Fig. 1C to F). Transcriptional readthrough was observed irrespective of whether the gene border contained overlapping transcription signals or signals that were separated by a short or a long IR. Additional transcripts that might represent readthrough from neighboring genes were also detected, such as VP40-GP and GP-VP30 bicistronic mRNAs (Fig. 1D, band 7, and E and F, bands 11). Moreover, hybridization with a VP40-specific probe revealed an mRNA that likely represents a VP40-GP readthrough mRNA terminated at the editing site within the GP gene (Fig. 1D, band 6). The editing site was suggested to be sporadically recognized by the filoviral polymerase as a GE signal leading to polyadenylation and termination of mRNA synthesis (11). Interestingly, transcripts corresponding to a putative VP24-L mRNA, which would hybridize with the VP24-specific probe and be 8,398 nt in length [without the poly(A) tail], were not detected, suggesting that VP24-L readthrough mRNAs are not synthesized to any significant level (Fig. 1E and F).
Our results show that the EBOV polymerase occasionally fails to recognize the highly conserved GE signals during transcription. Even the close proximity of the GE to the GS signal in the overlapping gene borders did not inhibit readthrough transcription.
Transcription of the VP24 gene in EBOV-infected cells is primarily terminated at the first GE signal.
The lack of VP24-L readthrough mRNA might be due to the remarkable structure of the EBOV VP24 gene: it contains two consecutive GE signals (Fig. 2A). The first GE signal (VP24 GE-1) is separated from the GS signal of the L gene (L GS) by an IR of 4 nt, whereas the second GE signal (VP24 GE-2) directly overlaps the L GS signal (Fig. 2A). Since we were not able to detect VP24-L readthrough mRNA in EBOV-infected cells, we hypothesized that the two consecutive GE signals might be used to ensure transcription termination at the VP24 gene and, consequently, efficient reinitiation at the L GS signal. If this were the case, VP24 GE-1 would be preferentially used to terminate VP24 transcription, while VP24 GE-2 would serve as a backup GE signal to terminate VP24 GE-1 readthrough transcripts. We therefore identified the 3′ ends of VP24 transcripts from EBOV-infected cells. Of 13 analyzed clones, all contained VP24 GE-1, indicating that the EBOV polymerase primarily stops at the first GE signal it encounters during transcription of the VP24 gene (Fig. 2B).
The structure of the gene border affects downstream gene expression in a bicistronic minigenome.
To analyze if the long IR has unique regulatory functions, as implied by a previous study (12), we compared mRNA synthesis at the short NP-VP35 IR and the long VP30-VP24 IR. To assess the impact of IR length on transcriptional activity independently of the locations of the gene borders on the viral genome, the GS and GE signals, IRs, and parts of the adjacent untranslated regions (UTRs) of the two gene borders were cloned into Bicis (Fig. 3A). As reporter genes, we used a truncated and therefore inactive form of the CAT gene (ΔCAT), as well as an intact version of the CAT gene, separated by the gene borders of interest (5). While mRNAs transcribed from both genes can be detected with a single CAT-specific probe by Northern hybridization, only the intact CAT ORF in the second gene mediates reporter gene activity. Transcription of the Bicis was expected to result in the synthesis of three different transcripts that could be discriminated by their sizes (Fig. 3B and C).
FIG 3.

Structure of bicistronic minigenomes and transcribed mRNAs. (A) Scheme of Bicis containing the gene borders between the VP30 and VP24 genes (Bici VP30/VP24-wt IR 144nt) and the NP and VP35 genes (Bici NP/VP35-wt IR 5nt). The Bicis and sequences are shown in negative-sense orientation (not to scale). Regulatory sequences are illustrated as in Fig. 1A. The black boxes in Bici VP30/VP24-wt IR 144nt represent the ORFs of the reporter genes. The Bicis differ only in the gene border regions and the 3′- and 5′-UTR sequences flanking the gene borders. (B) Scheme of the three different mRNAs transcribed from a Bici template. (C) Calculated lengths of the mRNAs synthesized from the different Bicis, excluding the poly(A) tails.
We first assessed the transcriptional activity of the bicistronic templates in EBOV-infected cells. As shown by Northern blotting, all expected mRNAs (mRNA 1, mRNA 2, and readthrough mRNA) were detected for both Bicis, indicating that they were accepted as surrogate genomes by the EBOV polymerase (Fig. 4A). Analysis of mRNAs from cells infected with Bici-containing supernatants demonstrated that the Bicis were not only transcribed and replicated, but also packaged into infectious virions (Fig. 4B). In both experiments, we detected mRNA 2 levels lower than those of mRNA 1 for the Bici containing the long IR (Bici VP30/VP24-wt IR 144nt) (Fig. 4, lanes 2) compared to the Bici with the short IR (Bici NP/VP35-wt IR 5nt) (Fig. 4, lanes 4). This effect was also observed when the transcriptional activities of the two Bicis were assessed in an EBOV minigenome system, suggesting that the length of the IR might have a regulatory role for the expression of the gene located downstream (Fig. 4C).
FIG 4.

Transcriptional activities of Bicis containing a short or a long IR. (A) Transcripts synthesized from Bicis in EBOV-infected cells. Huh-T7 cells were transfected with plasmid DNA carrying the indicated Bicis, along with a T7 RNA polymerase expression plasmid. At 12 h posttransfection, the cells were infected with EBOV at an MOI of 5 or left uninfected. Poly(A)+ mRNA was harvested at 48 h p.i. and analyzed by Northern hybridization with CAT-specific riboprobes. As a positive control, monocistronic minigenome 3E-5E encoding CAT was transfected (4). As a negative control, nontransfected cells infected with EBOV were used (lane 7). Shown is a representative result of three independent experiments. A short exposure (top) and a long exposure (bottom) of the X-ray film revealing readthrough mRNA are shown. The mRNA products are labeled as for Fig. 3B. (B) Transcripts synthesized in cells infected with Bici-containing viral supernatants. Supernatants of cells transfected and infected as described for panel A were collected at 72 h p.i. and used to infect Huh-T7 cells. At 48 h p.i., poly(A)+ mRNA was purified and analyzed as described for panel A. The experiment was repeated twice with similar results. (C) Transcriptional activities of Bicis in an EBOV minigenome system. BSR-T7/5 cells were transfected with Bici-carrying plasmids, along with expression plasmids for the EBOV polymerase complex. Cells were harvested at 48 h posttransfection and subjected to Northern hybridization. Shown is a representative result of four independent experiments.
The long intergenic region is not essential for transcriptional activity, but its length affects the transcription reinitiation frequency.
The long IR between the VP30 and VP24 genes has previously been proposed to play an important role in the control of EBOV gene expression (12). To investigate the role of the 144-nt-long IR in transcriptional activity in more detail, we performed a mutational analysis of the IR using the EBOV minigenome system (Fig. 5A). First, we tested a mutant Bici VP30/VP24 in which the complete IR of 144 nt was deleted. The expression rates of the first gene (mRNA 1) were not substantially affected by the deletion of the IR, even though we detected a slight increase of readthrough mRNA (Fig. 5B). Furthermore, although mRNA 2 amounts were reduced, they were still detectable by Northern hybridization, while CAT activity, reflecting mRNA 2 expression, was decreased to 71% ± 20% of wild-type levels (Fig. 5C). These results indicate that the IR is not essential for transcription activity at this gene border but that it influences expression of the downstream gene.
FIG 5.

The length of the intergenic region regulates reinitiation efficiency at the VP30-VP24 gene border. (A) Scheme of mutant VP30/VP24 Bicis. Bici VP30/VP24-wt IR 144nt is depicted as in Fig. 3A. The complete sequence of the wild-type IR is shown underneath. The arrows mark nucleotides mutated to create a restriction site for cloning of Bici VP30/VP24 IR 293nt. The lines illustrate IR sequences in the respective truncation mutants. (B to G) Bicis containing the VP30-VP24 gene border were tested in the EBOV minigenome system and analyzed by Northern hybridization (B, D, F, and G) or by quantitative CAT assay (C and E). Shown are representative results of at least 3 independent experiments. The quantified CAT activity (+ standard deviation [SD]) shown in panels C and E was normalized to the Bici containing the wild-type gene border (wt IR 144nt; black bars). Expression levels from each Bici were compared to wild-type values using a one-sample t test (***, P < 0.001; **, P < 0.01; *, P < 0.05). (G) mRNA 2/mRNA 1 ratios of quantified Northern blots. mRNA ratios (+SD) of the truncation mutants (a representative blot is shown in panel F) were normalized to the Bici containing the wild-type gene border (wt IR 144nt; black bar). Expression levels for each Bici were compared to wild-type values using a one-sample t test (***, P < 0.001; **, P < 0.01; *, P < 0.05; no asterisk, not significant). (B and C) Deletion of the IR. (D and E) Elongation of the IR. (F and G) Truncation of the IR. The length of the IR is indicated for each mutant. CAM, chloramphenicol; ac CAM, acetylated CAM. Note that different exposure times were used for the blots shown in panels B and D, with the blot in panel D having the longer exposure time to show the presence of mRNA 2. (H) Analysis of the transcriptional activity of a Bici with an inhibitory IR in cells infected with EBOV. Huh-T7 cells were transfected with Bici DNA containing the wild-type VP30-VP24 gene border or a mutated gene border with a truncated IR (IR 20nt) and subsequently infected with EBOV as described in the legend to Fig. 4A. CAT-specific mRNA was analyzed by Northern hybridization. The experiment was repeated twice with the same outcome.
Since the VP30-VP24 IR is already significantly longer than most IRs of other NNS RNA viruses, we tested whether further extension of this region would still be tolerated by the EBOV polymerase. We elongated the 144-nt-long IR in Bici VP30/VP24 to 293 nt by amplifying and inserting the wt IR into a cloning site introduced into the central region of the IR (Fig. 5A). Although transcription reinitiation of the mutant was significantly decreased, mRNA 2 and corresponding CAT activities were still detectable, demonstrating that the EBOV polymerase is able to access a GS signal located several hundred nucleotides downstream of the GE signal, albeit with greatly reduced efficiency (Fig. 5D and E). As elongation of the IR led to a decrease in mRNA 2 expression, the question arose as to whether a shorter IR at this gene border would consequently result in increased transcription rates. To test this hypothesis, we truncated the long IR stepwise to 131, 111, 51, 46, 40, 30, 20, 10, and 5 nt, starting from the center of the IR and leaving the nucleotides directly adjacent to the transcription signals unchanged (Fig. 5A). We analyzed the reinitiation efficiencies of the different minigenomes at the VP24 GS by calculating the mRNA 2/mRNA 1 ratio. Due to the sequential nature of NNS RNA virus transcription, reinitiation at the GS signal of mRNA 2 occurs only after termination of mRNA 1. The reinitiation efficiency, therefore, reflects the frequency of reinitiation events at the downstream GS signal after transcription termination at the upstream GE signal. Surprisingly, successively shorter IRs did not result in a corresponding gradual increase of mRNA 2 expression, and a direct correlation between the length of the IR and the reinitiation efficiency was not observed (Fig. 5F and G). While shortening the IR to 5 nt did not substantially affect the mRNA 2/mRNA 1 ratio, the truncation to 10, 20, or 30 nt significantly inhibited reinitiation of the downstream gene. Compared to the wild type, mRNA 2/mRNA 1 ratios decreased to about 25% in minigenomes with an IR of 10, 20, or 30 nt (Fig. 5G).
To exclude the possibility that the observed inhibition of reinitiation was an artifact of the EBOV minigenome system, one of the constructs showing pronounced inhibition of reinitiation (Bici VP30/VP24 IR 20nt) was tested in EBOV-infected cells. Consistent with the results using the minigenome system, we observed a striking decrease in mRNA 2 levels transcribed from Bici VP30/VP24 IR 20nt, confirming that specific IR lengths inhibit reinitiation (Fig. 5H).
The length, but not the sequence, of the IR affects reinitiation.
Since our results revealed that IRs of specific lengths inhibit transcription reinitiation, we next analyzed if the observed inhibitory effect was sequence specific. The 10-nt-long IR of mutant Bici VP30/VP24 IR 10nt was partially or completely replaced (Fig. 6A). CAT assay and Northern hybridization consistently showed that neither partial nor complete replacement of the inhibitory IR resulted in a significant change in reinitiation efficiencies (Fig. 6B to D). This indicates that the length rather than the sequence of the IR mediates the observed reduction of downstream gene expression.
FIG 6.

The inhibitory effects of an IR of 10 nt are sequence independent. (A) Sequences of the mutant Bicis containing the VP30-VP24 gene border with an IR of 10 nt. Substituted nucleotides are underlined. The sequences are shown in negative-sense orientation. (B to D) Mutated Bicis were tested in the EBOV minigenome system. (B and C) Representative results of Northern blot analysis of three independent experiments and quantification of the mRNA 2/mRNA 1 ratio. (D) Analysis of CAT gene expression from mRNA 2 by quantitative CAT assay. The quantified CAT activity (D) or mRNA 2/mRNA 1 ratio of quantified Northern blots (C) (+ SD) was normalized to the Bici containing the wild-type gene border (wt IR 144nt; black bars). Expression levels from each Bici were compared to the wild-type value with a one-sample t test (**, P < 0.01; *, P < 0.05). The mRNA ratios and CAT activities of the Bicis containing an IR of 10 nt were not significantly different from each other, as confirmed by one-way ANOVA (P = 0.7537 and 0.2686, respectively).
Truncation of the long intergenic region in recombinant EBOVs inhibits viral replication.
The observation that truncation of the long IR from 144 nt to 5 nt did not significantly affect transcribed mRNA levels in a minigenome system was surprising, as it poses the question of why such a remarkably long IR would be retained in all filovirus species if the same level of transcriptional regulation could be achieved by an IR of 5 nt. To analyze the role of the long IR in the viral replication cycle, we generated recombinant EBOVs in which the IR was shortened to 5 or 10 nt, based on our findings using the minigenome system (Fig. 7A). Since shortening the long IR to 5 nt in the Bici mutants did not alter the transcription efficiency, we hypothesized that it also would not affect virus replication. In contrast, since an IR of 10 nt led to a dramatic drop in the transcription activity of the downstream gene in the minigenome system (Fig. 5F and G), we expected to observe an attenuated phenotype for recEBOV-IR 10nt caused by a substantial decrease in expression of the VP24 and L genes.
Of five attempts to rescue mutant recEBOV-IR 10nt, only two were successful. We did not observe any CPE before day 22 after passaging of the supernatant from the transfected cells, whereas recEBOV-IR 5nt showed CPE 8 days after supernatant passage and was rescued in all five attempts (Fig. 7B). Sequencing of the rescued viruses after p1 revealed that one of the recEBOV-IR 10nt isolates acquired a substitution in the trailer region (U18903 to C) and an insertion within a 5-A stretch in the 5′ UTR of the NP gene (A2851–2855). In the second rescued isolate, we found a substitution within the GP gene (U7388 to G, leading to a Ser451Arg mutation), suggesting that rescue of variant recEBOV-IR 10nt might not be possible without the accumulation of additional mutations. The Ser451Arg substitution within the GP ORF of the second isolate is located within the highly glycosylated and disordered mucin-like domain of GP1 that was shown to be dispensable for attachment and entry of pseudotyped viruses (39–41). Therefore, we decided to use this recEBOV-IR 10nt isolate in the subsequent experiments.
First, we examined whether the effects on transcriptional reinitiation observed for the Bicis would be recapitulated using the recombinant EBOVs. Huh7 cells were infected with recombinant mutant or wild-type virus, and the relative expression rate of VP24 mRNA (equivalent to mRNA 2) compared to VP30 mRNA (equivalent to mRNA 1) was determined by qRT-PCR at 24 and 48 h p.i. (Fig. 7A). While there was no substantial difference in the ratio of VP24/VP30 transcripts between wild-type virus and variant recEBOV-IR 5nt, relative VP24 expression levels decreased to about 40% with recEBOV-IR 10nt compared to wild-type virus at both time points (Fig. 7C). This is consistent with our data obtained with the Bicis (Fig. 5G). Next, we compared the replication kinetics of the recombinant viruses in Huh7 cells. Surprisingly, both viruses with a truncated IR showed a pronounced replication defect late in infection, starting at 4 days p.i. (Fig. 7D). At earlier time points p.i., we did not observe statistically significant differences in viral titers between the mutants and the wild-type virus. In addition, both mutants showed a small-focus phenotype compared to wild-type virus, confirming the observed late-growth defect (Fig. 7E). Mutant recEBOV-IR 10nt formed the smallest foci, followed by mutant recEBOV-IR 5nt, with intermediate-size foci, and the wild-type virus, with substantially larger foci. This indicates that shortening the long IR to 5 nt has a negative effect on viral replication, even if the genes located downstream are expressed at levels comparable to those of the wild-type virus.
Taken together, both viruses lacking the largest part of the long IR showed significant growth defects late in infection that could not be entirely attributed to a change in gene expression.
DISCUSSION
In this report, we examined the role of the diverse EBOV gene borders in the regulation of viral gene expression. Similar to other NNS RNA viruses with diverse gene borders, such as RSV, EBOV produces readily detectable amounts of readthrough mRNAs during infection, suggesting that gene border diversity might hamper the recognition of the GE signals by the viral polymerase (25, 42–44). This hypothesis is supported by data obtained for VSVIndiana, a virus with conserved IR sequences in which all gene borders but one direct very low readthrough levels, indicating that the gene borders are optimized for highly efficient transcription termination (6, 22, 45). Readthrough transcription in EBOV-infected cells was observed at IRs, as well as gene overlaps, indicating that the filovirus-specific overlaps are not used as regulatory signals to prevent polymerase readthrough. However, as our analysis of readthrough transcripts is exclusively qualitative, further studies are needed to address whether termination efficiencies are modulated by the different gene borders. Notably, our data suggest that the presence of two consecutive GE signals at the last gene border between the VP24 and L genes inhibits readthrough transcription. This is consistent with the previous observation that mutation of either VP24 GE signal in a minigenome resulted in a significant decrease in downstream reporter expression, an effect that would be expected when readthrough levels are increased (12). Since our data show that the first GE signal is primarily used for mRNA termination during EBOV infection, we propose that the second GE signal serves as a backup mechanism to ensure effective termination of VP24 transcription in the rare event of polymerase readthrough at the first GE signal, thereby facilitating efficient expression of the most promoter-distal gene encoding L.
We further focused our analysis on unraveling the role of the 144-nt-long IR that separates the EBOV VP30 and VP24 genes and found that considerable changes in this IR, including deletion, elongation, substitution, and truncation, were tolerated without abrogating downstream gene expression, although some of the mutations led to a significant decrease in transcription reinitiation. Along the same lines, stepwise shortening of the long IR did not lead to successively enhanced downstream gene expression, indicating that there is no linear correlation between the length of the IR and the reinitiation rate at the following gene. Similar observations have been reported for RSV and SV5, suggesting that the variable IRs are not major regulators of transcription reinitiation (46–50). In contrast to EBOV and RSV, the highly conserved IRs in VSVIndiana and SeV play an essential role in both transcription termination and reinitiation (8, 45, 51–55). Furthermore, when analyzed independently of their role in termination, nucleotides within the GE signal and the IR of SeV and VSVIndiana were shown to be required for optimal reinitiation at the GS signal, indicating an overlap of function of the signals at the conserved gene borders (51–54). Deletion of the IR in a VSVIndiana Bici completely abrogated reinitiation (45). This was also the case for a RABV Bici lacking an IR, even though RABV contains variable IRs, similar to EBOV (56). The contrasting results might be explained by differences within the GS signals. While RABV, VSV, and SeV GS signals begin with a uridine residue and the IR might be essential to separate this signal from the upstream U tract of the GE, the GS signals of EBOV and RSV start with a cytidine, possibly facilitating discrimination of the adjoining transcription signals in the absence of an IR. Together, these data reflect fundamental differences in the control of polymerase behavior by cis-acting sequences between viruses with conserved and variable gene borders and suggest an important role of conserved IRs in transcription regulation, while the function of variable IRs remains less clear.
While there was no profound change in transcription reinitiation with most truncated IRs, shortening the 144-nt-long IR to 10, 20, or 30 nt significantly reduced expression of the downstream gene. Our results indicate that this inhibitory effect was caused by the IR length rather than the sequence, as reinitiation rates remained low even when the inhibitory IR of 10 nt was partially or completely replaced. It has been reported for VSV that the polymerase inefficiently reinitiates transcription at suboptimal GS signals within extended IRs, a process that transits the polymerase past the genuine GS signal, leading to reduced transcription levels of the downstream gene (8, 51, 53, 55). Although a similar scenario to explain the inhibitory effect of the 20- and 30-nt-long IRs cannot be ruled out, it seems unlikely, as the long IR was truncated by successive deletions from the center, preserving the GE- and GS-proximal sequences across the mutants and allowing only minimal sequence variations at the junction sites of the truncations (Fig. 5A).
An IR length-specific inhibition of transcription reinitiation is unique to EBOV and, to our knowledge, has not yet been observed for any other NNS RNA virus. For these, either a direct relationship between IR length and reinitiation ability has been reported (8, 51–54, 57, 58), or a large range of IR lengths was tolerated with no or only minimal impact on transcription of the following gene (46, 47, 49). Length-dependent mechanisms causing the observed inhibition might be (i) the involvement of secondary structures adopted by the IR; (ii) a shift in the NP encapsidation phase of the GS signal, leading to failure of the viral polymerase to recognize the GS signal; or (iii) spatial constraints inhibiting recognition of the GS signal.
It has been suggested in a previous study that secondary-structure formation in the IR might play a regulatory role (12). However, we did not observe any obvious correlation between predicted secondary structures and the function of the IRs (data not shown).
Another possible explanation for reduced reinitiation mediated by specific IR lengths is that EBOV GS signals are recognized with respect to a certain encapsidation phase imposed by the NP molecules. Phasing-dependent recognition of cis-acting transcription signals was postulated for paramyxoviruses that follow the “rule of six,” i.e., whose genome lengths have to be divisible by six to be efficiently replicated (13, 59), and was shown to influence reinitiation in SeV (60). Although none of the sequenced filoviral genomes are multiples of six, cryo-electron microscopy studies of EBOV nucleocapsids suggest an encapsidation unit of 6 nt per NP molecule (61). In addition, the bipartite EBOV replication promoter depends on a hexamer phase (62, 63). However, there is no indication of conserved phasing of the GS signals within the EBOV genome. As nonfunctional and functional IRs do not exhibit distinct NP phases in the tested Bicis and recombinant EBOVs, either, it seems unlikely that NP phasing of the GS signal is involved in the observed inhibitory effects.
Finally, it is also possible that distances of 10, 20, or 30 nt between the GE and the following GS signal are sterically unfavorable for polymerase recognition. As EBOV L might form oligomers (64), it is conceivable that transcription signals separated by short IRs are recognized by one L molecule, whereas at gene borders containing a long IR, one molecule recognizes the GE signal while the other recognizes the GS signal, a process that might be inhibited by specific IR lengths. However, further studies are needed to gain a more detailed insight into the structures of both the NP-RNA template and the filovirus polymerase engaged in the process of transcription. Taken together, the mechanism resulting in the observed inhibition of downstream gene expression caused by specific IR lengths remains elusive.
While the initial comparison of Bicis with a short IR (5 nt; NP-VP35) and with a long IR (144 nt; VP30-VP24) resulted in lower mRNA 2 expression rates for the latter construct, mutational analysis revealed that the long IR was not sufficient to explain this effect. Truncation of the long IR to 5 nt did not significantly increase mRNA 2 expression levels. Thus, the bordering UTRs are likely involved in the regulation of mRNA levels at these two gene borders. This is in accordance with previous findings showing that the UTRs of EBOV genes affect reporter gene expression (12). Our observation that deletion of the long IR in a Bici affects downstream but not upstream gene expression is in contrast to a previous study, in which deletion of this IR nearly abrogated the reporter activities of both the upstream and downstream genes (12). The conflicting results might be due to variations in the included bordering UTRs of the Bici constructs. A regulatory role in the transcription of genes and translation of mRNAs has also been reported for the long UTRs in the genomes of other NNS RNA viruses (65–69). It will therefore be of interest to determine whether the observed inhibitory effects of certain IR lengths are specific to the gene border containing the long IR or if they can be recapitulated at other EBOV gene borders.
A recombinant EBOV in which the VP30-VP24 IR was shortened to 10 nt not only showed reduced expression of VP24 mRNA, but also exhibited a small-focus phenotype and was substantially inhibited in viral growth. In contrast to recEBOV-IR 5nt, rescue efficiency was low, and all recovered recEBOV-IR 10nt viruses contained additional mutations. We speculate that these might have been acquired to overcome the severe loss of fitness associated with the inhibitory IR. The filovirus-unique protein VP24 is an essential inhibitor of interferon signaling in EBOV, negatively regulates replication and transcription, and is involved in the maturation of functional nucleocapsids (reviewed in references 3 and 70). It has also been shown to be essential for genome packaging and particle infectivity (71, 72). Rescue of VP24-deficient EBOV by expression of VP24 in trans has not yet been successful, indicating that subtle regulation of VP24 expression might be crucial (73). Downregulation of VP24 expression in recEBOV-IR 10nt is therefore expected to negatively affect virus replication. Due to the sequential nature of transcription, expression levels of L in cells infected with the virus would also be reduced by the introduced mutation, which together might account for the observed attenuation.
Unexpectedly, we also found a pronounced reduction in viral titers and small-focus morphology with the recEBOV-IR 5nt mutant, although transcription of VP24 was not substantially affected by the IR truncation. Sequencing of this mutant throughout the experiment showed that it developed a mutation within the editing site of the GP gene from 7 U to 8 U. In a previous study, this mutation was shown to lead to faster replication kinetics and larger plaque morphology (74). Therefore, it is assumed that the slower replication kinetics and the small-focus phenotype of recEBOV-IR 5nt can be attributed to the truncation of the IR. The observed effects are expected to be even more severe without the additional mutation in the GP editing site superimposed.
Similar to our observations, changes in the lengths of specific IRs in recombinant RSV and NDV led to reduced plaque sizes and, in the case of NDV, to decreased pathogenicity (46, 58). While these effects were attributed to increasing genome length in the case of RSV and changes in the transcription level of the downstream gene for NDV, we observed unaffected transcription levels upon truncation of the IR to 5 nt. This suggests that the long IR might be involved not only in EBOV transcriptional regulation, but also in later steps of the replication cycle essential for viral spread. Sequences residing within the long IR might also be required to ensure correct encapsidation by the nucleoprotein and the formation of fully functional nucleocapsids. The long IR is not only present in the genome and antigenome, it is also expressed as part of the VP30-VP24 readthrough mRNA in infected cells (Fig. 1E and F). Since readthrough mRNAs are not encapsidated, they might serve as regulatory RNAs or could give rise to small regulatory RNAs comprising IR sequences. There is growing evidence for virus-derived noncoding and small RNAs that are involved in regulatory processes (75–78). However, this concept remains speculative at this point, and further analyses are required to pursue this intriguing possibility.
Taken together, the present work corroborates the notion that regulation of RNA synthesis in the highly pathogenic filoviruses is more sophisticated than previously acknowledged and differs not only from the NNS RNA prototype viruses VSVIndiana and SeV, but also from other viruses with variable gene borders, including RSV, RABV, and NDV. Specific IR lengths not found naturally within any filovirus IR substantially inhibit downstream gene expression. This observation might help to develop strategies to modulate viral gene expression rates, allowing the rational design of recombinant attenuated EBOVs. Our findings also provide evidence that the exceptionally long IR within the filovirus genomes might be essential for late steps in the viral replication cycle, in addition to transcriptional regulation. This is, to our knowledge, unprecedented among NNS RNA viruses.
ACKNOWLEDGMENTS
We are indebted to H. Feldmann (LV, NIAID, NIH), F. Feldmann (RMVB, NIAID, NIH), and members of the Laboratory of Virology for training of K.B. in the high-containment laboratory and support for conducting BSL-4 experiments at the Rocky Mountain Laboratories, NAID, NIH, Hamilton, MT. We thank T. Takimoto, Y. Kawaoka, K. K. Conzelmann, and V. Gaussmüller for providing material. We are grateful to J. Pacheco and J. Taylor (BU) and L. Banadyga (LV, NIAID, NIH) for help with experiments and excellent technical assistance and to A. Hume (BU) for critically reading the manuscript.
This work was supported by NIH grants AI057159 (New England Regional Center of Excellence-Kasper, subaward 149047-0743) and U01-AI082954 (to E.M.), by startup funds from Boston University (to E.M.), by funds from the Deutsche Forschungsgemeinschaft (SFB 535; to E.M.) and the Cusanuswerk (to K.B.), and by the Intramural Research Program of the NIH, NIAID (to Y.T., T.H., and H.E.).
Footnotes
Published ahead of print 20 August 2014
REFERENCES
- 1.Feldmann H, Geisbert TW. 2011. Ebola haemorrhagic fever. Lancet 377:849–862. 10.1016/S0140-6736(10)60667-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Whelan SPJ, Barr JN, Wertz GW. 2004. Transcription and replication of nonsegmented negative-strand RNA viruses. Curr. Top. Microbiol. Immunol. 283:61–119. [DOI] [PubMed] [Google Scholar]
- 3.Mühlberger E. 2007. Filovirus replication and transcription. Future Virol. 2:205–215. 10.2217/17460794.2.2.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mühlberger E, Weik M, Volchkov VE, Klenk HD, Becker S. 1999. Comparison of the transcription and replication strategies of Marburg virus and Ebola virus by using artificial replication systems. J. Virol. 73:2333–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weik M, Modrof J, Klenk H-D, Becker S, Mühlberger E. 2002. Ebola virus VP30-mediated transcription is regulated by RNA secondary structure formation. J. Virol. 76:8532–8539. 10.1128/JVI.76.17.8532-8539.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iverson LE, Rose JK. 1981. Localized attenuation and discontinuous synthesis during vesicular stomatitis virus transcription. Cell 23:477–484. 10.1016/0092-8674(81)90143-4. [DOI] [PubMed] [Google Scholar]
- 7.Shabman RS, Hoenen T, Groseth A, Jabado O, Binning JM, Amarasinghe GK, Feldmann H, Basler CF. 2013. An upstream open reading frame modulates Ebola virus polymerase translation and virus replication. PLoS Pathog. 9:e1003147. 10.1371/journal.ppat.1003147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stillman EA, Whitt MA. 1998. The length and sequence composition of vesicular stomatitis virus intergenic regions affect mRNA levels and the site of transcript initiation. J. Virol. 72:5565–5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schubert M, Keene JD, Herman RC, Lazzarini RA. 1980. Site on the vesicular stomatitis virus genome specifying polyadenylation and the end of the L gene mRNA. J. Virol. 34:550–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barr JN, Whelan SPJ, Wertz GW. 1997. cis-Acting signals involved in termination of vesicular stomatitis virus mRNA synthesis include the conserved AUAC and the U7 signal for polyadenylation. J. Virol. 71:8718–8725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sanchez A, Kiley MP, Holloway BP, Auperin DD. 1993. Sequence analysis of the Ebola virus genome: organization, genetic elements, and comparison with the genome of Marburg virus. Virus Res. 29:215–240. 10.1016/0168-1702(93)90063-S. [DOI] [PubMed] [Google Scholar]
- 12.Neumann G, Watanabe S, Kawaoka Y. 2009. Characterization of Ebolavirus regulatory genomic regions. Virus Res. 144:1–7. 10.1016/j.virusres.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kolakofsky D, Pelet T, Garcin D, Hausmann S, Curran J, Roux L. 1998. Paramyxovirus RNA synthesis and the requirement for hexamer genome length: the rule of six revisited. J. Virol. 72:891–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feldmann H, Mühlberger E, Randolf A, Will C, Kiley MP, Sanchez A, Klenk H-D. 1992. Marburg virus, a filovirus: messenger RNAs, gene order, and regulatory elements of the replication cycle. Virus Res. 24:1–19. 10.1016/0168-1702(92)90027-7. [DOI] [PubMed] [Google Scholar]
- 15.Piyaratna R, Tollefson SJ, Williams JV. 2011. Genomic analysis of four human metapneumovirus prototypes. Virus Res. 160:200–205. 10.1016/j.virusres.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Collins PL, Olmsted RA, Spriggs MK, Johnson PR, Buckler-White AJ. 1987. Gene overlap and site-specific attenuation of transcription of the viral polymerase L gene of human respiratory syncytial virus. Proc. Natl. Acad. Sci. U. S. A. 84:5134–5138. 10.1073/pnas.84.15.5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McWilliam SM, Kongsuwan K, Cowley JA, Byrne KA, Walker PJ. 1997. Genome organization and transcription strategy in the complex GNS-L intergenic region of bovine ephemeral fever rhabdovirus. J. Gen. Virol. 78:1309–1317. [DOI] [PubMed] [Google Scholar]
- 18.Teninges D, Bras F, Dezélée S. 1993. Genome organization of the sigma rhabdovirus: six genes and a gene overlap. Virology 193:1018–1023. 10.1006/viro.1993.1219. [DOI] [PubMed] [Google Scholar]
- 19.Wang Y, McWilliam SM, Cowley JA, Walker PJ. 1994. Complex genome organization in the GNS-L intergenic region of Adelaide River rhabdovirus. Virology 203:63–72. 10.1006/viro.1994.1455. [DOI] [PubMed] [Google Scholar]
- 20.Schneemann A, Schneider PA, Kim S, Lipkin WI. 1994. Identification of signal sequences that control transcription of Borna disease virus, a nonsegmented, negative-strand RNA virus. J. Virol. 68:6514–6522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gupta KC, Kingsbury DW. 1985. Polytranscripts of Sendai virus do not contain intervening polyadenylate sequences. Virology 141:102–109. 10.1016/0042-6822(85)90186-2. [DOI] [PubMed] [Google Scholar]
- 22.Masters PS, Samuel CE. 1984. Detection of in vivo synthesis of polycistronic mRNAs of vesicular stomatitis virus. Virology 134:277–286. 10.1016/0042-6822(84)90297-6. [DOI] [PubMed] [Google Scholar]
- 23.Tsurudome M, Bando H, Kawano M, Matsumura H, Komada H, Nishio M, Ito Y. 1991. Transcripts of simian virus-41 (Sv41) matrix gene are exclusively dicistronic with the fusion gene which is also transcribed as a monocistron. Virology 184:93–100. 10.1016/0042-6822(91)90825-V. [DOI] [PubMed] [Google Scholar]
- 24.Rassa JC, Parks GD. 1998. Molecular basis for naturally occurring elevated readthrough transcription across the M-F junction of the paramyxovirus SV5. Virology 247:274–286. 10.1006/viro.1998.9266. [DOI] [PubMed] [Google Scholar]
- 25.Collins PL, Wertz GW. 1983. cDNA cloning and transcriptional mapping of nine polyadenylylated RNAs encoded by the genome of human respiratory syncytial virus. Proc. Natl. Acad. Sci. U. S. A. 80:3208–3212. 10.1073/pnas.80.11.3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cattaneo R, Rebmann G, Schmid A, Baczko K, ter Meulen V, Billeter MA. 1987. Altered transcription of a defective measles virus genome derived from a diseased human brain. EMBO J. 6:681–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bousse T, Takimoto T, Murti KG, Portner A. 1997. Elevated expression of the human parainfluenza virus type 1 F gene downregulates HN expression. Virology 232:44–52. 10.1006/viro.1997.8524. [DOI] [PubMed] [Google Scholar]
- 28.Spriggs MK, Collins PL. 1986. Human parainfluenza virus type 3: messenger RNAs, polypeptide coding assignments, intergenic sequences, and genetic map. J. Virol. 59:646–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wong TC, Hirano A. 1987. Structure and function of bicistronic RNA encoding the phosphoprotein and matrix protein of measles virus. J. Virol. 61:584–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barr JN, Whelan SPJ, Wertz GW. 2002. Transcriptional control of the RNA-dependent RNA polymerase of vesicular stomatitis virus. Biochim. Biophys. Acta 1577:337–353. 10.1016/S0167-4781(02)00462-1. [DOI] [PubMed] [Google Scholar]
- 31.Schultz DE, Honda M, Whetter LE, McKnight KL, Lemon SM. 1996. Mutations within the 5′ nontranslated RNA of cell culture-adapted hepatitis A virus which enhance cap-independent translation in cultured African green monkey kidney cells. J. Virol. 70:1041–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Buchholz UJ, Finke S, Conzelmann K-K. 1999. Generation of bovine respiratory syncytial virus (BRSV) from cDNA: BRSV NS2 is not essential for virus replication in tissue culture, and the human RSV leader region acts as a functional BRSV genome promoter. J. Virol. 73:251–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mühlberger E, Lötfering B, Klenk HD, Becker S. 1998. Three of the four nucleocapsid proteins of Marburg virus, NP, VP35, and L, are sufficient to mediate replication and transcription of Marburg virus-specific monocistronic minigenomes. J. Virol. 72:8756–8764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yanai H, Hayashi Y, Watanabe Y, Ohtaki N, Kobayashi T, Nozaki Y, Ikuta K, Tomonaga K. 2006. Development of a novel Borna disease virus reverse genetics system using RNA polymerase II promoter and SV40 nuclear import signal. Microbes Infect. 8:1522–1529. 10.1016/j.micinf.2006.01.010. [DOI] [PubMed] [Google Scholar]
- 35.Ghanem A, Kern A, Conzelmann K-K. 2012. Significantly improved rescue of rabies virus from cDNA plasmids. Eur. J. Cell Biol. 91:10–16. 10.1016/j.ejcb.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 36.Szewczyk E, Nayak T, Oakley CE, Edgerton H, Xiong Y, Taheri-Talesh N, Osmani SA, Oakley BR, Oakley B. 2006. Fusion PCR and gene targeting in Aspergillus nidulans. Nat. Protoc. 1:3111–3120. 10.1038/nprot.2006.405. [DOI] [PubMed] [Google Scholar]
- 37.Hoenen T, Shabman RS, Groseth A, Herwig A, Weber M, Schudt G, Dolnik O, Basler CF, Becker S, Feldmann H. 2012. Inclusion bodies are a site of ebolavirus replication. J. Virol. 86:11779–11788. 10.1128/JVI.01525-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ebihara H, Takada A, Kobasa D, Jones S, Neumann G, Theriault S, Bray M, Feldmann H, Kawaoka Y. 2006. Molecular determinants of Ebola virus virulence in mice. PLoS Pathog. 2:e73. 10.1371/journal.ppat.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jeffers SA, Sanders DA, Sanchez A. 2002. Covalent modifications of the Ebola virus glycoprotein. J. Virol. 76:12463–12472. 10.1128/JVI.76.24.12463-12472.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Manicassamy B, Wang J, Jiang H, Rong L. 2005. Comprehensive analysis of Ebola virus GP1 in viral entry. J. Virol. 79:4793–4805. 10.1128/JVI.79.8.4793-4805.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang Z-Y, Duckers HJ, Sullivan NJ, Sanchez A, Nabel EG, Nabel GJ. 2000. Identification of the Ebola virus glycoprotein as the main viral determinant of vascular cell cytotoxicity and injury. Nat. Med. 6:886–889. 10.1038/78645. [DOI] [PubMed] [Google Scholar]
- 42.Fearns R, Collins PL. 1999. Role of the M2-1 transcription antitermination protein of respiratory syncytial virus in sequential transcription. J. Virol. 73:5852–5864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hardy RW, Wertz GW. 1998. The product of the respiratory syncytial virus M2 gene ORF1 enhances readthrough of intergenic junctions during viral transcription. J. Virol. 72:520–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hardy RW, Harmon SB, Wertz GW. 1999. Diverse gene junctions of respiratory syncytial virus modulate the efficiency of transcription termination and respond differently to M2-mediated antitermination. J. Virol. 73:170–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barr JN, Whelan SPJ, Wertz GW. 1997. Role of the intergenic dinucleotide in vesicular stomatitis virus RNA transcription. J. Virol. 71:1794–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bukreyev A, Murphy BR, Collins PL. 2000. Respiratory syncytial virus can tolerate an intergenic sequence of at least 160 nucleotides with little effect on transcription or replication in vitro and in vivo. J. Virol. 74:11017–11026. 10.1128/JVI.74.23.11017-11026.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kuo L, Fearns R, Collins PL. 1996. The structurally diverse intergenic regions of respiratory syncytial virus do not modulate sequential transcription by a dicistronic minigenome. J. Virol. 70:6143–6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moudy RM, Sullender WM, Wertz GW. 2004. Variations in intergenic region sequences of human respiratory syncytial virus clinical isolates: analysis of effects on transcriptional regulation. Virology 327:121–133. 10.1016/j.virol.2004.06.013. [DOI] [PubMed] [Google Scholar]
- 49.Rassa JC, Parks GD. 1999. Highly diverse intergenic regions of the paramyxovirus simian virus 5 cooperate with the gene end U tract in viral transcription termination and can influence reinitiation at a downstream gene. J. Virol. 73:3904–3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.He B, Lamb RA. 1999. Effect of inserting paramyxovirus simian virus 5 gene junctions at the HN/L gene junction: analysis of accumulation of mRNAs transcribed from rescued viable viruses. J. Virol. 73:6228–6234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barr JN, Tang X, Hinzman EE, Shen R, Wertz GW. 2008. The VSV polymerase can initiate at mRNA start sites located either up or downstream of a transcription termination signal but size of the intervening intergenic region affects efficiency of initiation. Virology 374:361–370. 10.1016/j.virol.2007.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hinzman EE, Barr JN, Wertz GW. 2008. Selection for gene junction sequences important for VSV transcription. Virology 380:379–387. 10.1016/j.virol.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hinzman EE, Barr JN, Wertz GW. 2002. Identification of an upstream sequence element required for vesicular stomatitis virus mRNA transcription. J. Virol. 76:7632–7641. 10.1128/JVI.76.15.7632-7641.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Plattet P, Strahle L, le Mercier P, Hausmann S, Garcin D, Kolakofsky D. 2007. Sendai virus RNA polymerase scanning for mRNA start sites at gene junctions. Virology 362:411–420. 10.1016/j.virol.2006.12.033. [DOI] [PubMed] [Google Scholar]
- 55.Stillman EA, Whitt MA. 1997. Mutational analyses of the intergenic dinucleotide and the transcriptional start sequence of vesicular stomatitis virus (VSV) define sequences required for efficient termination and initiation of VSV transcripts. J. Virol. 71:2127–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Finke S, Conzelmann KK. 1997. Ambisense gene expression from recombinant rabies virus: random packaging of positive- and negative-strand ribonucleoprotein complexes into rabies virions. J. Virol. 71:7281–7288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Finke S, Cox JH, Conzelmann KK. 2000. Differential transcription attenuation of rabies virus genes by intergenic regions: generation of recombinant viruses overexpressing the polymerase gene. J. Virol. 74:7261–7269. 10.1128/JVI.74.16.7261-7269.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yan Y, Samal SK. 2008. Role of intergenic sequences in Newcastle disease virus RNA transcription and pathogenesis. J. Virol. 82:1323–1331. 10.1128/JVI.01989-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kolakofsky D, Roux L, Garcin D, Ruigrok RWH. 2005. Paramyxovirus mRNA editing, the “rule of six” and error catastrophe: a hypothesis. J. Gen. Virol. 86:1869–1877. 10.1099/vir.0.80986-0. [DOI] [PubMed] [Google Scholar]
- 60.Cordey S, Roux L. 2007. Further characterization of a paramyxovirus transcription initiation signal: search for required nucleotides upstream and importance of the N phase context. J. Gen. Virol. 88:1555–1564. 10.1099/vir.0.82701-0. [DOI] [PubMed] [Google Scholar]
- 61.Bharat TAM, Noda T, Riches JD, Kraehling V, Kolesnikova L, Becker S, Kawaoka Y, Briggs JAG. 2012. Structural dissection of Ebola virus and its assembly determinants using cryo-electron tomography. Proc. Natl. Acad. Sci. U. S. A. 109:4275–4280. 10.1073/pnas.1120453109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Weik M, Enterlein S, Schlenz K, Mühlberger E. 2005. The Ebola virus genomic replication promoter is bipartite and follows the rule of six. J. Virol. 79:10660–10671. 10.1128/JVI.79.16.10660-10671.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Enterlein S, Schmidt KM, Schümann M, Conrad D, Krähling V, Olejnik J, Mühlberger E. 2009. The Marburg virus 3′ noncoding region structurally and functionally differs from that of Ebola virus. J. Virol. 83:4508–4519. 10.1128/JVI.02429-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Trunschke M, Conrad D, Enterlein S, Olejnik J, Brauburger K, Mühlberger E. 2013. The L-VP35 and L-L interaction domains reside in the amino terminus of the Ebola virus L protein and are potential targets for antivirals. Virology 441:135–145. 10.1016/j.virol.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Anderson DE, Castan A, Bisaillon M, von Messling V. 2012. Elements in the canine distemper virus M 3′ UTR contribute to control of replication efficiency and virulence. PLoS One 7:e31561. 10.1371/journal.pone.0031561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bousse T, Matrosovich T, Portner A, Kato A, Nagai Y, Takimoto T. 2002. The long noncoding region of the human parainfluenza virus type 1 F gene contributes to the read-through transcription at the M-F gene junction. J. Virol. 76:8244–8251. 10.1128/JVI.76.16.8244-8251.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hino K, Sato H, Sugai A, Kato M, Yoneda M, Kai C. 2013. Downregulation of Nipah virus N mRNA occurs through interaction between its 3′ untranslated region and hnRNP D. J. Virol. 87:6582–6588. 10.1128/JVI.02495-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Poenisch M, Wille S, Staeheli P, Schneider U. 2008. Polymerase read-through at the first transcription termination site contributes to regulation of Borna disease virus gene expression. J. Virol. 82:9537–9545. 10.1128/JVI.00639-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rennick LJ, Duprex WP, Rima BK. 2007. Measles virus minigenomes encoding two autofluorescent proteins reveal cell-to-cell variation in reporter expression dependent on viral sequences between the transcription units. J. Gen. Virol. 88:2710–2718. 10.1099/vir.0.83106-0. [DOI] [PubMed] [Google Scholar]
- 70.Zhang APP, Abelson DM, Bornholdt ZA, Liu T, Woods VL, Saphire EO. 2012. The ebolavirus VP24 interferon antagonist: know your enemy. Virulence 3:440–445. 10.4161/viru.21302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hoenen T, Groseth A, Kolesnikova L, Theriault S, Ebihara H, Hartlieb B, Bamberg S, Feldmann H, Ströher U, Becker S. 2006. Infection of naive target cells with virus-like particles: implications for the function of Ebola virus VP24. J. Virol. 80:7260–7264. 10.1128/JVI.00051-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Watt A, Moukambi F, Banadyga L, Groseth A, Callison J, Herwig A, Ebihara H, Feldmann H, Hoenen T. 2014. A novel life cycle modeling system for Ebola virus shows a genome length-dependent role of VP24 on virus infectivity. J. Virol. 88:10511–10524. 10.1128/JVI.01272-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mateo M, Carbonnelle C, Martínez MJ, Reynard O, Page A, Volchkova VA, Volchkov VE. 2011. Knockdown of Ebola virus VP24 impairs viral nucleocapsid assembly and prevents virus replication. J. Infect. Dis. 204(Suppl 3):S892–S896. 10.1093/infdis/jir311. [DOI] [PubMed] [Google Scholar]
- 74.Volchkova VA, Dolnik O, Martínez MJ, Reynard O, Volchkov VE. 2011. Genomic RNA editing and its impact on Ebola virus adaptation during serial passages in cell culture and infection of guinea pigs. J. Infect. Dis. 204(Suppl3):S941–S946. 10.1093/infdis/jir321. [DOI] [PubMed] [Google Scholar]
- 75.Roby JA, Pijlman GP, Wilusz J, Khromykh AA. 2014. Noncoding subgenomic flavivirus RNA: multiple functions in West Nile virus pathogenesis and modulation of host responses. Viruses 6:404–427. 10.3390/v6020404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Iseni F, Garcin D, Nishio M, Kedersha N, Anderson P, Kolakofsky D. 2002. Sendai virus trailer RNA binds TIAR, a cellular protein involved in virus-induced apoptosis. EMBO J. 21:5141–5150. 10.1093/emboj/cdf513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Perez JT, Zlatev I, Aggarwal S, Subramanian S, Sachidanandam R, Kim B, Manoharan M, ten Oever BR. 2012. A small-RNA enhancer of viral polymerase activity. J. Virol. 86:13475–13485. 10.1128/JVI.02295-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tremaglio CZ, Noton SL, Deflubé LR, Fearns R. 2013. Respiratory syncytial virus polymerase can initiate transcription from position 3 of the leader promoter. J. Virol. 87:3196–3207. 10.1128/JVI.02862-12. [DOI] [PMC free article] [PubMed] [Google Scholar]