

ABSTRACT
The serine-arginine-specific protein kinase SRPK1 is a common binding partner of the E1^E4 protein of diverse human papillomavirus types. We show here for the first time that the interaction between HPV1 E1^E4 and SRPK1 leads to potent inhibition of SRPK1 phosphorylation of host serine-arginine (SR) proteins that have critical roles in mRNA metabolism, including pre-mRNA processing, mRNA export, and translation. Furthermore, we show that SRPK1 phosphorylates serine residues of SR/RS dipeptides in the hinge region of the HPV1 E2 protein in in vitro kinase assays and that HPV1 E1^E4 inhibits this phosphorylation. After mutation of the putative phosphoacceptor serine residues, the localization of the E2 protein was altered in primary human keratinocytes; with a significant increase in the cell population showing intense E2 staining of the nucleolus. A similar effect was observed following coexpression of E2 and E1^E4 that is competent for inhibition of SRPK1 activity, suggesting that the nuclear localization of E2 is sensitive to E1^E4-mediated SRPK1 inhibition. Collectively, these data suggest that E1^E4-mediated inhibition of SRPK1 could affect the functions of host SR proteins and those of the virus transcription/replication regulator E2. We speculate that the novel E4 function identified here is involved in the regulation of E2 and SR protein function in posttranscriptional processing of viral transcripts.
IMPORTANCE The HPV life cycle is tightly linked to the epithelial terminal differentiation program, with the virion-producing phase restricted to differentiating cells. While the most abundant HPV protein expressed in this phase is the E4 protein, we do not fully understand the role of this protein. Few E4 interaction partners have been identified, but we had previously shown that E4 proteins from diverse papillomaviruses interact with the serine-arginine-specific protein kinase SRPK1, a kinase important in the replication cycles of a diverse range of DNA and RNA viruses. We show that HPV1 E4 is a potent inhibitor of this host cell kinase. We show that E4 inhibits SRPK1 phosphorylation, not only of cellular SR proteins involved in regulating alternative splicing of RNA but also the viral transcription/replication regulator E2. Our findings reveal a potential E4 function in regulation of viral late gene expression through the inhibition of a host cell kinase.
INTRODUCTION
Human papillomaviruses (HPVs) cause hyperproliferative warts and papillomas of the squamous epithelium at different body sites. Infection of the anogenital tract and oropharynx can lead to benign and malignant disease. There are 13 HPV types defined as causative agents of cancers at these sites, the most common being HPV 16 (HPV16) and HPV18. Of the viruses that infect cutaneous surfaces, some, such as HPV1, cause only benign warts, while infection with others, such as HPV5 and HPV8, can, in immunocompromised individuals, cause the formation of lesions that are at risk of malignant conversion.
Despite the heterogeneity in pathogenesis, HPVs show a high degree of conservation in their infectious cycles (1). The virion-producing phase of the infectious cycle—viral DNA amplification, capsid protein expression, and the assembly of new progeny—occurs in suprabasal keratinocytes. While high levels of the viral protein E4 are expressed during this stage of the life cycle, E4's role is an enigmatic one. Genetic knockdown of E4 expression in papillomavirus life cycle models can result in aberrations in the productive cycle, including reduced viral DNA amplification and decreased viral late gene expression (2–5). These studies indicated that E4 function(s) are important at multiple stages of virion production, although there may be differences in the role of E4 between genotypes (6). Overexpression of E4 proteins leads to a G2/M arrest of the cell cycle, a function common to HPV types with different epithelial tropisms (7–9). However, abrogation of this function in an HPV18 replication model did not affect viral genome amplification or expression of viral late genes (10). Reorganization of the keratin networks is another function that is conserved between virus types and, while it is predicted to compromise the structural integrity of the superficial cells to aid release of newly synthesized virus, confirmation of this role has yet to obtained (11–13).
During HPV infection, E4 is first synthesized as an E1^E4 fusion protein from spliced E1^E4 transcripts, such that the first few amino acids of the E4 protein are derived from the N terminus of E1 (14). The number of E4 species expressed in the virus life cycle, however, is expanded by phosphorylation and proteolysis (15, 16). Posttranslational modification of E4 likely serves to regulate E4 function during the different late stages of the infectious cycle (9, 17–20). As well as E4 being a substrate for a range of different protein kinases, the primarily cytoplasmic protein can also interfere with the cellular distribution of some kinases. Several studies have shown, in cells grown in monolayer cell culture, that the E4 proteins of HPV16 and HPV18 sequester cyclin-dependent kinase 1 (CDK1) and CDK2 in complex with their cyclin partners to cytoplasmic E4-cytokeratin structures (10, 21, 22). The retention of the active CDK2-cyclin A and CDK1-cyclin B complexes in the cytoplasm has been linked to the G2-M cell cycle arrest function of E4 (10, 21, 22). The serine-arginine (SR)-specific kinase SRPK1 is a binding partner of the E4 proteins of HPV1, -16, and -18 (23). Overexpression of HPV1 E4 in cultured keratinocytes induces the sequestration of SRPK1 to cytoplasmic E4 inclusion bodies. That the cellular kinase is also present in E4 inclusions formed in the upper layers of HPV1 induced palmar warts is a strong indication that the association is of physiological relevance (23).
SRPK1 is one of a family of serine-threonine kinases (SRPK1a, SRPK1 to -3), which specifically phosphorylate serine residues in serine-arginine/arginine-serine (SR/RS) dipeptide motifs (24, 25). The most studied substrates of these kinases have been the RNA-binding SR-rich proteins involved in multiple stages of mRNA maturation, including constitutive and alternative splicing, mRNA transport from the nucleus, and mRNA translation. SRPKs have a predominantly cytoplasmic localization, where they phosphorylate SR proteins and facilitate their nuclear import (26). The kinases can translocate into the nucleus following stress, or other signals, and during the cell cycle (25, 27, 28). Here they are known to target substrates found in nuclear speckles causing their release and redistribution to the cytoplasm (29). They have also been shown to associate with small nuclear ribonucleoproteins involved in spliceosome assembly (30). Furthermore, SRPKs have also been shown to phosphorylate the RS domains present in proteins that are not directly linked to pre-mRNA metabolism, suggesting that the kinases have diverse functions in the cell (25).
Notably, SRPKs have been shown to be essential factors in the replication cycle of several DNA and RNA viruses. For example, the ICP27 protein of herpes simplex virus 1 associates with SRPK1 to induce hypophosphorylation of SR proteins, leading to impairment in spliceosome assembly and inhibition of splicing (31). SRPK1 and SRPK2 have been shown to phosphorylate the core protein of hepatitis B virus (HBV) (32), and this phosphorylation is required for multiple steps in HBV DNA synthesis (33). Interestingly, HBV hijacks and repurposes SRPKs to perform as chaperones to facilitate HBV genome packaging (34, 35), demonstrating that SRPKs have multiple roles in HBV replication. In fact, inhibition of SRPK activity using the selective isonicotinamide compounds (e.g., SRPIN340) leads to the suppression of replication of various RNA viruses, including hepatitis C virus and human immunodeficiency virus (36, 37).
In this study, we investigated the effect of E4 on SRPK1 kinase activity and show for the first time that HPV1 E4 is a potent inhibitor of SRPK1 phosphorylation of host SR proteins. Furthermore, we show that SRPK1 phosphorylates SR/RS dipeptides in the hinge region of the transcription/replication regulator E2 and that E4 can inhibit this phosphorylation. We provide evidence to suggest that E4 regulates the subnuclear distribution of E2 by inhibiting SRPK1 phosphorylation of the hinge region.
MATERIALS AND METHODS
Cell culture.
Cells from the human cervical carcinoma cell line C33a and the non-small-cell lung carcinoma cell line H1299 were cultured in Dulbecco's modified Eagle medium with HEPES modification (Sigma-Aldrich Co.), supplemented with 4 mM glutamine and 10% (vol/vol) fetal calf serum. Primary human foreskin keratinocytes (HFK) were isolated from neonate foreskins collected with informed written parental consent (ethical approval no. 06/Q1702/45) and maintained in SFM keratinocyte growth media (Invitrogen, Paisley, Scotland, United Kingdom) as previously described (3).
Bacterial expression of recombinant proteins.
The expression of His-SRPK1 and glutathione S-transferase (GST)-tagged proteins of HPV1 E1^E4, Δ44-48, Δ49-53, HPV16 E1^E4, and HPV18 E1^E4 has been described in our previous study (23). HPV5 E1^E4 was cloned into the EcoRI site of pGEX-2T (GE Healthcare, Little Chalfont, United Kingdom) following amplification of the HPV5b total genome (a gift from Ethel de Villiers, Heidelberg, Germany) with the primer pair 5′-GCGCGAATTCATACGGATCCTAATCCTAAAGCTCCACGCCTCCAGGGTC-3′ and 5′-GCGCGAATTCTTACTGGGGGGTCGCGAGCTTCTTCC-3′.
HPV1 E2 and E2 hinge sequences (residues 198 to 323) were PCR amplified from pSG1aE2 (a gift from Saleem Khan, University of Pittsburgh, Pittsburgh, PA) using the following primer pairs: E2 full-length, 5′-GCGCGAATTCATATGGAAAACCTCAGCAGTCGC-3′ and 5′-GCGCGAATTCTTAAGACCCATTAAACTGTCC-3′; and E2 hinge region, 5′-GCGCGAATTCATGTTATGTCTTCCACTAGCTCC-3′ and 5′-GCGCGAATTCTTATACACAGACCACGGGTGG-3′. The PCR fragments were cloned into the EcoRI site of pGex-2T. The generation of GST-tagged mutant E2 proteins was performed using QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). The oligonucleotide primers used to generate the S265AS267A and S281A mutations were 5′-CCACCTCTGGGAGCTCTGGCTCTTCGCCGTCC-3′ and 5′-TCCGGGTGTCCTAGCGGGCGTTGATTCTC-3′, respectively (the nucleotide changes are underlined). The triple mutation, S265AS267AS281A, was generated using the S265AS267A template with the S281A mutagenic primer. Mutations were confirmed by bidirectional DNA sequencing of the complete insert.
SRSF1, SRSF3, SRSF4, and SRSF7 were PCR amplified using the following primer pairs: SRSF1, 5′-GCGCGGATCCATGTCGGGAGGTGGTGTGATTCGTG-3′ and 5′-GCGCGGATCCTTATGTACGAGAGCGAGATCTGCT-3′ (template pEGFP-ASF/SF2, a gift from Bettina Heinrich, University of Erlangen); SRSF3, 5′-GCGCGGATCCATGCATCGTGATTCCTGTCC-3′ and 5′-GCGCGAATTCCTATTTCCTTTCATTTGACCTAG-3′ (template IMAGE clone 3049167); SRSF4, 5′-GCGCGGATCCATGCCGCGGGTGTACATCGGC-3′ and 5′-GCGCGAATTCTTAGGACCTTGAGTGGGACC-3′ (template IMAGE clone 3619538); and SRSF7, 5′-GCGCGGATCCATGTCGCGTTACGGGCGGTAC-3′ and 5′-GCGCGGATCCTCAGTCCATTCTTTCAGGACT-3′ (template IMAGE clone 2967417). IMAGE clones were obtained from MRC Geneservice, Cambridge, United Kingdom).
The amplified sequences were cloned into appropriately prepared pGEX-2T and transformed into Escherichia coli BL21 CodonPlus cells (Agilent Technologies, Wokingham, United Kingdom). Overnight cultures were used to inoculate 200 ml of Luria-Bertani (LB) medium and grown at 37°C with shaking. After 2 h, IPTG (isopropyl-β-d-thiogalactopyranoside; Sigma-Aldrich) was added to a final concentration of 1 mM, and the cultures were grown for 4 h at 30°C. Proteins were purified as described previously (23).
In vitro and in vivo kinase assays.
In vitro kinase assays were performed essentially as described previously (23). A 1-μg portion of His-SRPK1 was added to 1 to 5 μg of substrate protein (GST-SR proteins). For the inhibition studies, 0.3 to 10 μg of the inhibitory protein (GST and GST-E1^E4 proteins) was added to the reaction mixture. The reaction mixture was then incubated at 30°C for 30 min and either stopped by the addition of Laemmli loading buffer (2×) and resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) or stopped by the addition of 20% (wt/vol) trichloroacetic acid (TCA) and 0.2 mg of bovine serum albumin. After centrifugation at 16,100 × g for 5 min, the pellet was washed with 10% (wt/vol) TCA three times, resuspended in acetone, and centrifuged at 16,100 × g for 5 min. The pellet was allowed to air dry and resuspended in scintillation cocktail (OptiPhase HiSafe 3; Perkin-Elmer). The amount of phosphate incorporated was determined by using a scintillation counter (Packard Tri-Carb liquid scintillation counter) and calculated using the following equation: pmol incorporated = scintillation count/count 1 pmol {count 1 pmol = 1 μCi count/([32P]/specific activity)}, where a 1-μCi count was determined by measuring the scintillation count of 1 μCi of radioisotope. The concentration of the radioisotope [32P] and the specific activity of the batch of radioisotopes were both determined by using the Perkin-Elmer decay calculator (http://www.perkinelmer.co.uk/tools/RadCalculator).
For the E2-containing in vitro kinase assays, 1 μg of His-SRPK1 was added to 1 to 5 μg of GST-tagged wild-type or mutant E2 protein (with triple [E2-FLS265-267-281A and E2-HS256-267-281A], double [E2-FLS265-267A and E2-HS265-267A], or single [E2-FLS281A and E2-HS281A] substitutions). For inhibition analysis, increasing amounts of GST or GST-HPV1 E1^E4 proteins were added. After a 30-min incubation at 30°C, the reaction was stopped and resolved by SDS-PAGE. Quantification of the level of E2 phosphorylation was obtained from the dried gels using a Storm 860 PhosphorImager combined with ImageQuant 5.0 software (GE Healthcare, Chalfont St Giles, United Kingdom).
For the in vivo kinase assays, H1299 cells were transfected with plasmids encoding Flag-SRPK1 or Flag-MCM7, along with plasmids expressing the wild-type HPV1 E1^E4 protein or the SRPK1 binding-defective E1^E4 mutant Δ44-48 (23) using Lipofectamine LTX (Invitrogen) according to the manufacturer's instructions.
The cells were washed with ice-cold saline and lysed in kinase lysis buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1% [vol/vol] Triton X-100, 1 mM phenylmethylsulfonyl fluoride, 5 mM dithiothreitol). After incubation on ice for 30 min, the lysate was cleared by centrifugation at 16,100 × g for 30 min. One milligram of protein lysate was mixed with 1 μg of mouse anti-Flag antibody (M2; Sigma-Aldrich) for 30 min at 4°C and then added to protein G-Sepharose and rotated at 4°C for 1 h. Beads were washed twice with kinase lysis buffer and then twice with 50 mM Tris-HCl (pH 7.5)–150 mM NaCl. The beads were then resuspended in 50 μl of 50 mM Tris-HCl (pH 7.5)–150 mM NaCl, and 1 μg of GST-SRSF1 was added, followed by 5 μCi of [γ-32P]ATP (Perkin-Elmer) in 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 10 mM MgCl2, and 20 μM ATP. After 30 min at 30°C, the reaction was stopped by the addition of 50 μl of 2× Laemmli loading buffer and resolved by SDS-PAGE.
In vitro coprecipitations.
To remove the GST tag from GST-HPV1 E1^E4, the fusion protein was expressed in 200 ml of LB medium and isolated on glutathione-agarose beads (23). The beads were incubated with 5 U of thrombin protease (GE Healthcare) in phosphate-buffered saline (PBS) per 100-μl bed volume of beads overnight at 30°C. The cleaved protein was loaded onto a pre-equilibrated benzamidine FF HiTrap column (GE Healthcare) and washed with 50 mM Tris-HCl (pH 7.5)–0.5 M NaCl, followed by 50 mM Tris-HCl (pH 7.5)–1 M NaCl. The protein was collected in the high-salt wash, and the salt concentration was reduced to 200 mM by dialysis. To “pulldowns” containing GST-SRSF1, 10 μg of the cleaved HPV1 E1^E4 was added. After washing, the beads were resuspended in Laemmli loading buffer (2×), resolved by SDS-PAGE, and analyzed by Western blotting.
Generation of hemagglutinin (HA)-tagged wild-type and mutant HPV1 E2 proteins.
A cassette containing a multiple cloning region and the sequence of the HA tag was created using the primers 5′-AGCTTATGTACCCATACGATGTTCCAGATTACGCTGGATCCCCGGGAATTCC-3′ and 5′-TCGAGGAATTCCCGGGGATCCAGCGTAATCTGGAACATCGTATGGGTACATA-3′. The primers were resuspended to 100 μM, and 10 μl of each primer was mixed with 2 μl of restriction buffer M (Roche), heated to 95°C for 2 min, and then cooled slowly to room temperature. The cassette mixture was diluted to 50 nM and ligated into the plasmid pcDNA3 digested with HindIII and XhoI to form plasmid pcDNA3-HA.
HPV1 E2 sequences were isolated from the pGEX2T-HPV1E2FL plasmid by EcoRI digestion and cloned into EcoRI-digested pcDNA3-HA. The correct orientation of the insert was identified by bidirectional DNA sequencing. Mutant HA-tagged HPV1 E2 proteins were generated by site-directed mutagenesis using mutagenic primers as described for the generation of mutant GST-HPV1 E2 proteins.
Western blotting and immunofluorescence microscopy.
Resolved proteins were transferred onto a Biotrace NT nitrocellulose membrane (VWR International, Lutterworth, United Kingdom) and blocked in 2% (wt/vol) dried skimmed milk in PBS. HPV1 E4 was detected using mouse antibody 4.37 (38) at a dilution of 1/150, and SRPK1 was detected using mouse anti-SRPK1 (clone 12; BD Transduction Labs) at a dilution of 1/1,000. Epitope tags were detected using a rabbit anti-Flag antibody (Sigma-Aldrich), a mouse anti-His antibody (Sigma-Aldrich), a goat anti-GST antibody (GE Healthcare), and a mouse anti-HA antibody (Covance); all of these antibodies were used at a dilution of 1/1,000. Secondary antibodies conjugated to horseradish peroxidase were used at a dilution of 1/3,000 and were obtained from Sigma-Aldrich (mouse and goat) and Dako (rabbit). Membranes were developed by using enhanced chemiluminescence (Amersham ECL Western blotting detection reagents; GE Healthcare).
HFK were grown on collagen-coated glass coverslips and transfected with plasmid DNA by using Lipofectamine 2000 as described by the manufacturer. After 24 h, the cells were fixed in 3.6% formaldehyde (EM-Grade; TAAB Laboratories, Aldermaston, Berks, United Kingdom) for 20 min and permeabilized using 0.2% Triton X-100 for 5 min.
Cells were stained for E4 using antibody 4.37 (38) at a dilution of 1/200. HA-tagged E2 proteins were detected by using a rabbit anti-HA antibody at a 1/200 dilution (Abcam, Cambridge, United Kingdom). Nucleoli were visualized by using an antibody specific to C23 (Santa Cruz, catalog no. sc-8031) at a 1/100 dilution. Immune complexes were then detected by using species-specific Alexa Fluor (Molecular Probes, Invitrogen) conjugates, and nuclei were counterstained with DAPI (4′,6′-diamidino-2-phenylindole). Imaging was performed on a Nikon E600 microscope fitted with epifluorescence detection; images were acquired by using a DXM1200F digital camera and assembled in Adobe Photoshop CS5.
Quantification of the different immunofluorescent staining patterns of HPV1 E2 in primary keratinocytes was performed blind by two investigators (C.L.B. and S.R.). The data were taken from a minimum of three independent experimental repeats.
RESULTS
HPV1 E1^E4 inhibits SRPK1 phosphorylation of cellular SR proteins.
An in vitro kinase assay system was used to establish whether SRPK1 binding to HPV1 E1^E4 alters the activity of the kinase. The assay contained purified forms of bacterially expressed His-tagged SRPK1 and a GST fusion of the SRPK1 cellular substrate SRSF1. GST or GST-HPV1 E1^E4 proteins were titrated into the assay and, after 30 min, half of the reaction mixture was resolved by SDS-PAGE, and the dried gel was exposed to X-ray film. The level of phosphate incorporated into GST-SRSF1 was quantitated by scintillation counting of TCA-precipitated proteins isolated from the remaining half of the reaction. GST-SRSF1 was phosphorylated by His-tagged SRPK1 (Fig. 1A, lane a), and the amount of phosphate incorporation into the SR protein did not vary upon addition of increasing amounts of GST protein to the in vitro reactions (Fig. 1A). In contrast, in the presence of the GST-HPV1 E1^E4 protein, phosphorylation of GST-SRSF1 decreased in a dose-responsive manner. At the maximum amount of GST-HPV1 E1^E4 added to the assay, the phosphate incorporated into GST-SRSF1 was reduced by 65% compared to an equivalent amount of GST protein (P < 0.001) (Fig. 1A).
FIG 1.

HPV1 E1^E4 inhibits SRPK1 kinase activity in vitro. (A) GST-HPV1 E1^E4 was added in increasing amounts (0.15, 0.3, 0.6, 1.2, 2.5, or 5 μg) to an in vitro kinase assay containing 1 μg of GST-SRSF1 and 1 μg of His-SRPK1. Similar amounts of GST protein (0.3, 0.6, 1.2, 2.5, 5, or 10 μg) were added to separate reactions. Phosphorylation of GST-SRSF1 (P-GST-SRSF1) was shown by exposure of the Coomassie blue-stained gel to X-ray film (32P autoradiograph). Lane a contains only GST-SRSF1 and His-SRPK1, and lane b contains only GST-SRSF1. The level of phosphate incorporation into GST-SRSF1 is shown in the graph as the percentage of phosphorylation normalized to the level of phosphorylation in the absence of GST proteins (lane a). (B) GST-tagged HPV1, HPV5, HPV16, and HPV18 E1^E4 proteins were added in increasing amounts (2.5, 5, and 10 μg) to an in vitro kinase assay containing 3 μg of GST-SRSF1 and 1 μg of His-SRPK1. Quantification of phosphate incorporation from four independent experiments is given in the bar graph as the mean percentages of phosphorylation (± the standard deviations). Statistical significance was determined by using Student t test.
Previously, we have shown that SRPK1 was also an in vitro binding partner of the E1^E4 proteins of Alphapapillomavirus HPV16 and HPV18, (23) and of the Betapapillomavirus HPV5 (39). To determine whether these E1^E4 proteins affected SRPK1 activity, GST proteins encoding E1^E4 of HPV5, HPV16, and HPV18 were also added to the SRPK1 kinase assay. Here, the level of phosphate incorporation into GST-SRSF1 was not altered significantly by the presence of these E1^E4 proteins compared to the control reactions (Fig. 1B). Therefore, we conclude that only the type 1 E1^E4 protein is an inhibitor of SRPK1 in vitro.
To establish that HPV1 E1^E4-induced inhibition of SRPK1 activity is valid for more than one SRPK1 substrate, GST-HPV1 E1^E4 was added to in vitro kinase assays containing GST-tagged fusion proteins of SRSF3, SRSF4, and SRSF7. HPV1 E1^E4 inhibited SRPK1 phosphorylation of all the SR proteins tested (Fig. 2A). Although the level of inhibition by GST-HPV1 E1^E4 in comparison to the GST protein alone was significant in all cases, there was a degree of variation in the level of inhibition between substrates; SRSF4 showed the greatest reduction in phosphorylation (86% ± 2.3%) and SRSF3 the lowest (58% ± 0.02%) (Fig. 2B).
FIG 2.

HPV1 E1^E4 inhibits SRPK1 phosphorylation of multiple SR proteins. (A) Five micrograms of GST (lanes 1 and 2), GST-SRSF1 (lanes 3 to 5), GST-SRSF4 (lanes 6 to 8), GST-SRSF3 (lanes 9 to 11), or GST-SRSF7 (lanes 12 to 14) was added to an in vitro kinase assay containing His-SRPK1. Five micrograms of GST (lanes 1, 4, 7, 10, and 13) or 5 μg of GST-HPV1 E1^E4 (lanes 2, 5, 8, 11, and 14) was added to the individual kinase assays. Additional control reaction mixtures containing the GST-tagged SR proteins alone are shown in lanes 3, 6, 9, and 12. Left panels, autoradiographs; right panels, Coomassie blue-stained gels. (B) Quantification of the level of phosphorylation, as determined by phosphorimaging analysis, shown from three independent experiments as the mean percentages of phosphorylation normalized to the level of phosphorylation in the presence of GST protein (± the standard deviations). Statistical analysis was determined by using paired Student t test. *, P < 0.05; **, P < 0.01; ***, P < 0.0005.
An interaction between HPV1 E1^E4 and SRPK1 is necessary for the inhibition of kinase activity.
To confirm that an interaction between HPV1 E1^E4 and SRPK1 is required for inhibition of kinase activity, two E1^E4 deletion mutants were tested in the kinase assay. Deletion of residues 44 to 48 from the HPV1 E1^E4 protein (Δ44-48) has been shown previously to disrupt the binding of HPV1 E1^E4 to SRPK1, whereas deletion of the amino acids 49 to 53 (Δ49-53) does not abrogate this interaction (23). These mutants were bacterially expressed as GST fusion proteins and included in the assay with His-SRPK1 and GST-SRSF1. The E1^E4 mutant Δ44-48 did not inhibit the phosphorylation of GST-SRSF1, whereas the mutant competent for SRPK1 binding, Δ49-53, reduced GST-SRSF1 phosphorylation to a level comparable to that of the wild-type protein (Fig. 3A).
FIG 3.
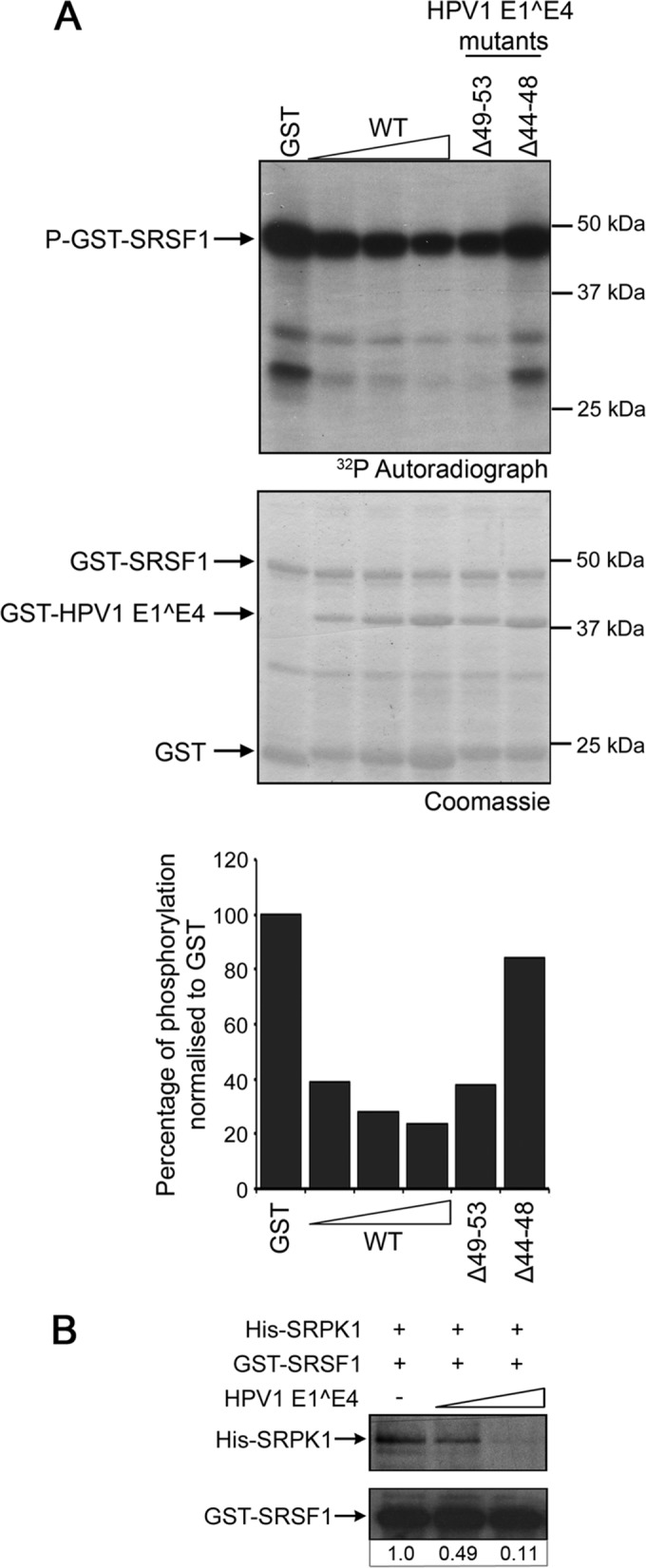
HPV1 E1^E4 inhibitory action is dependent on an interaction with SRPK1 and E1^E4 blocks binding between the kinase and the substrate SRSF1. (A) GST-HPV1 E1^E4 protein (WT, 2.5, 5, and 10 μg) and deletion mutants Δ49-53 (5 μg, SRPK1-binding mutant) and Δ44-48 (5 μg, non-SRPK1-binding mutant) were added in an in vitro kinase assay containing GST-SRSF1 and His-SRPK1. Quantification of phosphate incorporation is expressed in the graph as the percentage of phosphorylation normalized to the level of phosphorylation in the presence of GST protein. The data shown are from one of two experimental repeats. (B) GST-SRSF1 was bound to glutathione-agarose beads and incubated with His-SRPK1 alone or in the presence of increasing amounts of bacterially expressed HPV1 E1^E4 cleaved from the GST tag. After resolution of the washed beads by SDS-PAGE, the gels were blotted with anti-SRPK1 and anti-GST antibodies. The amount of His-SRPK1 coprecipitated with GST-SRSF1 in the presence of HPV1 E1^E4 relative to that bound in the absence of E1^E4 is shown below the blot.
Whether the interaction between E1^E4 and SRPK1 blocks binding between the kinase and substrate was tested in an in vitro binding assay. The association between His-SRPK1 and GST-SRSF1 was reduced in the presence of increasing amounts of recombinant HPV1 E1^E4 protein that had been cleaved from the GST tag (Fig. 3B). Together, these findings suggest that inhibition of SRPK kinase activity requires binding between the kinase and the E1^E4 protein and that this interaction is sufficient to block binding of SRPK1 to the substrate. There was no evidence of an association between HPV1 E1^E4 and the SR proteins themselves following pulldown experiments using the different GST-SR proteins and purified HPV1 E1^E4 protein (data not shown).
HPV1 E1^E4 inhibits SRPK1 activity in cells.
To establish whether HPV1 E1^E4 inhibits the activity of SRPK1 in cells, the Flag-SRPK1 expression plasmid was cotransfected into H1299 cells with plasmids expressing E1^E4 or the deletion mutant Δ44-48, which is unable to bind or inhibit SRPK1. A plasmid expressing Flag-tagged MCM7 (minichromosome maintenance complex component 7), which is known to interact with HPV1 E1^E4 (40), was also added to control for nonspecific effects. After transfection, the cells were harvested after 48 h and immunoprecipitations performed on the cell lysates with an anti-Flag antibody. The immunoprecipitated proteins were then added to an in vitro kinase assay containing GST-SRSF1 and 32P-labeled γ-ATP. After resolution by SDS-PAGE, 32P incorporation was quantified by phosphorimaging. A low level of phosphorylation of GST-SRSF1 was observed with Flag-MCM7 immunoprecipitates that was most likely due to nonspecific sticking of the kinase to the beads and was therefore considered to be the background level of phosphorylation (Fig. 4). In the presence of the HPV1 E1^E4 protein, the immunoprecipitated Flag-SRPK1 showed a significant (P < 0.05) reduction in phosphorylation of GST-SRSF1 compared to immunoprecipitates from cells transfected with the deletion mutant Δ44-48 (Fig. 4).
FIG 4.
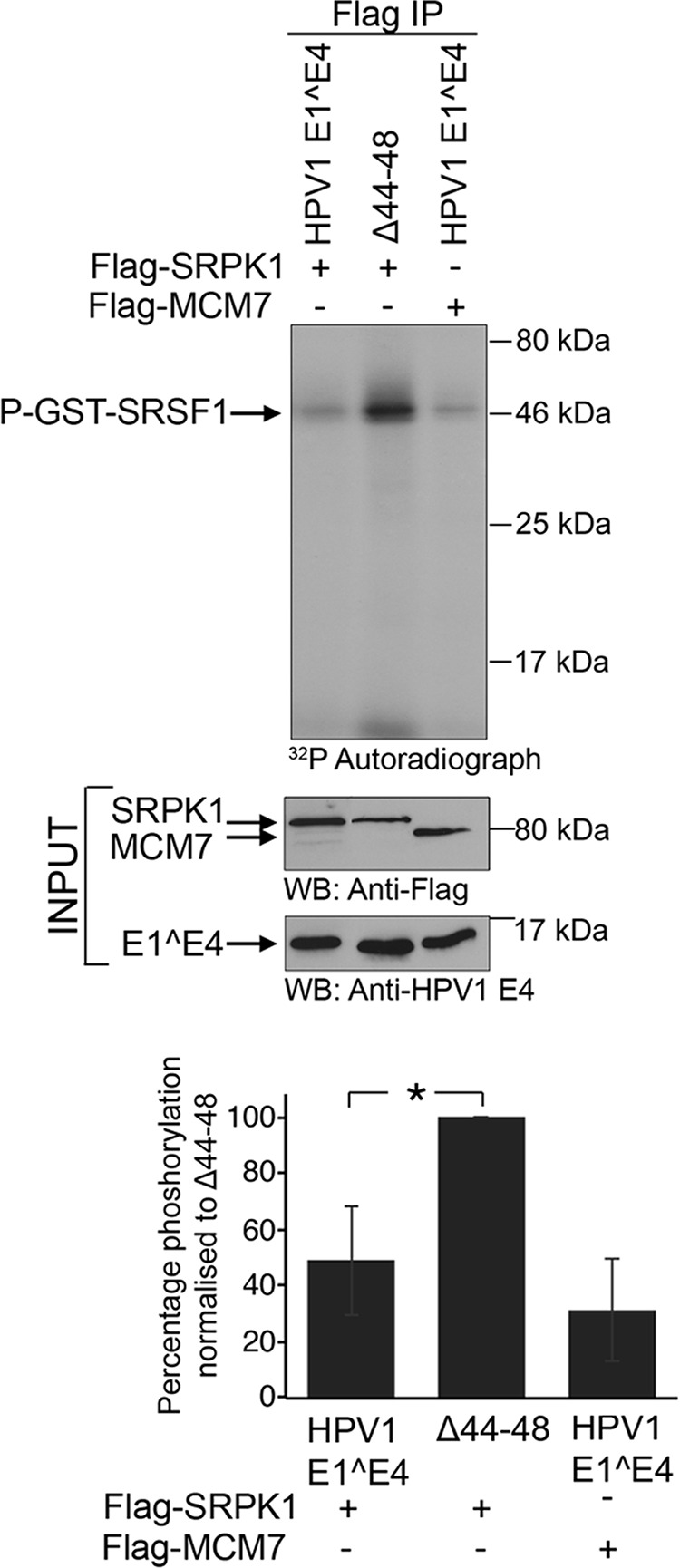
HPV1 E1^E4 inhibits SRPK1 activity in cells. A plasmid expressing HPV1 E1^E4 was transfected into cells, along with Flag-tagged SRPK1 or Flag-tagged MCM7, or the cells were cotransfected with Flag-SRPK1 and the HPV1 E1^E4 deletion mutant Δ44-48 that is unable to bind to SRPK1. The proteins were subjected to immunoprecipitation with an anti-Flag antibody and added to an in vitro kinase assay with GST-SRSF1 and, after SDS-PAGE, exposed to X-ray film (top panel). One-twentieth of the input lysate was Western blotted with anti-Flag and E4 antibodies. The graph shows the normalized data for phosphorylation of GST-SRSF1 as the means (± the standard deviations) from three experimental replicates (*, P < 0.05 [Student t test]).
When considered together, the data given in Fig. 1 to 4 indicate that HPV1 E1^E4 is a potent inhibitor of the protein kinase SRPK1 both in vitro and in vivo.
HPV1 E1^E4 inhibits SRPK1 phosphorylation of the HPV1 E2 protein.
Several unrelated viruses encode proteins with similar characteristics to cellular SR proteins (41). Most notable are E2 proteins of HPV types within the Betapapillomavirus genus (http://pave.niaid.nih.gov); the hinge regions, separating the amino-terminal transactivation and carboxy-terminal DNA binding and dimerization domains, contain multiple RS/SR repeats. For example, the HPV5 E2 hinge region contains 27 RS/SR motifs. The HPV5 E2 protein has been shown to be a substrate for SRPK1 in vitro, and evidence suggests that this phosphorylation is restricted to the hinge region (42, 43). Examination of the sequence of the HPV1 E2 hinge region identified four RS/SR dipeptide motifs, with the serines at positions 265, 267, 281, and 306 (Fig. 5A). Therefore, to determine whether HPV1 E2 is a substrate for SRPK1, the HPV1 E2 protein (E2-FL) and the hinge region only (E2-H, residues 198 to 323) were expressed as GST fusion proteins and included in an in vitro kinase assay containing His-SRPK1. Both E2-FL and E2-H were phosphorylated by His-SRPK1 (Fig. 5B and C).
FIG 5.

HPV1 E1^E4 inhibits SRPK1 phosphorylation of RS dipeptides in the hinge region of HPV1 E2. (A) Amino acid sequence of HPV1 E2 hinge region (residues 198 to 323). The RS/SR dipeptides are underlined, and serine residues altered to alanines are boxed. The triple (T), double (D), and single (S) alanine substitutions are as shown. (B and C) GST-HPV1 E2 full-length (FL) or GST-HPV1-E2 hinge (H) wild-type and mutant proteins were added to in vitro kinase reaction mixtures containing His-SRPK1. SRSF1 was added as a positive control. Bar graphs expressing the quantification of phosphorylation levels, as determined from phosphorimager analysis, show the means (± the standard deviations) from three experimental replicates, and a Student t test was used to determine the significance (**, P < 0.01; ***, P < 0.001) in comparison to the wild-type E2 protein (WT). (D) Increasing amounts of GST-HPV1 E1^E4 or GST alone were added to in vitro kinase assays containing GST-HPV1 E2 FL and His-SRPK1. The data shown represent one of two experimental replicates.
To establish whether the serine residues within the RS/SR dipeptides are SRPK1 phospho-acceptors, GST-tagged mutant full-length and hinge region E2 proteins containing alanine replacements of three of the four serine residues (triple [T], S265AS267A281A) were tested in the SRPK1 phosphorylation assay. SRPK1 mediated phosphorylation of the mutant full-length protein was markedly reduced in comparison to the wild-type E2 protein (by >80%, Fig. 5B). A similar level of reduction in phosphorylation occurred upon inclusion of a double mutant (D, S265A267A) in the in vitro assay, while substitution of serine 281 alone (S, S281A) had a less dramatic effect on SRPK1 phosphorylation (Fig. 5B, >50%). These data suggest that one or both of the serine residues at 265 and 267 are major SRPK1 phospho-acceptors and that serine 281 is not a significant target site of the kinase acceptors. These mutations when expressed in the hinge region alone produced a similar profile of SRPK1 phosphorylation (Fig. 5C). Since mutation of serines 265 and 267 was sufficient to block SRPK1 phosphorylation, the serine in the fourth RS motif (306-307) was judged not to be a relevant SRPK1 target.
Having established that the HPV1 E2 protein is phosphorylated by SRPK1, we then examined whether E1^E4 affects SRPK1 phosphorylation of the E2 protein. Increasing amounts of GST-HPV1 E1^E4 were added to in vitro kinase assays containing His-SRPK and GST-E2-FL proteins. The level of SRPK1 mediated phosphorylation of the full-length E2 protein was markedly reduced in the presence of HPV1 E1^E4 in a dose-dependent fashion, whereas the addition of increasing amounts of the GST protein alone had no effect on E2 phosphorylation (Fig. 5D).
Mutation of the SRPK phospho-acceptor residues within the E2 hinge region affects the subnuclear localization of the E2 protein.
We next wanted to identify a role for the SRPK1 phospho-acceptor residues in HPV1 E2 function. The hinge region of E2 is thought to be an unstructured flexible hinge between the transactivation and DNA-binding domains, although it is not required for the function of these two domains (44). However, it has been shown that E2 hinge sequences influence the nuclear distribution of the protein (45, 46) and that phosphorylation of hinge residues can lead to changes in E2 protein turnover (45, 47, 48). Therefore, the cellular localization of the HPV1 E2 protein, and the triple- and double-mutant proteins was examined in primary human foreskin keratinocytes (HFK) by immunofluorescence staining. The wild-type HA-tagged E2 protein was primarily restricted to the nucleus but showed variation in subnuclear distribution (Fig. 6A). The patterns of nuclear staining were classified as either diffuse throughout the nucleoplasm (Fig. 6Ai), excluded from the nucleolus (Fig. 6Aii), or concentrated in the nucleolus (Fig. 6Aiii to vi), with nucleolar staining ranging from faint (Fig. 6Aiii) through to very intense staining (Fig. 6Avi). Nucleolar localization was confirmed by costaining with the nucleolar protein C23 (Fig. 6A, right-hand panel). Small E2-positive nuclear foci were also observed in some of the cells (Fig. 6Av) and a very small subset of cells showed very bright cytoplasmic staining (data not shown). The staining patterns of both the triple and the double mutants were similar to wild-type protein (Fig. 6A, HA-E2-T and HA-E2-D, respectively). However, quantification of the different phenotypes formed by the mutant E2 proteins showed that the population of both triple- and double-mutant E2-positive cells with intense nucleolar staining significantly increased in comparison to cells expressing the wild-type protein and, furthermore, there was a concomitant significant decrease in cells with less intense nucleolar staining (Fig. 6Bi).
FIG 6.

Serine residues of the RS dipeptide motifs have a role in the subnuclear localization of the HPV1 E2 protein in primary keratinocytes. (A) Primary human foreskin keratinocytes were transfected with plasmids expressing HA-tagged wild-type or mutant HPV1 E2 proteins for 24 h, and the various immunofluorescent staining patterns are shown (E2, green; nucleus, blue). In the right-hand panels, localization of E2 to the nucleolus was confirmed by costaining with anti-C23 antibody (red). Scale bar, 10 μm. (B) Quantification of the patterns of E2 staining as “nuclear with faint to moderate nucleolar staining” (Fig. 6Biii and iv), “nuclear with intense nucleolar staining” (Fig. 6Bv and vi), and “other” (i.e., including the pattern categories “diffuse throughout the nucleus,” “nuclear but excluded from the nucleolus,” and “cytoplasmic”). The data represent the means (± the standard deviations) in three bar graphs, and the Student t test was used to determine significance (*, P < 0.05; **, P < 0.01) in comparison to the wild-type E2 protein (E2WT). The data are taken from a minimum of three experimental repeats, and the profile of the staining patterns was the same in all of the experiments. The number of cells counted for each transfection falls in the range of 60 to 425. (i) Effect of mutations (double, D; triple, T) within the hinge region of E2 on localization compared to E2WT; (ii) effect of coexpression of E4 on E2WT that is competent for SRPK1 inhibition (E4WT) or that lacks this function (E4Δ44-48); (iii) effect of coexpression of E4WT on the distribution of the mutant E2-T. (C) Western blot of equivalent amounts of cell lysate harvested from C33a keratinocytes transfected for 24 h with plasmids expressing HA-tagged wild-type or triple-mutant E2 proteins. Equivalent amounts of lysate from three independent transfections were blotted with anti-HA antibody. GAPDH (glyceraldehyde-3-phosphate dehydrogenase) protein levels were used as loading control. (D) Distribution of HA-tagged E2 (green) and E1^E4 (red) in HFK (nucleus, blue). Scale bar, 10 μm.
Because the transfection efficiency of the E2 expressing plasmids in HFK was very low (<5%), it was not possible to determine by Western blotting whether the mutation of the serine residues in the hinge affected E2 protein turnover. However, we observed little variation in the steady-state expression levels of the mutants compared to the wild-type E2 protein after transient expression in C33a keratinocytes or of the wild-type E2 protein following cotransfection with a Flag-tagged SRPK1 expression plasmid (Fig. 6C and data not shown).
The data described above indicated that the serine residues of the RS dipeptide motifs have a role in determining the subnuclear localization of E2 and suggest that SRPK1 phosphorylation may regulate the distribution of E2 in the nucleus. However, coexpression of Flag-tagged SRPK1 and full-length HPV1 E2 in HFK did not affect the distribution of the wild-type E2 between the different staining phenotypes (data not shown). This observation may reflect an existing high level of endogenous SRPK1 activity in the primary cells and so silencing SRPK1 protein expression using small interfering RNAs (siRNAs) transfected into primary cells transiently expressing E2 was also examined. Western blot analysis of total cell lysates confirmed efficient knockdown of SRPK1 protein expression; however, immunofluorescence staining of these cells for E2 and SRPK1 showed that there was no silencing of SRPK1 expression in cells positive for E2 expression (data not shown). We have no evidence that E2 upregulates expression of SRPK1 in primary keratinocytes. All of which leads us to conclude that E2 is able to overcome siRNA-mediated silencing of SRPK1 expression by some unexplained mechanism.
If SRPK1 phosphorylation of the E2 hinge residues is important in the subnuclear distribution of the viral protein and E4 can inhibit this activity, then we would expect E4 to affect the distribution of E2. To investigate this, HFK were cotransfected with plasmids expressing E2 and E1^E4 and the different E2 staining phenotypes quantified. Notably, in the presence of E4, the population of wild-type E2 positive cells with intense nucleolar staining increased significantly compared to cells expressing E2 alone (Fig. 6Bii). However, there was no change in this level of this population in cells coexpressing E2 and the E1^E4 deletion protein Δ44-48 that is unable to bind SRPK1 (Fig. 6Bii). The distribution of the triple mutant E2 protein is unaffected by coexpression of E1^E4 (Fig. 6Biii). We have reported previously that HPV1 E4 is present in the nucleoli of simian virus 40-immortalized keratinocytes (49); however, this was not the case in primary keratinocytes (Fig. 6D). In addition, unlike the effect of HPV16 E4 on E2 (50), there was no relocalization of the E2 protein to the cytoplasm in primary cells, even in cells in which E4 had assembled into cytoplasmic inclusions (Fig. 6D). Collectively, these data show that HPV1 E1^E4 protein affects the subnuclear distribution of E2 and that this activity is linked to the SRPK1-binding function of E4.
DISCUSSION
While our data have shown that SRPK1 is a conserved host target of E1^E4 proteins of HPV types representing diverse genotypes (Alpha, Beta, and Mu), only the E1^E4 protein of the Mupapillomavirus HPV1 inhibited SRPK1 activity. The interaction between HPV1 E1^E4 and SRPK1, which is mediated by direct binding (23), was necessary for the inhibition of SRPK1 phosphorylation of the cellular SR protein SRSF1. The fact that this association was sufficient to prevent the binding of SRPK1 to SRSF1 suggests that E1^E4 blocks access to the binding site on the kinase, either by steric hindrance and/or by provoking a change in the conformation of the SRPK1 protein. For E1^E4 proteins that interact with the kinase but do not inhibit its activity, at least in the in vitro assay used here, they may have a different mode of binding to that of the type 1 protein, or they may require specific posttranslational modifications or additional cellular and/or viral factors in order to modulate SRPK1 activity. These differences may be a reflection of the different tropism and biology of the HPV types (1).
In addition to SRSF1, HPV1 E1^E4 also inhibited SRPK1 phosphorylation of SRSF3, SRSF4, and SRSF7. The phosphorylation of SRSF4 and SRSF7 was more susceptible to inhibition than the phosphorylation of substrates SRSF1 or SRSF3. These data may reflect limitations of the in vitro assay since some of the GST-SR fusion proteins may not have been expressed as full-length proteins. However, it has recently been shown that SRPK1 uses different modes of phosphorylation of SR proteins that is dependent on the lengths and the topography of the RS dipeptide repeats in the RS domain (51). Thus, SRPK1 activity toward SR proteins such as SRSF4 and SRSF7 with shorter RS dipeptide repeats might be more prone to E1^E4-mediated inhibition than when acting upon SRSF1 and SRSF3 which have longer RS repeats. The variation in potency of E1^E4 inhibition of SRPK1 activity on different cellular SR protein substrates may indicate that there is differential regulation of SR protein function in HPV1 infections.
During the HPV life cycle, the expression of the late genes involves tight regulation of alternative splicing and polyadenylation. Cellular SR proteins seem to have a pivotal role in these events and in fact E2 from the high-risk virus HPV16 has been shown to regulate the transcription of some of these factors (52–55). Selection of bovine and human papillomavirus late-specific splice sites has been shown to be controlled by SRSF3, with this cellular factor suppressing the activity of a key exonic splicing enhancer in the papillomavirus genomes (56). In natural infections and organotypic raft systems of papillomavirus replication, the major capsid protein L1 only occurred in superficial cells containing low levels of the SRSF3 splicing factor (56). Multiple exonic enhancers in the HPV16 genome that bind to SRSF1 are also linked to suppression of late gene expression (55), and binding of SRSF1 to the negative regulatory element in the viral genome may be necessary to relieve repression of this element on late gene expression (57). In contrast, SRSF9 stimulates splicing of the late HPV16 viral transcripts (54). In HPV1 infections, the pattern of L1 and L2 expression is highly divergent; L1 is detected in the lower suprabasal cell layers, only a few cell layers after the initiation of E4 expression, while L2 protein appears in the higher, differentiated cell layers (58, 59). The analysis of capsid encoding transcripts confirmed that L1-specific transcripts were highly abundant in the lower and middle suprabasal cells but less abundant in the more differentiated cells, where L2-specific transcripts were preferentially expressed (58). Our previous study showed that SRPK1 was sequestered to the E4 inclusions formed in the more differentiated cells and not to those present in the lower epithelial cells (23). Therefore, it is tempting to speculate that in the superficial cell layers, E4-mediated inhibition of SRPK1 activity toward cellular SR proteins leads to an alteration in the production of RNA splice isoforms to one(s) more suitable for L2 protein production.
Binding between SRPK1 and E1^E4 may influence E4 function itself in the upper layers of the infection. Indeed, in our previous study we had shown that the HPV1 E1^E4 protein was a substrate for SRPK1 in the in vitro assay (23). However, in the experiments reported here, this phosphorylation was not observed. The most likely explanation for this anomaly is that the amounts of SRPK1 and HPV1 E1^E4 used in the assays described here were much lower than those used previously due to the cellular SR proteins being superior substrates for SRPK1 than E1^E4.
We have also shown SRPK1 phosphorylates RS dipeptide motifs in the hinge region of the HPV1 E2 protein and that E1^E4 inhibits this phosphorylation. The hinge regions of the Betapapillomavirus E2 proteins are particularly rich in RS/SR dipeptides motifs, and they have been shown to mediate interactions between E2 and cellular SR proteins (60). Interestingly, the HPV16 E2 protein, whose hinge region does not contain RS dipeptide motifs, also associates with multiple SR proteins (61). The interaction with cellular splicing factors may contribute to E2-mediated posttranscriptional regulation of cellular and/or viral gene expression during the virus life cycle (60–62). However, in the case of HPV1 E2, we have been unable to identify interactions between SR proteins (HA-tagged forms of SRSF1 to -4 and SRSF7) and unphosphorylated or SRPK1 phosphorylated forms of the E2 protein (M. Hartley, E. L. Prescott, and S. Roberts, unpublished data).
In the present study, mutation of the SRPK1 phospho-acceptor residues in the hinge region of E2 led to an alteration in the subnuclear localization of the protein in primary keratinocytes, specifically enhancing localization of E2 to the nucleolus. This suggests that SRPK1 phosphorylation may have a role in regulating the cellular localization of HPV1 E2. Indeed, SRPK1 activity is associated with the release of HPV5 E2 from nuclear speckles and transport to the cytoplasm (42, 43). Since E2 stability is sensitive to phosphorylation (44), an alternative explanation is that SRPK1 phosphorylation of the E2 hinge regulates the stability of the fraction of E2 protein that is located within the nucleolus. Surprisingly, overexpression of SRPK1 did not affect the distribution of HPV1 E2, perhaps because of high endogenous levels of SRPK1. However, coexpression of HPV1 E4 enhanced E2's nucleolar association but did not do so following coexpression of an E4 mutant that cannot inhibit SRPK1, suggesting that the cellular localization of E2 is sensitive to E4-mediated SRPK1 inhibition.
The biological significance of the nucleolar localization of HPV1 E2 is unclear. The HPV8 E2 protein locates to the nucleolus following the removal of sequences required for localization of the protein to nuclear speckles, and the hinge sequences alone are sufficient for localization to this subnuclear compartment (45). This may indicate an association between HPV8 E2 and nucleolar proteins and/or RNA or reflect attachment to ribosomal DNA loci, which are sequestered in the nucleolus during interphase; associations that are regulated by phosphorylation of hinge residues (63). Interestingly, the chromatin binding sequence present in the hinge region of the Betapapillomavirus E2 proteins is highly conserved in the HPV1 E2 hinge region and partially conserved in the other Mupapillomavirus HPV63 (Fig. 7). Notably, the sequence most conserved between the mu- and betapapillomaviruses covers the RS dipeptide repeat (HPV1, 264RSRS267). Phosphorylation of this HPV8 sequence by protein kinase A stabilizes E2 and promotes chromatin binding (45, 64). Within the HPV1 sequence a potential PKA recognition motif overlaps with the RS dipeptide repeat (262RRRS265) (Fig. 7). Although we have been unable to show that HPV1 E2 is phosphorylated by PKA in cells treated with the activator forskolin and using antibodies that detect PKA phosphorylated substrates (C. L. Brimacombe, unpublished data), it is possible that the function of this region in HPV1 is regulated by multiple host kinases, including SRPK1, and that there is differential regulation of this domain by these kinases during the HPV1 life cycle.
FIG 7.

Homology between the hinge regions of the betapapillomaviruses HPV5 and HPV8 and the mupapillomaviruses HPV1 and HPV63. The amino acid sequences of the E2 hinge regions show conservation of the chromosome binding regions of HPV5 and HPV8 in the HPV1 E2 hinge domain and partial homology with HPV63. The regions of homology are shaded. Serine 253 in HPV8 that is phosphorylated by PKA and serines 265 and 267 in the RS dipeptide repeat in HPV1 that is targeted by SRPK1 are underlined. The sequences were obtained from the National Institutes of Health (http://pave.niaid.nih.gov).
The E2 proteins of cutaneous and mucosal HPV types have been shown to induce late gene expression through inhibition of the early polyadenylation site in the papillomavirus genomes (62). This function of E2 is unlikely to be relevant during viral genome amplification since the presence of E1 inhibited E2's induction of late gene expression (62). However, since several studies have suggested that there may be a functional relationship between the E4 and E2 proteins (50, 65), it is reasonable to speculate that if SRPK1 activity on E2 has a role to play in HPV1 genome replication, then E4 inhibition of SRPK1 activity in the upper layers of an infection could switch E2 function to one that induces late gene expression. Although the resolution of the biological function of HPV1 E1^E4 inhibition of SRPK1 activity is hampered by the lack of suitable cell-based models of the HPV1 infectious cycle, loss of E1^E4 expression in models of high-risk HPV replication repressed late gene expression (2–4).
Collectively, our data suggest that inhibition of SRPK1 by E4 in the HPV1 life cycle could affect the functions of both host cell SR proteins and those of the virus transcription/replication regulator E2. While the work described here has primarily focused on the type 1 virus that causes highly productive palmar and plantar warts, the interaction between E4 and SRPK1 is conserved between diverse genotypes, including high-risk HPV types. Therefore, it will be important in future investigations to establish the role of SRPK1 in high-risk infections since selective inhibition of this kinase using chemical compounds can inhibit the replication of other clinically relevant viruses (36, 37, 66).
ACKNOWLEDGMENTS
We thank Joseph Spitzer and his patients for the collection and the donation of foreskin tissue for the isolation of primary foreskin keratinocytes. We are grateful to Ethel de Villiers (Heidelberg, Germany) for the gift of the HPV5b genome, to Bettina Heinrich (University of Erlangen) for the SRSF1 cDNA, and to Saleem Khan (University of Pittsburgh) for the gift of the HPV1 E2-containing plasmid. We also acknowledge the helpful comments from members of the Roberts laboratory and the technical assistance of Jonathon Rushton. We are grateful to our coworkers Roger Grand and Joanna Parish (University of Birmingham) for their invaluable comments on the study.
Cancer Research UK (C427/A3919 and C427/A11976) and the University of Birmingham (College of Medical and Dental Sciences) supported this study.
Footnotes
Published ahead of print 20 August 2014
REFERENCES
- 1.Doorbar J, Quint W, Banks L, Bravo IG, Stoler M, Broker TR, Stanley MA. 2012. The biology and life-cycle of human papillomaviruses. Vaccine 30(Suppl 5):F55–F70. 10.1016/j.vaccine.2012.06.083. [DOI] [PubMed] [Google Scholar]
- 2.Wilson R, Fehrmann F, Laimins LA. 2005. Role of the E1-E4 protein in the differentiation-dependent life cycle of human papillomavirus type 31. J. Virol. 79:6732–6740. 10.1128/JVI.79.11.6732-6740.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wilson R, Ryan GB, Knight GL, Laimins LA, Roberts S. 2007. The full-length E1^E4 protein of human papillomavirus type 18 modulates differentiation-dependent viral DNA amplification and late gene expression. Virology 362:453–460. 10.1016/j.virol.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 4.Nakahara T, Peh WL, Doorbar J, Lee D, Lambert PF. 2005. Human papillomavirus type 16 E1^E4 contributes to multiple facets of the papillomavirus life cycle. J. Virol. 79:13150–13165. 10.1128/JVI.79.20.13150-13165.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peh WL, Brandsma JL, Christensen ND, Cladel NM, Wu X, Doorbar J. 2004. The viral E4 protein is required for the completion of the cottontail rabbit papillomavirus productive cycle in vivo. J. Virol. 78:2142–2151. 10.1128/JVI.78.4.2142-2151.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fang L, Budgeon LR, Doorbar J, Briggs ER, Howett MK. 2006. The human papillomavirus type 11 E1^E4 protein is not essential for viral genome amplification. Virology 351:271–279. 10.1016/j.virol.2006.01.051. [DOI] [PubMed] [Google Scholar]
- 7.Nakahara T, Nishimura A, Tanaka M, Ueno T, Ishimoto A, Sakai H. 2002. Modulation of the cell division cycle by human papillomavirus type 18 E4. J. Virol. 76:10914–10920. 10.1128/JVI.76.21.10914-10920.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davy CE, Jackson DJ, Wang Q, Raj K, Masterson PJ, Fenner NF, Southern S, Cuthill S, Millar JB, Doorbar J. 2002. Identification of a G2 arrest domain in the E1^E4 protein of human papillomavirus type 16. J. Virol. 76:9806–9818. 10.1128/JVI.76.19.9806-9818.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Knight GL, Grainger JR, Gallimore PH, Roberts S. 2004. Cooperation between different forms of the human papillomavirus type 1 E4 protein to block cell cycle progression and cellular DNA synthesis. J. Virol. 78:13920–13933. 10.1128/JVI.78.24.13920-13933.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Knight GL, Pugh AG, Yates E, Bell I, Wilson R, Moody CA, Laimins LA, Roberts S. 2011. A cyclin-binding motif in human papillomavirus type 18 (HPV18) E1^E4 is necessary for association with CDK-cyclin complexes and G2/M cell cycle arrest of keratinocytes, but is not required for differentiation-dependent viral genome amplification or L1 capsid protein expression. Virology 412:196–210. 10.1016/j.virol.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Doorbar J, Ely S, Sterling J, McLean C, Crawford L. 1991. Specific interaction between HPV-16 E1-E4 and cytokeratins results in collapse of the epithelial cell intermediate filament network. Nature 352:824–827. 10.1038/352824a0. [DOI] [PubMed] [Google Scholar]
- 12.Roberts S, Ashmole I, Johnson GD, Kreider JW, Gallimore PH. 1993. Cutaneous and mucosal human papillomavirus E4 proteins form intermediate filament-like structures in epithelial cells. Virology 197:176–187. 10.1006/viro.1993.1578. [DOI] [PubMed] [Google Scholar]
- 13.McIntosh PB, Laskey P, Sullivan K, Davy C, Wang Q, Jackson DJ, Griffin HM, Doorbar J. 2010. E1-E4-mediated keratin phosphorylation and ubiquitylation: a mechanism for keratin depletion in HPV16-infected epithelium. J. Cell Sci. 123:2810–2822. 10.1242/jcs.061978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nasseri M, Hirochika R, Broker TR, Chow LT. 1987. A human papillomavirus type 11 transcript encoding an E1-E4 protein. Virology 159:433–439. 10.1016/0042-6822(87)90482-X. [DOI] [PubMed] [Google Scholar]
- 15.Grand RJ, Doorbar J, Smith KJ, Coneron I, Gallimore PH. 1989. Phosphorylation of the human papillomavirus type 1 E4 proteins in vivo and in vitro. Virology 170:201–213. 10.1016/0042-6822(89)90367-X. [DOI] [PubMed] [Google Scholar]
- 16.Doorbar J, Campbell D, Grand RJ, Gallimore PH. 1986. Identification of the human papillomavirus-1a E4 gene products. EMBO J. 5:355–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roberts S, Ashmole I, Gibson LJ, Rookes SM, Barton GJ, Gallimore PH. 1994. Mutational analysis of human papillomavirus E4 proteins: identification of structural features important in the formation of cytoplasmic E4/cytokeratin networks in epithelial cells. J. Virol. 68:6432–6445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khan J, Davy CE, McIntosh PB, Jackson DJ, Hinz S, Wang Q, Doorbar J. 2011. Role of calpain in the formation of human papillomavirus type 16 E1^E4 amyloid fibers and reorganization of the keratin network. J. Virol. 85:9984–9997. 10.1128/JVI.02158-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McIntosh PB, Martin SR, Jackson DJ, Khan J, Isaacson ER, Calder L, Raj K, Griffin HM, Wang Q, Laskey P, Eccleston JF, Doorbar J. 2008. Structural analysis reveals an amyloid form of the human papillomavirus type 16 E1-E4 protein and provides a molecular basis for its accumulation. J. Virol. 82:8196–8203. 10.1128/JVI.00509-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Q, Kennedy A, Das P, McIntosh PB, Howell SA, Isaacson ER, Hinz SA, Davy C, Doorbar J. 2009. Phosphorylation of the human papillomavirus type 16 E1-E4 protein at T57 by ERK triggers a structural change that enhances keratin binding and protein stability. J. Virol. 83:3668–3683. 10.1128/JVI.02063-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davy CE, Jackson DJ, Raj K, Peh WL, Southern SA, Das P, Sorathia R, Laskey P, Middleton K, Nakahara T, Wang Q, Masterson PJ, Lambert PF, Cuthill S, Millar JB, Doorbar J. 2005. Human papillomavirus type 16 E1 E4-induced G2 arrest is associated with cytoplasmic retention of active Cdk1/cyclin B1 complexes. J. Virol. 79:3998–4011. 10.1128/JVI.79.7.3998-4011.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davy CE, Ayub M, Jackson DJ, Das P, McIntosh P, Doorbar J. 2006. HPV16 E1-E4 protein is phosphorylated by Cdk2/cyclin A and relocalizes this complex to the cytoplasm. Virology 349:230–244. 10.1016/j.virol.2006.02.024. [DOI] [PubMed] [Google Scholar]
- 23.Bell I, Martin A, Roberts S. 2007. The E1^E4 protein of human papillomavirus interacts with the serine-arginine-specific protein kinase SRPK1. J. Virol. 81:5437–5448. 10.1128/JVI.02609-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gui JF, Tronchere H, Chandler SD, Fu XD. 1994. Purification and characterization of a kinase specific for the serine- and arginine-rich pre-mRNA splicing factors. Proc. Natl. Acad. Sci. U. S. A. 91:10824–10828. 10.1073/pnas.91.23.10824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Giannakouros T, Nikolakaki E, Mylonis I, Georgatsou E. 2011. Serine-arginine protein kinases: a small protein kinase family with a large cellular presence. FEBS J. 278:570–586. 10.1111/j.1742-4658.2010.07987.x. [DOI] [PubMed] [Google Scholar]
- 26.Yeakley JM, Tronchere H, Olesen J, Dyck JA, Wang HY, Fu XD. 1999. Phosphorylation regulates in vivo interaction and molecular targeting of serine/arginine-rich pre-mRNA splicing factors. J. Cell Biol. 145:447–455. 10.1083/jcb.145.3.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ding JH, Zhong XY, Hagopian JC, Cruz MM, Ghosh G, Feramisco J, Adams JA, Fu XD. 2006. Regulated cellular partitioning of SR protein-specific kinases in mammalian cells. Mol. Biol. Cell 17:876–885. 10.1091/mbc.E05-10-0963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhong XY, Ding JH, Adams JA, Ghosh G, Fu XD. 2009. Regulation of SR protein phosphorylation and alternative splicing by modulating kinetic interactions of SRPK1 with molecular chaperones. Genes Dev. 23:482–495. 10.1101/gad.1752109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gui JF, Lane WS, Fu XD. 1994. A serine kinase regulates intracellular localization of splicing factors in the cell cycle. Nature 369:678–682. 10.1038/369678a0. [DOI] [PubMed] [Google Scholar]
- 30.Mathew R, Hartmuth K, Mohlmann S, Urlaub H, Ficner R, Luhrmann R. 2008. Phosphorylation of human PRP28 by SRPK2 is required for integration of the U4/U6-U5 tri-snRNP into the spliceosome. Nat. Struct. Mol. Biol. 15:435–443. 10.1038/nsmb.1415. [DOI] [PubMed] [Google Scholar]
- 31.Sciabica KS, Dai QJ, Sandri-Goldin RM. 2003. ICP27 interacts with SRPK1 to mediate HSV splicing inhibition by altering SR protein phosphorylation. EMBO J. 22:1608–1619. 10.1093/emboj/cdg166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Daub H, Blencke S, Habenberger P, Kurtenbach A, Dennenmoser J, Wissing J, Ullrich A, Cotten M. 2002. Identification of SRPK1 and SRPK2 as the major cellular protein kinases phosphorylating hepatitis B virus core protein. J. Virol. 76:8124–8137. 10.1128/JVI.76.16.8124-8137.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lewellyn EB, Loeb DD. 2011. Serine phosphoacceptor sites within the core protein of hepatitis B virus contribute to genome replication pleiotropically. PLoS One 6:e17202. 10.1371/journal.pone.0017202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen C, Wang JC, Zlotnick A. 2011. A kinase chaperones hepatitis B virus capsid assembly and captures capsid dynamics in vitro. PLoS Pathog. 7:e1002388. 10.1371/journal.ppat.1002388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zheng Y, Fu XD, Ou JH. 2005. Suppression of hepatitis B virus replication by SRPK1 and SRPK2 via a pathway independent of the phosphorylation of the viral core protein. Virology 342:150–158. 10.1016/j.virol.2005.07.030. [DOI] [PubMed] [Google Scholar]
- 36.Karakama Y, Sakamoto N, Itsui Y, Nakagawa M, Tasaka-Fujita M, Nishimura-Sakurai Y, Kakinuma S, Oooka M, Azuma S, Tsuchiya K, Onogi H, Hagiwara M, Watanabe M. 2010. Inhibition of hepatitis C virus replication by a specific inhibitor of serine-arginine-rich protein kinase. Antimicrob. Agents Chemother. 54:3179–3186. 10.1128/AAC.00113-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fukuhara T, Hosoya T, Shimizu S, Sumi K, Oshiro T, Yoshinaka Y, Suzuki M, Yamamoto N, Herzenberg LA, Hagiwara M. 2006. Utilization of host SR protein kinases and RNA-splicing machinery during viral replication. Proc. Natl. Acad. Sci. U. S. A. 103:11329–11333. 10.1073/pnas.0604616103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Doorbar J, Evans HS, Coneron I, Crawford LV, Gallimore PH. 1988. Analysis of HPV-1 E4 gene expression using epitope-defined antibodies. EMBO J. 7:825–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Prescott E. 2012. Regulation of serine-arginine protein kinase 1 functions by human papillomavirus. Ph.D. thesis. University of Birmingham, Birmingham, United Kingdom. [Google Scholar]
- 40.Roberts S, Kingsbury SR, Stoeber K, Knight GL, Gallimore PH, Williams GH. 2008. Identification of an arginine-rich motif in human papillomavirus type 1 E1^E4 protein necessary for E4-mediated inhibition of cellular DNA synthesis in vitro and in cells. J. Virol. 82:9056–9064. 10.1128/JVI.01080-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lai MC, Peng TY, Tarn WY. 2009. Functional interplay between viral and cellular SR proteins in control of posttranscriptional gene regulation. FEBS J. 276:1517–1526. 10.1111/j.1742-4658.2009.06894.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lai MC, Lin RI, Huang SY, Tsai CW, Tarn WY. 2000. A human importin-beta family protein, transportin-SR2, interacts with the phosphorylated RS domain of SR proteins. J. Biol. Chem. 275:7950–7957. 10.1074/jbc.275.11.7950. [DOI] [PubMed] [Google Scholar]
- 43.Wang WS, Lee MS, Tseng CE, Liao IH, Huang SP, Lin RI, Li C. 2009. Interaction between human papillomavirus type 5 E2 and polo-like kinase 1. J. Med. Virol. 81:536–544. 10.1002/jmv.21404. [DOI] [PubMed] [Google Scholar]
- 44.McBride AA. 2013. The papillomavirus E2 proteins. Virology 445:57–79. 10.1016/j.virol.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sekhar V, Reed SC, McBride AA. 2010. Interaction of the betapapillomavirus E2 tethering protein with mitotic chromosomes. J. Virol. 84:543–557. 10.1128/JVI.01908-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zou N, Lin BY, Duan F, Lee KY, Jin G, Guan R, Yao G, Lefkowitz EJ, Broker TR, Chow LT. 2000. The hinge of the human papillomavirus type 11 E2 protein contains major determinants for nuclear localization and nuclear matrix association. J. Virol. 74:3761–3770. 10.1128/JVI.74.8.3761-3770.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Penrose KJ, McBride AA. 2000. Proteasome-mediated degradation of the papillomavirus E2-TA protein is regulated by phosphorylation and can modulate viral genome copy number. J. Virol. 74:6031–6038. 10.1128/JVI.74.13.6031-6038.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McBride AA, Bolen JB, Howley PM. 1989. Phosphorylation sites of the E2 transcriptional regulatory proteins of bovine papillomavirus type 1. J. Virol. 63:5076–5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roberts S, Hillman ML, Knight GL, Gallimore PH. 2003. The ND10 component promyelocytic leukemia protein relocates to human papillomavirus type 1 E4 intranuclear inclusion bodies in cultured keratinocytes and in warts. J. Virol. 77:673–684. 10.1128/JVI.77.1.673-684.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Davy C, McIntosh P, Jackson DJ, Sorathia R, Miell M, Wang Q, Khan J, Soneji Y, Doorbar J. 2009. A novel interaction between the human papillomavirus type 16 E2 and E1-E4 proteins leads to stabilization of E2. Virology 394:266–275. 10.1016/j.virol.2009.08.035. [DOI] [PubMed] [Google Scholar]
- 51.Aubol BE, Jamros MA, McGlone ML, Adams JA. 2013. Splicing kinase SRPK1 conforms to the landscape of its SR protein substrate. Biochemistry 52:7595–7605. 10.1021/bi4010864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mole S, Milligan SG, Graham SV. 2009. Human papillomavirus type 16 E2 protein transcriptionally activates the promoter of a key cellular splicing factor, SF2/ASF. J. Virol. 83:357–367. 10.1128/JVI.01414-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mole S, McFarlane M, Chuen-Im T, Milligan SG, Millan D, Graham SV. 2009. RNA splicing factors regulated by HPV16 during cervical tumour progression. J. Pathol. 219:383–391. 10.1002/path.2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Somberg M, Li X, Johansson C, Orru B, Chang R, Rush M, Fay J, Ryan F, Schwartz S. 2011. Serine/arginine-rich protein 30c activates human papillomavirus type 16 L1 mRNA expression via a bimodal mechanism. J. Gen. Virol. 92:2411–2421. 10.1099/vir.0.033183-0. [DOI] [PubMed] [Google Scholar]
- 55.Somberg M, Schwartz S. 2010. Multiple ASF/SF2 sites in the human papillomavirus type 16 (HPV-16) E4-coding region promote splicing to the most commonly used 3′-splice site on the HPV-16 genome. J. Virol. 84:8219–8230. 10.1128/JVI.00462-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jia R, Liu X, Tao M, Kruhlak M, Guo M, Meyers C, Baker CC, Zheng ZM. 2009. Control of the papillomavirus early-to-late switch by differentially expressed SRp20. J. Virol. 83:167–180. 10.1128/JVI.01719-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McPhillips MG, Veerapraditsin T, Cumming SA, Karali D, Milligan SG, Boner W, Morgan IM, Graham SV. 2004. SF2/ASF binds the human papillomavirus type 16 late RNA control element and is regulated during differentiation of virus-infected epithelial cells. J. Virol. 78:10598–10605. 10.1128/JVI.78.19.10598-10605.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Egawa K, Iftner A, Doorbar J, Honda Y, Iftner T. 2000. Synthesis of viral DNA and late capsid protein L1 in parabasal spinous cell layers of naturally occurring benign warts infected with human papillomavirus type 1. Virology 268:281–293. 10.1006/viro.1999.0174. [DOI] [PubMed] [Google Scholar]
- 59.Peh WL, Middleton K, Christensen N, Nicholls P, Egawa K, Sotlar K, Brandsma J, Percival A, Lewis J, Liu WJ, Doorbar J. 2002. Life cycle heterogeneity in animal models of human papillomavirus-associated disease. J. Virol. 76:10401–10416. 10.1128/JVI.76.20.10401-10416.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lai MC, Teh BH, Tarn WY. 1999. A human papillomavirus E2 transcriptional activator: the interactions with cellular splicing factors and potential function in pre-mRNA processing. J. Biol. Chem. 274:11832–11841. [DOI] [PubMed] [Google Scholar]
- 61.Bodaghi S, Jia R, Zheng ZM. 2009. Human papillomavirus type 16 E2 and E6 are RNA-binding proteins and inhibit in vitro splicing of pre-mRNAs with suboptimal splice sites. Virology 386:32–43. 10.1016/j.virol.2008.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Johansson C, Somberg M, Li X, Backstrom Winquist E, Fay J, Ryan F, Pim D, Banks L, Schwartz S. 2012. HPV-16 E2 contributes to induction of HPV-16 late gene expression by inhibiting early polyadenylation. EMBO J. 31:3212–3227. 10.1038/emboj.2012.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Poddar A, Reed SC, McPhillips MG, Spindler JE, McBride AA. 2009. The human papillomavirus type 8 E2 tethering protein targets the ribosomal DNA loci of host mitotic chromosomes. J. Virol. 83:640–650. 10.1128/JVI.01936-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sekhar V, McBride AA. 2012. Phosphorylation regulates binding of the human papillomavirus type 8 E2 protein to host chromosomes. J. Virol. 86:10047–10058. 10.1128/JVI.01140-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tan CL, Gunaratne J, Lai D, Carthagena L, Wang Q, Xue YZ, Quek LS, Doorbar J, Bachelerie F, Thierry F, Bellanger S. 2012. HPV-18 E2Ê4 chimera: 2 new spliced transcripts and proteins induced by keratinocyte differentiation. Virology 429:47–56. 10.1016/j.virol.2012.03.023. [DOI] [PubMed] [Google Scholar]
- 66.Szekelyhidi Z, Pato J, Waczek F, Banhegyi P, Hegymegi-Barakonyi B, Eros D, Meszaros G, Hollosy F, Hafenbradl D, Obert S, Klebl B, Keri G, Orfi L. 2005. Synthesis of selective SRPK-1 inhibitors: novel tricyclic quinoxaline derivatives. Bioorg. Med. Chem. Lett. 15:3241–3246. 10.1016/j.bmcl.2005.04.064. [DOI] [PubMed] [Google Scholar]