

ABSTRACT
Epstein-Barr virus (EBV) fusion with an epithelial cell requires virus glycoproteins gHgL and gB and is triggered by an interaction between gHgL and integrin αvβ5, αvβ6, or αvβ8. Fusion with a B cell requires gHgL, gp42, and gB and is triggered by an interaction between gp42 and human leukocyte antigen class II. We report here that, like alpha- and betaherpesviruses, EBV, a gammaherpesvirus, can mediate cell fusion if gB and gHgL are expressed in trans. Entry of a gH-null virus into an epithelial cell is possible if the epithelial cell expresses gHgL, and entry of the same virus, which phenotypically lacks gHgL and gp42, into a B cell expressing gHgL is possible in the presence of a soluble integrin. Heat is capable of inducing the fusion of cells expressing only gB, and the proteolytic digestion pattern of gB in virions changes in the same way following the exposure of virus to heat or to soluble integrins. It is suggested that the Gibbs free energy released as a result of the high-affinity interaction of gHgL with an integrin contributes to the activation energy required to cause the refolding of gB from a prefusion to a postfusion conformation.
IMPORTANCE The core fusion machinery of herpesviruses consists of glycoproteins gB and gHgL. We demonstrate that as in alpha- and betaherpesvirus, gB and gHgL of the gammaherpesvirus EBV can mediate fusion and entry when expressed in trans in opposing membranes, implicating interactions between the ectodomains of the proteins in the activation of fusion. We further show that heat and exposure to a soluble integrin, both of which activate fusion, result in the same changes in the proteolytic digestion pattern of gB, possibly representing the refolding of gB from its prefusion to its postfusion conformation.
INTRODUCTION
Epstein-Barr virus (EBV), one of the two known human oncogenic gammaherpesviruses, primarily infects B lymphocytes and epithelial cells. An infection by the orally transmitted virus, which eventually occurs in more than 95% of the global population, is never cleared (1). Instead, a reservoir of latent infection is established in long-lived memory B cells with sporadic terminal differentiation of infected cells into plasma cells, leading to the reactivation of virus replication and cell death (2). Virus released from B cells is thought to be amplified in epithelial cells for the replenishment of the latent reservoir or shedding in saliva for transmission to a new host (3, 4).
As with all herpesviruses, EBV enters cells as its envelope fuses with a membrane of the cell. Fusion with B cells requires endocytosis and occurs in a low pH compartment, although low pH is not required (5). Fusion with epithelial cells occurs at neutral pH and, in at least some cell lines, is thought to take place at the cell surface (5). Both events require the activation of the herpesvirus core fusion machinery, comprised of the conserved herpesvirus glycoproteins gB and gHgL (6). The crystal structure of EBV gB, which is a type I single-pass membrane protein, reveals that it exists as a trimer (7). The trimer resembles the postfusion structure of the class III fusion proteins vesicular stomatitis virus glycoprotein G and baculovirus gp64, both of which are necessary and sufficient for fusion. A similar structure has been solved for herpes simplex virus glycoprotein gB (8), and the gB homologues of both viruses are thought to be the final executors of fusion. However, in contrast to the vesicular stomatitis virus and baculovirus fusion proteins, none of the herpesvirus gB homologues can mediate fusion in the absence of gHgL.
Like gB, gH is a single-pass type I membrane protein, whereas gL is a peripheral membrane protein from which the signal peptide is cleaved. The crystal structure of the EBV gHgL dimer has a linear arrangement of 4 domains, and membrane distal domain I is formed by both gH and gL (9). The dimer resembles no known fusion protein, and its role generally is thought to be that of a regulator required to initiate the fusion-driving conversion of gB from a prefusion to a postfusion structure, although, to this point, there has been no experimental confirmation that EBV gB undergoes refolding (10).
The events responsible for the activation of a potential regulator such as gHgL differ for B cells and epithelial cells. The activation of fusion in the endosome of a B cell occurs as a result of an interaction between human leukocyte antigen class II (HLA class II) and a fourth gammaherpesvirus-specific glycoprotein, gp42 (11). Glycoprotein gp42, as a cleaved type II membrane protein, forms a tripartite complex with gHgL by virtue of its ability to bind to a region of gHgL which overlaps domain II (12, 13). The binding of gp42 to HLA class II changes its conformation, and this conformational change probably contributes to the activation of fusion (14). The activation of fusion with an epithelial cell, however, involves the direct interaction of gHgL with one of three integrins, αvβ5, αvβ6, or αvβ8 (15, 16). There is a KGD motif in domain II of gHgL which is responsible for this interaction. Integrin binding results in a conformational shift between domain I and domain II which, like the change in gp42 caused by HLA class II binding, may contribute to the activation of fusion (16, 17).
Because gp42 and integrins bind to overlapping sites in domain II of gHgL, the presence of gp42 in a complex with gHgL precludes the ability of the complex to interact with an integrin. Thus, EBV virions carry trimers of gHgLgp42 and dimers of gHgL in order to infect both B cells, which lack the necessary αv integrins, and epithelial cells, which do not constitutively express HLA class II (12). The ratios of the two types of complex vary according to the cell in which the virus is made. Virus made in a B cell loses three-part complexes as a result of trafficking with HLA class II to the proteolytic antigen-processing compartment. Emerging virions are enriched for gHgL dimers and are approximately 5-fold better able to infect epithelial cells than virus made in an epithelial cell. In contrast, virions made in an HLA class II-negative epithelial cell are enriched for trimers of gHgLgp42 and can be as much as a hundred-fold more infectious for B cells than virus made in a B cell. This switching of tropism is thought to facilitate the cycling of virus between B cells and epithelial cells (18).
Recent studies of herpes simplex virus (HSV) (19) and human cytomegalovirus (HCMV) fusion (20, 21) have revealed that the gB and gHgL homologues of these viruses can mediate cell fusion (19, 21) and virus entry (20) if they are expressed in opposing membranes. Although the fusion in trans mediated by HSV proteins, which were expressed from plasmid vectors, was not as efficient as fusion in cis, fusion in trans mediated by HCMV proteins, which were expressed at high levels from nonreplicating adenovirus vectors, was equally as efficient as fusion in cis. This implies that the ectodomains of the proteins are of paramount importance to the execution of fusion. We report here that EBV gB and gHgL likewise can mediate not only fusion but also entry in trans into either an epithelial cell or into a B cell after exposure to a soluble integrin.
Heat, as a source of free energy, can act as a surrogate trigger of transitions from prefusion to postfusion conformations of the paramyxovirus F proteins (22–25) and the influenza virus hemagglutinin (26, 27). We similarly found that the exposure of cells expressing EBV gB to heat in the absence of gHgL resulted in fusion. The proteolytic digestion pattern of gB was changed in the same way when exposed to heat as when triggered by an integrin in the presence of gHgL, discriminating between what may be the prefusion and postfusion conformations of EBV gB and supporting the hypothesis that the protein undergoes a conformational change during fusion, possibly aided by the Gibbs free energy that would be released by high-affinity binding of gHgL to an integrin.
MATERIALS AND METHODS
Cells and virus.
Akata-GFP, a Burkitt lymphoma-derived cell line carrying a recombinant EBV in which the thymidine kinase gene is interrupted with a double cassette expressing neomycin resistance and green fluorescence protein (GFP), Akata-gH-null, carrying a recombinant EBV in which the BXLF2 open reading frame encoding gH is interrupted with the same cassette (28), and EBV-negative Akata cells, which have lost EBV episomes (29), were grown in RPMI 1640 (Sigma). AGS, human gastric carcinoma cells (American Type Culture Collection) that have been cured of parainfluenza type 5 infection by treatment with ribavirin, and CHO cells were grown in Ham's F-12 medium (Sigma). 293 cells, 293T14 cells expressing T7 RNA polymerase (30), and 293-B8 AVAP cells, which secrete truncated αvβ8 conjugated to alkaline phosphatase (31), were grown in Dulbecco's modified Eagle medium (DMEM) (Sigma) supplemented with nonessential amino acids (Gibco). HaCaT cells (American Type Culture Collection) were grown in DMEM-low glucose (HyClone). Media for all mammalian cells also were supplemented with 10% heat-inactivated fetal bovine serum (Gibco). Insect Sf9 cells were grown in Sf900 II medium (Invitrogen) and infected at a multiplicity of infection of 3 with baculovirus expressing a truncated form of gH together with gL (gHtgL) (32). Akata virus made in B cells was harvested from the spent culture media of cells that had been induced by treatment with anti-human immunoglobulin as described previously (28). Akata virus made in epithelial cells was harvested from spent culture media of AGS cells 5 days after induction with 12-O-tetradecanoylphorbol-13-acetate (Sigma) and 2.5 mM sodium butyrate (Calbiochem). In both cases, virus was concentrated by centrifugation as described previously (28).
Antibodies.
Antibodies used included monoclonal antibodies CL59 to gH (28), CL55 to gB (33), 37E1 to αvβ8 (31), 10D5 to αvβ6, and P1F6 to αvβ5 (Chemicon), a rabbit polyclonal antibody (RgB) made to residues 754 to 857 of gB fused to glutathione-S-transferase, goat anti-rabbit IRDye 800 CW (LiCor Biosciences), goat anti-mouse antibody coupled to Alexa Fluor 647 (Life Technologies), sheep anti-mouse coupled to fluorescein isothiocyanate (FITC) (MP Biomedics), and sheep anti-mouse coupled to phycoerythrin (Jackson ImmunoResearch). Antibodies CL59, CL55, 37E1, and RgB were purified by affinity chromatography on protein A coupled with agarose (RepliGen).
Integrin and gHtgL purification.
Soluble truncated gHtgL and integrin αvβ8 were purified from spent culture media as previously described (15, 16).
Fusion assays.
Fusion was scored as previously described (15) either visually, in cells stained for glycoproteins by antibody CL55 or CL59, or in a luciferase-based assay in which one population of cells transfected with a plasmid expressing luciferase under the control of a T7 promoter was overlaid with 293T14 cells expressing T7 polymerase. To compare fusion in trans to fusion in cis, either cells were transfected with various combinations of plasmids pCAGGS, pCAGGS-gH, pCAGGS-gL, and pCAGGS-gB in cis or two separate populations of cells were transfected with combinations of plasmids. Twenty-four h later, cells were trypsinized and reseeded together. Fusion was measured after a further 24 h of incubation. Fusion of AGS cells supported by soluble gHtgL was obtained by transfecting cells with pCAGGS-gB for 24 h, adding 40 nM soluble gHtgL, a saturation concentration (34), or 40 nM soluble gHtgL preincubated with 6 nM soluble integrin αvβ8. Fusion was scored visually after an additional 24 h and expressed as a percentage of fusion achieved when gHgL and gB were expressed in cis. The induction of fusion by heat was done by transfecting AGS cells on culture slides with pCAGGS-gH and pCAGGS-gL or with pCAGGS-gB. Thirty-six h later, when cells reached approximately 90% confluence, the slides were placed in a preheated hybridization oven with the temperature maintained with a thermostat and additionally monitored with a digital thermometer (Fisher) with an accuracy of ±0.2°C. Following exposure to heat for a set time, cool media were added to adjust the temperature back to 37°C, and the slides were incubated at 37°C for a further 16 h. Fusion was scored visually and compared to fusion achieved in cells transfected with pCAGGS-gH, pCAGGS-gL, and pCAGGS-gB and incubated at 37°C throughout.
Cell infection.
Five hundred thousand 293 cells were seeded in 6-well plates, and 5 h later they were transfected with pCAGGS-gH and pCAGGS-gL or with pCAGGS-gB. After a further 36 h, cells were infected with equal genome copy numbers of either Akata-GFP virus or Akata-gH-null virus. Both viruses were bound at 37°C 1 h. Unbound virus was removed, and cells were washed and incubated at 37°C for 48 h. Cells were stained with CL59 to gH or CL55 to gB and anti-mouse antibody coupled to Alexa Fluor 647. Glycoprotein expression and infection were measured by flow cytometry. CHO cells were transfected with pCAGGS-CR2 (35) or with pCAGGS-CR2, pCAGGS-gH, and pCAGGS-gL. Twenty-four h later they were infected with Akata-GFP virus or Akata-gH-null virus as described above. At 72 h postinfection, cells were examined visually for GFP expression. Two million EBV-negative Akata cells were nucleofected with pCAGGS-gH and pCAGGS-gL or with pCAGGS alone using an Amaxa nucleofector 4-D and program CA137. Thirty-six h later, cells were infected with Akata GFP virus or Akata-gH-null virus. Virus was bound for 30 min in the cold, unbound virus was removed, and cells and bound virus were overlaid with 100 μl media or media containing soluble αvβ8 at 37°C. Two h later, medium was added and cells were incubated for 48 h. Cells were stained with CL59 to gH or CL55 to gB and anti-mouse antibody coupled to Alexa Fluor 647. Glycoprotein expression and infection were measured by flow cytometry.
EBV digestion with proteinase K and Western blotting.
Concentrated Akata-GFP virus was either preincubated at temperatures between 37°C and 45°C for 30 min, preincubated with 0.6 nM to 6 nM αvβ8 at room temperature, or kept at room temperature without treatment. Samples were left undigested or were digested with proteinase K (10 μg/ml) for 10 min at room temperature before analysis by SDS-PAGE on a 12.5% reducing gel. Proteins were transferred to a nitrocellulose membrane (Bio-Rad) and reacted with RgB to the cytoplasmic tail of gB and goat anti-rabbit IRDye 800 CW secondary antibody. Fluorescence was read using an Odyssey infrared imager (LiCor Biosciences).
RESULTS
Epithelial cell fusion occurs when gB and gHgL are expressed in trans.
To determine whether EBV gB and gHgL can mediate fusion in trans, we first used a cell-based fusion assay in which fusion is scored visually. Three separate dishes of AGS cells were transfected with plasmids expressing gB, gHgL, or both gB and gHgL. Twenty-four h later, cells were overlaid or trypsinized and reseeded in groups that consisted of either 50% of cells expressing gB and gHgL and 50% of cells transfected with empty vector or 50% of cells expressing gB and 50% of cells expressing gHgL. More than 80% of the level of fusion seen if gHgL and gB were expressed in cis was seen when proteins were expressed in trans (Fig. 1A). Similar experiments were done in which AGS cells were transfected with plasmids expressing gB or gHgL and gB and reseeded with CHO cells (Fig. 1B) or HaCaT cells (Fig. 1C) transfected with empty vector or with plasmids expressing gHgL. Once again, fusion was seen if glycoproteins were provided in cis or in trans, although the levels were lower, particularly in the case of the CHO cells, than those seen when all cells were AGS cells. This may reflect in part the fact that CHO cells do not express a human integrin with which EBV gHgL can productively interact. Fusion in trans also was achieved in a luciferase assay in which plasmids expressing gHgL were expressed in CHO cells and CHO cells were overlaid with 293T14 cells expressing gB (Fig. 1D).
FIG 1.
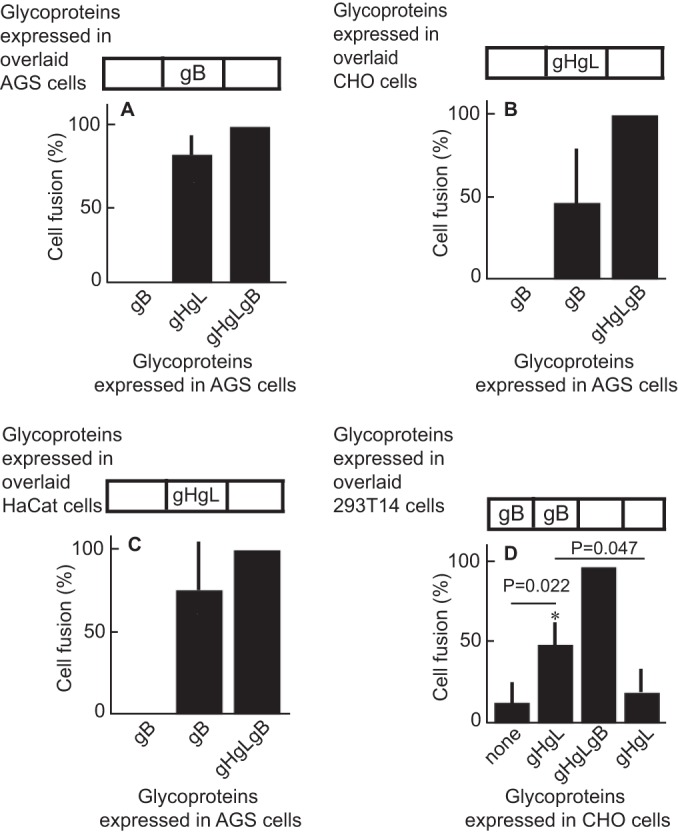
Glycoproteins gHgL and gB can trigger fusion of epithelial cells when expressed in trans. (A to C) AGS cells were transfected with plasmids expressing gHgL and gB in cis or with gHgL alone or gB alone and overlaid with AGS (A), CHO (B), or HaCat (C) cells transfected with empty vector, gHgL, or gB as indicated. Fusion was measured 24 h after cells were overlaid by staining with monoclonal antibody CL55 to gB and counting the percentage of stained cells that contained 4 or more nuclei. Fusion in trans was expressed as the percentage of fusion in cis. The values for fusion in cis were 24% ± 1.8% (A), 28% ± 5.3% (B), and 25% ± 2.7% (C). (D) CHO cells were transfected with a plasmid expressing luciferase under the control of a T7 promoter and with either empty vector, gHgL and gB in cis, or gHgL or gB alone and overlaid with 293T14 cells transfected with empty vector or gB as indicated. Fusion was measured in terms of luciferase activity. The error bars are the standard deviations from 4 experiments.
Cells expressing gHgL can be infected with a gH-null virus.
Cell-based fusion assays are subject to the criticism that the transfer of membrane from one cell to another might occur and allow transfer of glycoproteins, putting them back in cis instead of in trans. This concern is somewhat ameliorated by the observation that in HCMV cell-based fusion assays, fusion is as efficient when proteins are expressed in trans as in cis (21). Even more compelling, however, is the finding that entry of HCMV virions lacking gB can be effected if gB is expressed in target cells (20). An EBV virus lacking gB is not currently available. Therefore, we explored whether Akata-gH-null virus, which is phenotypically null for gHgL and gp42 (28), could infect cells expressing gHgL. The first experiments were done with CHO cells transfected with plasmids expressing the EBV receptor CR2 together with gHgL. These cells were chosen because, as noted above, they do not express any of the human integrins to which EBV gHgL can bind; thus, they presumably would not be subject to the receptor interference, which has been reported to occur when cells are transfected with HCMV gHgL (20). Fusion was triggered by the addition of soluble integrin αvβ8, which can trigger cell-to-cell fusion of CHO cells transfected with gB and gHgL in cis (15). Although only low levels of infection were seen, they were as high as those seen with Akata-GFP virus, which carries a full complement of glycoproteins (Fig. 2). To improve on this result, we attempted the infection of 293 cells, which naturally express CR2 and can be infected to higher levels with Akata-GFP virus than can CHO cells. 293 cells do express αvβ5 and αvβ8, although very little αvβ6; thus, they might be subject to receptor interference. However, after transfection of plasmids expressing gHgL, the expression levels of αv integrin were essentially unchanged (Fig. 3). Therefore, cells were transfected with plasmids expressing gHgL or gB and infected with Akata-gH-null virus. Glycoprotein expression and infection were measured by flow cytometry (Fig. 4). No infection by Akata-gH-null virus was seen in cells transfected with empty vector (Fig. 4A) or cells expressing gB (Fig. 4B). However, almost 10% of cells transfected with gHgL were infected with Akata-gH-null virus (Fig. 4C). This represented slightly more than 20% of the untreated cells that could be infected with Akata-GFP virus (Fig. 4D), which carries all virus glycoproteins.
FIG 2.
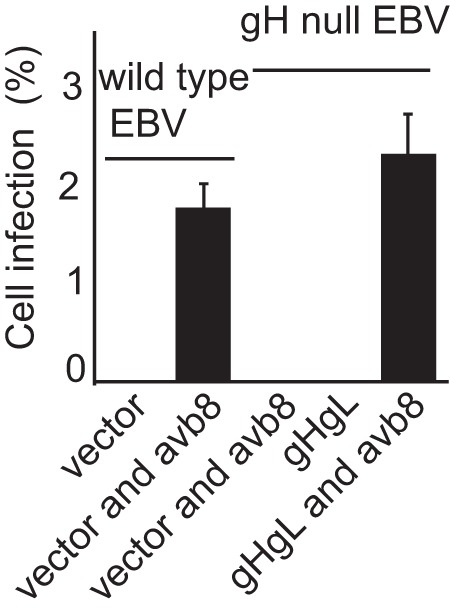
Integrins can induce infection of CHO cells expressing CR2 and gHgL with a gH-null virus. CHO cells were transfected with a plasmid expressing CR2 and empty vector or plasmids expressing CR2 and gHgL. Twenty-four h later, cells were infected with Akata-gH-null virus. Virus was bound for 2 h, and cells and virus then were overlaid with soluble αvβ8 as indicated. Two hours later additional medium was added, and GFP expression was determined visually at 3 days. The errors bars are the standard deviations from 6 experiments.
FIG 3.
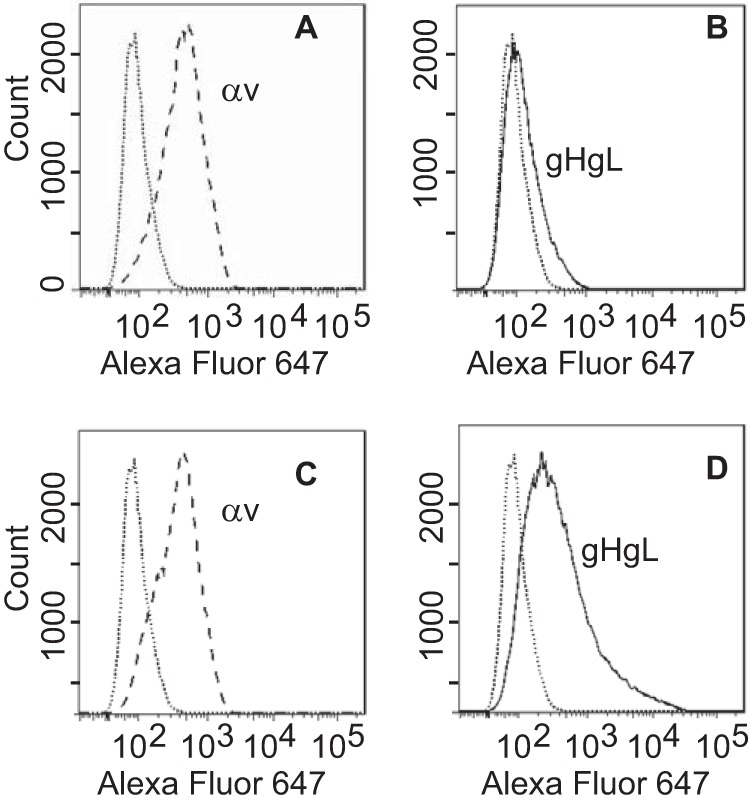
Expression of gHgL does not reduce αv integrin expression. 293 cells were transfected with an empty vector (A and B) or with plasmids expressing gHgL (C and D), and 48 h later they were stained either with monoclonal antibody CL59 to gH (B and D) or with monoclonal antibody L230 to αv integrin (A and C), followed by goat anti-mouse antibody coupled to Alexa Fluor 647. Isotype controls are indicated by dotted lines.
FIG 4.
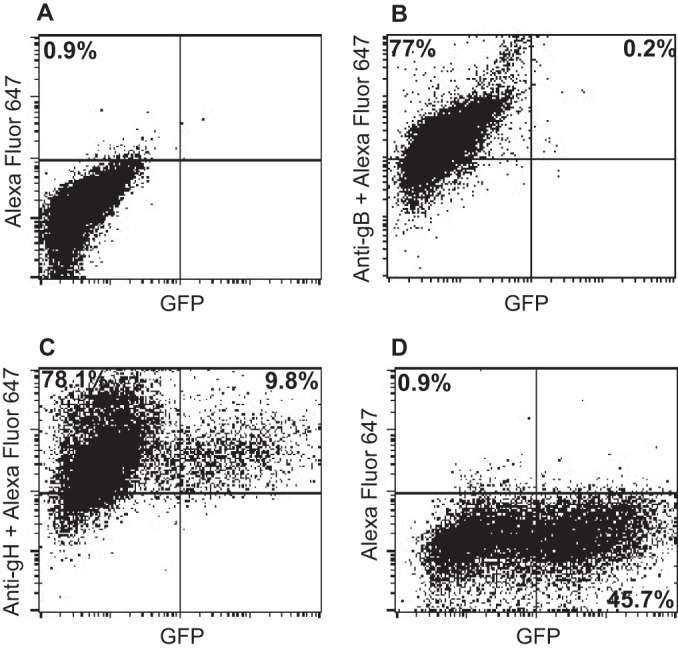
gH-null virus can infect 293 cells expressing gHgL but not cells expressing gB. 293 cells were transfected with empty vector (A and D) or with plasmids expressing gB (B) or gHgL (C), and 36 h later they were infected with Akata-gH-null virus (A, B, and C) or Akata-GFP virus (D). At 48 h postinfection, glycoprotein expression was detected using monoclonal antibody CL55 to gB (B) or monoclonal antibody CL59 to gHgL (C) and goat anti-mouse antibody conjugated to Alexa Fluor 647 (A to D), and infection was monitored in terms of GFP expression.
Since fusion and virus entry could be mediated by the expression of gB and gHgL in opposing membranes, we also asked whether soluble EBV gHtgL mediates fusion, as can, at low levels, soluble herpes simplex virus gHgL (19). AGS cells were transfected with plasmids expressing gB, and soluble gHtgL was added 24 h later. Only around 14% of fusion achieved by expressing gB and gHgL in cis was seen (Fig. 5). However, this low level was eliminated if the gHtgL were preincubated with soluble αvβ8, supporting its specificity.
FIG 5.
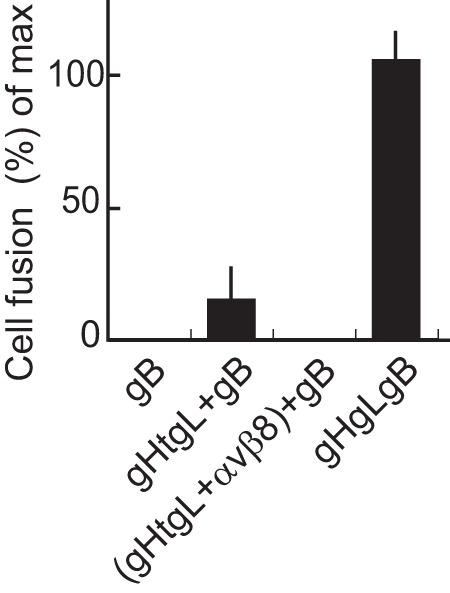
Soluble gHtgL is capable of supporting low levels of fusion. AGS cells were transfected with plasmids expressing gB or gB and gHgL, and 24 h later they were overlaid as indicated with soluble gHtgL or gHtgL that had been preincubated with soluble αvβ8. After a further 24 h, cells were fixed and stained with the monoclonal antibody CL55 to gB and FITC-conjugated goat anti-mouse antibody. Fusion was calculated as the percentage of stained cells containing 4 or more nuclei and is expressed as a percentage of that seen in cells transfected with gB and gHgL. The errors bars are standard deviations from 4 experiments.
Mutations in EBV gH have been shown to alter fusion with epithelial cells and B cells differentially (17, 36), raising the possibility that there is a fundamental difference between the two events. The availability of soluble integrins and the knowledge that fusion can proceed with proteins expressed in trans gave us the opportunity to confirm that elements of the process are shared. EBV fusion and entry of a B cell ultimately requires gB and gHgL, although the triggering of the core fusion machinery here requires an interaction between gp42 and HLA class II. We asked whether the Akata-gH-null virus, lacking gp42, could be triggered by a soluble integrin to enter a B cell expressing only gHgL. As expected, no gHgL-null virus could enter cells transfected with empty vector (Fig. 6A), no gHgL-null virus could enter cells expressing gB with or without the addition of a soluble integrin (Fig. 6C and D), and no gHgL-null virus could enter cells expressing gHgL in the absence of a soluble integrin (Fig. 6E). However, low levels of infection of cells expressing gHgL were clearly seen in the presence of a soluble integrin (Fig. 6F), reaching approximately 7% of the number of untransfected cells that could be infected with the same DNA copy number of wild-type Akata virus (Fig. 6B).
FIG 6.
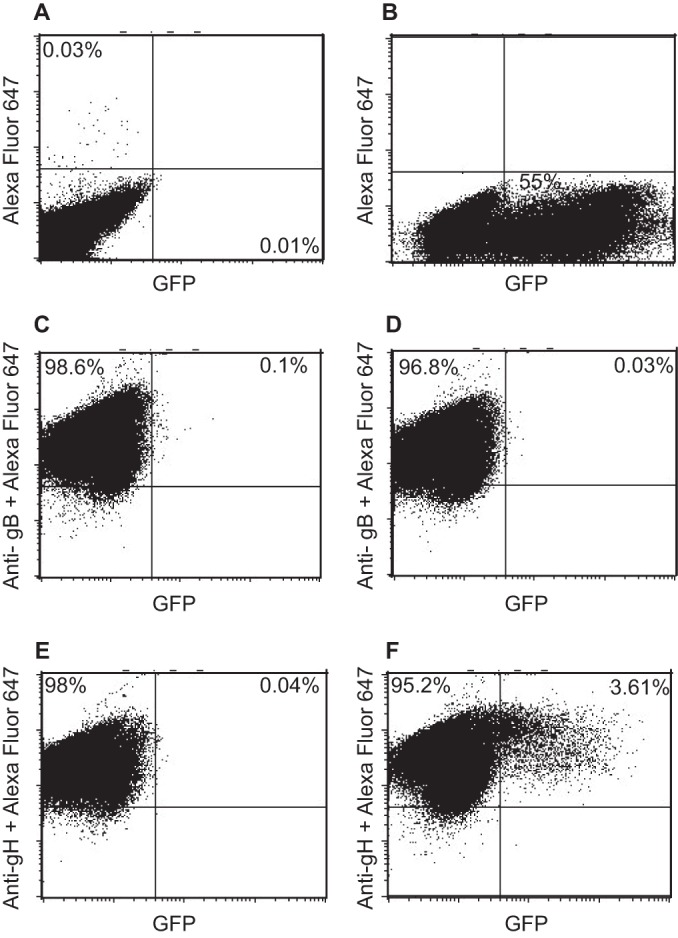
gHgL null virus can infect B cells expressing gHgL if triggered with soluble αvβ8. EBV-negative Akata B cells were nucleoporated with empty vector (A and B), with a plasmid expressing gB (C and D), or with plasmids expressing gH and gL (E and F). After 36 h, cells were bound to Akata-GFP virus (B) or Akata-gH-null virus (A and C to F) on ice for 30 min and then overlaid with medium alone (A, B, C, and E) or medium containing soluble αvβ8 (D and F). At 48 h postinfection, cells were stained with monoclonal antibody CL55 to gB (C and D), monoclonal antibody CL59 to gH (E and F), and goat anti-mouse antibody conjugated to Alexa Fluor 647 (A to F). Glycoprotein and GFP expression were measured by flow cytometry.
Exposure of cells expressing gB to transient heat shock can trigger fusion.
The ability of gB and gHgL to mediate fusion in trans implicates the ectodomains of the proteins in the process. Integrins bind gHgL with very high affinity. The thermodynamic KD (equilibrium dissociation constant) values for the interactions are between 2 × 10−9 and 6 × 10−9 M (16). Therefore, we considered whether the Gibbs free energy released by such a high-affinity interaction contributes to the process, possibly by helping to overcome an activation energy barrier needed to support the large conformational change proposed to occur as the prefusion form of gB adopts its postfusion conformation (10). Heat has been shown to act as a surrogate form of energy capable of destabilizing metastable virus fusion proteins (22–27). Therefore, we looked at the effect of heat on the ability of gB to mediate fusion. Cells expressing gB alone or gHgL alone were exposed for 20 min to different temperatures. Cells expressing gHgL alone showed no evidence of fusion (Fig. 7A), but cells transfected with gB alone supported low levels of fusion that plateaued at a temperature of 45°C and at approximately 30% of that seen in control cells transfected with gB and gHgL and maintained at 37°C throughout (Fig. 7B). Holding the cells for longer than 30 min at 45°C did not lead to any further increase in fusion (Fig. 7C). Cells expressing gB together with gHgL fused maximally at 37°C. Exposing such cells to 48°C for 20 min did not increase fusion (Fig. 7D).
FIG 7.
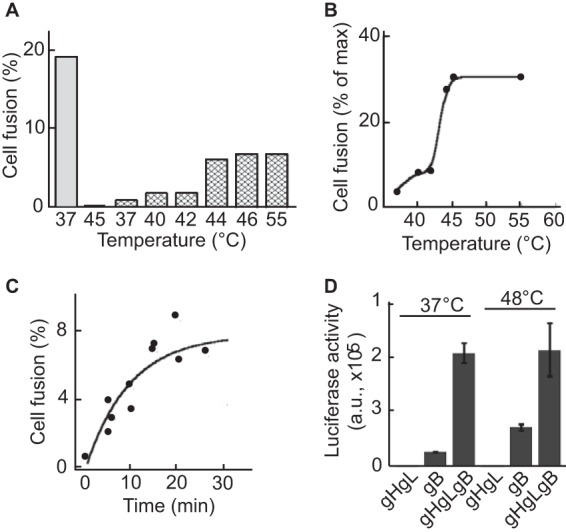
Increase of temperature triggers fusion of AGS cells expressing gB. (A) AGS cells were transfected with plasmids expressing gB, gH, and gL (gray bar), gH and gL alone (second bar from the left), or gB alone (hatched bars). Twenty-four h later, cells were incubated at the indicated temperature for 20 min, cooled, and returned to 37°C. After a further 16 h, cells were fixed and stained with monoclonal antibody CL55 to gB and FITC-conjugated goat anti-mouse antibody. Fusion was calculated as the percentage of stained cells containing 4 or more nuclei. (B) Graphical representation of data shown in panel A, where the maximum fusion (max) is that seen in cells transfected with plasmids expressing gB, gH, and gL and maintained at 37°C. (C) Kinetics of fusion achieved by exposing cells to 45°C for different times as indicated. (D) CHO cells were transfected with plasmids expressing gH and gL, gB alone, or gB, gH, and gL as indicated together with a plasmid expressing luciferase under the control of the T7 promoter. Twenty-four h later, cells were trypsinized and mixed with an equal number of 293T14 cells for 150 min. Cells then either were maintained at 37°C or warmed to 48°C for 20 min before the addition of cool medium and a return to 37°C. Twenty-four h later, cells were lysed and luciferase activity was measured in a luminometer. Error bars represent the standard deviations from 4 replicates, and the experiment is representative of three independent experiments. a.u., arbitrary units.
Exposure of virus to heat or triggering by exposure to soluble integrins changes the proteolytic digestion pattern of gB in the same way.
Proteolysis has been used to monitor conformational changes in protein structure (25). When plasmids expressing gB are transfected into cells, a significant portion of the protein that is made is found within the cell as well as at the cell surface; thus, it is unavailable to receive a trigger of conformational change via an integrin and gHgL. Therefore, the proteinase K digestion patterns of gB were analyzed in untreated virions, virions heated to 45°C, and virions exposed to 6 nM soluble integrin αvβ8. CL55, the only available monoclonal antibody to gB, does not react in Western blotting, and the only robust polyclonal antibody that we had available was raised against the gB cytoplasmic tail. This antibody, however, could recognize by Western blotting both the full-length uncleaved gB, which is found in virions (37), and the carboxy-terminal fragment of gB, which is cleaved by furins in the cell in which it is made (38) (Fig. 8A, lane 1). After proteolytic digestion, only a few small fragments of gB could be seen (Fig. 8A, lane 2), presumably because only these fragments contained pieces of the cytoplasmic tail. However, in virions exposed to heat, an additional larger fragment was visible, indicating a change in accessibility of protease digestion sites (Fig. 8A, lane 3). The same fragment was seen in virions that had been exposed to the soluble integrin αvβ8 (Fig. 8A, lane 4). Pre- and postfusion forms of the class III fusion proteins baculovirus gp64, vesicular stomatitis virus G protein, and HSV gB have been proposed to exist in an equilibrium, as at least some conformational changes induced by low pH can be reversed if the pH is raised (39–42). Therefore, we determined whether the change in the proteolytic digestion pattern seen after heating could be reversed by returning the protein to 20°C. Although the larger fragments were slightly less prominent in the sample that had been returned to 37°C, there was no convincing indication of reversibility in this or repeat experiments (Fig. 8A, lanes 5 and 6). We also determined at what temperature changes in the digestion pattern become apparent. The larger fragments seen when virus was exposed to 45°C could not be seen at lower temperatures but were faintly apparent at 42°C (Fig. 8B). Likewise, a titration of the soluble integrin used to trigger the conformational change indicated that whereas no change in gB could be elicited with 0.6 nM αvβ8, a small fraction of gB changed conformation in response to 1.75 nm and more again at 3 and 6 nM (Fig. 8C).
FIG 8.
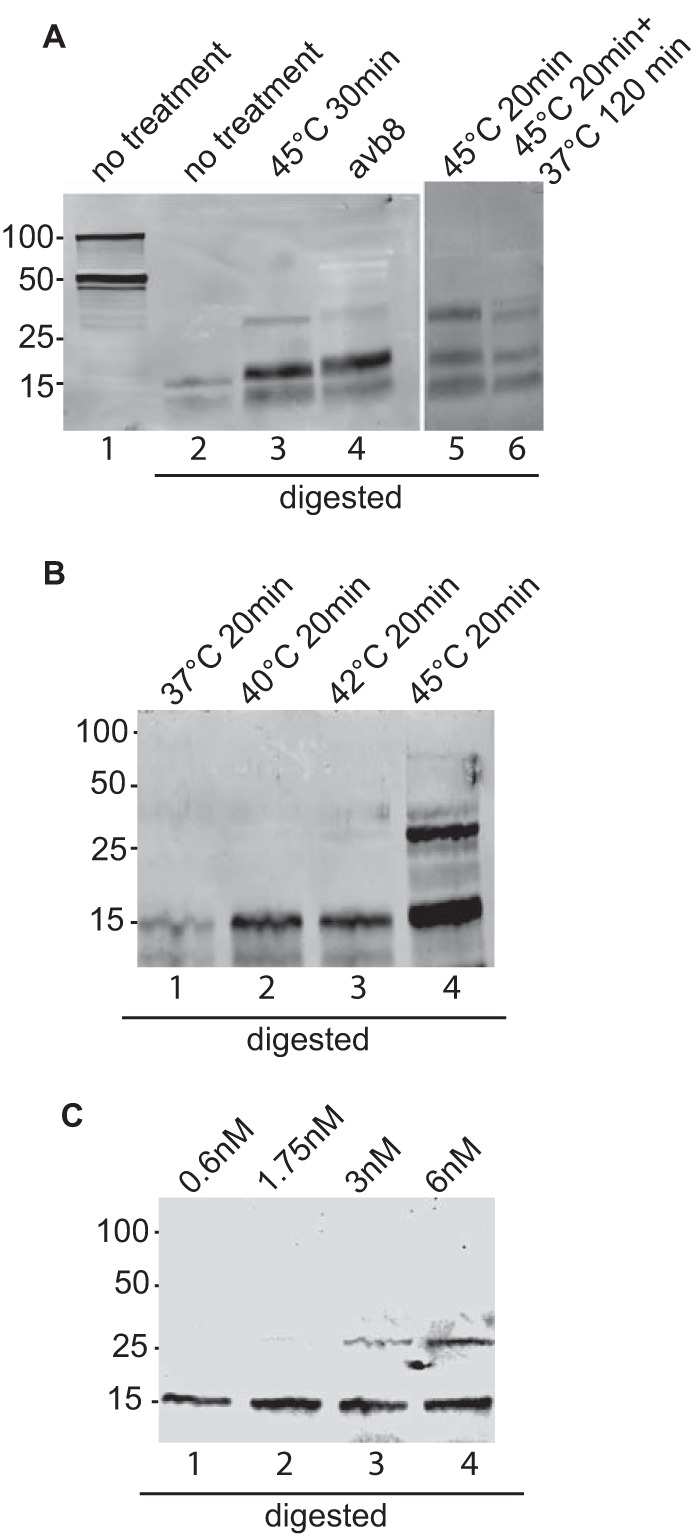
Proteolytic digestion pattern of gB in virus incubated with soluble αvβ8 or treated with heat. (A, left) Aliquots of virus were held at room temperature, incubated at 45°C for 30 min, or incubated at room temperature with 6 nM soluble αvβ8. All but one of the aliquots held at room temperature were digested with 10 μg/ml proteinase K for 10 min, and all aliquots were analyzed by SDS-PAGE and Western blotting with antibody to gB. The blot is representative of those obtained in three independent experiments. (Right) Two aliquots of virus were incubated at 45°C for 20 min. One was digested immediately, and the other was incubated at 37°C for 120 min before digestion. The blot is representative of 5 independent experiments. (B) Four aliquots of virus were incubated for 30 min at the temperatures indicated, digested with proteinase K, and analyzed by SDS-PAGE and Western blotting with antibody to gB. The blot is representative of 4 independent experiments. (C) Four aliquots of virus were incubated at room temperature with soluble αvβ8 at the concentrations indicated, digested with proteinase K, and analyzed by SDS-PAGE and Western blotting with antibody to gB. The blot is representative of 2 independent experiments.
DISCUSSION
The initial observations that EBV glycoproteins gB and gHgL can mediate fusion when expressed in trans in opposing cells were not unexpected, given previous work done with HSV and HCMV (19, 21). Fusion between several different epithelial cell lines was quite robust; in some cases, at least 80% of that obtained when glycoproteins were expressed in cis. These values are almost as high as those reported for the expression of HCMV gB and gHgL, where fusion in trans in cell-based assays was as efficient as fusion in cis (21), and they are considerably higher than those reported for HSV, where cells transfected with plasmids expressing gB were mixed with those transfected with plasmids expressing gHgL, gD, and nectin (19). However, fusion of cells in which HSV gB and gHgL were expressed in cis and only gD was expressed in trans also were lower, reaching levels less than half of those seen when all proteins were expressed in cis, so this may at least in part reflect the greater complexity of the HSV than the EBV and HCMV models.
Of perhaps greater interest were the observations on entry in trans made with the gH-null virus, phenotypically negative for gHgL and gp42 (28), which contrasted with those made for both HSV and HCMV. Infection with HSV lacking either gHgL or gB could not be achieved with either of the missing proteins expressed in trans (20), and although HCMV lacking gB could enter a cell expressing gB, a virus lacking gHgL was unable to enter a cell expressing gHgL. It is not clear why HSV entry was not seen with proteins expressed in trans. However, the failure of the HCMV particles lacking gHgL to infect a cell expressing gHgL was interpreted as reflecting the need for the gHgL complex, but not gB, to be oriented toward a cell membrane carrying receptors. The identities of such putative receptors are not yet known, but experimental evidence for their existence has been provided by interference studies (43, 44). Receptors for EBV gHgL have been definitively identified as αvβ5, αvβ6, and αvβ8 (15, 16), but the observation that soluble forms of these proteins can trigger EBV fusion (15) implies that the importance of the integrin-gHgL interaction, at least during the initial stages of entry, does not depend on a canonical receptor engagement, including additional signaling or coordinated events. Thus, CHO cells lacking human integrins but expressing gHgL could be infected, albeit at low levels, with the EBV gH-null virus in the presence of a soluble integrin. The infection of human 293 cells expressing both integrins capable of interacting with gHgL and gHgL was substantial. Receptor interference, reported to occur in cells expressing HCMV gHgL, was not an issue, perhaps because in contrast to the HCMV studies, where adenovirus vectors were used to transduce glycoproteins at high levels, the work described here used plasmid vectors which do not express high levels of protein. Certainly no loss of the expression of αv integrins was observed. Beyond this, however, there is a need to explain how gHgL opposing the virus and not another cell could have been triggered to initiate a fusion cascade, as is currently envisioned for herpesviruses. The EBV that was used was made in a B cell that lacks expression of αvβ5, αvβ6, and αvβ8 and expresses no other gHgL receptors capable of triggering fusion (45, 46), so gHgL could not have been triggered by any cell molecules incorporated in the virus envelope. Thus, we assume that the trigger came either from an integrin expressed in the same cell as gHgL or from an integrin expressed on an adjacent cell. We cannot currently distinguish between these two possibilities.
The ability of the gH-null virus to infect a gHgL-expressing B cell in the presence of a soluble integrin is perhaps easier to envision spatially, although it is interesting that, to this point, it has never been certain that a combination of gHgL and an integrin could substitute functionally for gHgLgp42 and HLA class II for B cell entry. There are clear differences between B cell and epithelial cell fusion that are reflected in the differential behavior of gH mutants (17, 33, 36), and the levels of infection seen here were not high. However, they are consistent with at least some of the events involving gHgL and fusion being common to both cell types. In our hands, a soluble form of gHgL, gHtgL, also was able to support low but reproducible levels of epithelial cell fusion, which contrasts with work done by others with EBV (47, 48). The reasons for these discrepancies at the limits of detection are not clear but may reflect protein expression levels and variability in cell types and assays used. In one case soluble gHgL was made, as in the current study, from a baculovirus construct, but the cells used were CHO cells and not AGS (47). In another case the soluble gHgL was expressed from a plasmid vector in CHO cells. A significant amount of the protein formed aggregates, and fusion was measured only in a B cell context of fusion, with gp42 coexpressed with the soluble gHgL and CHO cells expressing HLA class II as the target (48). The presence of soluble gHgL also decreased the amounts of gp42 expressed, a characteristic that the authors suggested contributed to a reduced level of fusion seen in its presence. The fusion achieved here with soluble gHtgL is, however, consistent with work done with HSV (19), where fusion levels also were reproducible, although, at 2.1% of fusion mediated by full-length membrane anchored protein, the levels were even lower than those seen here. Function is clearly only residual when gHgL is not membrane anchored, perhaps in part reflecting a spatial flexibility inconsistent with optimal functional interactions.
Current models of herpesvirus entry favor gB as the primary fusogen, and the crystal structures of HSV gB and EBV gB (7, 8) generally are interpreted as representing the postfusion forms which have already undergone the large conformational change thought likely to be required for fusion to occur (10). Such a conformational change would require activating energy to allow refolding. Consistent with previous demonstrations that heat can provide such activation energy for a virus fusogen (22–27), we found that as much as 30% of the fusion seen when gHgL and gB were expressed in cis could be achieved by heating cells expressing gB alone to 45°C. This temperature is lower than the 62°C reported to be optimal for a pre- to postfusion conformation change in the class I fusion protein, influenza virus hemagglutinin, where low pH and elevated temperature can be used interchangeably as triggers of fusion (26, 27). It is closer to the 50°C optimum reported for the class II fusion proteins of many paramyxoviruses (23–25). Of even greater interest was the fact that the proteolytic digestion patterns of gB in virus to which heat was applied and in virus triggered by exposure to soluble integrins were the same. This is consistent with a change representing a refolding of gB from a prefusion to a postfusion conformation and, together with observations made with liposomes that HSV gB can directly interact with lipids via its fusion loops while gHgL cannot (49), provides support for the model in which gB is the primary executor of fusion. Somewhat disappointingly, we were unable to reverse the conformational change by returning virus to 37°C and mirror the reversibility of other class III fusion proteins, which undergo a pH-dependent conformational change (50). Whether this reflects a fundamental difference in the thermodynamic stability of gB or simple experimental failure will need further exploration. However, to a limited extent we were able to monitor the appearance of the conformational change by increasing the temperature in small increments or by exposing virus to gradually increasing amounts of soluble integrin. We speculate that at least part of the contribution that the gHgL integrin interaction makes to activation of fusion by gB is by providing Gibbs free energy to facilitate its refolding.
ACKNOWLEDGMENTS
This work was supported by Public Health Service grant DE016669 (to L.M.H.-F.) from the National Institute of Dental and Craniofacial Research and grant 8P20GM103433 from the National Institute of General Medical Sciences.
We thank Stephen Nishimura (University of California, San Francisco) for 293 cells expressing αvβ8 and antibody 37E1, Andrew Morgan (University of Bristol, England) for baculovirus expressing gHtgL, and Richard Longnecker (Northwestern University) for 293T-14 cells.
Footnotes
Published ahead of print 20 August 2014
REFERENCES
- 1.Rickinson AB, Kieff E. 2007. Epstein-Barr virus, p 2655–2700 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Laichalk LL, Thorley-Lawson DA. 2005. Terminal differentiation into plasma cells initiated the replicative cycle of Epstein-Barr virus in vivo. J. Virol. 79:1296–1307. 10.1128/JVI.79.2.1296-1307.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hadinoto V, Shapiro M, Sun CC, Thorley-Lawson DA. 2009. The dynamics of EBV shedding implicate a central role for epithelial cells in amplifying viral output. PLoS Pathog. 7:e10000496. 10.1371/journal.ppat.1000496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jiang R, Scott RS, Hutt-Fletcher LM. 2006. Epstein-Barr virus shed in saliva is high in B cell tropic gp42. J. Virol. 80:7281–7283. 10.1128/JVI.00497-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miller N, Hutt-Fletcher LM. 1992. Epstein-Barr virus enters B cells and epithelial cells by different routes. J. Virol. 66:3409–3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spear PG, Longnecker R. 2003. Herpesvirus entry: an update. J. Virol. 77:10179–10185. 10.1128/JVI.77.19.10179-10185.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Backovic M, Longnecker R, Jardetzky TS. 2009. Structure of a trimeric variant of the Epstein-Barr virus glycoprotein B. Proc. Natl. Acad. Sci. U. S. A. 106:2880–2885. 10.1073/pnas.0810530106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heldwein EE, Lou H, Bender FC, Cohen GH, Eisenberg RJ, Harrison SC. 2006. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 313:217–220. 10.1126/science.1126548. [DOI] [PubMed] [Google Scholar]
- 9.Matsuura H, Kirschner AN, Longnecker R, Jardetzky TS. 2010. Crystal structure of the Epstein-Barr virus (EBV) glycoprotein H/glycoprotein L (gH/gL) complex. Proc. Natl. Acad. Sci. U. S. A. 107:22641–22646. 10.1073/pnas.1011806108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Connolly SA, Jackson JO, Jardetzky TS, Longnecker R. 2011. Fusing structure and function: a structural view of the herpesvirus entry machinery. Nat. Rev. Microbiol. 9:369–381. 10.1038/nrmicro2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li QX, Spriggs MK, Kovats S, Turk SM, Comeau MR, Nepom B, Hutt-Fletcher LM. 1997. Epstein-Barr virus uses HLA class II as a cofactor for infection of B lymphocytes. J. Virol. 71:4657–4662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang X, Kenyon WJ, Li QX, Mullberg J, Hutt-Fletcher LM. 1998. Epstein-Barr virus uses different complexes of glycoproteins gH and gL to infect B lymphocytes and epithelial cells. J. Virol. 72:5552–5558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen J, Rowe CL, Jardetzky TS, Longnecker R. 2012. The KGD motif of Epstein-Barr virus gH/gL is bifunctional, orchestrating infection of B cells and epithelial cells. mBio 3:e00290–11. 10.1128/mBio.00290-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kirschner AN, Sorem J, Longnecker R, Jardetzky TS. 2009. Structure of Epstein-Barr virus glycoprotein gp42 suggests a mechanism for triggering receptor-activated virus entry. Structure 17:223–233. 10.1016/j.str.2008.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chesnokova LS, Nishimura S, Hutt-Fletcher L. 2009. Fusion of epithelial cells by Epstein-Barr virus proteins is triggered by binding of viral proteins gHgL to integrins avb6 or avb8. Proc. Natl. Acad. Sci. U. S. A. 106:20464–20469. 10.1073/pnas.0907508106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chesnokova LS, Hutt-Fletcher LM. 2011. Fusion of EBV with epithelial cells can be triggered by αvβ5 in addition to αvβ6 and αvβ8 and integrin binding triggers a conformational change in gHgL. J. Virol. 85:13214–13223. 10.1128/JVI.05580-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen J, Jardetzky TS, Longnecker R. 2013. The large groove found in the gH/gL structure is an important functional domain for Epstein-Barr virus fusion. J. Virol. 87:3620–3627. 10.1128/JVI.03245-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Borza CM, Hutt-Fletcher LM. 2002. Alternate replication in B cells and epithelial cells switches tropism of Epstein-Barr virus. Nat. Med. 8:594–599. 10.1038/nm0602-594. [DOI] [PubMed] [Google Scholar]
- 19.Atanasiu D, Saw WT, Cohen GH, Eisenberg RJ. 2010. Cascade of events governing cell-cell fusion induced by herpes simplex virus glycoproteins gD, gH/gL, and gB. J. Virol. 84:12292–12299. 10.1128/JVI.01700-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wille T, Wisner TW, Ryckman BJ, Johnson DC. 2013. Human cytomegalovirus (HCMV) glycoprotein B promotes virus entry in trans acting as the viral fusion protein rather than as a receptor-binding protein. mBio 4:e00332–13. 10.1128/mBio.00332-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vanarsdall AL, Ryckman BJ, Chase MC, Johnson DC. 2008. Human cytomegalovirus glycoproteins gB and gH/gL mediate epithelial cell-cell fusion when expressed either in cis or in trans. J. Virol. 82:11837–11850. 10.1128/JVI.01623-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ader N, Brindley M, Avila M, Orvell C, Horvat B, Hiltensperger G, Schneider-Schaulies J, Vandevelde M, Zurbriggen A, Plemper RK, Plattet P. 2013. Mechanism for active membrane fusion triggered by morbillivirus attachment protein. J. Virol. 87:314–326. 10.1128/JVI.01826-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Connolly SA, Leser GP, Yin H-S, Jardetzky TS, Lamb RA. 2006. Refolding of a paramyxovirus F protein from prefusion to postfusion conformations observed by liposome binding and electron microscopy. Proc. Natl. Acad. Sci. U. S. A. 103:17903–17908. 10.1073/pnas.0608678103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chan Y-P, Lu M, Dutta S, Yan L, Barr J, Flora M, Feng Y-R, Xu K, Nikolov DB, Wang L-F, Skiniotis G, Broder CC. 2012. Biochemical, conformational, and immunogenic analysis of soluble trimeric forms of henipavirus fusion glycoproteins. J. Virol. 86:11457–11471. 10.1128/JVI.01318-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wharton SA, Skehel JJ, Wiley DC. 2000. Temperature dependence of fusion by Sendai virus. Virology 271:71–78. 10.1006/viro.2000.0280. [DOI] [PubMed] [Google Scholar]
- 26.Carr CM, Chaudhry C, Kim PS. 1997. Influenza hemagglutinin is spring-loaded by a metastable native conformation. Proc. Natl. Acad. Sci. U. S. A. 94:14306–14313. 10.1073/pnas.94.26.14306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ruigrok RWH, Martin SR, Wharton SA, Skehel JJ, Bayley PM, Wiley DC. 1986. Conformational changes in the hemagglutinin of influenza virus which accompany heat-induced fusion of virus with liposomes. Virology 155:484–497. 10.1016/0042-6822(86)90210-2. [DOI] [PubMed] [Google Scholar]
- 28.Molesworth SJ, Lake CM, Borza CM, Turk SM, Hutt-Fletcher LM. 2000. Epstein-Barr virus gH is essential for penetration of B cell but also plays a role in attachment of virus to epithelial cells. J. Virol. 74:6324–6332. 10.1128/JVI.74.14.6324-6332.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shimizu NA, Tanabe-Tochikura A, Kuroiwa Y, Takada K. 1994. Isolation of Epstein-Barr virus (EBV)-negative cell clones from the EBV-positive Burkitt's lymphoma (BL) line Akata: malignant phenotypes of BL cells are dependent on EBV. J. Virol. 68:6069–6073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Omerovic J, Lev L, Longnecker R. 2005. The amino terminus of Epstein-Barr virus glycoprotein gH is important for fusion with B cells and epithelial cells. J. Virol. 79:12408–12415. 10.1128/JVI.79.19.12408-12415.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mu D, Cambier S, Fjellbirkeland L, Baron JL, Munger JS, Kawakatsu H, Sheppard D, Braoaddus VC, Nishimura S. 2002. The integrin avb8 mediates epithelial homeostasis through MT-1-MMP-dependent activation of TGF-b1. J. Cell Biol. 157:493–507. 10.1083/jcb.200109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pulford DJ, Lowrey P, Morgan AJ. 1995. Co-expression of the Epstein-Barr virus BXLF2 and BKRF2 genes with a recombinant baculovirus produces gp85 on the cell surface with antigenic similarity to the native protein. J. Gen. Virol. 76:3145–3152. 10.1099/0022-1317-76-12-3145. [DOI] [PubMed] [Google Scholar]
- 33.Wu L, Borza CM, Hutt-Fletcher LM. 2005. Mutations of Epstein-Barr virus gH that are differentially able to support fusion with B cells or epithelial cells. J. Virol. 79:10923–10930. 10.1128/JVI.79.17.10923-10930.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chesnokova LS, Hutt-Fletcher L. 2011. Fusion of Epstein-Barr virus with epithelial cells can be triggered by αvβ5 in addition to αvβ6 and αvβ8, and integrin binding triggers a conformational change in glycoproteins gHgL. J. Virol. 85:13214–13223. 10.1128/JVI.05580-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Valencia SM, Hutt-Fletcher LM. 2012. Important but differential roles for actin in trafficking of Epstein-Barr virus in B cells and epithelial cells. J. Virol. 86:2–10. 10.1128/JVI.05883-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu L, Hutt-Fletcher LM. 2007. Point mutations in EBV gH that abrogate or differentially affect B cell and epithelial cell fusion. Virology 363:148–155. 10.1016/j.virol.2007.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johannsen E, Luftig M, Chase MR, Weicksel S, Cahir-McFarland E, Illanes D, Sarracino D, Kieff E. 2004. Proteins of purified Epstein-Barr virus. Proc. Natl. Acad. Sci. U. S. A. 101:16286–16291. 10.1073/pnas.0407320101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Backovic M, Leser GP, Lamb RA, Longnecker R, Jardetzky TS. 2007. Characterization of EBV gB indicates properties of both class I and class II fusion proteins. Virology 368:102–103. 10.1016/j.virol.2007.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gaudin Y, Tuffereau C, Segretain D, Knossow M, Flamand A. 1991. Reversible conformational changes and fusion activity of rabies virus fusion protein. J. Virol. 65:4853–4859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou J, Blissard GW. 2006. Mapping the conformational epitope of a neutralizing antibody (AcV1) directed against the AcMNPV GP64 protein. Virology 352:427–437. 10.1016/j.virol.2006.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Markovic JA, Pulyaeva H, Sokoloff A, Chernomordik LV. 1998. Membrane fusion mediated by baculovirus gp64 involves assembly of stable gp64 trimers into multiprotein aggregates. J. Cell Biol. 143:1155–1166. 10.1083/jcb.143.5.1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dollery SJ, Delboy MG, Nicola AV. 2010. Low pH-induced conformational change in herpes simplex virus glycoprotein B. J. Virol. 84:3759–3766. 10.1128/JVI.02573-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ryckman BJ, Chase MC, Johnson DC. 2008. HCMV gH/gL/UL128-131 interferes with virus entry into epithelial cells: evidence for cell type-specific receptors. Proc. Natl. Acad. Sci. U. S. A. 105:14118–14123. 10.1073/pnas.0804365105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vanarsdall AL, Chase MC, Johnson DC. 2011. Human cytomegalovirus glycoprotein O complexes with gH/gL, promoting interference with viral entry into human fibroblasts but not entry into epithelial cells. J. Virol. 85:11638–11645. 10.1128/JVI.05659-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Borza CM, Morgan AJ, Turk SM, Hutt-Fletcher LM. 2004. Use of gHgL for attachment of Epstein-Barr virus to epithelial cells compromises infection. J. Virol. 78:5007–5014. 10.1128/JVI.78.10.5007-5014.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang X, Hutt-Fletcher LM. 1998. Epstein-Barr virus lacking glycoprotein gp42 can bind to B cells but is not able to infect. J. Virol. 72:158–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kirschner AN, Omerovic J, Popov B, Longnecker R, Jardetzky TS. 2006. Soluble Epstein-Barr virus glycoproteins gH, gL, and gp42 form a 1:1:1 stable complex that acts like soluble gp42 in B-cell fusion but not in epithelial cell fusion. J. Virol. 80:9444–9454. 10.1128/JVI.00572-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rowe CL, Connolly SA, Chen J, Jardetzky TS, Longnecker R. 2013. A soluble form of Epstein-Barr virus gH/gL inhibits EBV-induced membrane fusion and does not function in fusion. Virology 436:118–126. 10.1016/j.virol.2012.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hannah BP, Cairns TM, Bender FC, Whitbeck JC, Lou H, Eisenberg RJ, Cohen GH. 2009. Herpes simplex virus glycoprotein B associates with target membranes via its fusion loops. J. Virol. 83:6825–6836. 10.1128/JVI.00301-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Backovic M, Jardetzky TS. 2009. Class III viral membrane fusion proteins. Curr. Opin. Struct. Biol. 19:189–196. 10.1016/j.sbi.2009.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]