

ABSTRACT
Human immunodeficiency virus type 1 (HIV-1) and simian immunodeficiency virus (SIV) viral infectivity factor (Vif) form a CRL5 E3 ubiquitin ligase complex to suppress virus restriction by host APOBEC3 (A3) proteins. The primate lentiviral Vif complex is composed of the unique cofactor core binding factor β (CBF-β) and canonical ligase components Cullin 5 (CUL5), Elongin B/C (ELOB/C), and RBX2. However, the mechanism by which the Vif protein of the related lentivirus bovine immunodeficiency virus (BIV) overcomes its host A3 proteins is less clear. In this study, we show that BIV Vif interacts with Cullin 2 (CUL2), ELOB/C, and RBX1, but not with CBF-β or CUL5, to form a CRL2 E3 ubiquitin ligase and degrade the restrictive bovine A3 proteins (A3Z2Z3 and A3Z3). RNA interference-mediated knockdown of ELOB or CUL2 inhibited BIV Vif-mediated degradation of these A3 proteins, whereas knockdown of CUL5 or CBF-β did not. BIV Vif with mutations in the BC box (Vif SLQ-AAA) or putative VHL box (Vif YI-AA), which cannot interact with ELOB/C or CUL2, respectively, lost the ability to counteract bovine A3 proteins. Moreover, CUL2 and UBE2M dominant negative mutants competitively inhibited the BIV Vif-mediated degradation mechanism. Thus, although the general strategy for inhibiting A3 proteins is conserved between HIV-1/SIV and BIV, the precise mechanisms can differ substantially, with only the HIV-1/SIV Vif proteins requiring CBF-β as a cofactor, HIV-1/SIV Vif using CUL5-RBX2, and BIV Vif using CUL2-RBX1.
IMPORTANCE Primate lentivirus HIV-1 and SIV Vif proteins form a ubiquitin ligase complex to target host antiviral APOBEC3 proteins for degradation. However, the mechanism by which the nonprimate lentivirus BIV Vif inhibits bovine APOBEC3 proteins is unclear. In the present study, we determined the mechanism for BIV Vif-mediated degradation of bovine APOBEC3 proteins and found that it differs from the mechanism of HIV-1/SIV Vif by being CBF-β independent and requiring different ubiquitin ligase scaffolding proteins (CUL2-RBX1 instead of CUL5-RBX2). BIV Vif is the only known retroviral protein that can interact with CUL2. This information broadens our understanding of the distinct mechanisms by which the Vif proteins of different lentiviruses facilitate viral infection. This novel mechanism for assembly of the BIV Vif-APOBEC3 ubiquitin ligase complex advances our understanding of viral hijacking of host E3 ubiquitin ligases and illustrates the evolutionary flexibility of lentiviruses.
INTRODUCTION
All lentivirus genomes except the genome of equine infectious anemia virus (EIAV) encode the accessory protein viral infectivity factor (Vif), which is essential for viral replication and infection of the respective mammalian hosts (1–3). Human immunodeficiency virus type 1 (HIV-1), simian immunodeficiency virus (SIV), bovine immunodeficiency virus (BIV), maedi-visna virus (MVV), arthritis-encephalitis virus (CAEV), and feline immunodeficiency virus (FIV), which infect humans, monkeys, cattle, sheep, goats, and cats, respectively, all utilize Vif to neutralize the host APOBEC3 (A3) antiviral proteins (1, 4–11) HIV-1 Vif has been well studied because of its critical function in viral replication. The eukaryotic ubiquitin conjugation system is a main pathway of protein degradation and includes activating enzymes (E1s), conjugating enzymes (UBCs/E2s), and ubiquitin ligases (E3s) (1). The members of the Cullin (CUL) family of RING E3 ubiquitin ligases are modular enzymes that act as scaffolding to bring a specific substrate into close proximity with the E2 ubiquitin-conjugating enzyme, thereby facilitating ubiquitination and subsequent proteasomal degradation. There are seven known human Cullin proteins, Cullin 1 (CUL1), CUL2, CUL3, CUL4a, CUL4b, CUL5, and CUL7, with diverse cellular functions (2). Previously, HIV-1 Vif was shown to form a CUL5-Elongin B/C (ELOB/C) E3 ubiquitin ligase that targets selected A3 proteins for proteasomal degradation by recruiting the cellular factors CUL5, ELOB/C, and RBX (3–10). In 2012, core binding factor β (CBF-β) was identified to be a novel critical regulator of HIV-1 Vif activity (12–14). SIVagm Vif and SIVmac Vif also recruit CBF-β and ELOB/C to the CUL5-RBX2 complex to degrade their host's antiviral A3 proteins, but BIV Vif and FIV Vif do not require CBF-β for this activity (11, 15).
The functional domains of HIV-1 Vif have been well characterized by many groups. HIV-1 Vif binds ELOB/C through its BC box region (residues 144SLQYLA149), which mimics the conserved cellular interface of suppressor of cytokine signaling (SOCS) box proteins, and a conserved HX5CX17-18CX3-5H (HCCH) motif in the carboxyl-terminal region is used to recruit CUL5 (3–10). The amino-terminal region of HIV-1 Vif was determined to recognize various A3 proteins through different motifs (5–7, 16–18). The functional domains in HIV-1 Vif required for binding to CBF-β were recently identified (19–22),
Cullin-RING E3 ubiquitin ligases (CRLs) are essential for the turnover of a large number of proteins in mammalian cells and are also critical for virus replication. The Von Hippel-Lindau (VHL) tumor suppressor specifically interacts with endogenous CUL2-RBX1 in mammalian cells, whereas SOCS box proteins, such as HIV-1 Vif or ASB9, associate with CUL5-RBX2. VHL box and SOCS box domains determine the binding specificity for the CUL2-RBX1 and CUL5-RBX2 modules of each ubiquitin ligase complex. The VHL box (the BC box plus a downstream CUL2 box) and SOCS box (the BC box plus a downstream CUL5 box) thus appear to define two distinct types of complexes. In addition to VHL, two BC box proteins, LRR-1 and FEM1B, also contain a CUL2 box and bind CUL2 (23).
BIV is a lentiviral pathogen similar to HIV-1 that is causally associated with debilitating diseases in cattle (24). The genome of humans (Homo sapiens) encodes seven known A3 (hsA3) proteins (hsA3A, hsA3B, hsA3C, hsA3D, hsA3F, hsA3G, and hsA3H) (25, 26). In contrast, the genome of cattle (Bos taurus) encodes four A3 (btA3) proteins (btA3Z2Z3, btA3Z2, btA3Z3, and btA3Z1), the genome of sheep (Ovis aries) encodes four homologous A3 (oaA3) proteins (oaA3Z2Z3, oaA3Z2, oaA3Z3, and oaA3Z1), and the genome of the more distantly related pigs (Sus scrofa) encodes three A3 (ssA3) proteins (ssA3Z2Z3, ssA3Z2, and ssA3Z3) due to a loss of the Z1 type gene (12). However, despite the need for BIV Vif to overcome bovine A3 proteins, several details of the molecular mechanism are unclear.
In the present study, we investigated the mechanism by which BIV Vif inhibits bovine A3s and show that BIV Vif can form a CRL2-E3 ligase without CBF-β by recruiting CUL2, RBX1, and ELOB/C, but not CUL5-RBX2. RNA interference (RNAi)-mediated knockdown of ELOB or CUL2 inhibited the BIV Vif-mediated degradation of btA3Z2Z3, whereas knockdown of CUL5 or CBF-β did not. The VHL box in BIV Vif was characterized, and the BIV Vif SLQ-AAA and BIV Vif YI-AA mutants with mutations in the VHL box, which could not interact with ELOB/C or CUL2, also lost the ability to inhibit the antiviral activity of A3 proteins. Moreover, CUL2 and UBE2M dominant negative mutants competitively inhibited the BIV Vif-mediated degradation of btA3Z2Z3, demonstrating an essential role for CUL2 in the formation of the E3 ubiquitin ligase. Thus, CRL2-E3 ubiquitin ligase activity is required for BIV Vif function. These observations deepen our understanding of the different mechanisms by which the Vif accessory proteins of different lentiviruses facilitate infection and pathogenesis.
MATERIALS AND METHODS
Plasmid construction.
The Vif mutant infectious molecular clone (pNL4-3ΔVif) was obtained from the AIDS Research and Reference Reagents Program, Division of AIDS, National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH). The VR1012 vector was generously provided by Vical (San Diego, CA). BIV hemagglutinin (HA)-tagged Vif (Vif-HA), BIV Vif-Myc, and HIV Vif-HA have been described previously (17, 27). The BIV Vif mutant constructs derived from BIV Vif-HA and BIV Vif-Myc expression vectors were constructed by PCR-based mutagenesis using the following primers: for BIV VifY-A, forward primer 5′-GCATGTCTCTGTTGGGCCCCGTTAGGACG-3′ and reverse primer 5′-CGTCCTAACGGGGCCCAACAGAGACATGC-3′, and for BIV Vif I-A, forward primer 5′-CCCGTTAGGACGCGCCAACGACACCACCC-3′ and reverse primer 5′-GGGTGGTGTCGTTGGCGCGTCCTAACGGG-3′. Expression vectors for cattle A3 proteins, A3Z2Z3-HA, green fluorescent protein-tagged A3Z2Z3 (A3Z2Z3-GFP), A3Z2-GFP, A3Z3-GFP, and A3Z1-GFP, have been described previously (1, 28) CUL2ΔNedd8-HA, UBE2M-Flag (C111S), and UBE2F-Flag (C116S) dominant negative mutants were synthesized by Shanghai Generay Biotech Co. and constructed in the VR1012 vector. A3Z2Z3, A3Z3, BIV Vif wild type (WT), BIV Vif SLQ-AAA, and BIV Vif YI-AA were cloned into the mCherry-C1 vector with a C-terminal HA tag in order to obtain better expression in MDBK cells. Bos taurus ELOB (btELOB)-HA, Bos taurus CBF-β (btCBF-β)–Myc, Bos taurus CUL2 (btCUL2)-Myc, and Bos taurus CUL5 (btCUL5)-Myc were constructed by the PCR-based mutagenesis method using human ELOB (huELOB)-HA, human CBF-β (huCBF-β)–Myc, human CUL2 (huCUL2)-Myc, and human CUL5 (huCUL5)-Myc sequences as the templates. Each amplified DNA segment was sequenced in its entirety to ensure that no unintended mutations were introduced.
Antibodies and cell culture.
The following antibodies were used in this study: anti-p24 monoclonal antibody (MAb; catalog no. 1513; AIDS Research and Reference Reagents Program, Division of AIDS, NIAID, NIH), mouse anti-HA MAb (catalog no. MMS-101R-10000; Covance), mouse anti-Myc MAb (catalog no. M5546; Sigma), anti-CUL5 (catalog no. sc-13014; Santa Cruz), anti-CUL2 (catalog no. sc-10781; Santa Cruz), anti-ELOB (catalog no. sc-11447; Santa Cruz), anti-ELOC (catalog no. 610760; BD Biosciences), anti-CBF-β MAb (catalog no. sc-166142; Santa Cruz), anti-GFP (catalog no. A11001; Invitrogen), anti-human tubulin (catalog no. DKM9003; Tianjin Sanjian), and anti-human ribosomal P antigen (catalog no. HP0-0100; Immunovision).
MAGI-CCR5 cells were obtained through the NIH AIDS Research and Reference Reagents Program (catalog no. 3522) from Julie Overbaugh. MAGI-CCR5, HEK293T (catalog no. CRL-11268; ATCC), and MDBK (catalog no. CCL-22; ATCC) cells were maintained in Dulbecco's modified Eagle's medium (DMEM; Invitrogen) with 10% fetal bovine serum and penicillin-streptomycin (D-10 medium) and passaged upon confluence. The following reagents were purchased: MLN4924 (catalog no. M425100; Toronto Research Chemicals Inc.), MG132 (catalog no. C2211; Sigma), and N,N,N′-tetrakis-(2′-pyridylmethyl) ethylenediamine (TPEN; catalog no. P4413; Sigma).
Transfection and virus purification.
DNA transfection in HEK293T cells was carried out using the Lipofectamine 2000 reagent (Invitrogen) as recommended by the manufacturer. For degradation assays, HEK293T cells in 12-well plates were transfected with 1 μg of BIV Vif or 1 μg of empty vector and 0.3 μg of btA3 at a 3:1 ratio. For viral infectivity assays, HEK293T cells in T25 flasks were transfected with 2 μg of BIV Vif, 2 μg of Vif-deficient infectious clone NL4-3ΔVif, and 2 μg of btA3Z2Z3 at a 1:1:1 ratio in various combinations with empty-vector controls. For immunoprecipitation assays, HEK293T cells in T25 flasks were transfected with 5 μg of VR1012 or Vif. DNA transfection in MDBK cells was carried out using an electroporator (Gene Pulser Xcell; Bio-Rad). MDBK cells (2 × 106) were transfected with 2 μg of btA3Z2Z3 or btA3Z3 and 2.5 μg of BIV Vif WT or the BIV Vif SLQ-AAA or BIV Vif YI-AA mutant. The parameters used were 140 V, 500 μF, and maximum resistance (exponential decay mode). After electroporation, cells were cultured in 6-well plates for 48 h at 37°C in a non-CO2 incubator. The cell culture supernatants, which contained the viruses, were cleared of cellular debris by centrifugation at 3,000 rpm for 15 min in a Sorvall RT 6000B centrifuge and filtered through a 0.2-μm-pore-size membrane (Millipore). The virus particles were then concentrated by centrifugation through a 20% sucrose cushion by ultracentrifugation at 100,000 × g for 2 h at 4°C in a Sorvall Ultra80 ultracentrifuge. Viral lysates were analyzed by immunoblotting.
MAGI assays.
Virus infectivity was determined with the multinuclear activation of galactosidase indicator (MAGI) assay (13). MAGI-CCR5 cells were plated in 12-well plates in D-10 medium 1 day before infection to reach 30 to 40% confluence on the day of infection. Cells were infected by removing medium from each well and adding dilutions of virus in a total volume of 500 μl of D-10 medium with 20 μg/ml of DEAE-dextran. After a 2-h incubation at 37°C in a 5% CO2 incubator, 1 ml of D-10 medium was added to each well. After another 46 h of incubation under the same conditions, supernatants were removed and the cells were fixed for 5 min at room temperature in 800 μl of fixing solution (1% formaldehyde, 0.2% glutaraldehyde in phosphate-buffered saline [PBS]). The cells were then washed twice with PBS and incubated with staining solution (20 μl 0.2 M potassium ferrocyanide, 20 μl 0.2 M potassium ferricyanide, 2 μl 1 M MgCl2, 10 μl 40 mg/ml 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) for 2 h at 37°C in a non-CO2 incubator. Staining was stopped by removing the staining solution and thoroughly washing twice with PBS. Blue dots, which indicated a positive result, were counted manually, and viral infectivity was determined after normalizing the amount of virus input by measurement of the p24 antigen level by enzyme-linked immunosorbent assay (Perkin-Elmer).
RNAi.
RNAi against ELOB, CUL2, and CUL5 was carried out using a pool of three duplexed short interfering RNAs (siRNAs; Guangdong Ruibo) with the following sequences (where F indicates forward and R indicates reverse): for CUL2 siRNAs, the duplex 1 (siB10324102633) F1 sequence was CGAAAGAGCAACAUGGAAUdTdT and the duplex 1 R1 sequence was dTdTGCUUUCUCGUUGUACCUUA, the duplex 2 (siB10324102648) F2 sequence was GUGUACAGGUGAAGUUAAAdTdT and the duplex 2 R2 sequence was dTdTCACAUGUCCACUUCAAUUU, and the duplex 3 (siB10324102701) F3 sequence was GACUGAAACAGGAGAGUAUdTdT and the duplex 3 R3 sequence was dTdTCUGACUUUGUCCUCUCAUA; for CUL5 siRNAs, the duplex 1 (siG1110890928) F1 sequence was GAGCAAAUAGAGUGGCUAAdTdT and the duplex 1 R1 sequence was dTdTCUCGUUUAUCUCACCGAUU, the duplex 2 (siG000008065B) F2 sequence was GCUCUUGGUACUUAAGUAUdTdT and the duplex 2 R2 sequence was dTdTCGAGAACCAUGAAUUCAUA, and the duplex 3 (siG1110890949) F3 sequence was CUACUGAACUGGAGGACUUdTdT and the duplex 3 R3 sequence was dTdTGAUGACUUGACCUCCUGAA; and for ELOB siRNAs, the duplex 1 (siG1364191354) F1 sequence was GGACGUGUUCCUCAUGAUCdTdT and the duplex 1 R1 sequence was dTdTCCUGCACAAGGAGUACUAG, the duplex 2 (siG1364191411) F2 sequence was GCCACAAGACCACCAUCUUdTdT and the duplex 2 R2 sequence was dTdTCGGUGUUCUGGUGGUAGAA, and the duplex 3 (siG1364191429) F3 sequence was GGAAGCAGUGCCAAUGAACdTdT and the duplex 3 R3 sequence was dTdTCCUUCGUCACGGUUACUUG. HEK293T cells were transfected with the CUL2, CUL5, or ELOB siRNA pool at a total final concentration of 100 nM using the Lipofectamine 2000 reagent. Nontargeting control siRNA (siControl) no. 2 (Guangdong Ruibo) was used as a control. Expression of the CUL2, CUL5, or ELOB protein was monitored by immunoblotting 2 days after transfection. CBF-β silencing by small hairpin RNA (shRNA) was described previously (15).
RNA extraction and RT-qPCR.
For real-time quantitative PCR (RT-qPCR), cellular RNA was extracted from transfected HEK293T cells using the TRIzol reagent, and cDNA was generated using a High-Capacity cDNA reverse transcription kit (Applied Biosystems) and oligo(dT)18 primers according to the supplier's instructions. The sequences of the primers, designed using the CUL2 and CUL5 sequences, were as follows: CUL2 forward primer, 5′-TGCTTTATGTGTGGCCTATC-3′; CUL2 reverse primer, 5′-TTCTTCTGACTCCAAAACTCTC-3′; CUL5 forward primer, 5′-TGGCGACGTCTAATCTGTTA-3′; and CUL5 reverse primer 5′-CAAAGACAGACTGCATGCAC-3′ (Shanghai Shenggong). The SYBR green-based RT-qPCR was carried out on an Mx3005P instrument (Stratagene; Agilent Technologies) using the double-stranded DNA-binding dye method with SYBR green PCR master mix (Applied Biosystems). Each 20-μl reaction mixture contained 10 μl of SYBR premix, 0.2 μl (10 μM) each of the forward and reverse primers, 7.2 μl of double-distilled H2O, and 2 μl of cDNA template. Cycling conditions were as follows: 50°C for 2 min and then 95°C for 10 min, followed by 50 cycles consisting of 95°C for 15 s and 60°C for 1 min. The melting curve analysis was performed at 90°C for 1 min and then at 55°C for 30 s and 95°C for 30 s. The relative abundance of CUL2 and CUL5 was determined by using GAPDH (glyceraldehyde-3-phosphate dehydrogenase) as a control.
Immunoblot analysis.
HEK293T cells were collected at 48 h after transfection. Cell lysates and viral lysates were prepared as previously described (3). Samples were lysed in 1× loading buffer (0.08 M Tris, pH 6.8, with 2.0% SDS, 10% glycerol, 0.1 M dithiothreitol, and 0.2% bromophenol blue) and boiled for 5 min before the proteins were separated by SDS-PAGE. Membranes were probed with various primary antibodies against the proteins of interest. Secondary antibodies were alkaline phosphatase-conjugated antihuman, antirabbit, or antimouse (Jackson ImmunoResearch) antibodies, and staining was carried out with 5-bromo-4-chloro-3-indolylphosphate (BCIP) and nitroblue tetrazolium (NBT) solutions prepared from chemicals obtained from Sigma.
Immunoprecipitation.
For BIV Vif-HA immunoprecipitation, transfected HEK293T cells were harvested and washed twice with cold PBS, followed by disruption with lysis buffer (PBS containing 1% Triton X-100 and Complete protease inhibitor cocktail [Roche]) at 4°C for 30 min. Cell lysates were clarified by centrifugation at 10,000 × g for 30 min at 4°C. Anti-HA agarose beads (catalog no. 190-119; Roche) were mixed with the precleared cell lysates, and the mixture was incubated at 4°C for 3 h on an end-over-end rocker. The reaction mixtures were then washed six times with cold wash buffer (20 mM Tris HCl, pH 7.5, 100 mM NaCl, 0.1 mM EDTA, 0.05% Tween 20) and subsequently analyzed by immunoblotting.
Cycloheximide chase.
HEK293T cells were transfected with VR1012 carrying BIV Vif-Myc or the VR1012 empty vector control and cattle A3Z2Z3-HA at a 1:1 ratio. At 18 h after transfection, 100 μg/ml cycloheximide was added to the cells. At various time points, equal cell numbers were collected and samples were prepared by adding loading buffer. For chase experiments in the presence of proteasome inhibitor, 10 μM MG132 was added to the cells at 12 h before harvest. The proteins were separated by SDS-PAGE and analyzed by immunoblotting.
RESULTS
BIV Vif inhibits bovine A3Z2Z3 and neutralizes its antiviral activity via a proteasomal pathway.
Previous studies have shown that bovine A3Z2Z3 (btA3Z2Z3) can restrict HIV-1 infection (11, 28–30). Thus, we performed a series of single-cycle HIV-1 infectivity assays to test whether BIV Vif can overcome the antiviral activity of btA3Z2Z3. HEK293T cells were transfected with the infectious molecular clone of the Vif mutant NL4-3ΔVif plus VR1012 as a control vector or wild-type (WT) BIV Vif in the presence or absence of the bovine A3Z2Z3 expression vector. Virus was produced from transfected cells and tested for infectivity in a standard MAGI assay as previously described (13). The infectivity of NL4-3ΔVif was considered 100%, and expression of btA3Z2Z3 reduced the infectivity of NL4-3ΔVif in the absence of Vif (Fig. 1A). However, in the presence of BIV Vif, btA3Z2Z3 restriction activity was suppressed and NL4-3ΔVif infectivity was maintained compared to that in the absence of BIV Vif (Fig. 1A). These results show that BIV Vif efficiently inhibits btA3Z2Z3 activity, as reported previously (12, 14, 15). Subsequently, the stability of btA3Z2Z3 in virus producer cells and the incorporation of btA3Z2Z3 into virions were examined. In agreement with the infectivity data, we observed that BIV Vif reduced the intracellular level of btA3Z2Z3 (Fig. 1B, lane 3) compared to the effect of the VR1012 vector control (Fig. 1B, lane 2). The incorporation of btA3Z2Z3 was also inhibited by BIV Vif (Fig. 1B, lane 3) compared to the level of incorporation of the negative control (NC), VR1012 (Fig. 1B, lane 2).
FIG 1.

Influence of BIV Vif on the antiviral activity of btA3Z2Z3. (A) BIV Vif rescued the infectivity of NL4-3ΔVif in the presence of btA3Z2Z3. BIV Vif, the Vif-deficient infectious clone NL4-3ΔVif, and btA3Z2Z3 at a 1:1:1 ratio (2 μg each) were cotransfected in various combinations with empty vector controls into HEK293T cells in T25 flasks, as indicated. Viruses from the supernatants were cultured on MAGI-CCR5 indicator cells to test for infectivity. Values indicate the percent infectivity compared to that of virus produced in the absence of btA3Z2Z3. Error bars represent the SDs for triplicate wells. (B) BIV Vif reduced btA3Z2Z3 expression and inhibited btA3Z2Z3 virion packaging. Immunoblotting of lysates of HEK293T cells cotransfected as described in the legend to panel A showed that btA3Z2Z3 is degraded by BIV Vif. Ribosomal P protein (p19) was used as the sample loading control. btA3Z2Z3 was efficiently packaged into Vif-deficient HIV-1 in the absence of BIV Vif but not in the presence of BIV Vif. btA3Z2Z3 was measured after normalization to the structural Gag protein content (HIV-1 p24). (C) BIV Vif degraded btA3Z2Z3 via the ubiquitin-proteasome pathway. Cycloheximide (100 μg/ml) was added to the cells, and after various chase periods, equal numbers of cells were collected, separated by SDS-PAGE, and analyzed by immunoblotting and the amount of btA3Z2Z3 was quantified. The amount of btA3Z2Z3 at time zero was normalized to 100%. (D) BIV Vif polyubiquitinated btA3Z2Z3. HEK293T cells in T25 flasks were transfected with various combinations of expression vectors with 0.5 μg of BIV Vif-Myc, 2 μg of btA3Z2Z3-HA, and 2 μg of Myc-tagged ubiquitin. All experiments were performed in the presence of MG132. Cells were lysed and analyzed by immunoblot analysis using an anti-Myc, anti-HA, or antitubulin antibody. Cell lysates were immunoprecipitated with the anti-HA antibody and analyzed by immunoblotting with an anti-Myc antibody to detect ubiquitinated btA3Z2Z3 proteins. Unmodified btA3Z2Z3 proteins were detected with the anti-HA antibody. Ubn, ubiquitin; Tub, tubulin; IP, immunoprecipitation.
HIV-1 Vif has been shown to degrade human A3 proteins through polyubiquitination and proteasomal degradation (3). Therefore, we next asked whether proteolysis and polyubiquitination play a role in the effect of BIV Vif on the steady-state levels of btA3Z2Z3. HEK293T cells were transfected with BIV Vif-Myc or the VR1012 control vector plus btA3Z2Z3-HA. At 18 h posttransfection, 100 μg/ml cycloheximide was added to the cells to block protein synthesis. For chase experiments in the presence of proteasome inhibitor, 10 μM MG132 was added to the cells 12 h before harvest. In the absence of BIV Vif, btA3Z2Z3 was stable (Fig. 1C, No BIV Vif), but it was highly unstable in the presence of BIV Vif (Fig. 1C, +BIV Vif). The proteasome inhibitor MG132 decreased the degradation of btA3Z2Z3 in the presence of BIV Vif (Fig. 1C, +BIV Vif + MG132). To further confirm that BIV Vif can induce polyubquitination of bovine A3Z2Z3, we transfected HEK293T cells with various combinations of expression vectors for BIV Vif-Myc, btA3Z2Z3-HA, and Myc-tagged ubiquitin. To inhibit btA3Z2Z3 degradation, all transfected cells were treated with MG132. HA-tagged btA3Z2Z3 was immunoprecipitated by anti-HA antibody, followed by the detection of ubiquitinated btA3Z2Z3-HA using anti-Myc antibody. Polyubquitinated btA3Z2Z3-HA proteins were detected in the presence of Myc-tagged ubiquitin and were enhanced in the presence of BIV Vif (Fig. 1D, lane 4). Thus, BIV Vif induced polyubiquitination of btA3Z2Z3 in a fashion similar to that in which HIV-1 Vif induced the polyubiquitination of A3G (3, 31). These results show that BIV Vif inhibits the antiviral activity of btA3Z2Z3 via a polyubiquitination and proteasomal degradation mechanism.
BIV Vif requires cellular factors ELOB/C and CUL2, but not CUL5 and CBF-β, to form an active E3 ubiquitin ligase complex.
HIV-1 Vif has been shown to overcome human A3 proteins by inducing their degradation through the CUL5–ELOB/C–CBF-β E3 ubiquitin ligase complex (3, 32, 33). Our model for BIV Vif-induced degradation of btA3Z2Z3 predicts that BIV Vif also acts as a receptor to bring its target proteins to the E3 ubiquitin ligase, where they are polyubiquitinated and degraded via the proteasomal pathway. To examine whether BIV Vif interacts with cellular factors to form an E3 ubiquitin ligase, we transfected HEK293T cells with VR1012 as a control vector or with the BIV Vif-HA or HIV-1 Vif-HA expression vector. The interaction of cellular proteins with BIV or HIV-1 Vif-HA was examined by coimmunoprecipitation analysis. An anti-HA antibody conjugated to agarose beads was used to immunoprecipitate HA-tagged Vif from lysates of transfected HEK293T cells. As expected, CUL5, ELOB/C, and CBF-β coprecipitated with HIV-1 Vif-HA (Fig. 2A, lane 6) but not with the sample containing the empty vector, VR1012 (Fig. 2A, lane 4). In contrast, CUL5 and CBF-β failed to coprecipitate with BIV Vif-HA (Fig. 2A, lane 5). On the other hand, ELOB/C and CUL2 coprecipitated with BIV Vif-HA (Fig. 2A, lane 5). Amino acid sequence alignments showed that the homology rates for CUL2, CUL5, ELOB, ELOC, and CBF-β between humans and cattle are 99.6%, 99.6%, 97.5%, 100%, and 98.9%, respectively. We further compared the interactions of HIV-1 Vif and BIV Vif with btCUL2-Myc, btCUL5-Myc, btELOB-HA, and btCBF-β. The results showed that BIV Vif interacts with btELOB (Fig. 2B, lane 2) and btCUL2 (Fig. 2D, lane 2) but not with btCBF-β (Fig. 2C, lane 2) or btCUL5 (Fig. 2E, lane 2). Furthermore, we found that HIV-1 Vif also interacts with btELOB (Fig. 2B, lane 3), btCBF-β (Fig. 2C, lane 3), and btCUL5 (Fig. 2E, lane 3), but not btCUL2 (Fig. 2D, lane 3). These results demonstrate that BIV Vif does interact with the btELOB/C and btCUL2 factors and forms an E3 ligase in the bovine system. Silencing of ELOB and CUL2 with specific siRNAs targeted to the ELOB- and CUL2-coding regions, respectively, inhibited BIV Vif-mediated degradation of btA3Z2Z3 (Fig. 3A and C), but silencing of CUL5 or CBF-β did not (Fig. 3E and G). In contrast, silencing of CBF-β inhibited HIV-1 Vif-mediated degradation of hsA3G (Fig. 3H). To determine the extent of the ELOB, CUL2, and CUL5 knockdown, we used RT-qPCR to determine the relative abundance of ELOB, CUL2, CUL5, and CBF-β RNAs, using GAPDH as a control. The results showed that the ELOB, CUL2, CUL5, and CBF-β mRNA levels were lowered by >60% in the corresponding siRNA groups compared to those in the NC siRNA group (Fig. 3B, D, F, and I). Based on these findings, we concluded that ELOB and CUL2, but not CUL5, are essential for BIV Vif function.
FIG 2.

Interaction of BIV Vif with cellular factors. (A) BIV Vif assembled with endogenous CUL2 and ELOB/C but not with CUL5 or CBF-β. HEK293T cells in T25 flasks were transfected with 5 μg of HIV Vif-HA or BIV Vif-HA. Equal amounts of cell lysates were immunoprecipitated with anti-HA antibody conjugated to agarose and analyzed by immunoblotting with antibodies against HA, ELOB, ELOC, CBF-β, CUL5, and CUL2. BIV Vif assembled with exogenous btELOB (B) and btCUL2 (D) but not with btCBF-β (C) or btCUL5 (E). (B) HEK293T cells in T25 flasks were transfected with 2 μg of VR1012 as a negative-control vector or with BIV Vif-Myc or HIV Vif-Myc plus 2 μg of btELOB-HA at a 1:1 ratio. Equal amounts of cell lysates were immunoprecipitated with anti-Myc antibody conjugated to agarose and analyzed by immunoblotting with antibodies against HA, Myc, and tubulin. HEK293T cells in T25 flasks were transfected with 2 μg of VR1012 as a negative-control vector or with BIV Vif-HA or HIV Vif-HA plus 2 μg of btCBF-β-Myc (C), btCUL2-Myc (D), or btCUL5-Myc (E) at a 1:1 ratio. Equal amounts of cell lysates were immunoprecipitated with anti-HA antibody conjugated to agarose and analyzed by immunoblotting with antibodies against HA, Myc, and tubulin.
FIG 3.

ELOB and CUL2, but not CUL5 and CBF-β, are necessary for BIV Vif-mediated degradation. (A) Silencing of ELOB inhibited the BIV Vif-mediated degradation of btA3Z2Z3. (B) RT-qPCR analysis indicated that ELOB siRNA caused a >80% reduction in the ELOB RNA level. GAPDH was used as a control. Error bars indicate the SDs from three replicates within one experiment. (C) Silencing of CUL2 inhibited the BIV Vif-mediated degradation of btA3Z2Z3. (D) RT-qPCR analysis indicated that CUL2 siRNA caused a >60% reduction in the CUL2 RNA level. GAPDH was used as a control. Error bars indicate the SDs from three replicates within one experiment. (E) Silencing of CUL5 did not inhibit the BIV Vif-mediated degradation of btA3Z2Z3. (F) RT-qPCR analysis indicated that CUL5 siRNA caused a >60% reduction in the CUL5 RNA level. GAPDH was used as a control. Error bars indicate the SDs from three replicates within one experiment. (G) Silencing of CBF-β did not inhibit the BIV Vif-mediated degradation of btA3Z2Z3. (H) Silencing of CBF-β inhibited the HIV-1 Vif-mediated degradation of hsA3G. (I) RT-qPCR analysis indicated that CBF-β shRNA caused a >60% reduction in the CBF-βRNA level. GAPDH was used as a control. Error bars indicate the SDs from three replicates within one experiment. VR, VR1012 vector.
BIV Vif has a BC box and CUL2 box required for binding ELOB/C and CUL2, respectively.
CRL E3 ligase substrate receptors are divided into multiple classes. However, all substrate receptors interact with ELOC through a version of the BC box. Cellular substrate receptors select CUL2 or CUL5 on the basis of the presence of a CUL2 box or a CUL5 box. VHL, LRR-1, and FEM1B, which contain a VHL box, have all been identified to be CUL2-interacting proteins (23). By aligning the sequences of BIV Vif and other adaptor proteins (Fig. 4A), we found that the SLQ motif of BIV Vif and the downstream amino acids YxxxxI/X, which have been found to form a CUL2 box, have significant similarity to the VHL box (23). We therefore constructed two BIV Vif mutants with changes in these two motifs (Fig. 4A) and then used coimmunoprecipitation to examine the interactions between WT BIV Vif, mutant BIV Vif SLQ-AAA, or mutant BIV Vif YI-AA and endogenous CUL2 and ELOB/C in transfected HEK293T cells. As expected, CUL2 and ELOB/C could interact specifically with WT BIV Vif (Fig. 4B, lane 2) but not with the sample containing the empty vector, VR1012 (Fig. 4B, lane 1). However, the BIV Vif SLQ-AAA mutant lost the ability to interact with ELOB/C, while it maintained the interaction with CUL2 (Fig. 4B, lane 3). The BIV Vif YI-AA mutant could still interact with ELOB/C but lost the ability to interact with CUL2 (Fig. 4B, lane 4). These results indicate that the SLQ and YI motifs of BIV Vif are required for interactions with ELOB/C and CUL2, respectively.
FIG 4.

Mutations in the VHL box affect BIV Vif binding to other cellular factors. (A) Alignment of partial sequences of BIV Vif and other VHL box-containing proteins and the point mutations made in the BIV Vif VHL box. (B) BIV Vif SLQ-AAA and YI-AA mutants with mutations in the VHL box were defective for binding to ELOB/C and CUL2, respectively. HEK293T cells in T25 flasks were transfected with 5 μg of BIV Vif-HA, BIV Vif SLQ-AAA–HA, or BIV Vif YI-AA–HA. Equal amounts of cell lysates were immunoprecipitated with anti-HA antibody conjugated to agarose and analyzed by immunoblotting with antibodies against HA, ELOB, ELOC, and CUL2. (C) The VHL box in BIV Vif is required for BIV Vif-mediated degradation in MDBK cells. The SLQ-AAA and YI-AA mutants showed an impaired ability to degrade btA3Z2Z3. MDBK cells (2 × 106) were transfected with 2 μg of btA3Z2Z3-HA-cherry and 2.5 μg of BIV Vif WT-HA-cherry or the SLQ-AAA or YI-AA mutant. Cells were lysed and analyzed by immunoblotting with anti-HA or anti-tubulin antibody.
The BC box and CUL2 box in BIV Vif are required for suppression of btA3Z2Z3.
To investigate the contribution of the SLQ and YI motifs to the functions of BIV Vif, we evaluated the BIV Vif function in bovine MDBK cells. Immunoblot analysis of the transfected MDBK cell lysates showed that BIV Vif induced the degradation of btA3Z2Z3 (Fig. 4C, lane 2) but not in its absence (lane 1). However, the BIV Vif SLQ-AAA and BIV Vif YI-AA mutants, which are defective for E3 ubiquitin ligase assembly, were defective in mediating the degradation of btA3Z2Z3 (Fig. 4C, lanes 3 and 4). The results of this assay demonstrated that BIV Vif also forms an E3 ligase in the bovine system in order to degrade the bovine host's A3 proteins.
The degradation of btA3Z2Z3 induced by BIV Vif (Fig. 5A, lane 3) was also observed in HEK293T cells. Again, the degradation of btA3Z2Z3 was not seen with BIV Vif SLQ-AAA or BIV Vif YI-AA (Fig. 5A, lanes 4 and 5). The level of incorporation of btA3Z2Z3 into HIV-1ΔVif virions in the presence of the BIV Vif SLQ-AAA or BIV Vif YI-AA mutant (Fig. 5B, lanes 4 and 5) was higher than that in the presence of WT BIV Vif (Fig. 5B, lane 3). The functionality of the BIV Vif SLQ-AAA and BIV Vif YI-AA mutants was then tested in the MAGI assay. Virus infectivity in the absence of btA3Z2Z3 was set to 100%. BIV Vif suppressed the antiviral activity of btA3Z2Z3, whereas the expression of the BIV Vif SLQ-AAA or BIV Vif YI-AA protein did not (Fig. 5C). These results suggest that the mechanism by which BIV Vif overcomes the antiviral activity of btA3Z2Z3 requires both the BC box and the YI motif, which interact with ELOB/C and CUL2, respectively.
FIG 5.

The VHL box in BIV Vif is required for BIV Vif function. (A) The BIV Vif SLQ-AAA and YI-AA mutants showed an impaired ability to degrade btA3Z2Z3. The btA3Z2Z3 expression vector (2 μg) was cotransfected with 2 μg of NL4-3ΔVif or VR1012 (as a control vector), with or without 2 μg of WT BIV Vif or a BIV Vif mutant (SLQ-AAA or YI-AA), into HEK293T cells in T25 flasks, as indicated. The stability of btA3Z2Z3 was analyzed by SDS-PAGE of equal amounts of cell lysate, followed by immunoblotting with antibodies against Myc-tagged BIV Vif and the mutants, HA-tagged btA3Z2Z3, p24, and ribosomal P protein. Ribosomal P protein was used to ensure equal loading. (B) Virion packaging of btA3Z2Z3 was not inhibited by the BIV Vif SLQ-AAA or YI-AA mutant. HEK293T cells were cotransfected with vectors as described in the legend to panel A, and viruses from the supernatants were analyzed by immunoblotting with antibodies against HA and p24. (C) BIV Vif VHL box mutants could not rescue the infectivity of NL4-3ΔVif in the presence of btA3Z2Z3. Viruses from the supernatants were analyzed as described in the legend to Fig. 1A. Errors bars represent the SDs for triplicate wells within one experiment.
CCHC mutants impair BIV Vif-mediated degradation of btA3Z2Z3.
A novel HCCH zinc binding motif in HIV-1 Vif which is highly conserved in primate lentiviral Vif proteins is critical for the interaction of Vif with CUL5 (34). To determine whether BIV Vif requires this motif to interact with Cullin proteins, we first tested the effect of the membrane-permeant zinc chelator TPEN on BIV Vif function. In the absence of TPEN, BIV Vif induced the degradation of btA3Z2Z3, but this effect was abolished by the addition of 2.5 μM TPEN (Fig. 6A, lanes 2 and 4). TPEN had no effect on btA3Z2Z3 expression in the absence of BIV Vif (Fig. 6A, lane 3). The observed effect of zinc chelation on btA3Z2Z3 degradation implied the existence of a zinc binding motif in BIV Vif that may be critical for function. We therefore searched for an analogous zinc binding motif by aligning the sequences of BIV Vif and various primate Vif proteins. The alignment revealed a putative zinc HCCHCH motif located upstream of the BC box in BIV Vif. We then constructed a series of BIV Vif mutants (Fig. 6B) and examined their function. The results revealed that the H102L and H149L mutations did not affect BIV Vif-mediated btA3Z2Z3 degradation (Fig. 6C, lanes 3 and 8), but mutants containing the C111S, C113S, H115L, or C134S mutation could no longer induce the degradation of btA3Z2Z3 (Fig. 6C, lanes 4 to 7). The level of incorporation of btA3Z2Z3 into HIV-1ΔVif virions in the presence of the BIV Vif C111S, C113S, H115L, or C134S mutant (Fig. 6C, lanes 4 to 7) was greater than that in the presence of WT BIV Vif or the H102L or H149L mutant (Fig. 6C, lanes 2, 3, and 8). We then tested the functionality of the BIV Vif mutants in the MAGI assay. Virus infectivity in the absence of btA3Z2Z3 was set to 100%. Expression of the BIV Vif WT suppressed the antiviral activity of btA3Z2Z3, but expression of the BIV Vif C111S, C113S, H115L, or C134S mutant did not (Fig. 6D). These biochemical and genetic requirements strongly suggest that BIV Vif has a zinc binding motif that is required for CUL2 recruitment and bovine A3 degradation.
FIG 6.

The CCHC motif in BIV Vif is required for BIV Vif function. (A) TPEN inhibited BIV Vif-mediated btA3Z2Z3 degradation. HEK293T cells in 12-well plates were cotransfected with 0.3 μg btA3Z2Z3-HA plus 1 μg of VR1012 or BIV Vif-HA, and cells were treated 24 h later with 2.5 μM dimethyl sulfoxide (DMSO) or TPEN for 24 h. The cells were then harvested for immunoblot analysis with the indicated antibodies. (B) Alignment of various Vif proteins and BIV Vif mutants with mutations in the HCCHCH motif constructed in this study. (C) CCHC mutations in BIV Vif inhibited BIV Vif-mediated btA3Z2Z3 degradation. HEK293T cells in 12-well plates were transfected with 0.3 μg btA3Z2Z3 plus 1 μg of VR1012, WT BIV Vif, or one of its mutants (the H102L, C111S, C113S, H115L, or C134S mutant). At 48 h after transfection, cells were harvested for immunoblotting with anti-HA and antitubulin antibodies. (D) CCHC mutations in BIV Vif could not rescue the infectivity of NL4-3ΔVif in the presence of btA3Z2Z3. Viruses from supernatants were analyzed as described in the legend to Fig. 1A. Errors bars represent the SDs for triplicate wells within one experiment.
A CUL2ΔNedd mutant blocks BIV Vif-mediated degradation of bovine A3Z2Z3.
Neddylation of Cullin proteins is critical for E3 ubiquitin ligase activity (35, 36). To study the role of CUL2 in the BIV Vif-induced degradation of btA3Z2Z3, we used a dominant negative CUL2ΔNedd mutant to inhibit CUL2 function. CUL2ΔNedd is able to interact with Vif but is defective in E3 ligase activity, resulting in inhibition of Vif activity. When HEK293T cells were cotransfected with btA3Z2Z3 and NL4-3ΔVif along with BIV Vif or VR1012 (as a control vector), with or without the CUL2ΔNedd mutant, we found that the presence of the dominant negative CUL2 mutant inhibited BIV Vif-induced btA3Z2Z3 degradation (Fig. 7A, lane 3), suggesting that CUL2 plays a role in the BIV Vif-induced turnover of btA3Z2Z3. The ability of BIV Vif to exclude btA3Z2Z3 virion packaging was inhibited by CUL2ΔNedd (Fig. 7B, lane 3). We then asked whether disruption of the CUL2 function could influence the ability of BIV Vif to suppress btA3Z2Z3's antiviral activity. Supernatants were collected from transfected cells and used to infect the MAGI-CCR5 cell line. The results showed that the infectivity of virus produced in the presence of CUL2ΔNedd was dramatically reduced (Fig. 7C) compared to that of virus produced in the absence of CUL2ΔNedd, indicating that CUL2 activity is required for BIV Vif-mediated suppression of btA3Z2Z3.
FIG 7.
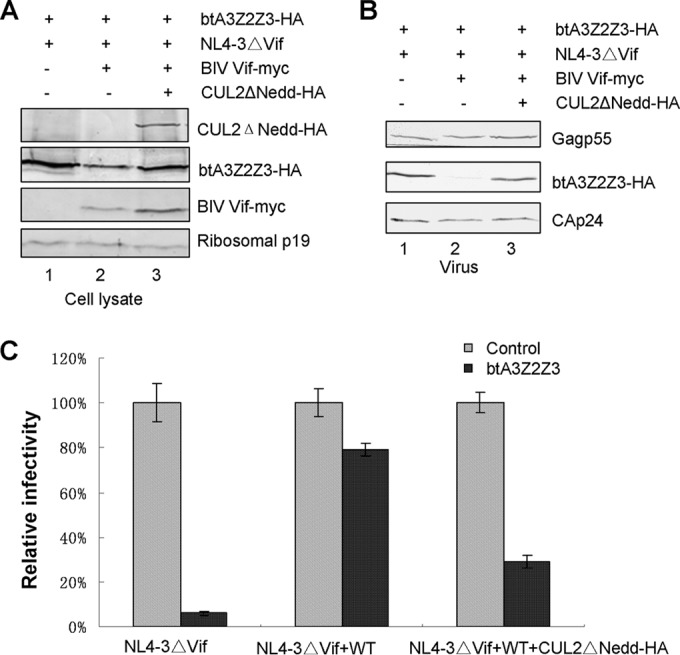
The CUL2 dominant negative mutant CUL2ΔNedd inhibits the function of BIV Vif. (A) The CUL2ΔNedd mutant blocked the BIV Vif-induced degradation of btA3Z2Z3. HEK293T cells in T25 flasks were cotransfected with 2 μg btA3Z2Z3 and 2 μg NL4-3ΔVif, along with 2 μg BIV Vif or a control vector (VR1012) and with or without 2 μg of the CUL2ΔNedd mutant. Cell lysates from transfected cells were prepared and separated by SDS-PAGE, transferred to nitrocellulose membranes, and probed with the indicated antibody. Ribosomal P protein was used as the sample loading control. (B) The CUL2ΔNedd mutant could not inhibit btA3Z2Z3 virion packaging. Viruses from supernatants were analyzed by immunoblotting with antibodies against HA and p24. (C) The CUL2ΔNedd mutant could inhibit the infectivity of NL4-3ΔVif virus in the presence of BIV Vif and btA3Z2Z3. Viruses from the supernatants were analyzed as described in the legend to Fig. 1A. Errors bars represent the SDs for triplicate wells within one experiment.
MLN4924 impairs BIV Vif-mediated degradation of the btA3Z2Z3 protein.
MLN4924 is an investigational small-molecule inhibitor of the NEDD8-activating enzyme (NAE) that is currently in phase I clinical trials (37). NAE inhibition prevents the ubiquitination and proteasomal degradation of substrates for CRL E3 ligases (35). Therefore, we examined the ability of MLN4924 to inhibit the neddylation of the BIV Vif-CRL E3 complex. HEK293T cells were transfected with btA3Z2Z3-HA and VR1012 or BIV Vif-HA. Twenty-four hours later, 300 nM MLN4924 was added to the cell culture. After another 24 h, the cells were harvested for immunoblotting with an HA antibody to detect HA-tagged btA3Z2Z3 and BIV Vif. The results showed that MLN4924 strongly impaired the ability of BIV Vif to induce btA3Z2Z3 degradation (Fig. 8A, lanes 2 and 4), functioning as well as the specific proteasome inhibitor MG132.
FIG 8.
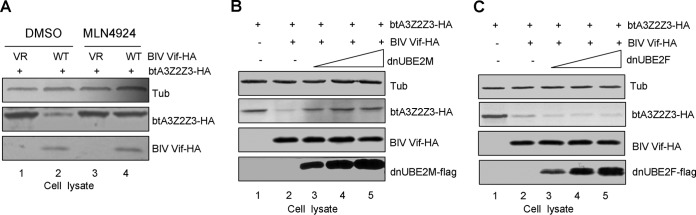
Effects of MLN4924 and UBE2M on BIV Vif-mediated degradation of btA3Z2Z3. (A) MLN4924 inhibited the BIV Vif-mediated degradation of btA3Z2Z3 by blocking the neddylation activation of BIV Vif-CRL E3 ligase. HEK293T cells in 12-well plates were cotransfected with 0.3 μg btA3Z2Z3-HA plus 1 μg of VR1012 or BIV Vif-HA, and cells were treated 24 h later with 300 nM DMSO or MLN4924 for 24 h. The cells were then harvested for immunoblot analysis with the indicated antibodies. (B) A UBE2M mutant inhibited BIV Vif-mediated btA3Z2Z3 degradation in a dose-dependent manner. HEK293T cells in 12-well plates were cotransfected with 0.3 μg btA3Z2Z3-HA and 1 μg of VR1012 or BIV Vif-HA plus increasing amounts (0.2 μg, 0.6 μg, and 1.8 μg in lanes 3 to 5, respectively) of UBE2M (C111S). At 48 h after transfection, cell lysates were harvested for immunoblot analysis with the indicated antibodies. (C) A UBE2F (C116S) mutant did not inhibit BIV Vif-mediated btA3Z2Z3 degradation. dn, dominant negative mutant.
Neddylation is required for the activation of CRLs (38, 39). Two well-known pathways exist for the neddylation system: RBX1-UBE2M and RBX2-UBE2F (35). We next evaluated the role of neddylation in the ability of BIV Vif to counteract the effects of the btA3Z2Z3 protein. As expected, we found that the expression of the UBE2M (UBC12) dominant negative mutant (C111S) in HEK293T cells impaired BIV Vif-mediated btA3Z2Z3 degradation in a dose-dependent manner (Fig. 8B, lanes 3 to 5). However, the expression of the UBE2F dominant negative mutant (C116S) did not affect the function of BIV Vif (Fig. 8C, lanes 3 to 5). These data are consistent with prior reports demonstrating a requirement for RBX2-UBE2F for HIV-1 Vif-mediated degradation of hsA3G (40) and observations from the ubiquitin field indicating exclusive associations between CUL5-RBX2 and CUL2-RBX1 (3, 23, 25, 26, 41).
BIV Vif selectively degrades and inhibits various bovine A3 proteins.
Bovine cells encode four A3 proteins: btA3Z2Z3, btA3Z2, btA3Z3, and btA3Z1. We next asked whether BIV Vif can degrade these A3 proteins and inhibit their antiviral activity. HEK293T cells were cotransfected with GFP, btA3Z2Z3-GFP, btA3Z2-GFP, or btA3Z1-GFP and NL4-3ΔVif plus BIV Vif. We found that BIV Vif could specifically degrade btA3Z2Z3, btA3Z2, and btA3Z3 (Fig. 9A; compare lanes 2 and 3, 5 and 6, and 8 and 9) but not btA3Z1 (Fig. 9A, lanes 17 and 18). We also found that the packaging of btA3Z2Z3, btA3Z2, and btA3Z3 into HIV virions in the presence of BIV Vif was inefficient compared to packaging in the absence of BIV Vif (Fig. 9A, lanes 2 and 3, 5 and 6, and 8 and 9), but btA3Z1 could be packaged into the virions whether or not BIV Vif was present (Fig. 9A, lanes 17 and 18). To further examine the effect of BIV Vif inhibition on the antiviral activity of btA3 proteins, we used the supernatants from transfected HEK293T cells to infect MAGI-CCR5 reporter cells. The viral infectivity in the absence of BIV Vif was set to 100%. BIV Vif could efficiently inhibit the antiviral activity of btA3Z2Z3 and btA3Z3 against the NL4-3ΔVif virus compared to their antiviral activity in the absence of BIV Vif; however, btA3Z2 and btA3Z1 could not inhibit the infectivity of NL4-3ΔVif, regardless of whether BIV Vif was present or not (Fig. 9B). Moreover, coimmunoprecipitation assays demonstrated that BIV Vif also interacts with btA3Z1 (Fig. 9C, lane 4), indicating that the interaction between Vif and APOBEC3 proteins is not sufficient for Vif-mediated degradation. A similar observation has been reported for Cullin-based E3-mediated substrate degradation (28). These results suggest that bovine A3 proteins have different antiviral activities and BIV Vif selectively degrades only the restrictive A3 proteins of its bovine host, consistent with prior reports (12, 14, 15).
FIG 9.

BIV Vif can selectively degrade bovine A3 proteins and overcome their antiviral activity. (A) HEK293T cells in T25 flasks were cotransfected with 2 μg of an infectious clone of the Vif mutant virus NL4-3ΔVif plus 2 μg of VR1012 or BIV Vif-Myc plus 2 μg of a control vector (GFP) or an expression vector encoding a GFP-tagged bovine A3 protein, as indicated. At 48 h after transfection, cell lysates were harvested for immunoblot analysis with the indicated antibodies. Supernatants were centrifuged for virion packaging analysis. (B) BIV Vif selectively inhibited the antiviral activity of btA3Z2Z3 and btA3Z3. The infectivity of the resulting virus was assayed by infecting MAGI-CCR5 cells. Virus infectivity in the absence of A3 was set to 100%. Error bars represent the SDs for triplicate wells within one experiment. (C) BIV Vif cannot degrade btA3Z1 but still interacts with btA3Z1. HEK293T cells in T25 flasks were transfected with 2 μg of VR1012 as a negative-control vector, BIV Vif-HA plus 2 μg of btA3Z2Z3-GFP as a positive control, or btA3Z1-GFP. All experiments were performed in the presence of MG132. Cells were lysed and analyzed by immunoblotting with an anti-GFP, anti-HA, or antitubulin antibody. Cell lysates were immunoprecipitated with the anti-HA antibody and analyzed by immunoblotting with an anti-GFP antibody and an anti-HA antibody.
DISCUSSION
Recent studies have shown that BIV Vif efficiently degrades the restrictive A3 proteins of its bovine host (14), but the precise mechanism had not been investigated. Here, we show that BIV Vif forms a CRL E3 ubiquitin ligase by recruiting CUL2, ELOB/C, and RBX1. Remarkably, unlike the strong HIV-1/SIV requirement for CBF-β as a cofactor (12, 13, 31, 32, 36, 37), BIV Vif functions without CBF-β. These requirements parallel those for VHL, which also forms an E3 ubiquitin ligase with ELOB/C and CUL2 (27, 42), also without CBF-β (33). Additional studies will be necessary to address whether BIV Vif requires a different cofactor or whether it truly uses a more minimal ligase complex to counteract the A3 proteins of its bovine host (Fig. 10).
FIG 10.
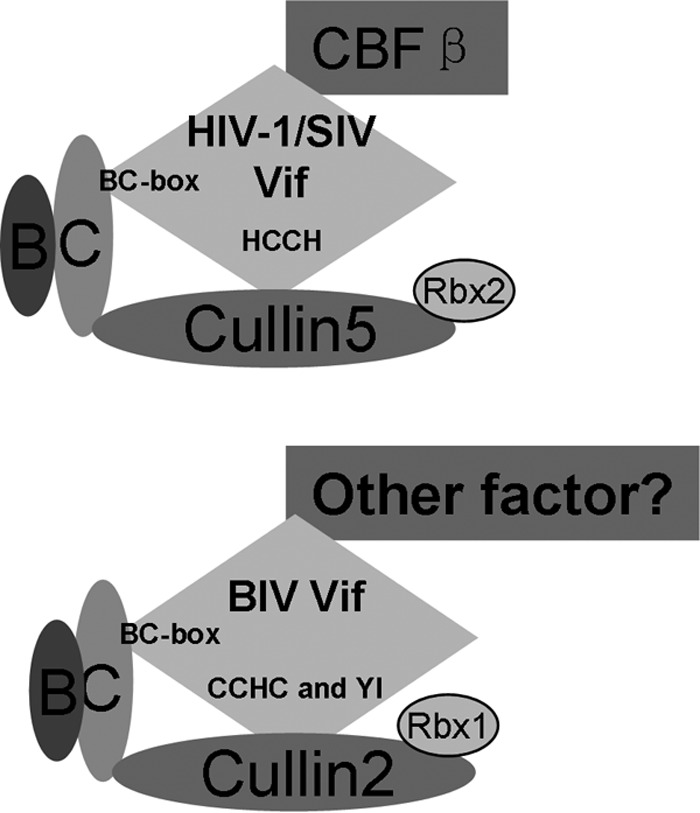
Models for HIV-1/SIV Vif and BIV Vif recruitment of various CRL E3 ligase complexes. (Top) HIV-1/SIV Vif recruits CUL5, ELOB/C, CBF-β, and RBX2 to form an A3 targeting CRL5 E3 ligase. HIV-1/SIV Vif utilized the BC box to interact with ELOB/C and HCCH to interact with CUL5. (Bottom) BIV Vif recruits CUL2, ELOB/C, and RBX1, but not CUL5 or CBF-β, to form an active CRL2 E3 ligase. BIV Vif utilized the BC box to interact with ELOB/C and CCHC as well as YI motifs to interact with CUL2.
Both CUL2 and CUL5 can interact with ELOB/C to form E3 ubiquitin ligase complexes. The VHL box or SOCS box located in the substrate adaptor specifically determines the association with the CUL2-RBX1 or CUL5-RBX2 module to form CRL complexes, and these interactions are determined by the presence of a domain known as the CUL2 or CUL5 box. By comparison of the sequence of BIV Vif with the sequences of other VHL, LRR-1, and FEM1B proteins that have been identified to be CUL2-RBX1-interacting proteins, we found that BIV Vif also contains a VHL box that determines the interaction of BIV Vif with CUL2 but not with CUL5 (23).
In the HIV-1 Vif recruitment of the E3 ubiquitin complex, HIV-1 Vif has been proposed to bind ELOB/C at its C terminus, followed by CBF-β at its N terminus, inducing structural changes at both termini, and, consequently, to bind CUL5, which requires residues in both the N and C termini of the protein to assemble a functional ubiquitin ligase (29). Here, BIV Vif seems to have different steps for assembling a functional ubiquitin ligase with ELOB/C and CUL2. The BIV Vif SLQ-AAA mutant still maintained the ability to interact with CUL2 (Fig. 3), which shows that BIV Vif does not require ELOB/C or CBF-β in order to bind CUL2. However, HIV Vif SLQ-AAA or SIVagm Vif, which cannot bind to ELOB/C, also lacks the ability to interact with CUL5 (17, 27). While CBF-β maintains a regulatory function in the assembly of the HIV-1 Vif CRL5 E3 ubiquitin ligase, BIV Vif may or may not use an alternative factor to facilitate formation of the CRL2 E3 ligase (Fig. 10).
The eukaryotic ubiquitination system involves a ubiquitin-activating enzyme, E1; a ubiquitin-conjugating enzyme, E2; and, often, a substrate-specific ubiquitin-protein ligase, E3 (30). In addition, ubiquitin-like proteins, such as NEDD8, require their own E1s and E2s (UBE2M and UBE2F) for attachment (1). UBE2M and UBE2F have been identified to be putative NEDD8-conjugating enzymes (E2) which link NEDD8 to a conserved Lys in the C-terminal domain of Cullin proteins, including CUL2, CUL1, and CUL5, followed by activation of the Cullin-RING ligase (E3). The E2s have distinct functions, with UBE2M/CUL2-RBX1 and UBE2F/CUL5-RBX2 displaying different target Cullin specificities (35, 36). We showed that a dominant negative UBE2M mutant inhibited BIV-mediated degradation of btA3Z2Z3, but a dominant negative UBE2F mutant did not. These data indicate that BIV Vif recruits CUL2-RBX1 to the CRL E3 ligase through a UBE2M E2-conjugating enzyme.
BIV Vif-mediated degradation of btA3Z2Z3 could be detected in human HEK293T cells as well as in bovine MDBK cells. Homology analyses of various bovine and human proteins contained in the CUL2-ELOB/C E3 ligase, including btCUL2 and hsCUL2, btELOB/C and hsELOB/C, and btCBF-β and hsCBF-β, were performed. The homologies between all these proteins exceeded 97%. Consistent with such high identities, we also observed a selective interaction of BIV Vif with btCUL2 but not with btCUL5 (Fig. 2). In contrast, HIV-1 Vif selectively interacted with btCUL5 but not btCUL2 (Fig. 2). Thus, lentiviral Vif proteins have evolved to use different Cullin molecules to assemble CRL E3 ubiquitin ligases and to inactivate host restriction APOBEC3 proteins (Fig. 10). To our knowledge, this study is the first to reveal the mechanism by which BIV Vif overcomes its bovine host A3 defensive factors and to show that this mechanism is different from that of primate lentiviruses. These findings provide useful information for further understanding of the functional mechanisms of different lentiviral Vif accessory proteins. It also suggests that, depending on the mechanism involved, strategies to leverage the A3 pathway in humans against HIV-1 may not be useful for treating nonprimate lentiviral Vif infections.
ACKNOWLEDGMENTS
We thank Chunyan Dai for critical reagents and Deborah McClellan for editorial assistance.
This work was supported by funding from the National Natural Science Foundation of China (no. 31270202), the Chinese Ministry of Science and Technology (2012CB911100 and 2013ZX10001-005), the Chinese Ministry of Education (IRT1016), and the Key Laboratory of Molecular Virology, Jilin Province (20102209). The contributions of the R. S. Harris lab were supported by funding from the NIH (R01 AI064046).
Footnotes
Published ahead of print 20 August 2014
REFERENCES
- 1.Jones D, Crowe E, Stevens TA, Candido EP. 2002. Functional and phylogenetic analysis of the ubiquitylation system in Caenorhabditis elegans: ubiquitin-conjugating enzymes, ubiquitin-activating enzymes, and ubiquitin-like proteins. Genome Biol. 3:RESEARCH0002. 10.1186/gb-2001-3-1-research0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Petroski MD, Deshaies RJ. 2005. Function and regulation of cullin-RING ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 6:9–20. 10.1038/nrm1547. [DOI] [PubMed] [Google Scholar]
- 3.Yu X, Yu Y, Liu B, Luo K, Kong W, Mao P, Yu XF. 2003. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science 302:1056–1060. 10.1126/science.1089591. [DOI] [PubMed] [Google Scholar]
- 4.Conticello SG, Harris RS, Neuberger MS. 2003. The Vif protein of HIV triggers degradation of the human antiretroviral DNA deaminase APOBEC3G. Curr. Biol. 13:2009–2013. 10.1016/j.cub.2003.10.034. [DOI] [PubMed] [Google Scholar]
- 5.Marin M, Rose KM, Kozak SL, Kabat D. 2003. HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat. Med. 9:1398–1403. 10.1038/nm946. [DOI] [PubMed] [Google Scholar]
- 6.Mehle A, Strack B, Ancuta P, Zhang C, McPike M, Gabuzda D. 2004. Vif overcomes the innate antiviral activity of APOBEC3G by promoting its degradation in the ubiquitin-proteasome pathway. J. Biol. Chem. 279:7792–7798. 10.1074/jbc.M313093200. [DOI] [PubMed] [Google Scholar]
- 7.Sheehy AM, Gaddis NC, Malim MH. 2003. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat. Med. 9:1404–1407. 10.1038/nm945. [DOI] [PubMed] [Google Scholar]
- 8.Stopak K, de Noronha C, Yonemoto W, Greene WC. 2003. HIV-1 Vif blocks the antiviral activity of APOBEC3G by impairing both its translation and intracellular stability. Mol. Cell 12:591–601. 10.1016/S1097-2765(03)00353-8. [DOI] [PubMed] [Google Scholar]
- 9.Mehle A, Goncalves J, Santa-Marta M, McPike M, Gabuzda D. 2004. Phosphorylation of a novel SOCS-box regulates assembly of the HIV-1 Vif-Cul5 complex that promotes APOBEC3G degradation. Genes Dev. 18:2861–2866. 10.1101/gad.1249904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu Y, Xiao Z, Ehrlich ES, Yu X, Yu XF. 2004. Selective assembly of HIV-1 Vif-Cul5-ElonginB-ElonginC E3 ubiquitin ligase complex through a novel SOCS box and upstream cysteines. Genes Dev. 18:2867–2872. 10.1101/gad.1250204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hultquist JF, Binka M, LaRue RS, Simon V, Harris RS. 2012. Vif proteins of human and simian immunodeficiency viruses require cellular CBFbeta to degrade APOBEC3 restriction factors. J. Virol. 86:2874–2877. 10.1128/JVI.06950-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jonsson SR, Hache G, Stenglein MD, Fahrenkrug SC, Andresdottir V, Harris RS. 2006. Evolutionarily conserved and non-conserved retrovirus restriction activities of artiodactyl APOBEC3F proteins. Nucleic Acids Res. 34:5683–5694. 10.1093/nar/gkl721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chackerian B, Long EM, Luciw PA, Overbaugh J. 1997. Human immunodeficiency virus type 1 coreceptors participate in postentry stages in the virus replication cycle and function in simian immunodeficiency virus infection. J. Virol. 71:3932–3939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Larue RS, Lengyel J, Jonsson SR, Andresdottir V, Harris RS. 2010. Lentiviral Vif degrades the APOBEC3Z3/APOBEC3H protein of its mammalian host and is capable of cross-species activity. J. Virol. 84:8193–8201. 10.1128/JVI.00685-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Han X, Liang W, Hua D, Zhou X, Du J, Evans SL, Gao Q, Wang H, Viqueira R, Wei W, Zhang W, Yu XF. 2014. Evolutionarily conserved requirement for core binding factor beta in the assembly of the human immunodeficiency virus/simian immunodeficiency virus Vif-Cullin 5-RING E3 ubiquitin ligase. J. Virol. 88:3320–3328. 10.1128/JVI.03833-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang W, Chen G, Niewiadomska AM, Xu R, Yu XF. 2008. Distinct determinants in HIV-1 Vif and human APOBEC3 proteins are required for the suppression of diverse host anti-viral proteins. PLoS One 3:e3963. 10.1371/journal.pone.0003963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.He Z, Zhang W, Chen G, Xu R, Yu XF. 2008. Characterization of conserved motifs in HIV-1 Vif required for APOBEC3G and APOBEC3F interaction. J. Mol. Biol. 381:1000–1011. 10.1016/j.jmb.2008.06.061. [DOI] [PubMed] [Google Scholar]
- 18.Chen G, He Z, Wang T, Xu R, Yu XF. 2009. A patch of positively charged amino acids surrounding the human immunodeficiency virus type 1 Vif SLVx4Yx9Y motif influences its interaction with APOBEC3G. J. Virol. 83:8674–8682. 10.1128/JVI.00653-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo Y, Dong L, Qiu X, Wang Y, Zhang B, Liu H, Yu Y, Zang Y, Yang M, Huang Z. 2014. Structural basis for hijacking CBF-beta and CUL5 E3 ligase complex by HIV-1 Vif. Nature 505:229–233. 10.1038/nature12884. [DOI] [PubMed] [Google Scholar]
- 20.Fribourgh JL, Nguyen HC, Wolfe LS, Dewitt DC, Zhang W, Yu XF, Rhoades E, Xiong Y. 2014. Core binding factor beta plays a critical role by facilitating the assembly of the Vif-cullin 5 E3 ubiquitin ligase. J. Virol. 88:3309–3319. 10.1128/JVI.03824-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou X, Han X, Zhao K, Du J, Evans SL, Wang H, Li P, Zheng W, Rui Y, Kang J, Yu XF. 2014. Dispersed and conserved hydrophobic residues of HIV-1 Vif are essential for CBFβ recruitment and A3G suppression. J. Virol. 88:2555–2563. 10.1128/JVI.03604-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matsui Y, Shindo K, Nagata K, Io K, Tada K, Iwai F, Kobayashi M, Kadowaki N, Harris RS, Takaori-Kondo A. 2014. Defining HIV-1 Vif residues that interact with CBFbeta by site-directed mutagenesis. Virology 449:82–87. 10.1016/j.virol.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kamura T, Maenaka K, Kotoshiba S, Matsumoto M, Kohda D, Conaway RC, Conaway JW, Nakayama KI. 2004. VHL-box and SOCS-box domains determine binding specificity for Cul2-Rbx1 and Cul5-Rbx2 modules of ubiquitin ligases. Genes Dev. 18:3055–3065. 10.1101/gad.1252404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gonda MA, Oberste MS, Garvey KJ, Pallansch LA, Battles JK, Pifat DY, Bess JW, Jr, Nagashima K. 1990. Development of the bovine immunodeficiency-like virus as a model of lentivirus disease. Dev. Biol. Stand. 72:97–110. [PubMed] [Google Scholar]
- 25.Mahrour N, Redwine WB, Florens L, Swanson SK, Martin-Brown S, Bradford WD, Staehling-Hampton K, Washburn MP, Conaway RC, Conaway JW. 2008. Characterization of Cullin-box sequences that direct recruitment of Cul2-Rbx1 and Cul5-Rbx2 modules to Elongin BC-based ubiquitin ligases. J. Biol. Chem. 283:8005–8013. 10.1074/jbc.M706987200. [DOI] [PubMed] [Google Scholar]
- 26.Querido E, Blanchette P, Yan Q, Kamura T, Morrison M, Boivin D, Kaelin WG, Conaway RC, Conaway JW, Branton PE. 2001. Degradation of p53 by adenovirus E4orf6 and E1B55K proteins occurs via a novel mechanism involving a Cullin-containing complex. Genes Dev. 15:3104–3117. 10.1101/gad.926401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iwai K, Yamanaka K, Kamura T, Minato N, Conaway RC, Conaway JW, Klausner RD, Pause A. 1999. Identification of the von Hippel-Lindau tumor-suppressor protein as part of an active E3 ubiquitin ligase complex. Proc. Natl. Acad. Sci. U. S. A. 96:12436–12441. 10.1073/pnas.96.22.12436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang W, Huang M, Wang T, Tan L, Tian C, Yu X, Kong W, Yu XF. 2008. Conserved and non-conserved features of HIV-1 and SIVagm Vif mediated suppression of APOBEC3 cytidine deaminases. Cell. Microbiol. 10:1662–1675. 10.1111/j.1462-5822.2008.01157.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Evans SL, Schon A, Gao Q, Han X, Zhou X, Freire E, Yu XF. 2014. HIV-1 Vif N-terminal motif is required for recruitment of Cul5 to suppress APOBEC3. Retrovirology 11:4. 10.1186/1742-4690-11-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pickart CM. 2004. Back to the future with ubiquitin. Cell 116:181–190. 10.1016/S0092-8674(03)01074-2. [DOI] [PubMed] [Google Scholar]
- 31.Kobayashi M, Takaori-Kondo A, Miyauchi Y, Iwai K, Uchiyama T. 2005. Ubiquitination of APOBEC3G by an HIV-1 Vif-Cullin5-Elongin B-Elongin C complex is essential for Vif function. J. Biol. Chem. 280:18573–18578. 10.1074/jbc.C500082200. [DOI] [PubMed] [Google Scholar]
- 32.Jager S, Kim DY, Hultquist JF, Shindo K, LaRue RS, Kwon E, Li M, Anderson BD, Yen L, Stanley D, Mahon C, Kane J, Franks-Skiba K, Cimermancic P, Burlingame A, Sali A, Craik CS, Harris RS, Gross JD, Krogan NJ. 2012. Vif hijacks CBF-beta to degrade APOBEC3G and promote HIV-1 infection. Nature 481:371–375. 10.1038/nature10693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang W, Du J, Evans SL, Yu Y, Yu XF. 2012. T-cell differentiation factor CBF-beta regulates HIV-1 Vif-mediated evasion of host restriction. Nature 481:376–379. 10.1038/nature10718. [DOI] [PubMed] [Google Scholar]
- 34.Luo K, Xiao Z, Ehrlich E, Yu Y, Liu B, Zheng S, Yu XF. 2005. Primate lentiviral virion infectivity factors are substrate receptors that assemble with cullin 5-E3 ligase through a HCCH motif to suppress APOBEC3G. Proc. Natl. Acad. Sci. U. S. A. 102:11444–11449. 10.1073/pnas.0502440102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wada H, Yeh ET, Kamitani T. 2000. A dominant-negative UBC12 mutant sequesters NEDD8 and inhibits NEDD8 conjugation in vivo. J. Biol. Chem. 275:17008–17015. 10.1074/jbc.275.22.17008. [DOI] [PubMed] [Google Scholar]
- 36.Huang DT, Ayrault O, Hunt HW, Taherbhoy AM, Duda DM, Scott DC, Borg LA, Neale G, Murray PJ, Roussel MF, Schulman BA. 2009. E2-RING expansion of the NEDD8 cascade confers specificity to cullin modification. Mol. Cell 33:483–495. 10.1016/j.molcel.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, Brownell JE, Burke KE, Cardin DP, Critchley S, Cullis CA, Doucette A, Garnsey JJ, Gaulin JL, Gershman RE, Lublinsky AR, McDonald A, Mizutani H, Narayanan U, Olhava EJ, Peluso S, Rezaei M, Sintchak MD, Talreja T, Thomas MP, Traore T, Vyskocil S, Weatherhead GS, Yu J, Zhang J, Dick LR, Claiborne CF, Rolfe M, Bolen JB, Langston SP. 2009. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 458:732–736. 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- 38.Liu J, Furukawa M, Matsumoto T, Xiong Y. 2002. NEDD8 modification of CUL1 dissociates p120(CAND1), an inhibitor of CUL1-SKP1 binding and SCF ligases. Mol. Cell 10:1511–1518. 10.1016/S1097-2765(02)00783-9. [DOI] [PubMed] [Google Scholar]
- 39.Morimoto M, Nishida T, Honda R, Yasuda H. 2000. Modification of cullin-1 by ubiquitin-like protein Nedd8 enhances the activity of SCF(skp2) toward p27(kip1). Biochem. Biophys. Res. Commun. 270:1093–1096. 10.1006/bbrc.2000.2576. [DOI] [PubMed] [Google Scholar]
- 40.Stanley DJ, Bartholomeeusen K, Crosby DC, Kim DY, Kwon E, Yen L, Cartozo NC, Li M, Jager S, Mason-Herr J, Hayashi F, Yokoyama S, Krogan NJ, Harris RS, Peterlin BM, Gross JD. 2012. Inhibition of a NEDD8 cascade restores restriction of HIV by APOBEC3G. PLoS Pathog. 8:e1003085. 10.1371/journal.ppat.1003085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kamura T, Koepp DM, Conrad MN, Skowyra D, Moreland RJ, Iliopoulos O, Lane WS, Kaelin WG, Jr, Elledge SJ, Conaway RC, Harper JW, Conaway JW. 1999. Rbx1, a component of the VHL tumor suppressor complex and SCF ubiquitin ligase. Science 284:657–661. 10.1126/science.284.5414.657. [DOI] [PubMed] [Google Scholar]
- 42.Kibel A, Iliopoulos O, DeCaprio JA, Kaelin WG., Jr 1995. Binding of the von Hippel-Lindau tumor suppressor protein to Elongin B and C. Science 269:1444–1446. 10.1126/science.7660130. [DOI] [PubMed] [Google Scholar]