

ABSTRACT
Human herpesvirus 6B (HHV-6B) is a ubiquitous pathogen causing lifelong infections in approximately 95% of humans worldwide. To persist within its host, HHV-6B has developed several immune evasion mechanisms, such as latency, during which minimal proteins are expressed, and the ability to disturb innate and adaptive immune responses. The primary cellular targets of HHV-6B are CD4+ T cells. Previous studies by Flamand et al. (L. Flamand, J. Gosselin, I. Stefanescu, D. Ablashi, and J. Menezes, Blood 85:1263–1271, 1995) reported on the capacity of HHV-6A as well as UV-irradiated HHV-6A to inhibit interleukin-2 (IL-2) synthesis in CD4+ lymphocytes, suggesting that viral structural components could be responsible for this effect. In the present study, we identified the HHV-6B U54 tegument protein (U54) as being capable of inhibiting IL-2 expression. U54 binds the calcineurin (CaN) phosphatase enzyme, causing improper dephosphorylation and nuclear translocation of NFAT (nuclear factor of activated T cells) proteins, resulting in suboptimal IL-2 gene transcription. The U54 GISIT motif (amino acids 293 to 297), analogous to the NFAT PXIXIT motif, contributed to the inhibition of NFAT activation.
IMPORTANCE Human herpesvirus 6A (HHV-6A) and HHV-6B are associated with an increasing number of pathologies. These viruses have developed strategies to avoid the immune response allowing them to persist in the host. Several studies have illustrated mechanisms by which HHV-6A and HHV-6B are able to disrupt host defenses (reviewed in L. Dagna, J. C. Pritchett, and P. Lusso, Future Virol. 8:273–287, 2013, doi:10.2217/fvl.13.7). Previous work informed us that HHV-6A is able to suppress synthesis of interleukin-2 (IL-2), a key immune growth factor essential for adequate T lymphocyte proliferation and expansion. We obtained evidence that HHV-6B also inhibits IL-2 gene expression and identified the mechanisms by which it does so. Our work led us to the identification of U54, a virion-associated tegument protein, as being responsible for suppression of IL-2. Consequently, we have identified HHV-6B U54 protein as playing a role in immune evasion. These results further contribute to our understanding of HHV-6 interactions with its human host and the efforts deployed to ensure its long-term persistence.
INTRODUCTION
Herpesviruses are among the most successful viral pathogens infecting humans. Following the primary infection, a lifelong relationship is established, with the virus residing in a state of dormancy (latency) with episodic reactivation, which can lead to severe complications depending on the immune status of the individual. Two human herpesviruses increasingly recognized as medically relevant pathogens are human herpesvirus 6A (HHV-6A) and HHV-6B. Due to biological, epidemiological, and disease association differences (1), the International Committee on Taxonomy of Viruses recently classified HHV-6A and HHV-6B, belonging to the subfamily Betaherpesvirinae, as two distinct viruses (2, 3). HHV-6A was first isolated from AIDS patients and subjects with lymphoproliferative disorders (4), while HHV-6B was first isolated from a healthy subject from Zaire (5). HHV-6B is the etiological agent of the sixth infantile eruptive disease, known as roseola (6), and is an increasingly recognized problem in hematopoietic stem cell transplantation, where HHV-6B reactivation is often linked with encephalitis, especially when the source of stem cells is cord blood (7). The epidemiology and disease associations of HHV-6A are less clear.
HHV-6A and HHV-6B primary target cells are CD4+ T lymphocytes (8, 9). The immunomodulatory impacts of HHV-6 infection on T cell functions have been studied in some detail. HHV-6 infection is associated with downregulation of CD3 expression (10), de novo induction of CD4 expression on CD8+ T cells (11) through activation of the CD4 promoter (12), induction of cytokines (IL-10) (13) and chemokines (RANTES) (14), inhibition of interferon beta production (15) and type 1 interferon signaling (16), induction of T-regulatory type 1 cells (13), inhibition of the T-cell-lymphoproliferative response (17, 18), and IL-2 synthesis (17). Some of these in vitro effects have also been observed under in vivo conditions. For example, in patients who received allogeneic bone marrow transplantation, active HHV-6 infection, as revealed by the presence of plasma viremia, was associated with lymphocytopenia and defective T cell proliferation to recall antigens (19).
The development of a specific and efficient T cell response is key for the generation of robust immunity against any virus. The clonal expansion of T cells in response to T cell receptor (TCR) engagement is intimately linked to the cell's ability to synthesize, secrete, and consume IL-2, the main T cell growth factor (20–22). TCR signaling induces AP-1 and increases the levels of active NF-κB p65/rel and calcium, resulting in calmodulin activation followed by calcineurin (CaN)-mediated dephosphorylation of NFAT, promoting its translocation into the nucleus. NFAT, in conjunction with constitutive factors such as OCT-1, binds to specific sites of the IL-2 promoter in a cooperative fashion, resulting in IL-2 gene transcription (23–25). Introduction of mutations that abolish NFAT binding to the two high-affinity NFAT-binding sites results in a dramatic reduction in promoter activity (26). Furthermore, low doses of the immunosuppressants cyclosporine (CsA) and FK506, which inhibit the phosphatase activity of CaN and thus the nuclear translocation of NFATs, also block IL-2 gene expression. These results show that the induction of the IL-2 gene transcription in T cells depends critically on the activity of NFAT factors (27–29).
Flamand et al. first reported that HHV-6A infection of T cells is associated with IL-2 gene transcription inhibition (17). Their results clearly showed that viral infectivity was dispensable for IL-2 inhibition, indicating that viral attachment to the cellular receptors or an immunosuppressive protein present within the virion could be responsible for the observed effects. In the present study, we analyzed the ability of HHV-6B at inhibiting IL-2 gene transcription. As reported for HHV-6A, we observed that HHV-6B efficiently suppresses IL-2 gene expression in T cells. The effects of tegument proteins on IL-2 expression were monitored and indicated that U54 but not U11 was responsible for IL-2 promoter inhibition and IL-2 mRNA production. By physically interacting with CaN, U54 prevents the dephosphorylation of NFAT, blocking its nuclear translocation and the subsequent IL-2 promoter activation. The GISIT (amino acids [aa] 293 to 297) motif within U54, analogous to the PXIXIT motif present within NFAT family members and the site where calcineurin docks (30–32), is partially accountable for U54's inhibitory effects.
MATERIALS AND METHODS
Cell lines.
Human embryonic kidney 293T (293T) and HeLa cell lines were obtained from the ATCC and cultured in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (HI-FBS), HEPES, nonessential amino acids (Sigma-Aldrich, Saint Louis, MO), sodium pyruvate (Wisent, Montreal, Quebec, Canada), and Plasmocin (5 μg/ml) (Invivogen, San Diego, CA). The J-Jhan and Jurkat human T cell lines were cultured in RPMI 1640 supplemented with 10% HI-FBS.
Plasmids.
The coding sequences for the U54 tegument proteins of HHV-6B strain Z29 and HHV-6A strain GS were amplified by PCR from viral DNA using the primers U54 forward (5′-TACAAGTCCGGACTCAGATCTATGCAACCCGCCACTCTA-3′) and U54 reverse (5′-GTTATCTAGATCCGGTGTCAATGGTGATGGTGATGATG-3′). After PCR, the U54 1.6-kbp bands were isolated and digested with BamHI/XhoI and inserted into the BamHI/XhoI sites of the pcDNA4TO/Myc-His A (4TO) (Life Technologies, Burlington, Ontario, Canada) vector in frame with a C-terminal Myc tag to generate the pcDNA4TO-U54-Myc vector (4TO-U54 and 4TO-U54A). Site-directed mutagenesis was used to mutate aa 296 and 297 (IT) of U54 to AA to yield the 4TO-U54mut vector. U54 and U54mut of HHV-6B were subcloned into the pcDNA4TO-mCherry (4TO-mCherry) vector using the BamHI/XhoI sites to generate the pcDNA4TO-U54-mCherry (U54-mCherry) and pcDNA4TO-U54mut-mCherry vectors. The sequence coding for the U11 tegument protein of HHV-6B was PCR amplified from viral DNA using U11 forward (5′-GGATCCGATTTGAAAGCGCAGTCGATCCCG-3′) and U11 reverse (5′-CTCGAGTCACGACGCGATCACTGACTTGTC-3′) primers. The PCR product was digested with BamHI/XbaI and cloned into BamHI/XbaI-digested pENTR4-FLAG vector (Addgene plasmid 17423) (33) to generate the pENTR4-U11-FLAG vector. pENTR4-U11-FLAG was digested with BamHI/EcoRV, and the insert was blunted with Klenow and subcloned into EcoRV-digested 4TO vector to generate the pcDNA4TO-U11-FLAG vector (4TO-U11). The sequence coding for the DNA polymerase p41 (U27) of HHV-6 GS strain was PCR amplified from viral DNA using primers p41 forward (5′-AAGCTTATGTGTTGGTCATTTCATTTG-3′) and p41 reverse (5′-CCGCGGGACGACGCATGTCTGCCTCTTG-3′). The PCR amplicon was digested with HindIII/SacII and cloned into the HindIII/SacII-digested pcDNA4TO/Myc-His B (Life Technologies) vector in frame with a C-terminal Myc tag to generate the pcDNA4TO-p41-Myc vector (4TO-p41). The pcDNA His-Max A-IE2 (IE2) (34) and pZVH14 (35) were described previously. The pAP-1-Luc, pNF-κB-Luc, and pCRE-Luc plasmids were obtained from Stratagene (Mississauga, Ontario, Canada); pCLN15deltaCX (IL-2-Luc) and p-NFAT-Luc (NFAT-Luc) were described previously (36). Plasmids pREP4-NFAT1 (REP-NFAT1) and pREP4-NFAT2 (REP-NFAT2), containing NFAT1 or NFAT2 coding sequences, were described in previous work (37). To generate the peGFP-NFAT1 vector, the NFAT1 coding sequence was amplified from REP-NFAT1 using primers NFAT1 forward (5′-GTACCGCGGGCCCGGATGAACGCCCCCGAGCGG-3′) and NFAT1 reverse (5′-GTTATCTAGATCCGGTGTTACGTCTGATTTCTGGCAGGAGGTC-3′). The PCR product was cloned into the BamHI site of the peGFP-C3 vector (Clontech) using the Gibson assembly mix (Life Technologies). Plasmid peGFP-C1-NFAT2 (NFAT2-GFP), coding for NFAT2-GFP, was obtained from Novus Biologicals (Oakville, Ontario, Canada).
Viral infection.
HHV-6A and HHV-6B were produced and titrated as described previously (16). J-Jhan T cells were infected (15 × 106 cells per condition) with HHV6-A (GS strain) and HHV6-B (Z29 strain) at a multiplicity of infection (MOI) of 1.
Drugs.
Cyclosporine (CsA) (LC Laboratories, Woburn, MA) is used as a calcineurin inhibitor. Ionomycin (Sigma-Aldrich) is a Ca2+ ionophore used as a mobile ion carrier for Ca2+. TPA (12-O-tetradecanoylphorbol-13-acetate) (Sigma-Aldrich) is a phorbol ester that binds and activates protein kinase C, which subsequently targets and activates AP-1 and NF-κB transcription factors.
Luciferase assays.
293T cells were seeded at 1.25 × 105 cells per well in 12-well plates (Sigma-Aldrich). The next day, cells were transfected with 4TO, 4TO-U54, 4TO-U11, 4TO-U54mut, 4TO-mCherry, 4TO-U54-mCherry, 4TO-U54mut-mCherry, REP-NFAT1, REP-NFAT2, NFAT-Luc, and IL-2-Luc using Lipofectamine 2000 (Life Technologies) and incubated at 37°C for 48 h. Cells were then stimulated with 25 ng/ml TPA and 0.5 μM ionomycin for 24 h before being lysed with 1× lysis buffer, and cell lysates were assayed for luciferase activity using a luminometer (Dynex Technologies, Chantilly, VA). Bicinchoninic acid (BCA) protein assays (Thermo Scientific, Waltham, MA) were used for normalization.
Western blot analysis.
293T cells were transfected with 4TO, 4TO-U54, 4TO-U11, 4TO-U54mut, 4TO-p41, REP-NFAT1, and REP-NFAT2 plasmids using Lipofectamine 2000. To evaluate protein expression, cells were lysed in 2× Laemmli lysis buffer and boiled for 5 min before centrifugation for 1 min at 13,000 rpm. U54, U54mut, and p41 proteins were detected using a mouse monoclonal mouse anti-Myc (clone 9E10). U11 protein was detected with a mouse monoclonal anti-D-tag (Flag) antibody (ABM, Richmond, British Columbia, Canada), NFAT1 was detected with a mouse monoclonal anti-NFAT1 antibody (25A10.D6.D2) (Novus Biologicals), NFAT2 was detected with a mouse monoclonal anti-NFAT2 antibody (SC-7294) (Santa Cruz, Santa Cruz, CA), and β-actin was detected with a mouse monoclonal anti-β-actin antibody (Sigma-Aldrich).
Densitometry.
Densitometric analysis of NFAT1 and coimmunoprecipitation immunoblots were performed using a Molecular Imager Gel Doc XR+ system (Bio-Rad, Mississauga, Ontario, Canada) and analyzed with Image Lab software (Bio-Rad). β-Actin expression was used for normalization.
Coimmunoprecipitation assays.
293T cells were seeded at 1.25 × 105 cells per well in 12-well plates and maintained in DMEM as described above for 24 h. Cells were transfected with 4TO, 4TO-U54, 4TO-U11, 4TO-U54mut, 4TO-p41, and REP-NFAT1 plasmids using Lipofectamine 2000 for 48 h. Cells were then stimulated with 25 ng/ml TPA and 0.5 μM ionomycin for 10 min before being lysed with 500 μl of radioimmunoprecipitation assay (RIPA) lysis buffer as described by Gravel et al. (38) supplemented with a cocktail of protease inhibitors (Thermo Scientific, Ottawa, Ontario, Canada) for 30 min at 4°C. Lysates were centrifuged at 10,000 rpm at 4°C for 10 min, and 450 μl of supernatants was collected for the next step, while 50 μl was added to 2× Laemmli buffer as total extract to evaluate expression of proteins of interest. Supernatants were incubated with anti-Myc, anti-FLAG, or anti-CaN (Novus Biologicals) monoclonal antibodies for 1 h at 4°C with rotation before addition of protein G agarose beads (Pierce, Rockford, IL) for an overnight incubation at 4°C on a rotary platform. Beads were centrifuged at 10,000 rpm at 4°C, washed with RIPA buffer four times, and resuspended in 50 μl of 2× Laemmli lysis buffer for Western blot analysis.
Microscopy. (i) Fluorescence microscopy assay.
HeLa cells were plated in 35-mm2 dishes at 1 × 105 cells per dish and maintained in DMEM as described above at 37°C for 24 h. Cells were transfected with 4TO-mCherry, U54-mCherry, U54mut-mCherry, eGFP, NFAT2-GFP, and NFAT1-GFP plasmids using Lipofectamine 2000 for 48 h. Then cells were stimulated with 25 ng/ml TPA and 0.5 μM ionomycin for 10 min. Images were captured by a CoolSNAP HQ camera mounted on an Olympus BX-51 upright microscope using a 40× or 60× UPlan Apo objective and processed with ImagePro 4.5.1 software (Media Cybernetics, Silver Spring, MD) before being analyzed.
(ii) Live-cell assay.
HeLa cells were plated and transfected as described above. mCherry cells were put inside an environmental LiveCell chamber (Pathology Devices, Westminster, MD) containing 5% CO2 at 37°C. Images were captured with a confocal spinning-disc WaveFX microscope (Quorum Technologies, Guelph, Ontario) with an electron-multiplying charge-coupled device (EM-CCD) ImageEM C9100-13 camera (Hamamatsu Photonics, Boston, MA) using a 20× HC Plan Apo 20×/0.70 Ph2 objective (Leica, Allendale, NJ). Images (green fluorescent protein [GFP] and mCherry) were captured before and every 2 min for 1 h after treatment with 25 ng/ml TPA and 0.5 μM ionomycin. Images were analyzed using Volocity 5.2.1 software (PerkinElmer, Woodbridge, Ontario, Canada) to make a movie with the obtained images as well as for the quantification of nuclear GFP fluorescence.
Reverse transcription-PCR (RT-PCR) assay.
J-Jhan cells were transfected (5 × 106 cells per condition) with 4TO or 4TO-U54 vectors using the T-008 program of the Amaxa Nucleofector system (Lonza, Mississauga, Ontario, Canada) and maintained in RPMI 1640 medium at 37°C for 48 h. Then cells were stimulated with 25 ng/ml TPA and 0.5 μM ionomycin for 6 h before being harvested, and RNA was extracted using the Qiazol lysis reagent (Qiagen, Gaithersburg, MD). Similarly, RNA was extracted from chronically HHV-6A- and HHV-6B-infected J-Jhan cells. Retrotranscription (RT) into cDNA was carried out as described by Jaworska et al. (15). One-tenth of the cDNA was used for real-time PCR as described by Jaworska et al. (16). The primers used for IL-2 detection were IL-2 forward (5′-GAA TCC CAA ACT CAC CAG GAT GCT C-3′ and IL-2 reverse (5′-TAG CAC TTC CTC CAG AGG TTT GAG T-3′). The primers used for U65-U66 detection were U65-U66 forward (5′-GAC AAT CAC ATG CCT GGA TAA TG-3′) and U65-U66 reverse (5′-TGT AAG CGT GTG GTA ATG GAC TAA-3′). 293T or Jurkat cells were harvested, and RNA was extracted and retrotranscribed as described above. The primers used for NFAT1, NFAT2, and NFAT3 detection were NFAT1 forward (5′-CGA AGA AGA GCC GAA TGC AC-3′) and NFAT1 reverse (5′-AGA AAC TTC TGC GGC CCT AC-3′), NFAT2 forward (5′-CAC TCC TGC TGC CTT ACA CA-3′) and NFAT2 reverse (5′-AAG ATG CGA GCA TGC GAC TA-3′), and NFAT3 forward (5′-CGG CCT CTA AGA GAG GTT GA-3′) and NFAT3 reverse (5′-CCT CCT TTT CCT CCC CGA AC-3′). The GAPDH primers used for normalization were described previously (15).
Statistical analysis.
Experimental groups were compared using a one-way analysis of variance (ANOVA) followed by Bartlett's test to show if the variances differed significantly, followed by Bonferroni's multiple-comparison test. Results were considered significantly different when P was <0.05. Experimental groups were also compared using a two-way ANOVA followed by Bonferroni's multiple-comparison test. Results were considered significantly different when P was <0.05.
RESULTS
Identification of HHV-6B as an inhibitor of IL-2 gene transcription.
Following observations made by Flamand et al. demonstrating that IL-2 gene expression is inhibited during HHV6-A (GS strain) infection (17), we evaluated the capacity of HHV6-B (Z29 strain) to exert the same effect. Uninfected, HHV-6A-infected, and HHV-6B-infected J-Jhan T cells were stimulated with TPA-ionomycin for 6 h, after which the IL-2 mRNA was measured by quantitative RT-PCR assay. Stimulation of uninfected cells with TPA-ionomycin led to a 12-fold increase in IL-2 mRNA expression (P < 0.0001). Under similar conditions, the IL-2 mRNA expression in HHV-6A- and HHV-6B-infected cells was reduced by 70% and 80%, respectively (P < 0.0001) (Fig. 1). Infection was confirmed by RT-PCR assay for the presence of U65-U66 viral mRNA in J-Jhan T cells (data not shown). These results confirm that like HHV6-A, HHV-6B is capable of inhibiting IL-2 gene transcription in T cells.
FIG 1.
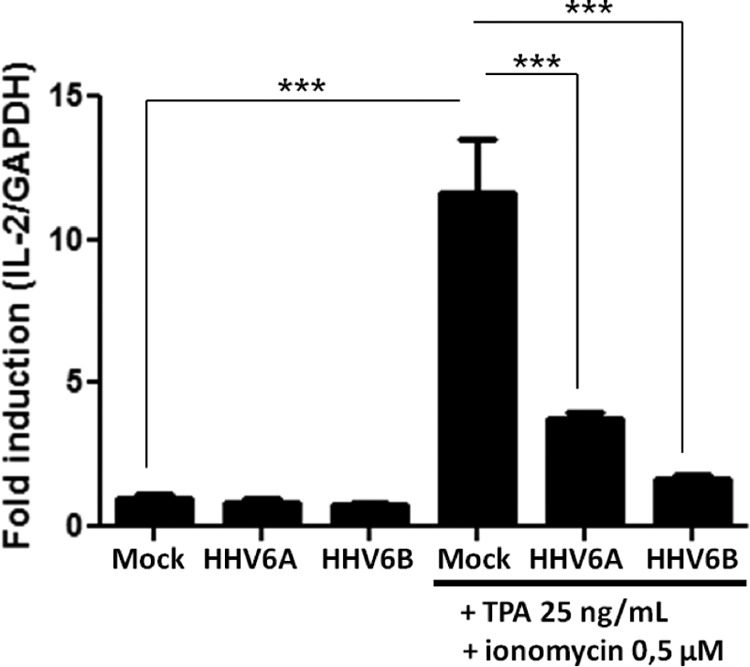
HHV-6B and HHV-6A inhibit IL-2 gene transcription in T cells. J-Jhan cells were mock infected or infected with HHV-6B or HHV-6A and were stimulated for 6 h with TPA-ionomycin or left unstimulated. Then IL-2 mRNA levels were determined by RT-PCR assay and normalized with GAPDH (n = 4).
Characterization of U54 tegument protein as an inhibitor of IL-2 gene transcription.
Previous results suggested that HHV-6A virion components, such as tegument proteins, were likely to be responsible for the inhibition of IL-2 gene expression observed following infection (17). We tested whether HHV-6B U11 and U54, two such tegument proteins, could inhibit the expression of a luciferase reporter construct whose expression is dependent on the activation of NFAT proteins, transcription factors essential for IL-2 gene transcription (39). 293T cells were transfected with the NFAT-Luc reporter and control 4TO, 4TO-U54, and 4TO-U11 expression vectors. Forty-eight hours later, cells were stimulated with TPA-ionomycin for an additional 24 h, after which the luciferase activity was determined. TPA-ionomycin treatment activated endogenous NFAT, resulting in a 7.5-fold increase (P < 0.0001) in luciferase activity (Fig. 2A). When cells were transfected with the U54 expression vector, luciferase activity was reduced by more than 95%, equivalent to levels on nonactivated cells (P < 0.0001). Expression of U11, a second HHV-6 tegument protein, had no significant effect on luciferase activity. Expression of both U54 and U11 proteins was monitored by Western blot analysis. To determine if this effect of U54 is specific for NFAT, we tested reporters driven by NF-κB, AP-1, and cyclic AMP-responsive elements (CRE). Cells were transfected with the U54 expression vector or known HHV-6 transactivators, including the immediate early 2 protein (IE2) and plasmid pZVH14, containing the U30 and U31 open reading frames (ORFs) (also encoding tegument proteins). In contrast to NFAT-regulated promoters that are inhibited by U54, reporters responsive to AP-1 (Fig. 2B), CRE (Fig. 2C), and NF-κB (Fig. 2D) were activated 2.5-fold to 5-fold by U54. Proteins encoded by the pZVH14 and IE2 vectors activated these reporters very efficiently (50- to 400-fold). These results suggest that U54 interferes specifically with the expression of genes that are regulated through NFAT transcription factors.
FIG 2.
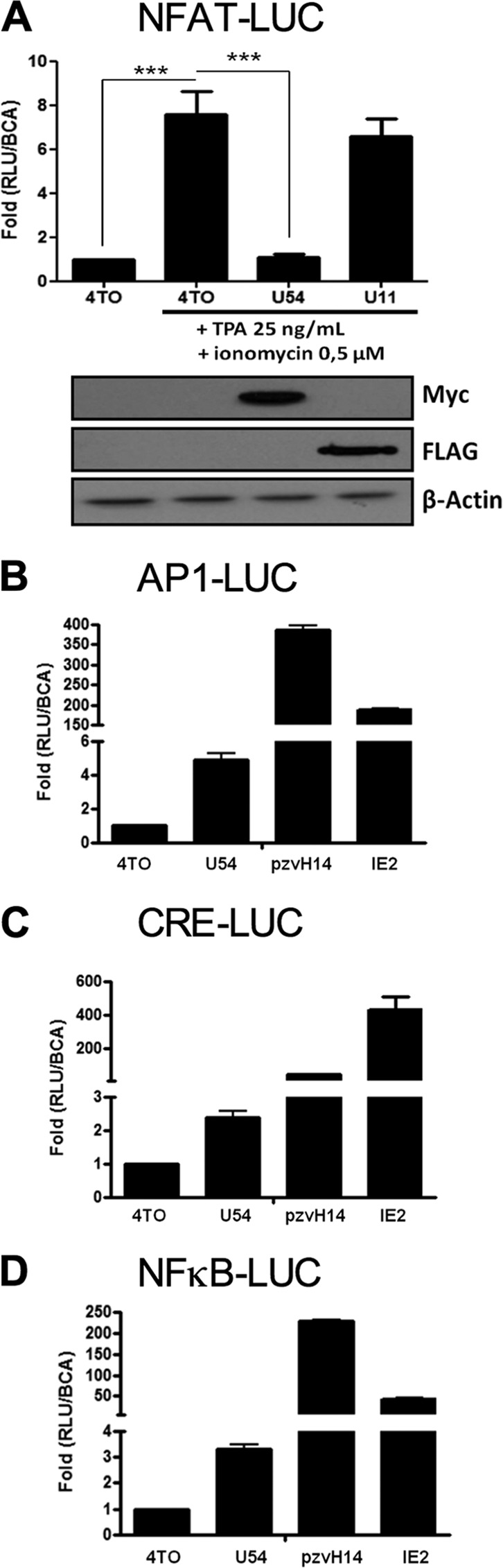
U54 inhibits NFAT transcriptional activity. (A) 293T cells were transfected with 4TO, 4TO-U54, 4TO-U11 and NFAT-Luc reporter plasmids. Cells were stimulated for 24 h or left unstimulated, and luciferase activity was determined and normalized to protein content (n = 9). Western blot analysis confirmed the expression of proteins of interest for each condition tested. U54-Myc and U11-FLAG proteins were expressed in cells that had been transfected with 4TO-U54 and 4TO-U11 plasmids (top). Beta-actin was included as a loading control. (B to D) Transcriptional activities of AP-1 (B), CRE (C), and NF-κB (D) were evaluated in 293T cells by luciferase assay. 293T cells were transfected with 4TO, 4TO-U54, pZVH14, IE2, and either pAP1-Luc, pCRE-Luc, or pNF-κB-Luc. Luciferase activity was determined by luciferase assay and normalized to a BCA assay (n = 3).
Considering that 293T cells are nonlymphoid cells expressing mostly NFAT3 while lacking appreciable expression of NFAT1 and NFAT2 (Table 1) (40), the main transcription factors associated with IL-2 gene transcription, we repeated these experiments in the presence of ectopically expressed NFAT1 and NFAT2. As presented in Fig. 2B, in the absence of NFAT1 or NFAT2 expression, the NFAT-Luc reporter is minimally activated by TPA-ionomycin treatment. Such an increase in NFAT-Luc activity is abrogated by U54 expression (P < 0.0001) (Fig. 3). When NFAT1 or NFAT2 was expressed, the NFAT-Luc activity increased more than 900-fold in response to TPA-ionomycin treatment (P < 0.0001). In the absence of TPA-ionomycin treatment, the NFAT-Luc reporter was minimally activated (data not shown). When NFAT1 or NFAT2 was coexpressed with U54, luciferase activity was reduced by more than 90% (P < 0.0001). Western blot analysis indicated that all proteins were efficiently expressed and that U54 had no significant impact on NFAT1 or NFAT2 expression.
TABLE 1.
Analysis of NFAT1, NFAT2, and NFAT3 expression in HEK293T and Jurkat T cells
| Protein | Expression ina: |
|
|---|---|---|
| 293T cells | Jurkat cells | |
| NFAT1 | − | +++ |
| NFAT2 | ± | +++ |
| NFAT3 | + | ++ |
−, absence of expression; ±, marginal expression; +, basal expression level; ++, moderate expression level; +++, high expression level.
FIG 3.
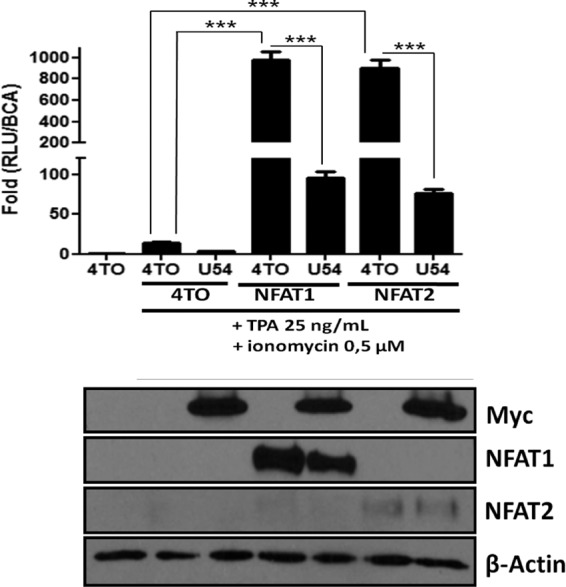
U54 inhibits ectopically expressed NFAT1 and NFAT2 transcriptional activity. (Top) Transcriptional activity of ectopically expressed NFAT1 and NFAT2 factors was evaluated in 293T cells by luciferase assay. 293T cells were transfected with 4TO, 4TO-U54, 4TO-U11, REP-NAFT1, REP-NFAT2, and NFAT-Luc reporter plasmids. Cells were then stimulated for 24 h or left unstimulated, and luciferase activity was determined and normalized to protein content (n = 9). (Bottom) Western blot analysis confirmed the expression of proteins of interest for each condition tested. U54-Myc or U11-FLAG proteins were expressed in cells that had been transfected with 4TO-U54 or 4TO-U11 plasmids (upper panels), and NFAT1 or NFAT2 proteins were expressed in those transfected with REP-NFAT1 or REP-NFAT2 plasmids (middle panels). Beta-actin was included as a loading control.
We next repeated the experiment whose results are presented in Fig. 3 using a luciferase reporter construct driven by the more complex IL-2 promoter. As for NFAT-Luc, in the absence of NFAT1 or NFAT2 expression, the IL-2 promoter activity was minimal (Fig. 4). In the presence of ectopically expressed NFAT1 or NFAT2, the IL-2-Luc reporter was activated approximately 15-fold following TPA-ionomycin treatment (P < 0.0001). In the presence of U54, the IL-2-Luc reporter activity was significantly inhibited (P < 0.0001). Western blot analysis indicates that all proteins were efficiently expressed and that U54 had no significant impact on NFAT1 or NFAT2 expression.
FIG 4.
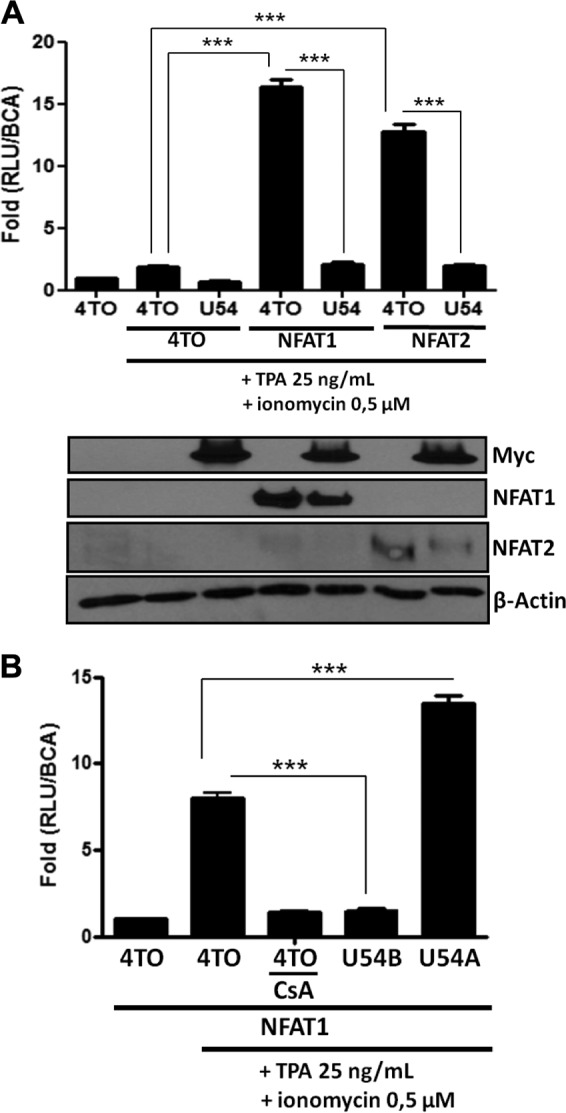
U54 inhibits NFAT1 and NFAT2 activity on the IL-2 promoter. Transcriptional activity of ectopically expressed NFAT1 and NFAT2 factors was evaluated in 293T cells by luciferase assay. (A) 293T cells were transfected with 4TO, 4TO-U54, 4TO-U11, REP-NFAT1, REP-NFAT2, and IL-2-Luc reporter plasmids. Cells were stimulated for 24 h or left unstimulated, and then luciferase activity was determined and normalized to protein content (n = 9). Western blot analysis confirmed the expression of proteins of interest for each condition tested. U54-Myc or U11-FLAG proteins were expressed in cells transfected with 4TO-U54 or 4TO-U11 (upper panels), and NFAT1 or NFAT2 proteins were expressed in those transfected with REP-NFAT1 or REP-NFAT2 (middle panels). Beta-actin was included as a loading control. (B) 293T cells were transfected with 4TO, 4TO-U54, 4TO-U54A, REP-NFAT1, and IL-2-Luc reporter plasmids. Cells were stimulated for 24 h or left unstimulated, and luciferase activity was determined by luciferase assay and normalized to protein content (n = 6).
Having identified U54 of HHV-6B as a suppressor of IL-2 promoter activation, we enquired whether U54 from HHV-6A would behave similarly. Unlike the NFAT-Luc reporter activity, which was inhibited by HHV-6B U54 (P < 0.0001), U54 from HHV-6A activated NFAT-Luc in a dose-response manner (P < 0.0001) (data not shown). These results were confirmed using the IL-2-Luc reporter, with >90% inhibition by HHV-6B U54 (P < 0.0001) and a 2-fold increase in reporter activity by HHV-6A U54 (P < 0.0001) (Fig. 4B). Despite their similarities at the amino acid level (80% amino acid identity), these results indicate that HHV-6B U54 is capable of inhibiting IL-2 promoter activation while HHV-6A U54 is not. Another HHV-6A protein is therefore likely responsible for this effect.
HHV-6B U54 protein interacts with calcineurin phosphatase, which regulates NFAT activity.
We analyzed the amino acid sequence of U54 and identified the GISIT motif, which resembles the conserved PXIXIT motif (where X represents polar residues) present within all NFAT family members. Considering that a peptide with the sequence VIVIT acts as a potent inhibitor of NFAT activation (31), we surmised that the U54293–297 GISIT motif could be responsible for NFAT inhibition. Furthermore, knowing that the PXIXIT motif represents the CaN docking site (30–32), we hypothesized that U54 might physically interact with CaN. To verify this, cells were transfected with a control or Myc-tagged 4TO-U54B expression vector. In parallel, cells were transfected with REP-NFAT1 and FLAG-tagged 4TO-U11 expression vectors as controls. After 48 h, cells were stimulated with TPA-ionomycin for 10 min or left unstimulated, and then endogenous CaN and U54 were immunoprecipitated using anti-CaN and anti-Myc antibodies and protein G agarose beads followed by Western blot analysis using anti-Myc and anti-CaN antibodies, respectively. The results obtained indicate that U54 coimmunoprecipitated with CaN independently of CaN activation (Fig. 5A and B) while U11 did not (Fig. 5C). We next tested whether U54 might interact with NFAT1. To do this, NFAT1 was expressed in the absence or in the presence of U54 or U11, as a control. We individually immunoprecipitated U54 (anti-Myc), CaN (anti-CaN), and U11 (anti-FLAG) and carried out Western blot analysis using anti-NFAT1 antibodies. As shown, U54 and U11 did not interact with NFAT1 (Fig. 5D). In contrast, CaN efficiently precipitated NFAT1 following TPA-ionomycin stimulation (Fig. 5D).
FIG 5.
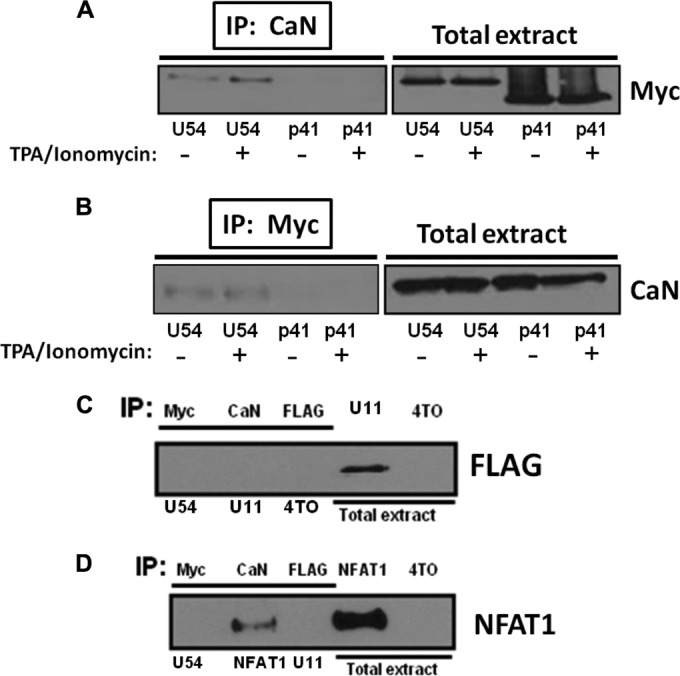
U54 interacts with CaN phosphatase enzyme. Interaction between U54 and CaN was evaluated in 293T cells by coimmunoprecipitation (co-IP) assay. 293T cells were transfected with 4TO, 4TO-U54, and 4TO-p41 plasmids. Thereafter, co-IP was performed by using antibodies against CaN (A) or Myc (B) and then by blotting with the anti-Myc or anti-CaN antibodies, respectively. Total extracts were used as control to verify expression of U54. (C) Interaction between U11 and CaN was evaluated in 293T cells by co-IP. 293T cells were transfected by the 4TO, 4TO-U54, and 4TO-U11 plasmids. Forty-eight hours later, co-IP was performed using anti-Myc (U54), anti-FLAG (U11), and anti-CaN antibodies. Control lysate (4TO) was immunoprecipitated with anti-FLAG antibodies. The immunoprecipitated material was blotted using anti-FLAG antibodies. Total extracts were used as controls to verify expression of U11. (D) Interaction between NFAT1 and CaN, U54, or U11 was evaluated in 293T cells by co-IP. 293T cells were transfected with 4TO, 4TO-U54, 4TO-U11, and REP-NFAT1 plasmids. co-IP was performed by immunoprecipitating U54, CaN, or U11 before blotting with anti-NFAT1 antibodies. Total extracts were used as controls to verify the expression of NFAT1.
U54 inhibits phosphatase activity of calcineurin on NFAT.
To migrate to the nucleus, NFAT proteins must be dephosphorylated by CaN. Considering that U54 interacts with CaN, we tested whether U54 might interfere with NFAT dephosphorylation upon cellular activation. Cells were transfected with the NFAT1 expression vector together with a 4TO control or a 4TO-U54 expression vector. Half the cultures were treated with TPA-ionomycin for 10 min. Cells pretreated with CsA were used as a positive control for inhibition of NFAT dephosphorylation. Under resting conditions, NFAT1 was hyperphosphorylated, as expected. In control-transfected cells (4TO), TPA-ionomycin induced the dephosphorylation of NFAT1. In contrast, in the presence of U54 or CsA, NFAT1 remained hyperphosphorylated (Fig. 6A). Using densitometric analysis, we could determine that activation led to a 75% decrease in phosphorylated forms of NFAT1 in the control (4TO), while in cells treated with CsA or expressing U54, NFAT1 remained hyperphosphorylated (Fig. 6B).
FIG 6.
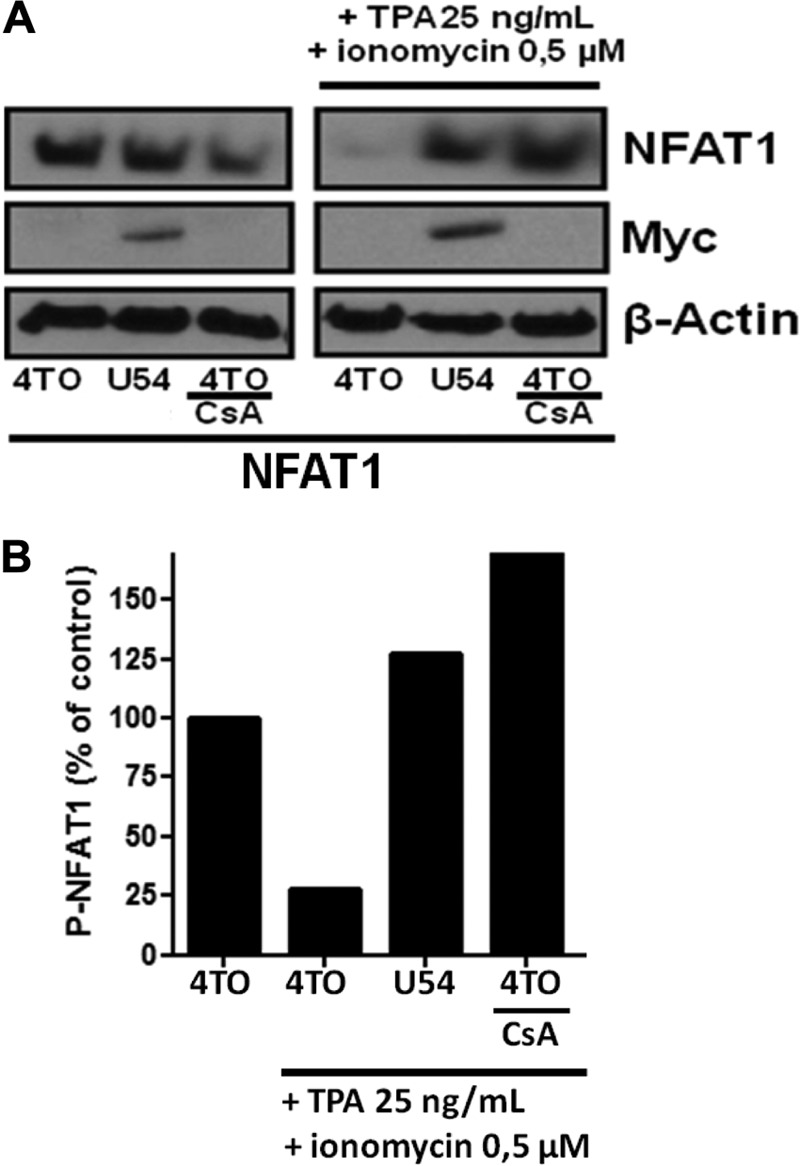
U54 inhibits the dephosphorylation of NFAT1 protein by CaN. NFAT1 dephosphorylation was evaluated in 293T cells by Western blotting assay. (A) 293T cells were transfected with 4TO, 4TO-U54, and REP-NFAT1 plasmids. As a positive control, we pretreated 4TO-transfected cells with CsA. Cells were stimulated for 10 min, and then Western blot analysis was performed using anti-NFAT1 antibodies specific for phosphorylated forms of NFAT1 (top). Western blot analysis confirmed the expression of U54 using anti-Myc antibodies (middle). Beta-actin was included as a loading control. (B) Densitometric analysis of P-NFAT1 was performed following Western blot analysis. The P-NFAT1 level in resting cells (4TO) was set at 100%. Following TPA-ionomycin stimulation, P-NFAT1 levels were compared to those of the respective resting controls (results are from one representative experiment of three independent experiments).
Involvement of U54 GISIT motif in the interaction with CaN and inhibition of phosphatase activity.
Considering that U54 interacted with CaN, we tested whether the U54 GISIT motif could be responsible for this interaction. We mutated the GISIT motif to GISAA (Fig. 7A), based on previous work indicating that mutation of the IT residues within the VIVIT motif was sufficient to abrogate the inhibitory properties of the VIVIT peptide (31). To compare the NFAT-inhibitory activity of U54mut relative to U54, we performed a dose-response experiment. Cells were transfected with a suboptimal quantity of the REP-NFAT1 expression vector (a smaller amount of this vector was used to avoid saturating the system), an optimal quantity of the 4TO control or NFAT-Luc reporter, and various amounts of 4TO-U54 and 4TO-U54mut expression vectors. Twenty-four hours after TPA-ionomycin stimulation, the luciferase activity was determined. In cells transfected with the 4TO control vector, a 7.5-fold increase in NFAT-Luc activity was observed following stimulation. Transfection of increasing quantities of U54 led to a dose-dependent inhibition of NFAT-Luc activity (Fig. 7B). U54mut also led to a dose-dependent reduction of NFAT-Luc activity, but the activity of the mutant was always significantly less than that obtained with U54 (P < 0.001 and P < 0.01). A gross estimate indicated that U54mut is half as potent as U54. Tegument proteins U54 and U54mut were expressed at roughly similar levels, as verified by Western blot analysis (Fig. 7C). These results suggest that the U54293–297 GISIT motif plays a role in the inhibition of NFAT activation but that another U54 domain(s) also likely participates.
FIG 7.
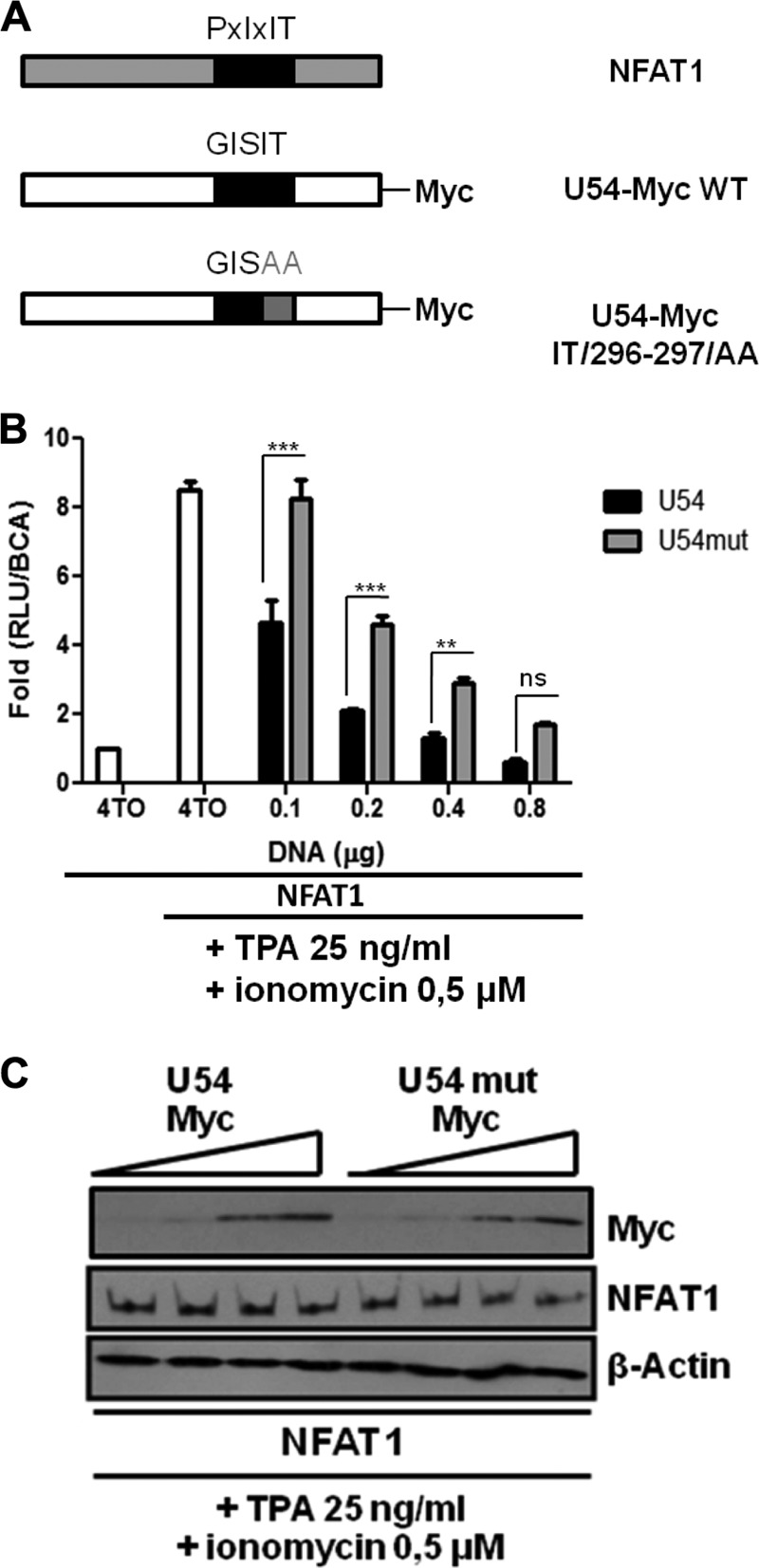
The U54 GISIT motif is important for the inhibition of NFAT activity. (A) Schematic representation of CaN docking sites within NFAT1, U54, and U54mut proteins. (B) 293T cells were transfected with 4TO, 4TO-U54, 4TO-U54mut, REP-NAFT1, and NFAT-Luc reporter plasmids (adjusted to a total of 0.8 μg/well with 4TO empty vector) for 48 h before being stimulated with TPA-ionomycin for an additional 24 h or left unstimulated. Then luciferase activity was determined by luciferase assay and normalized to protein content (n = 9). (C) Western blot analysis confirmed the expression of proteins of interest for each condition tested. U54-Myc or U54mut-Myc proteins were expressed in 4TO-U54- or 4TO-U54mut-transfected cells (top), and NFAT1 protein was expressed in REP-NFAT1-transfected cells (middle). Beta-actin was included as a loading control.
U54mut cannot inhibit NFAT dephosphorylation as efficiently as U54.
We next determined the capacity of U54mut to interact with CaN. Cells were transfected with the control vector and both Myc-tagged 4TO-U54 or 4TO-U54mut expression vectors. After 48 h, endogenous CaN was immunoprecipitated using anti-CaN antibodies and protein G agarose beads, followed by Western blot analysis using anti-Myc antibodies. The results indicate that U54 coimmunoprecipitated with CaN as expected. U54mut also interacted with CaN, albeit less than U54, explaining the partial NFAT-inhibitory activity of U54mut (Fig. 8A and B). Lastly, we tested the impact of U54mut on NFAT dephosphorylation. Cells were transfected with 4TO, 4TO-U54, or 4TO-U54mut expression vectors, and 48 h later, cells were stimulated with TPA-ionomycin for 10 min before lysis and Western blot analysis. NFAT1 was efficiently dephosphorylated in the presence of U54mut, while it remained hyperphosphorylated in the presence of U54 or following pretreatment with CsA (positive control) (Fig. 8C and D). These results indicate that the U54 GISIT motif plays an inhibitory role in CaN phosphatase activity.
FIG 8.
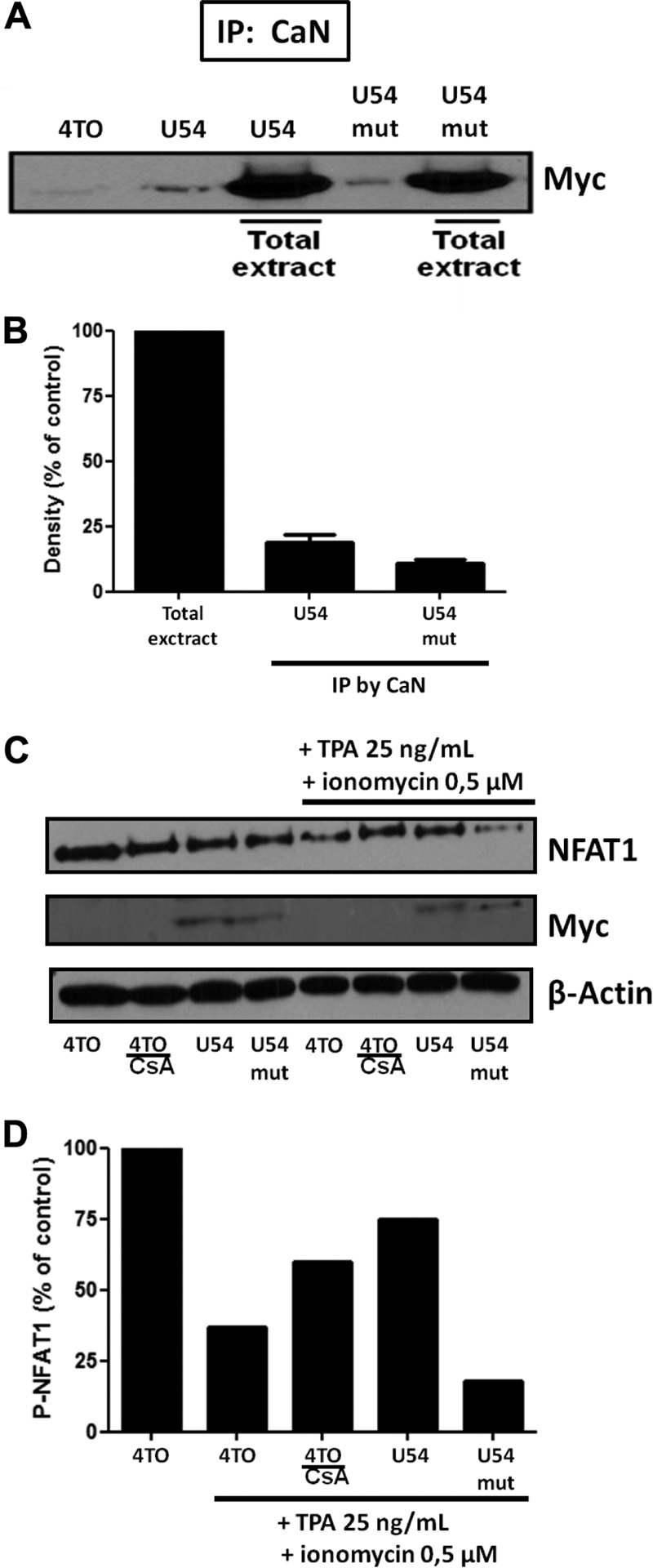
U54mut interacts with CaN but minimally compared to U54 and is not able to prevent NFAT dephosphorylation. Interaction of U54 or U54mut with CaN was evaluated in 293T cells by co-IP assay. (A) 293T cells were transfected with 4TO, 4TO-U54, and 4TO-U54mut plasmids. Thereafter, co-IP was performed by immunoprecipitating samples from cells under all conditions by anti-CaN antibody and then by blotting with anti-Myc antibody. Total extracts were used as control to verify the expression of U54 and U54mut proteins. (B) Quantitation of U54 or U54mut that coprecipitated with CaN. Results are expressed relative to the value for whole-cell extracts, set at 100%. (C) 293T cells were transfected with 4TO, 4TO-U54, 4TO-U54mut, and REP-NFAT1 plasmids. As a positive control, we pretreated 4TO-transfected cells with CsA. Western blot analysis was performed by blotting each sample with an anti-NFAT1 antibody specific for phosphorylated forms of NFAT1 at 140 kDa (top). Western blot analysis confirmed the expression of U54 and U54mut by anti-Myc blotting (middle). Beta-actin was included as a loading control. (D) P-NFAT1 was quantitated by densitometric analysis. P-NFAT1 levels in resting cells were set at 100% (results are from one representative experiment of three independent experiments).
U54 inhibits nuclear translocation of NFAT transcriptional factors.
Our results so far indicate that HHV-6B U54 can inhibit the expression of NFAT-regulated genes, such as the IL-2 gene. To understand how this might occur, we studied NFAT2-GFP and NFAT1-GFP nuclear translocation in the absence or in the presence of U54-mCherry. We first confirmed that U54-mCherry behaved similarly to U54-myc by demonstrating the inhibition of NFAT-Luc activity in NFAT2/U54-mCherry/NFAT-Luc-cotransfected cells (Fig. 9A). The results indicated that following TPA-ionomycin stimulation, 90% of NFAT2 activity was inhibited by U54-mCherry (P < 0.0001). Then we transfected HeLa cells with NFAT2-GFP and NFAT1-GFP and with mCherry, U54-mCherry, and U54mut-mCherry vectors to study NFAT2-GFP and NFAT1-GFP nuclear translocation by fluorescence microscopy. Under resting conditions, NFAT2-GFP and NFAT1-GFP were mainly cytoplasmic, as expected (Fig. 9B and data not shown). U54-mCherry was detected in both the cytoplasm and the nucleus. In cells transfected with mCherry and U54mut-mCherry, NFAT2-GFP and NFAT1-GFP efficiently translocated and accumulated in the nucleus in response to TPA-ionomycin activation. When U54-mCherry was coexpressed, NFAT2-GFP and NFAT1-GFP remained cytoplasmic following TPA-ionomycin stimulation (Fig. 9B and data not shown). As a positive control for inhibition of NFAT2-GFP and NFAT1-GFP translocation, cells were preincubated with CsA prior to stimulation. Under these conditions, NFAT2-GFP and NFAT1-GFP remained cytoplasmic, as expected. All these results were confirmed by live-cell experiments in which we cotransfected HeLa cells with mCherry control or U54-mCherry with NFAT2-GFP. Images and intranuclear GFP fluorescence measurements obtained using these assays (Fig. 9C and D) indicated that in the presence of mCherry, NFAT2-GFP efficiently translocated to the nucleus during the first 5 min following TPA-ionomycin stimulation, followed by relocalization to the cytoplasm. In contrast, in U54-mCherry-transfected cells, NFAT2-GFP remained cytoplasmic at all time points observed (Fig. 9C and D). These results indicate that HHV-6 U54 inhibits IL-2 gene activation by preventing nuclear translocation of NFAT1 and NFAT2 and that the GISIT motif was important for this effect.
FIG 9.

NFAT2-GFP nuclear translocation is inhibited in the presence of U54-mCherry. The capacity of U54-mCherry to abrogate NFAT2 transcriptional activity was evaluated in 293T cells by luciferase assay. (A) 293T cells were transfected with 4TO, 4TO-U54, 4TO-mCherry, U54-mCherry, U54mut-mCherry, REP-NFAT2, and NFAT-Luc reporter plasmids. Following stimulation for 24 h, luciferase activity was determined by luciferase assay and normalized to protein content (n = 3). (B) Nuclear translocation of NFAT2-GFP was tested in HeLa cells by fluorescence microscopy. HeLa cells were transfected with eGFP, 4TO-mCherry, NFAT2-GFP, U54-mCherry, and U54mut-mCherry plasmids. Cells were stimulated or left unstimulated, and green fluorescent protein (GFP) was evaluated using a laser with excitation at 460 to 500 nm and emission at 510 to 560 nm, while red fluorescence (mCherry) was determined using a laser with excitation at 525 to 555 nm and emission at 600 to 660 nm. (C) HeLa cells were transfected with NFAT2-GFP and 4TO-mCherry or U54-mCherry plasmids for 48 h. An image of the cell culture was taken as a control for cytoplasmic NFAT2-GFP location before cells were stimulated with TPA-ionomycin for an additional 1 h. During stimulation, images were taken every 2 min. (D) Images from both 4TO-mCherry/NFAT2-GFP and U54-mCherry/NFAT2-GFP experiments were analyzed with ImageJ software to quantify GFP fluorescence following TPA-ionomycin stimulation for 1 h. GFP fluorescence represents the intranuclear presence of NFAT2-GFP. Images were selected 3 min before stimulation and at 3, 15, and 30 min following the addition of TPA-ionomycin.
Inhibition of IL-2 gene transcription by U54 tegument protein in the J-Jhan T cell line.
Lastly, to validate the above results in a more physiological setting, we evaluated the ability of U54 to inhibit IL-2 gene expression in T lymphocytes. J-Jhan T cells were transfected with 4TO, 4TO-U54, 4TO-U11, and 4TO-U54mut expression vectors. Forty-eight hours later, cells were treated with TPA-ionomycin for 6 h before being harvested and lysed for the RT-PCR assay. As previously determined, TPA-ionomycin treatment efficiently activated endogenous IL-2 gene transcription, resulting in a 30-fold increase (P < 0.05) in IL-2 mRNA (Fig. 10A). When cells expressed U54, the IL-2 mRNA levels were reduced to those in resting cells (P < 0.05) and comparable to those in CsA-pretreated cells. These results confirmed the capacity of U54 to abrogate IL-2 gene expression in T cells, the primary HHV-6 target cell type. Western blot analysis indicated that all proteins were efficiently expressed.
FIG 10.
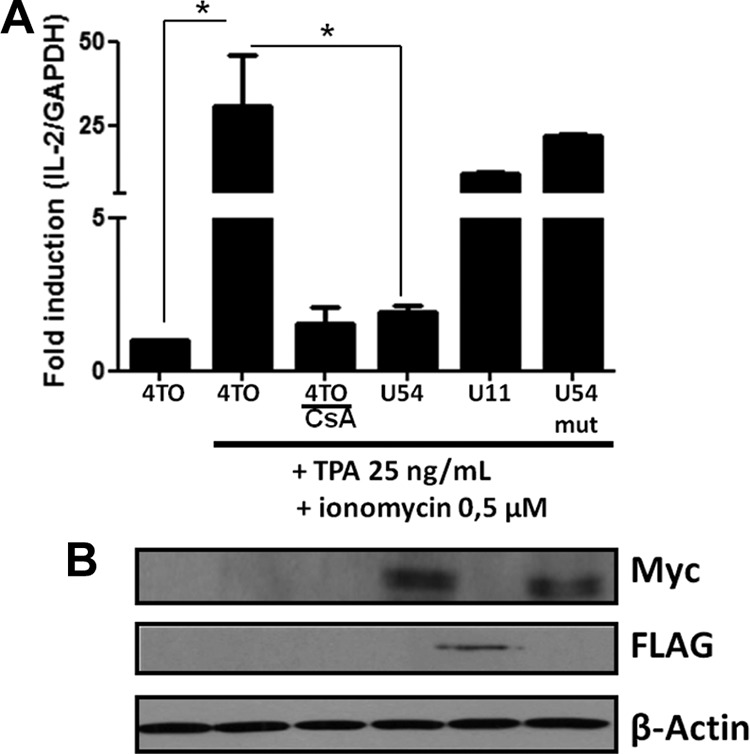
U54 abrogates IL-2 gene transcription in J-Jhan T cells. (A) J-Jhan cells were transfected with 4TO, 4TO-U54, 4TO-U11, and 4TO-U54mut plasmids for 48 h before being stimulated with TPA-ionomycin for an additional 6 h or left unstimulated. RNA was extracted, and IL-2 mRNA levels were determined by real-time RT-quantitative PCR (RT-qPCR). Western blot analysis confirmed the expression of U54 and U54mut proteins. Beta-actin was included as a loading control.
DISCUSSION
Following primary infection of a new host, HHV-6B, the etiologic agent of exanthem subitum, is able to persist for life, with occasional reactivation episodes. In healthy subjects, reactivations are controlled by the immune system, but in immunocompromised patients, these can lead to serious complications (41). Like most herpesviruses, HHV-6B has developed several immune evasion strategies aimed at disturbing the effectors responsible for the elimination of pathogens (see the introduction). Interleukin-2 is necessary for the growth, proliferation, and differentiation of T cells to become such effector cells. In addition, the fact that IL-2 also negatively affects HHV-6 replication in T cells (42) makes this cytokine a strategic HHV-6 target. Immune evasion mechanisms are likely to affect the anti-HHV-6 immune response but also that against other pathogens. In support of this, bone marrow transplant recipients with reactivated HHV-6 showed impaired T cell responses against cytomegalovirus (CMV) (19).
Flamand et al. described the effects of HHV-6A infection on IL-2 gene transcription and T cell proliferation (17). Similarly, Horvat et al. reported that HHV-6B efficiently inhibited T cell proliferation (18). In both studies, wild-type infectious and UV-irradiated HHV-6 were equally potent at suppressing T cell proliferation. These observations led us to generate the hypothesis that suppression of IL-2 gene transcription is caused by a structural component of HHV-6. The structure of all herpesvirus particles is made of three major components: an external envelope, a tegument, and the nucleocapsid containing the viral nucleic acid. Tegument proteins of CMV, another betaherpesvirus, were previously described as potent regulators for viral replication or immunosuppression (43, 44). In the current work, we provide evidence that the HHV-6B U54 tegument protein behaves as an inhibitor of IL-2 gene expression. Our study adds an additional mechanism by which the virus damps specific anti-HHV-6 immune responses, allowing it to persist in its host.
Following TCR activation, induced AP-1 increases NF-κB p65/rel and Ca2+ release in the cytoplasm. The serine/threonine calcineurin phosphatase forms a complex with calmodulin, binds to the cytoplasmic and hyperphosphorylated forms of NFAT (NFAT1-NFAT4) proteins, and induces their dephosphorylation. Subsequently, dephosphorylated NFATs translocate to the nucleus, specifically bind to DNA in association with other transcription factors, such as OCT-1, and initiate IL-2 gene transcription (27, 45). IL-2 gene regulation is well characterized, and because of its importance in immunity, several immunosuppressive therapeutic drugs designed to block IL-2 gene activation have been developed (45). Macrolides, such as CsA and FK506, that block substrate access to the active site of CaN represent very potent inhibitors of IL-2 gene expression (46). There are also peptides that bind calcineurin and inhibit its activity. These peptides include CaN docking site binders, which can even be more specific than CsA or FK506. Such peptides include CaN457–482-AID and CaN424–521-AID peptides (47, 48). Alternatively, some peptides, such as the mNFAT1106–121-SPRIEIT and VIVIT peptides, can bind CaN and block CaN-NFAT interaction in the cytoplasm (30, 31, 49). More recently, some pathogens have also been characterized as having mechanisms allowing them to disrupt the IL-2 pathway, such as the VacA protein of Helicobacter pylori, whose modes of action are not all CaN-NFAT dependent but which inhibit NFAT translocation (50). More interestingly, the A238L protein of African swine fever virus has been identified as a potent inhibitor of the CaN-NFAT pathway (51). First, the A238L protein can inhibit CREBbp/p300 transactivation, leading to the suppression of acetylation and transcriptional activation of NFAT1, NF-κB, and c-Jun (52). Second, A238L inhibits the activation of JNK (53, 54), and third, A238L contains the PKIIITG sequence, analogous to the PXIXIT CaN docking site (30, 31), which binds to CaN with high affinity and blocks NFAT dephosphorylation (55).
In this study, we highlighted the capacity of HHV-6B to inhibit IL-2 gene expression (Fig. 1) and showed that U54 from HHV-6 strain Z29 (HHV-6B) inhibited NFAT activity by causing improper NFAT dephosphorylation and nuclear translocation. We have identified the U54293–297 GISIT motif, analogous to the NFAT major CaN docking site PXIXIT motif (30, 31), as playing a role in the inhibition of IL-2 gene activation. Although U54 from HHV-6A and that from HHV-6B are 80% identical, the U54293–297 GISIT motif from HHV-6B differs slightly from that of HHV-6A, which has an isoleucine-to-threonine substitution at amino acid 296 (U54293–297 GISTT). Considering that the proteins behaved differently with regard to IL-2 promoter inhibition, it can be speculated that isoleucine 296 is critical for this effect. In support of this idea, mutating amino acids 296 and 297 abrogated the HHV-6B U54 inhibitory potential. A yet-to-be identified protein, other than U54, must therefore downmodulate IL-2 expression during HHV-6A infection. Our work clearly outlines the blockade provoked by U54 expression. By interacting with CaN, U54 likely prevents adequate interactions between CaN and NFAT, leading to insufficient dephosphorylation of NFAT. As a consequence, the NFAT nuclear import signal remains unexposed, and it remains cytoplasmic and unable to transactivate the IL-2 promoter. Despite having reduced inhibitory abilities, the U54 GISAA mutant was capable of interacting with CaN as efficiently as WT U54, suggesting that the U54 domain interacting with CaN differs from the GISIT inhibitory domain. The effects of U54 were observed in several cell types and, most importantly, in physiologically relevant T cells. In the presence of HHV-6B U54 protein, T cells are unable to synthesize IL-2 to an appreciable level. Since U54 is a tegument protein that is part of the virion, cells will be immediately exposed to U54 following viral entry. Whether this amount of U54 is sufficient to bind CaN and inhibit IL-2 synthesis remains to be determined, but considering that tegument proteins are among the most abundant proteins in viral particles, making up to 50% of the protein content (56, 57), it can be argued that a sufficient amount of U54 is present upon viral entry. Furthermore, U54 expression would be turned on during infection, resulting in an appreciable quantity of inhibitory protein.
In summary, our work describes a new mechanism utilized by HHV-6B to prevent the development of specific antiviral T cell expansion and likely favoring its persistence within the infected host. In view of this, it would be of interest to fully characterize the entire U54 motifs capable of inhibiting CaN-NFAT interaction, as was done for A238L protein, for which the inhibitory domain was mapped to amino acids 157 to 238, extending beyond the PKIIITG peptide. It would also be of interest to evaluate the impact of U54 on other cytokine genes whose expression is regulated by NFAT, such as those encoding Foxp3 (58), IL-21 (59), and IL-17 (60), among others (27). NFAT proteins are also key elements regulating numerous cell processes and in particular are involved in pathologies such as cancers (61). It could therefore be interesting and informative to test the impact of U54 expression in cancer cell lines and possibly observe NFAT-dependent anti-proliferative effects; such a study is reported in the accompanying article (62).
ACKNOWLEDGMENTS
This work was supported by a grant from the Canadian Institute of Health Research of Canada to L.F.
We thank Sachiko Sato and especially Julie-Christine Lévesque from the Bio-imagery Platform of the Centre de Recherche en Infectiologie, who performed live-cell experiments. We thank Marc Pouliot for kindly providing the mouse monoclonal anti-NFAT2 antibody.
Footnotes
Published ahead of print 13 August 2014
REFERENCES
- 1.Ablashi DV, Balachandran N, Josephs SF, Hung CL, Krueger GR, Kramarsky B, Salahuddin SZ, Gallo RC. 1991. Genomic polymorphism, growth properties, and immunologic variations in human herpesvirus-6 isolates. Virology 184:545–552. 10.1016/0042-6822(91)90424-A. [DOI] [PubMed] [Google Scholar]
- 2.Ablashi D, Agut H, Alvarez-Lafuente R, Clark DA, Dewhurst S, Diluca D, Flamand L, Frenkel N, Gallo R, Gompels UA, Hollsberg P, Jacobson S, Luppi M, Lusso P, Malnati M, Medveczky P, Mori Y, Pellett PE, Pritchett JC, Yamanishi K, Yoshikawa T. 2014. Classification of HHV-6A and HHV-6B as distinct viruses. Arch. Virol. 159:863–870. 10.1007/s00705-013-1902-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adams MJ, Carstens EB. 2010. Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2012). Arch. Virol. 157:1411–1422. 10.1007/s00705-012-1299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Salahuddin SZ, Ablashi DV, Markham PD, Josephs SF, Sturzenegger S, Kaplan M, Halligan G, Biberfeld P, Wong-Staal F, Kramarsky B, et al. 1986. Isolation of a new virus, HBLV, in patients with lymphoproliferative disorders. Science 234:596–601. 10.1126/science.2876520. [DOI] [PubMed] [Google Scholar]
- 5.Lopez C, Pellett P, Stewart J, Goldsmith C, Sanderlin K, Black J, Warfield D, Feorino P. 1988. Characteristics of human herpesvirus-6. J. Infect. Dis. 157:1271–1273. 10.1093/infdis/157.6.1271. [DOI] [PubMed] [Google Scholar]
- 6.Yamanishi K, Okuno T, Shiraki K, Takahashi M, Kondo T, Asano Y, Kurata T. 1988. Identification of human herpesvirus-6 as a causal agent for exanthem subitum. Lancet i:1065–1067. [DOI] [PubMed] [Google Scholar]
- 7.Scheurer ME, Pritchett JC, Amirian ES, Zemke NR, Lusso P, Ljungman P. 2013. HHV-6 encephalitis in umbilical cord blood transplantation: a systematic review and meta-analysis. Bone Marrow Transplant. 48:574–580. 10.1038/bmt.2012.180. [DOI] [PubMed] [Google Scholar]
- 8.Lusso P, Markham PD, Tschachler E, di Marzo Veronese F, Salahuddin SZ, Ablashi DV, Pahwa S, Krohn K, Gallo RC. 1988. In vitro cellular tropism of human B-lymphotropic virus (human herpesvirus-6). J. Exp. Med. 167:1659–1670. 10.1084/jem.167.5.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takahashi K, Sonoda S, Higashi K, Kondo T, Takahashi H, Takahashi M, Yamanishi K. 1989. Predominant CD4 T-lymphocyte tropism of human herpesvirus 6-related virus. J. Virol. 63:3161–3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lusso P, Malnati M, De Maria A, Balotta C, DeRocco SE, Markham PD, Gallo RC. 1991. Productive infection of CD4+ and CD8+ mature human T cell populations and clones by human herpesvirus 6. Transcriptional down-regulation of CD3. J. Immunol. 147:685–691. [PubMed] [Google Scholar]
- 11.Lusso P, De Maria A, Malnati M, Lori F, DeRocco SE, Baseler M, Gallo RC. 1991. Induction of CD4 and susceptibility to HIV-1 infection in human CD8+ T lymphocytes by human herpesvirus 6. Nature 349:533–535. 10.1038/349533a0. [DOI] [PubMed] [Google Scholar]
- 12.Flamand L, Romerio F, Reitz MS, Gallo RC. 1998. CD4 promoter transactivation by human herpesvirus 6. J. Virol. 72:8797–8805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang F, Yao K, Yin QZ, Zhou F, Ding CL, Peng GY, Xu J, Chen Y, Feng DJ, Ma CL, Xu WR. 2006. Human herpesvirus-6-specific interleukin 10-producing CD4+ T cells suppress the CD4+ T-cell response in infected individuals. Microbiol. Immunol. 50:787–803. 10.1111/j.1348-0421.2006.tb03855.x. [DOI] [PubMed] [Google Scholar]
- 14.Grivel JC, Ito Y, Faga G, Santoro F, Shaheen F, Malnati MS, Fitzgerald W, Lusso P, Margolis L. 2001. Suppression of CCR5- but not CXCR4-tropic HIV-1 in lymphoid tissue by human herpesvirus 6. Nat. Med. 7:1232–1235. 10.1038/nm1101-1232. [DOI] [PubMed] [Google Scholar]
- 15.Jaworska J, Gravel A, Fink K, Grandvaux N, Flamand L. 2007. Inhibition of transcription of the beta interferon gene by the human herpesvirus 6 immediate-early 1 protein. J. Virol. 81:5737–5748. 10.1128/JVI.02443-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jaworska J, Gravel A, Flamand L. 2010. Divergent susceptibilities of human herpesvirus 6 variants to type I interferons. Proc. Natl. Acad. Sci. U. S. A. 107:8369–8374. 10.1073/pnas.0909951107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Flamand L, Gosselin J, Stefanescu I, Ablashi D, Menezes J. 1995. Immunosuppressive effect of human herpesvirus 6 on T-cell functions: suppression of interleukin-2 synthesis and cell proliferation. Blood 85:1263–1271. [PubMed] [Google Scholar]
- 18.Horvat RT, Parmely MJ, Chandran B. 1993. Human herpesvirus 6 inhibits the proliferative responses of human peripheral blood mononuclear cells. J. Infect. Dis. 167:1274–1280. 10.1093/infdis/167.6.1274. [DOI] [PubMed] [Google Scholar]
- 19.Wang FZ, Larsson K, Linde A, Ljungman P. 2002. Human herpesvirus 6 infection and cytomegalovirus-specific lymphoproliferative responses in allogeneic stem cell transplant recipients. Bone Marrow Transplant. 30:521–526. 10.1038/sj.bmt.1703657. [DOI] [PubMed] [Google Scholar]
- 20.Gillis S, Smith KA. 1977. Long term culture of tumour-specific cytotoxic T cells. Nature 268:154–156. 10.1038/268154a0. [DOI] [PubMed] [Google Scholar]
- 21.Malek TR. 2008. The biology of interleukin-2. Annu. Rev. Immunol. 26:453–479. 10.1146/annurev.immunol.26.021607.090357. [DOI] [PubMed] [Google Scholar]
- 22.Morgan DA, Ruscetti FW, Gallo R. 1976. Selective in vitro growth of T lymphocytes from normal human bone marrows. Science 193:1007–1008. 10.1126/science.181845. [DOI] [PubMed] [Google Scholar]
- 23.Jain J, Loh C, Rao A. 1995. Transcriptional regulation of the IL-2 gene. Curr. Opin. Immunol. 7:333–342. 10.1016/0952-7915(95)80107-3. [DOI] [PubMed] [Google Scholar]
- 24.Serfling E, Avots A, Neumann M. 1995. The architecture of the interleukin-2 promoter: a reflection of T lymphocyte activation. Biochim. Biophys. Acta 1263:181–200. 10.1016/0167-4781(95)00112-T. [DOI] [PubMed] [Google Scholar]
- 25.Shaw J, Meerovitch K, Bleackley RC, Paetkau V. 1988. Mechanisms regulating the level of IL-2 mRNA in T lymphocytes. J. Immunol. 140:2243–2248. [PubMed] [Google Scholar]
- 26.Boise LH, Petryniak B, Mao X, June CH, Wang CY, Lindsten T, Bravo R, Kovary K, Leiden JM, Thompson CB. 1993. The NFAT-1 DNA binding complex in activated T cells contains Fra-1 and JunB. Mol. Cell. Biol. 13:1911–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hogan PG, Chen L, Nardone J, Rao A. 2003. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 17:2205–2232. 10.1101/gad.1102703. [DOI] [PubMed] [Google Scholar]
- 28.Macian F. 2005. NFAT proteins: key regulators of T-cell development and function. Nat. Rev. Immunol. 5:472–484. 10.1038/nri1632. [DOI] [PubMed] [Google Scholar]
- 29.Serfling E, Berberich-Siebelt F, Chuvpilo S, Jankevics E, Klein-Hessling S, Twardzik T, Avots A. 2000. The role of NF-AT transcription factors in T cell activation and differentiation. Biochim. Biophys. Acta 1498:1–18. 10.1016/S0167-4889(00)00082-3. [DOI] [PubMed] [Google Scholar]
- 30.Aramburu J, Garcia-Cozar F, Raghavan A, Okamura H, Rao A, Hogan PG. 1998. Selective inhibition of NFAT activation by a peptide spanning the calcineurin targeting site of NFAT. Mol. Cell 1:627–637. 10.1016/S1097-2765(00)80063-5. [DOI] [PubMed] [Google Scholar]
- 31.Aramburu J, Yaffe MB, Lopez-Rodriguez C, Cantley LC, Hogan PG, Rao A. 1999. Affinity-driven peptide selection of an NFAT inhibitor more selective than cyclosporin A. Science 285:2129–2133. 10.1126/science.285.5436.2129. [DOI] [PubMed] [Google Scholar]
- 32.Garcia-Cozar FJ, Okamura H, Aramburu JF, Shaw KT, Pelletier L, Showalter R, Villafranca E, Rao A. 1998. Two-site interaction of nuclear factor of activated T cells with activated calcineurin. J. Biol. Chem. 273:23877–23883. 10.1074/jbc.273.37.23877. [DOI] [PubMed] [Google Scholar]
- 33.Campeau E, Ruhl VE, Rodier F, Smith CL, Rahmberg BL, Fuss JO, Campisi J, Yaswen P, Cooper PK, Kaufman PD. 2009. A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS One 4:e6529. 10.1371/journal.pone.0006529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tomoiu A, Gravel A, Tanguay RM, Flamand L. 2006. Functional interaction between human herpesvirus 6 immediate-early 2 protein and ubiquitin-conjugating enzyme 9 in the absence of sumoylation. J. Virol. 80:10218–10228. 10.1128/JVI.00375-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Josephs SF, Salahuddin SZ, Ablashi DV, Schachter F, Wong-Staal F, Gallo RC. 1986. Genomic analysis of the human B-lymphotropic virus (HBLV). Science 234:601–603. 10.1126/science.3020691. [DOI] [PubMed] [Google Scholar]
- 36.Northrop JP, Ullman KS, Crabtree GR. 1993. Characterization of the nuclear and cytoplasmic components of the lymphoid-specific nuclear factor of activated T cells (NF-AT) complex. J. Biol. Chem. 268:2917–2923. [PubMed] [Google Scholar]
- 37.Hoey T, Sun YL, Williamson K, Xu X. 1995. Isolation of two new members of the NF-AT gene family and functional characterization of the NF-AT proteins. Immunity 2:461–472. 10.1016/1074-7613(95)90027-6. [DOI] [PubMed] [Google Scholar]
- 38.Gravel A, Gosselin J, Flamand L. 2002. Human herpesvirus 6 immediate-early 1 protein is a sumoylated nuclear phosphoprotein colocalizing with promyelocytic leukemia protein-associated nuclear bodies. J. Biol. Chem. 277:19679–19687. 10.1074/jbc.M200836200. [DOI] [PubMed] [Google Scholar]
- 39.Chow CW, Rincon M, Davis RJ. 1999. Requirement for transcription factor NFAT in interleukin-2 expression. Mol. Cell. Biol. 19:2300–2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Macian F, Garcia-Rodriguez C, Rao A. 2000. Gene expression elicited by NFAT in the presence or absence of cooperative recruitment of Fos and Jun. EMBO J. 19:4783–4795. 10.1093/emboj/19.17.4783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zerr DM, Corey L, Kim HW, Huang ML, Nguy L, Boeckh M. 2005. Clinical outcomes of human herpesvirus 6 reactivation after hematopoietic stem cell transplantation. Clin. Infect. Dis. 40:932–940. 10.1086/428060. [DOI] [PubMed] [Google Scholar]
- 42.Roffman E, Frenkel N. 1990. Interleukin-2 inhibits the replication of human herpesvirus-6 in mature thymocytes. Virology 175:591–594. 10.1016/0042-6822(90)90447-Y. [DOI] [PubMed] [Google Scholar]
- 43.Kalejta RF. 2008. Tegument proteins of human cytomegalovirus. Microbiol. Mol. Biol. Rev. 72:249–265. 10.1128/MMBR.00040-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tomtishen JP., III 2012. Human cytomegalovirus tegument proteins (pp65, pp71, pp150, pp28). Virol. J. 9:22. 10.1186/1743-422X-9-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sieber M, Baumgrass R. 2009. Novel inhibitors of the calcineurin/NFATc hub—alternatives to CsA and FK506? Cell Commun. Signal. 7:25. 10.1186/1478-811X-7-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fruman DA, Klee CB, Bierer BE, Burakoff SJ. 1992. Calcineurin phosphatase activity in T lymphocytes is inhibited by FK 506 and cyclosporin A. Proc. Natl. Acad. Sci. U. S. A. 89:3686–3690. 10.1073/pnas.89.9.3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Perrino BA. 1999. Regulation of calcineurin phosphatase activity by its autoinhibitory domain. Arch. Biochem. Biophys. 372:159–165. 10.1006/abbi.1999.1485. [DOI] [PubMed] [Google Scholar]
- 48.Sagoo JK, Fruman DA, Wesselborg S, Walsh CT, Bierer BE. 1996. Competitive inhibition of calcineurin phosphatase activity by its autoinhibitory domain. Biochem. J. 320:879–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roehrl MH, Kang S, Aramburu J, Wagner G, Rao A, Hogan PG. 2004. Selective inhibition of calcineurin-NFAT signaling by blocking protein-protein interaction with small organic molecules. Proc. Natl. Acad. Sci. U. S. A. 101:7554–7559. 10.1073/pnas.0401835101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gebert B, Fischer W, Weiss E, Hoffmann R, Haas R. 2003. Helicobacter pylori vacuolating cytotoxin inhibits T lymphocyte activation. Science 301:1099–1102. 10.1126/science.1086871. [DOI] [PubMed] [Google Scholar]
- 51.Miskin JE, Abrams CC, Goatley LC, Dixon LK. 1998. A viral mechanism for inhibition of the cellular phosphatase calcineurin. Science 281:562–565. 10.1126/science.281.5376.562. [DOI] [PubMed] [Google Scholar]
- 52.Granja AG, Perkins ND, Revilla Y. 2008. A238L inhibits NF-ATc2, NF-kappa B, and c-Jun activation through a novel mechanism involving protein kinase C-theta-mediated up-regulation of the amino-terminal transactivation domain of p300. J. Immunol. 180:2429–2442. 10.4049/jimmunol.180.4.2429. [DOI] [PubMed] [Google Scholar]
- 53.Granja AG, Nogal ML, Hurtado C, Vila V, Carrascosa AL, Salas ML, Fresno M, Revilla Y. 2004. The viral protein A238L inhibits cyclooxygenase-2 expression through a nuclear factor of activated T cell-dependent transactivation pathway. J. Biol. Chem. 279:53736–53746. 10.1074/jbc.M406620200. [DOI] [PubMed] [Google Scholar]
- 54.Matsuda S, Shibasaki F, Takehana K, Mori H, Nishida E, Koyasu S. 2000. Two distinct action mechanisms of immunophilin-ligand complexes for the blockade of T-cell activation. EMBO Rep. 1:428–434. 10.1093/embo-reports/kvd090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Miskin JE, Abrams CC, Dixon LK. 2000. African swine fever virus protein A238L interacts with the cellular phosphatase calcineurin via a binding domain similar to that of NFAT. J. Virol. 74:9412–9420. 10.1128/JVI.74.20.9412-9420.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Loret S, Guay G, Lippe R. 2008. Comprehensive characterization of extracellular herpes simplex virus type 1 virions. J. Virol. 82:8605–8618. 10.1128/JVI.00904-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Varnum SM, Streblow DN, Monroe ME, Smith P, Auberry KJ, Pasa-Tolic L, Wang D, Camp DG, II, Rodland K, Wiley S, Britt W, Shenk T, Smith RD, Nelson JA. 2004. Identification of proteins in human cytomegalovirus (HCMV) particles: the HCMV proteome. J. Virol. 78:10960–10966. 10.1128/JVI.78.20.10960-10966.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tone Y, Furuuchi K, Kojima Y, Tykocinski ML, Greene MI, Tone M. 2008. Smad3 and NFAT cooperate to induce Foxp3 expression through its enhancer. Nat. Immunol. 9:194–202. 10.1038/ni1549. [DOI] [PubMed] [Google Scholar]
- 59.Kim HP, Korn LL, Gamero AM, Leonard WJ. 2005. Calcium-dependent activation of interleukin-21 gene expression in T cells. J. Biol. Chem. 280:25291–25297. 10.1074/jbc.M501459200. [DOI] [PubMed] [Google Scholar]
- 60.Shen F, Hu Z, Goswami J, Gaffen SL. 2006. Identification of common transcriptional regulatory elements in interleukin-17 target genes. J. Biol. Chem. 281:24138–24148. 10.1074/jbc.M604597200. [DOI] [PubMed] [Google Scholar]
- 61.Muller MR, Rao A. 2010. NFAT, immunity and cancer: a transcription factor comes of age. Nat. Rev. Immunol. 10:645–656. 10.1038/nri2818. [DOI] [PubMed] [Google Scholar]
- 62.Iampietro M, Gravel A, Flamand L. 2014. Inhibition of breast cancer cell proliferation throguh disturbance of the calcineurin/NFAT pathway by human herpesvirus 6B U54 tegument protein. J. Virol. 88:12910–12914. 10.1128/JVI.02107-14. [DOI] [PMC free article] [PubMed] [Google Scholar]