

ABSTRACT
The RV144 vaccine trial implicated epitopes in the C1 region of gp120 (A32-like epitopes) as targets of potentially protective antibody-dependent cellular cytotoxicity (ADCC) responses. A32-like epitopes are highly immunogenic, as infected or vaccinated individuals frequently produce antibodies specific for these determinants. Antibody titers, as measured by enzyme-linked immunosorbent assay (ELISA) against these epitopes, however, do not consistently correlate with protection. Here, we report crystal structures of CD4-stabilized gp120 cores complexed with the Fab fragments of two nonneutralizing, A32-like monoclonal antibodies (MAbs), N5-i5 and 2.2c, that compete for antigen binding and have similar antigen-binding affinities yet exhibit a 75-fold difference in ADCC potency. We find that these MAbs recognize overlapping epitopes formed by mobile layers 1 and 2 of the gp120 inner domain, including the C1 and C2 regions, but bind gp120 at different angles via juxtaposed VH and VL contact surfaces. A comparison of structural and immunological data further showed that antibody orientation on bound antigen and the capacity to form multivalent antigen-antibody complexes on target cells were key determinants of ADCC potency, with the latter process having the greater impact. These studies provide atomic-level definition of A32-like epitopes implicated as targets of protective antibodies in RV144. Moreover, these studies establish that epitope structure and mode of antibody binding can dramatically affect the potency of Fc-mediated effector function against HIV-1. These results provide key insights for understanding, refining, and improving the outcome of HIV vaccine trials, in which relevant immune responses are facilitated by A32-like elicited responses.
IMPORTANCE HIV-1 Env is a primary target for antibodies elicited during infection. Although a small number of infected individuals elicit broadly neutralizing antibodies, the bulk of the humoral response consists of antibodies that do not neutralize or do so with limited breadth but may effect protection through Fc receptor-dependent processes, such as antibody-dependent cellular cytotoxicity (ADCC). Understanding these nonneutralizing responses is an important aspect of elucidating the complete spectrum of immune response against HIV-1 infection. With this report, we provide the first atomic-level definition of nonneutralizing CD4-induced epitopes in the N-terminal region of the HIV-1 gp120 (A32-like epitopes). Further, our studies point to the dominant role of precise epitope targeting and mode of antibody attachment in ADCC responses even when largely overlapping epitopes are involved. Such information provides key insights into the mechanisms of Fc-mediated function of antibodies to HIV-1 and will help us understand the outcome of vaccine trials based on humoral immunity.
INTRODUCTION
Antibodies contribute significantly to protection against HIV-1, but how they do so is only partially understood. Existing evidence suggests that protective antibody responses can involve neutralizing activity as well as Fc receptor-dependent processes, such as antibody-dependent cellular cytotoxicity (ADCC) (1–10). A role of Fc-mediated effector function by nonneutralizing antibodies (nnAbs) in blocking HIV-1 acquisition is supported by vaccine trials in nonhuman primates (4, 11, 12) and humans (3, 13, 14), as well as by a breast milk transmission study of mother-infant pairs (2). In contrast, unlike an early study of passive immunization against simian immunodeficiency virus (SIV) using polyclonal sera (15, 16), more recent passive immunization studies using well-defined monoclonal antibodies (MAbs) showed no protection against acquisition (17, 18). Postinfection control of viremia was observed in both studies, suggesting that nnAbs can impact the transmitted virus (17, 18) without blocking acquisition. Postinfection control is often seen in nonhuman primate (NHP) models when protective levels of anti-retroviral drugs (19) or MAbs (20, 21) are too low to block acquisition. If postinfection control and blocking acquisition are a continuum of protection, there is reason to believe that nnAbs could protect against infection in NHPs with the right MAb(s) or vaccine. Thus, an understanding of Fc-mediated effector function, including the epitopes and mechanisms by which potent antibodies mediate ADCC, is critical for clarifying the role of nnAbs in protection.
ADCC escape variants emerging in HIV-1-infected people (22) and ADCC responses correlating with reduced risk of infection in the RV144 vaccine trial (3, 13, 14) point to nonneutralizing epitopes in the C1 region of gp120 (A32-like epitopes) (23, 24) as relevant targets for potentially protective antibodies. The gp120 regions recognized by MAb A32 were also shown to be immunogenic during HIV-1 infection, as infected individuals frequently produce antibodies specific for these determinants (25–27). Antibody titers, as measured by enzyme-linked immunosorbent assay (ELISA) against these epitopes, however, do not consistently correlate with protection (3). This discordance between ADCC, antibody-binding responses, and protection suggests that ADCC responses to the A32-like epitopes (and ADCC epitopes in general) are governed by a mechanism(s) more complex than simply antibody binding. Here we define for the first time A32-like epitope footprints at the atomic level by X-ray crystallography and provide the structural basis for differences in ADCC responses to the A32-like epitopes. We believe that the epitope footprints are key to clarifying the question of whether antibody responses to the A32 epitope subregion are protective after vaccination in the RV144 trial and during postinfection of HIV-1. Further, these studies shed new light on the structural basis of ADCC and point to the importance of epitope fine specificity and the mode of antibody attachment as dominant elements affecting ADCC potency.
MATERIALS AND METHODS
Protein production and purification.
The A32-like human MAb 2.2c was derived by Epstein-Barr virus (EBV) transformation of peripheral blood B cells from an HIV-1-infected subject (RW/92/13) according to published methods (28). The B cell line was subsequently converted to a hybridoma (28) to improve MAb yields. MAb 2.2c was purified from hybridoma supernatant by protein A affinity chromatography. MAb N5-i5 was expressed and purified as described previously (27). N5-i5 and 2.2c Fabs were prepared from purified IgG (10 mg/ml) by proteolytic digestion with immobilized papain (Pierce, Rockford, IL) and LysC, respectively, and purified using protein A (GE Healthcare, Piscataway, NJ) followed by gel filtration chromatography on a Superdex 200 16/60 column (GE Healthcare, Piscataway, NJ). The d1d2 domain of CD4 (d1d2CD4) protein and gp120 glycoproteins were prepared as previously described (28–30). Deglycosylated gp120 was combined in a 20% molar excess with d1d2CD4 or the CD4-mimetic protein M48U1 (31) and purified by size exclusion chromatography. After concentration, the gp120:d1d2CD4 (or gp120:M48U1) complexes were mixed with a 20% molar excess of Fab, passed again through a size exclusion column, and concentrated to ∼10 mg/ml in 0.35 M NaCl, 2.5 mM Tris (pH 7.0), 0.02% NaN3 for crystallization experiments.
Complex formation and crystallization.
Multiple combinations of gp120 extended cores (corees) (29) of different clades (including clade A/E gp12093TH057 coree, clade B gp12089.6P coree, clade B gp120YU2 coree and clade A gp12092UG037 coree) with d1d2CD4 or the CD4-mimetic miniprotein M48U1 were used to prepare complexes of N5-i5 and 2.2c for crystallographic studies and tested for crystal formation. Only complexes forming diffraction-quality crystals were selected for further analysis. Initial screening for crystals was performed in robotic vapor diffusion sitting trials with sparse matrix screens (Hampton Crystal Screen; Hampton Research), precipitant Wizard screens (Emerald Biosystems), and Synergy screens (Emerald Biosystems). The screens were monitored periodically to identify potential hits, and initial crystals were then reproduced and optimized using the hanging-drop vapor diffusion method, with drops consisting of 0.5 μl protein and 0.5 μl precipitant solution equilibrated against a 500-μl reservoir volume at room temperature.
Crystals of 2.2c Fab were grown from 100 mM sodium acetate (NaOAc) (pH 4.5), 2% (vol/vol) isopropanol, 2 M Li2SO4, 100 mM MgSO4. Prior to freezing, crystals were transferred into a solution containing 100 mM NaOAc (pH 4.5), 1% (vol/vol) isopropanol, 2 M Li2SO4, 100 mM MgSO4, and 15% ethylene glycol, and cryoprotected in mineral oil (Hampton).
Crystals of N5-i5 Fab-gp12093TH057 coree-d1d2CD4 were grown from 11 to 13% polyethylene glycol 8000 and 100 mM Tris-HCl (pH 8.5) with 65 mM sodium chloride added to the reservoir after mixing of the drop. Prior to freezing, crystals were soaked briefly in 13% polyethylene glycol 8000 (PEG 8000), 100 mM Tris-HCl (pH 8.5), and 65 mM NaCl containing increasing concentrations of 2-methyl-2,4-pentanediol (MPD) (5, 10, and 15% vol/vol).
Crystals of 2.2c Fab-gp120YU2coree-M48U1 were grown from 10% MPD, 1.5 M lithium sulfate, and 100 mM imidazole (pH 8.0) with 65 mM sodium chloride added to the reservoir after mixing of the drop. Prior to freezing, crystals were transferred to a solution containing 10% 2R,3R-butanediol, 1.5 M lithium sulfate, and 100 mM imidazole (pH 8.0), and cryoprotected in mineral oil (Hampton).
Crystals of 2.2c Fab-gp12092UG037coree-M48U1 were grown from 14% PEG 3350, 1 M sodium formate, 0.1 M calcium chloride, and 100 mM sodium acetate (pH 4.5) with 65 mM sodium chloride added to the reservoir after mixing of the drop. Prior to freezing, crystals were soaked briefly in 20% PEG 3350, 1 M sodium formate, 0.1 M calcium chloride, and 100 mM sodium acetate (pH 4.5) containing 30% ethylene glycol.
Crystals of 2.2c Fab-gp12089.6PdV1V2-d1d2CD4 were grown from 10% PEG 5000 monomethyl ether (MME), 100 mM sodium acetate, and 100 mM Tris-HCl (pH 8.5) with 65 mM sodium chloride added to the reservoir after mixing of the drop. Prior to freezing, crystals were transferred to a cryoprotectant solution containing 15% PEG 5000 MME, 100 mM sodium acetate,100 mM Tris-HCl (pH 8.5), and 50% (wt/vol) xylitol.
Data collection, structure solution, and refinement.
Data for the N5-i5 Fab-gp12093TH057 coree-d1d2CD4 complexes were collected at the SSRL 12-2 beam line. Data for the 2.2c Fab and 2.2c Fab complexes were collected at the ID22 beamline (SER-CAT) at the Advanced Photon Source. Data were processed with HKL2000 (32) and the structure solved by molecular replacement using the program Phaser (33) from the CCP4i suite (34).
For the N5-i5 Fab-gp12093TH057 coree-d1d2CD4, 2.2c Fab, and 2.2c Fab-gp12089.6PdV1V2-d1d2CD4 complexes, initial refinement and model building were done with the programs CNS (35) and Refmac5 (36) and, at later stages, using the program PHENIX (37). For all other complexes, refinement was carried out using PHENIX (37). Refinement was coupled with manual refitting and rebuilding with COOT (38). Crystals for the 2.2c-bound complexes diffracted to resolutions between 3.5 and 4.3 Å. For these structures, rigid-body, translation-libration-screw (TLS) parameters, group B factor, and individual-site refinement was tightly restrained to favor the geometric component, and the structures were checked and validated by running MolProbity at every step of refinement and model building. Additionally, secondary structure restraints and reference models—the 2.1 Å resolution structure of 2.2c Fab and the 1.49 Å structure of YU2 gp120 bound to M48U1—were used to restrain refinement. For the Fab 2.2c-gp12092UG037-M48U1 structure, noncrystallographic symmetry (NCS) was used throughout refinement.
Structure validation and analysis and figures.
The quality of the final refined models was monitored using the program MolProbity (39). Structural alignments were performed using the Dali server and the program lsqkab from CCP4i suite (34). PISA (40) and PIC (41) webservers were used to determine contact surfaces and residues. All illustrations were prepared with the PyMol molecular graphic suite (DeLano Scientific, San Carlos, CA, USA).
ADCC assays.
ADCC assays were carried out using the rapid fluorescence ADCC (RFADCC) method (42), modified to reduce prozone effects (27). All ADCC studies used CEM-NKr-CCR5 target cells sensitized with recombinant gp120 from the HIV-1BaL isolate.
Binding of labeled MAbs to sensitized CEM-NKr-CCR5 target cells.
CEM-Nkr-CCR5+ target cells were sensitized with gp120 as described in reference 27 for our standard ADCC assay, and a saturation curve was developed for increasing concentrations of Alexa-Fluor 488-tagged N5-i5. The number of molecules of N5-i5 bound per cell was determined by measuring the number of dye molecules per molecule of N5-i5. A BDFortessa flow cytometer was used to determine the number of dye molecules per channel using Alexa-Fluor 488 calibration beads (Bangs Laboratories, Fishers, IN). This permitted the direct determination of the number of molecules of N5-i5 bound per cell. Half-maximal binding and saturation values were determined by nonlinear curve fitting using GraphPad Prism. Because 2.2c could not be labeled, we used a standard competition strategy to determine the number of molecules of N5-i5 and 2.2c bound per cell. A single fixed concentration of Alexa-Fluor 488-N5-i5 that corresponds to one concentration up from the first saturating concentration determined by nonlinear curve fitting was used. This concentration of labeled N5-i5 was mixed with unlabeled N5-i5 or 2.2c beginning at a 30-fold molar excess of the labeled N5-i5 and titrating downward with a log2 dilution interval. Each binding experiment was repeated independently four to seven times, and half-maximal binding and maximal binding (Bmax) values were pooled for statistical analysis.
Antibody binding to cell surface expressed envelope trimers.
293FS cells (Invitrogen) were transfected with gp160 Env glycoprotein from BaL HIV-1 clone C3 (Reitz; GenBank no. M68893) and maintained in 293 FreeStyle medium (Invitrogen). In order to maximize glycoprotein expression in human cells, codon sequences were “human optimized” using in-house codon optimization charts. Expression of human BaL (huBaL) gp160 glycoprotein was confirmed by gp120 ELISA and Western blotting using anti-gp120 antibodies targeting various gp120 epitopes. For reactions with sCD4, 10 mg sCD4 (d1d2 domain; Biogen) was added and reaction mixtures were incubated for 45 min with shaking. Antibodies tested for binding were labeled with a Zenon allophycocyanin-human IgG labeling kit (Invitrogen). Data were acquired with a FACSCalibur flow cytometer (Becton Dickinson, Franklin Lakes, NJ) and processed and analyzed using FlowJo software (Tree Star, Ashland, OR).
SPR analyses.
Surface plasmon resonance (SPR) analyses were performed with Biacore T100 at room temperature. Protein A was covalently bound to individual flow cell surfaces of a CM5 sensor chip by the amine-coupling method following the procedure of Steckbeck et al. (43). Antibodies were captured on protein A surfaces to a specific level at about 150 response units (RU). For kinetic measurement of the single-chain gp120BaL-CD4 complex (FLSC) (44) binding to immobilized antibody, sensorgrams were obtained by passing various concentrations of FLSC (0 to 200 nM). A buffer injection served as a negative control. Upon completion of each association-dissociation cycle, surfaces were pulsed with regeneration solution. Antibody association rates (ka), dissociation rates (kd), and affinity constants (KD) were calculated with BIAevaluation software.
Protein structure accession numbers.
Coordinates and structure factors have been deposited in the Protein Data Bank under accession numbers 4H8W, 4R4B, 4R4H, 4R4F, and 4R4N (see Table S1 in the supplemental material).
RESULTS
Antibodies N5-i5 and 2.2c recognize overlapping but distinct epitope surfaces within the C1 and C2 regions of gp120.
MAb N5-i5 is one of the most potent antibodies thus far identified; it is specific for an A32-like epitope, and it mediates ADCC against target cells sensitized with gp120 of the HIV-1BaL isolate with a 50% effective concentration (EC50) of 0.05 nM (Fig. 1) (27). MAb 2.2c shows weaker ADCC potency with a 75-fold higher EC50. MAbs N5-i5 and 2.2c were both expressed under identical conditions using 293T cells, and their variable region genes were cloned onto the same human IgG1 constant region, as described previously (27). This controls for isotype and possible glycoform heterogeneity, which could affect ADCC potency. Monoclonal antibodies N5-i5 and 2.2c are nonneutralizing in the TZM-bl assay and cross-compete with each other and with MAb A32 for binding to monomeric gp120, as tested by ELISA (27; J. E. Robinson, unpublished data).
FIG 1.
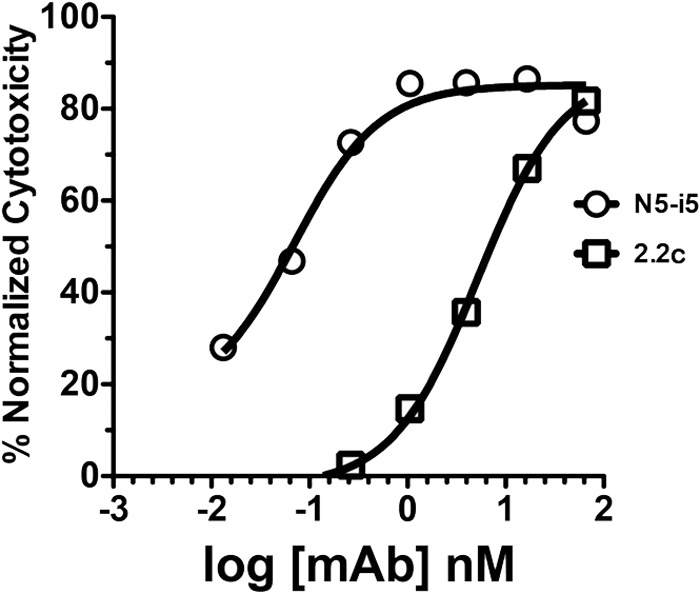
Representative ADCC curves for MAbs N5-i5 and 2.2c. ADCC assays were performed as described in Materials and Methods using CEM-NKr-CCR5 target cells sensitized with gp120 of the HIV-1BaL isolate.
We solved the crystal structures of N5-i5 and 2.2c antigen binding fragments (Fabs) complexed with the two-domain human CD4 (d1d2CD4)-triggered variable loop-deleted gp120 cores to determine whether epitope specificity could account for the observed differences in ADCC potency. A Fab fragment of N5-i5 was complexed with the clade A/E gp12093TH057 coree, whereas a Fab fragment of 2.2c was complexed with the clade B gp12089.6P coree. Structures were solved at 1.85-Å and 4.3-Å resolutions for N5-i5 and 2.2c complexes, respectively (PDB codes 4H8W, 4R4B, 4R4H, 4R4F, and 4R4N; see Table S1 in the supplemental material). 2.2c Fab fragments were also cocrystallized with clade B gp120YU2 and clade A gp12092UG037 corees using the CD4-mimetic miniprotein M48U1 (31). These structures were solved at 3.5 Å and 3.6 Å, respectively (see Table S1 in the supplemental material). The three different complexes with Fab revealed strong conservation in the mode of recognition of gp120 from different isolates within a clade and across clades by 2.2c (see Table S2 in the supplemental material). The root mean square deviation of gp120 core and antibody 2.2c in the different crystal complexes ranged from 0.7 to 0.75 Å and 1.1 to 2.1 Å, respectively. Unless otherwise noted, the highest-resolution structure of 2.2c Fab-gp120YU2 coree-M48U1 (3.5 Å) was used for structural comparisons.
As shown in Fig. 2, N5-i5 and 2.2c bind to overlapping as well as distinct elements of only the inner domain of gp120, proximal to the N- and C-terminal extensions. The structures reveal precise complementarity between positively charged Fab paratopes, contributed largely by heavy-chain complementarity-determining region 2 (CDRH2) (see Table S3 in the supplemental material) and electronegative and glycan-free surfaces of the inner domain (see Fig. S1A in the supplemental material). Five and six CDRs form the paratopes of 2.2c and N5-i5, respectively. The epitope terrains contributed by short CDRH3s of 10 residues and 11 residues for N5-i5 and 2.2c, respectively, are relatively flat in both cases (see Fig. S1B in the supplemental material). These short CDRH3s are distinct from the extended CDRH3s that many broadly neutralizing MAbs have (45, 46). Comparisons to the crystal structure of unbound N5-i5 (27) and 2.2c Fab also indicate that only marginal adjustment of the interactive side chains occurs within the CDRs upon gp120 corees binding (see Fig. S2 in the supplemental material), which is consistent with a lock-and-key model with minimal induced fit. It is also important to note that broadly neutralizing antibodies (bnAbs) typically exhibit high levels of somatic hypermutation (SHM) (47, 48), whereas variable domains of N5-i5 and 2.2c are mutated only modestly (6.6% and 8.9%, respectively) at the protein level (see Fig. S1B in the supplemental material). If the N5-i5 and 2.2c epitopes are targets of protective antibodies in vivo, these relatively low levels of SHM can be readily attained with standard vaccination regimens. In addition, the lack of significant differences in SHM of N5-i5 and 2.2c, which mediate ADCC with different potencies, indicates that there is no link between level of affinity maturation of A32-like antibodies and their ability to mediate Fc receptor function. This is in contrast to the positive correlation between somatic hypermutation and neutralization breadth observed for broadly neutralizing antibodies (47, 48).
FIG 2.

Crystal structures of the N5-i5 Fab-gp12093TH057 coree-d1d2CD4 and 2.2c Fab-gp12089.6P coree-d1d2CD4 complexes. The light/heavy chains of N5-i5 Fab and 2.2c Fab are shown in light blue/dark blue and light pink/dark pink, respectively, and the complementarity-determining regions (CDRs) are shown in black (CDR L1), brown (CDR L2), light blue (CDR L3), gray (CDR H1), green (CDR H2), and cyan (CDR H3). The gp120 inner domain is shown in yellow, and the outer domain is in orange. See also Fig. S1A in the supplemental material.
We analyzed MAb N5-i5 and MAb 2.2c footprints in the context of the layered inner domain architecture of gp120 (Fig. 3A and B). Layers (or loop-like extensions) of gp120 have been identified previously as topologically separate components of the gp120 inner domain acting as shape-changing spacers and facilitating movement in the CD4-triggered conformational transitions of gp120 (49–52). N5-i5 binds between layer 1 (β2̄ and β1̄ strands, α0 helix, and β2̄-α0 and β1̄-β0 connecting coils; residues 51 to 54, 56, 58 to 61, and 68 to 80) and layer 2 (α1 helix, β4 strand, and β4-β5 connecting coil; residues 103, 106 to 107, 110, 114, 217, 219, and 221) of the C1 and C2 regions (Fig. 3A and B; also, see Table S4 in the supplemental material). An average of 82% and 18% of the epitope surface maps to layer 1 and 2, respectively (see Table S3 in the supplemental material). In contrast, 2.2c targets mainly residues of layer 1 (residues 53, 57 to 61, and 71 to 79; 94% of contact surface area [see Tables S2 and S3 in the supplemental material]) with few contacts within the β4 strand of layer 2 (residues 218 to 221) at the junction of the 7-stranded β sandwich and layer 3 (residues 246 to 247). There are no contacts with the α1 helix of layer 2 (Fig. 3A and B; also, see Fig. S3 in the supplemental material). Most of residues forming the N5-i5 and 2.2c epitope footprints are highly conserved (Fig. 3C), suggesting that the gp120 region targeted by these antibodies is functionally relevant. Indeed, many of them, such as Leu52, Phe53, Trp69, His72, Ala73, and Asp107, have been shown to stabilize the unliganded Env trimer or contribute to layer 1 and 2 functions in conformational transitions upon CD4 receptor binding (49, 51, 52).
FIG 3.

N5-i5 and 2.2c epitopes. (A) N5-i5 and 2.2c epitope footprints on monomeric gp120. The Cα atoms of the gp120 residues involved in N5-i5 and 2.2c binding are represented by blue and pink balls, respectively, and displayed over the ribbon diagram of the gp120 inner domain. Antibodies' contact surfaces displayed over the gp120 molecular surface are shown in black (right). The “layered” architecture of the gp120 inner domain is shown, with the 7-stranded β sandwich in magenta, layer 1 in yellow, layer 2 in cyan, and layer 3 in light orange. The outer domain is shown in orange. (B) Mapping of the N5-i5 and 2.2c contact residues on the gp120 primary sequence of the gp120 inner domain of the isolates used in structural studies. The topology diagrams depicting a distribution of secondary structure elements as calculated with DSSP (66) is shown above the gp120 sequences. The gp120 residues involved in N5-i5 and 2.2c binding are highlighted in blue and pink, respectively. Residues contributing to the binding through H bonds and salt bridges are indicated by blue asterisks and green lowercase letters above and below the 93TH057 and 89.6P sequences, respectively. The gp120 layers are colored as in panel A. (C) Sequence conservation of N5-i5 and 2.2c epitopes. The height of the residue at each position is proportional to its frequency of distribution among the HIV-1 isolates, as deposited in the Los Alamos database (all clades are included). Residues are colored according to hydrophobicity: black, hydrophilic; green, neutral; blue, hydrophobic. Residues forming the N5-i5 and 2.2c epitope are indicated by blue and pink lines above the sequence, respectively.
Exposure of epitopes recognized by MAbs N5-i5 and 2.2c is dependent on the binding of trimeric Env to cell surface CD4.
Analysis of the N5-i5 and 2.2c epitope footprints in the context of the cryo-electron microscopy (cryo-EM) tomograms of native untriggered virion-associated HIV-1 trimers (53) and recently resolved crystallographic and cryo-EM structures of a cleaved, soluble BG505 SOSIP.664 gp140 trimer (54, 55, 69) indicate that they map to a region of gp120 that is buried within the trimer interface engaged directly in interactions with the gp41 subunit of the trimer (Fig. 4 and 5A). The gp41 contacts with the nascent N5-i5/2.2c epitope in the trimer involve residues 548 to 583 of prefusion gp41. The gp120-gp41 interaction in this region is rather loose, as indicated by the fact that a larger portion of the gp41 contact region is disordered (residues 548 to 568) and only partially resolved in the trimer structure determined at a 3.5-Å resolution (69). The rest of the contact surface is formed by the N terminus of the α7 helix of the 4-helix collar of prefusion gp41 (residues 571 to 583) (Fig. 4A). Interestingly, close examination of the assemblies of Fab complexes and soluble trimer revealed a striking similarity of the contact residues used by gp120 to interact with MAbs and gp41, respectively (Fig. 4A and B). This similarity is less for MAb 2.2c, as it does not bind the α1 helix (Fig. 3A; also, see Fig. S3 in the supplemental material). Although N5-i5 and 2.2c mimic the interaction of gp41 with gp120 within the trimer to some extent, structural alignments indicate differences in secondary structure of the gp120 region forming the nascent N5-i5/2.2c epitope within the trimer compared to the N5-i5/2.2c epitope on sCD4-triggered gp120 (Fig. 4B). These structural differences occur mainly in layer 1 and the α1 helix of layer 2, as shown by different distributions of secondary structure elements in layer 1 and α1-helix tilting (Fig. 4B). These findings indicate that N5-i5 and 2.2c MAbs target a gp120 region that is exposed on the Env spike by gp41 detachment followed by a substantial structural rearrangement post-CD4 binding.
FIG 4.

Colocalization of N5-i5 and 2.2c epitopes within the BG505 SOSIP.664 gp140 trimer. (A) N5-i5 and 2.2c epitope footprints are mapped onto the BG505 SOSIP.664 trimer structure solved by X-ray crystallography (69). The gp120 protomers are colored as in Fig. 3, and gp41 is shown in green. The spheres represent the Cα atoms of residues that contribute to formation of N5-i5 and 2.2c epitopes post-CD4 binding. The enlargements show regions of contact between the gp120 inner domain and the gp41 of the unliganded trimer on which N5-i5 Fab-gp12093TH057 coree-d1d2CD4 (top) and 2.2c Fab-gp12089.6P coree-d1d2CD4 complex (bottom) are aligned based on the gp120 outer domain. Residues contributing to both the interface with gp41 within the trimer and the interface with MAb within the complex are shown as spheres. Layers 1 and 2 of the gp120 inner domain of the gp120-gp41 and the gp120-MAb interface are in darker and lighter shades of yellow and cyan, respectively. Only variable parts of N5-i5 and 2.2c Fab are depicted, and CDRs involved in interfaces are shown. Many residues of gp120 involved in N5-i5/2.2c epitope post-CD4 binding also line the interface with the gp41 of the trimer. These include multiple residues of β2̄ and β1̄ strands and α0 helix of layer 1, all residues forming the N5-i5 epitope on the α1 helix, and multiple residues of the β4 strand and the β4 -β5 connecting loop of layer 2 (marked by black dots below the 93TH057 sequence on panel B). (B) Details of the conformational changes of layer 1 (left) and layer 2 (right) from the prefusion state of trimer (light yellow and light cyan for layers 1 and 2, respectively) to the N5-i5/2.2c-bound conformation of the CD4-triggered gp120 coree (red). A change in the distribution of secondary structure elements from unliganded to MAb-bound conformation is shown above the gp120 sequences on which residues involved in N5-i5 and 2.2c binding are highlighted in blue and pink, respectively. The CD4 triggering induces rearrangements of secondary structural elements in layer 1 of inner domain manifested primarily by formation of the α0 helix and unfolding of the α1̄ helix. The α1 helix of layer 2 is shortened and tilted.
FIG 5.

Exposure of N5-i5 and 2.2c epitopes on a viral trimer. (A) Colocalization of N5-i5 and 2.2c epitopes within the virion-associated untriggered HIV-1 trimers. The Cα atoms of gp120 residues involved in interaction with N5-i5 (blue balls) and 2.2c (pink balls) are mapped into the trimer structure derived by cryo-electron tomography (53) and are shown as blue and pink balls, respectively. gp120 molecules are shown as ribbon diagrams and colored as in Fig. 3. The views of trimers are from the side, with the viral membrane oriented toward the bottom (left) and rotated 90° about a horizontal axis with the viral membrane at the bottom (right). (B) Colocalization of N5-i5 and 2.2c epitopes within HIV-1 trimers triggered with the soluble CD4. N5-i5 and 2.2c epitope footprints were mapped in the structure derived by cryo-electron tomography of the gp120Bal-d1d2CD4 trimer (67). The mapping of N5-i5 and 2.2c epitope footprints in the tomograms confirms that they stay largely within the interface of d1d2CD4-triggered spike and are not available for antibody recognition, due to steric hindrance. See also Fig. S4 in the supplemental material. (C) The binding curves of MAb N5-i5 (red line) and 2.2c (green line) to surface-expressed HIV-1BaL trimers in the presence or absence of soluble d1-d4CD4 (sCD4). Experiments were performed as described in Materials and Methods. CD4i antibody 17b (cyan line) was used as a positive control. The enhancement of binding to sCD4-triggered HIV-1BaL trimers was observed only for coreceptor binding site MAb 17b. (D) MAb N5-i5 binding (solid lines) and 2.2c binding (dashed lines) to CEM-Nkr-CCR5+ cells (left) or CEM-Nkr-CCR5− cells (right). The rightmost curves (red) in each histogram overlay represent the binding of N5-i5 or 2.2c to virion-sensitized cells, whereas the leftmost curves (blue) represent the binding of these MAbs on cells not sensitized with virions.
In addition, our studies point to a critical role of cell surface CD4, over and above simply binding gp120, in trimer activation during viral entry. It was suggested previously, based on mutagenesis, that the A32 and C11 subregions of epitope cluster A remain buried at the interface of virion-associated HIV-1 trimers triggered with soluble CD4, despite the exposure of the coreceptor binding site (50). We have confirmed this prediction by mapping the epitope footprints of N5-i5 and 2.2c onto the cryo-EM tomograms of the gp120BaL-d1d2CD4 trimer (53) (Fig. 5B). In addition, fitting of N5-i5 and 2.2c Fab-bound complexes onto the same tomograms indicates that the Fv domains partially overlay and occupy the presumptive position of the gp41 within the trimer (see Fig. S4 in the supplemental material). Multiple clashes occur between the Fabs and gp120 in the trimer, suggesting that MAb N5-i5 and 2.2c should not be able to reach their cognate epitopes within the soluble CD4-triggered spikes. Since soluble CD4 does not expose the A32 subregion (Fig. 5C) (56), and since we know that it is exposed during cell-to-cell fusion and during viral entry (57), either binding to cell surface CD4 or binding of trimer-CD4 complexes to coreceptor exposes this region. We tested this hypothesis by spinoculating AT-2 inactivated HIV-1BaL virions onto the surfaces of CEM-Nkr and CEM-Nkr-CCR5+ cells and testing binding of MAbs N5-i5 and 2.2c. As shown in Fig. 5D, the A32 subregion was exposed on both cell types suggesting that additional energy to trimer unfolding after binding mediated by cell surface CD4 is required for effective exposure of the A32 subregion on the virus surface, although such exposure does not require formation of gp120-CD4-CCR5 complexes. It is noteworthy that the neutralizing monoclonal anti-CD4 MAb ibalizumab (58) does not block the binding of gp120 to CD4; rather, it binds an epitope at the d1d2 interface, preventing a conformational change in CD4 that is necessary for viral entry, further suggesting the requirement for additional changes effected by cell surface CD4 binding for HIV-1 entry.
Multivalent binding and orientation of the bound antibodies to antigen on target cells are key determinants of the differences in ADCC potency between N5-i5 and 2.2c.
Although MAb N5-i5 and 2.2c recognize largely overlapping areas of the gp120 surface their binding modes are quite different. They approach their cognate epitopes at different angles, and the VH and VL contact surfaces are reversed in orientation (Fig. 6A; also, see Fig. S5 in the supplemental material). In addition, five and six CDRs contribute to gp120 binding of MAbs 2.2c and N5-i5, respectively (see Table S3 in the supplemental material). Furthermore, MAb N5-i5 is involved in binding residues of both α0 and α1 helices of gp120, whereas MAb 2.2c forms its complex with no binding contacts to the α1 helix (Fig. 3A and B; also, see Fig. S3 in the supplemental material). Overall, the interactive surface that becomes buried due to the MAb 2.2c-gp120 coree interaction encompasses 1,768 Å2, compared to 1,890 Å2 buried at the MAb N5-i5-gp120 coree interface; thus, it is smaller by 122 Å2. These observations raised the question of whether the differences in antibody binding modes account for the observed differences in their ADCC potency. The differences could be due to differences in affinity for monomeric gp120, ability to cross-link antigen on the target cell surface, differences in orientation of the antibody CH2 domains in the immune complexes, or all three. SPR studies showed that N5-i5 and 2.2c bound with similar affinities to monomeric gp120-CD4 complexes (see Fig. S6A in the supplemental material). In contrast, binding to gp120-sensitized CEM-Nkr-CCR5+ target cells revealed an approximately 18-fold-greater binding of N5-i5 than 2.2c (Fig. 6B and C). Thus, N5-i5 has a greater ability to bind gp120-CD4 complexes on the cell surface than 2.2c. Surprisingly, N5-i5 saturated the gp120-sensitized target cells at approximately 25,000 molecules of IgG per cell, whereas 2.2c saturated at approximately half that value (Fig. 6B). Additionally, N5-i5 binding was linear in Scatchard plots, whereas 2.2c binding was curvilinear (Fig. 6C). These data suggest that there is a population of gp120-CD4 complexes on the cell surface that is poorly accessible to 2.2c.
FIG 6.

MAb N5-i5 and 2.2c binding to the gp120 antigen. (A) Superimposition of N5-i5 Fab-gp12093TH057 coree-d1d2CD4 and 2.2c Fab-gp12089.6P coree-d1d2CD4 complexes. Structures were aligned based on the gp120 molecule; a molecular surface is displayed over N5-i5 Fab, and 2.2c Fab is shown in a ribbon diagram. See also Fig. S5 in the supplemental material. (B) MAb N5-i5 and 2.2c binding kinetics to gp120-sensitized CEM-Nkr-CCR5+ target cells, as measured with an unlabeled MAb competition protocol. CEM-Nkr-CCR5+ target cells were sensitized with gp120, and a saturation curve was developed as described in Materials and Methods. (C) Scatchard plots of N5-i5 and 2.2c binding to CEM-Nkr-CCR5+ target cells were derived from the binding data using the standard equation (68) by nonlinear curve fitting (Prism; GraphPad, La Jolla, CA).
In addition, models of N5-i5 and 2.2c IgG1s binding to cell surface CD4 sensitized with gp120 (see Fig. S7 in the supplemental material) suggested that the binding mode of 2.2c may result in suboptimal positioning of the Fc domain for interaction with Fcγ receptors on the effector cell membrane. This difference in orientation is predicted to position the 2.2c CH2 domain toward the target cell surface, potentially obstructing its accessibility to Fcγ receptors. To test whether differences in Fc positioning due to the juxtaposed heavy and light chains contribute to ADCC potency, we swapped the VH and VL domains onto the opposite chains of IgG1s to generate hybrid variants with interconverted CH2 domain orientations, as schematically shown in Fig. 7A. No significant differences were found between the binding of the swapped versions and the wild-type MAbs to monomeric gp120-CD4 complexes by SPR (see Fig. S6B in the supplemental material) or to gp120-sensitized target cells (Fig. 7B). No significant difference in ADCC potency was found between wild-type and VH-VL-swapped versions of N5-i5, suggesting that the ADCC potency of N5-i5 is not dependent on the relative orientations of its light and heavy chains. In contrast, swapping the VH and VL domains of 2.2c improved its ADCC potency approximately 7-fold (Fig. 7C), which was statistically highly significant. This suggested that the ADCC potency directed to the 2.2c epitope that is poorly accessible on the target cell surface, as judged by cell surface binding and saturation studies, could be increased by changing the binding mode to more optimally position the CH2 domains for interaction with the Fc receptors on the effector cells. Thus, both the ability of an antibody to cross-link antigen on the target cell and the positioning of its CH2 domain for optimal Fcγ receptor binding contribute to ADCC potency.
FIG 7.
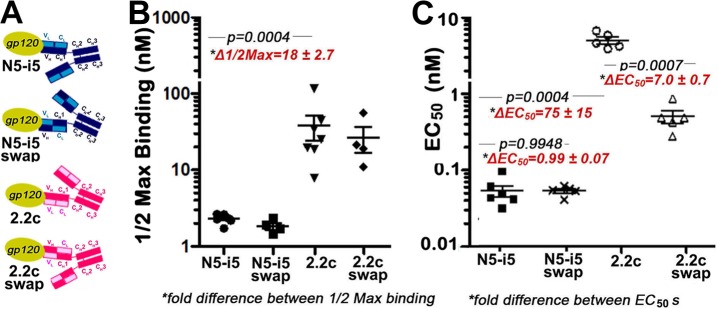
Fv-swapped versions of N5-i5 and 2.2c. (A) Design of Fv-swapped versions of N5-i5 and 2.2c. Swaps were made by moving the variable heavy (VH) domain onto the constant light (CL) domain and the variable light (VL) domain onto the constant heavy 1 (CH1) domain for each MAb to replicate the Fc domain orientations of counterpart. (B) Half-maximal binding of MAbs N5-i5 and 2.2c and their swapped versions to gp120-sensitized CEM-Nkr-CCR5+ target cells. Each binding experiment was repeated independently four to seven times as described in Materials and Methods, and half-maximal-binding and Bmax values were pooled for statistical analysis. (C) Cytotoxicity mediated by MAbs N5-i5 and 2.2c and their swapped versions on gp120-sensitized CEM-Nkr-CCR5+ target cells.
DISCUSSION
Natural history studies of the prevalence of ADCC responses (25–27) and ADCC escape (22) in infected individuals and the RV144 vaccine trial (3, 13, 14) implicate nonneutralizing antibody responses to the A32 subregion of gp120 in protective immunity to HIV-1. If these correlations are causal, targeting this region of gp120 by vaccination becomes a useful possibility, as this subregion is highly conserved and strongly immunogenic (25–27). This hypothesis is controversial, as there has been no successful demonstration from passive immunization studies using antibodies to this region to block infection of NHPs by simian/human immunodeficiency viruses (SHIVs). In the absence of such data, the atomic-level definition of the A32 epitope subregion may be key to determining whether A32-like antibodies contribute to protective immunity. Having the A32-like epitope footprints will make it possible to analyze if A32-like antibodies exert immunologic pressure during active and passive vaccination in NHPs as well as in vaccine and natural history studies of HIV-1-infected people. If these antibodies were truly protective, epitope escape at the key contact residues or at noncontact residues that indirectly affect epitope exposure would be expected to be apparent in sieving studies in both NHPs and humans.
Previously, the A32 epitope was mapped roughly to the C1 region by mutagenesis and antibody cross-competition studies (23, 24). Subsequent mutagenesis studies confirmed this assignment and showed additionally that exposure of this epitope is strongly affected by mutations in the 7-stranded β sandwich, mobile layers 1, 2, and 3, the outer domain, and the β20-β21 loop of gp120 (23, 51, 59) (see Fig. S8 in the supplemental material). Our studies define the contact regions of two A32-like antibodies and map them to the highly conserved surface of the gp41 reactive region of gp120, proximal to the N- and C-terminal extensions. These epitopes involve surfaces formed by inner domain layers 1 and 2 of C1-C2 region. Thus, our studies settle the localization of the A32 epitope subregion at the atomic level and allow accurate mapping of mutations under antibody selection to determine if the mutations affect the epitope directly by altering contacts or indirectly by affecting its exposure on infected cells.
Further, our studies confirm that the A32 epitope subregion is buried in trimers triggered with soluble CD4 as previously suggested (50). These studies indicate that this epitope region remains buried at the interface of virion-associated HIV-1 trimers triggered with soluble CD4, despite the exposure of the coreceptor binding site. This is consistent with previous findings regarding exposure of the MAb A32 epitope on virions in solution and using a cell-cell fusion system (56, 57, 59) and points to an additional energy component likely provided by a conformational change in the d3d4 region of cell surface CD4, which is required for trimer unfolding during viral entry.
The epitopes characterized here are predicted to be potent ADCC targets for antibody-mediated prevention of HIV-1 infection and postinfection control of viremia at two points in the virus replicative cycle. First, the A32-like epitopes become exposed during viral entry, where they remain long enough to sensitize a newly infected CD4+ T cell for killing by ADCC in the presence of cognate antibodies. This was predicted from our earlier studies imaging A32-epitope exposure during Env-mediated membrane fusion (57, 60), which we have confirmed experimentally (27). We have termed these epitopes “entry targets,” as they sensitize a newly infected target cell within minutes of virion binding to CD4, and remain exposed for several hours (61). Surprisingly, the importance of entry target epitopes during HIV-1 acquisition and antibody-mediated postinfection control of viremia has been largely unappreciated. As entry targets (27, 61), the A32-like epitopes will appear largely on virions entering target cells (59, 62, 63). Interestingly, they were shown to persist on freshly infected cell surfaces for extended periods of time postinfection and also to appear on the surfaces of budding virions (release targets), where they are targets for ADCC activity (25, 27). Exposure of A32-like epitope targets on budding virions largely depends on the levels of CD4 on the surface of the infected target cell; thus, their expression is modulated by viral effector proteins such as Nef and Vpu, which are capable of downregulating the levels of CD4 on the surface of the infected cell (59, 64). Furthermore, it was suggested recently that Nef and Vpu have evolved as viral defenses against the exposure of these highly conserved CD4i epitope targets during virion release and as a humoral immunity evasion mechanism preventing clearance of virally infected cells through antibody-mediated effector function (59, 64). It will be important to determine the contribution of this category of A32-like release targets to ADCC responses in vitro.
Our findings further indicate that the epitope region recognized by N5-i5 and 2.2c is buried in its nascent, alternatively folded conformation at the gp120-gp41 interface of in the untriggered Env spike. Analysis of the N5-i5 and 2.2c epitope footprints in context of the structure of the BG505 SOSIP.664 trimer (69) revealed differences in secondary structure of this region between the trimer and sCD4-triggered gp120 corees recognized by N5-i5 and 2.2c MAbs. These structural differences occur mainly in layer 1 and the α1 helix of layer 2. It was also revealed by hydrogen-deuterium exchange and oxidative labeling that SOSIP trimers undergo substantial reorganizations upon CD4 activation (65). The CD4 induced conformational changes occur within the inner domain layers 1 and 2, including α0 and α1 helices. Altogether, the findings indicate that N5-i5 and 2.2c MAbs target a gp120 region that is conformationally plastic and undergoes substantial structural rearrangements from the structure interacting with the gp41 within the untriggered trimer to the one induced by CD4 attachment.
Finally, our data provide the first insight at the atomic level into how antibody recognition influences ADCC potency. The weaker ADCC potency of 2.2c results from its poor ability to cross-link antigen on the target cell surface and from the orientation of its attachment to the Env target, which likely positions its CH2 domain in a less favorable orientation for Fcγ receptor interaction. Thus, it is possible that two highly similar/proximal epitopes could be equally accessible for monovalent MAb binding but differ significantly in multivalent cross-linking by MAb on the target cell surface. The latter is caused by either fine differences in epitope footprints, differences in the angles by which the epitope is approached, or both. To our knowledge, this is the first report of such a connection between precise antigen targeting and ADCC potency at the atomic level. Further, the ability of the CH2 domain to bind Fcγ receptor and activate ADCC is defined by orientations of antibody attachment to the antigen. It is not possible to read and distinguish between these variables by simply measuring epitope-specific binding titers by ELISA, as was done in the RV144 trial (3, 13, 14). Collectively our results provide a potential explanation for the discordance between A32-epitope specific ELISA binding assays and ADCC as correlates of reduced infection risk in RV144 by establishing that the magnitudes of HIV envelope binding versus Fc receptor-dependent antiviral function can be uncoupled by structural constraints within immune complexes. Elucidation of these constraints provides key insights for understanding, refining, and improving the outcome of HIV vaccine trials, in which relevant immune responses are facilitated by antibody-mediated protection. Furthermore, our data will be critical to the process of designing an effective immunogen to the A32-like region capable of eliciting MAbs acting through potent Fc-mediated function. Although epitope footprints of N5-i5 and 2.2c largely overlap, the atomic resolution information on their antigen complexes makes it possible to identify differences in their recognition, which could be used to optimize the antigenicity of potential immunogens.
Supplementary Material
ACKNOWLEDGMENTS
Support for this work was provided by the Intramural Research Program of the VRC, National Institute of Allergy and Infectious Diseases, NIH, by grant OPP1033109 from The Bill and Melinda Gates Foundation (to G.K.L.), by National Institute of Allergy and Infectious Diseases (NIAID)/NIH grants R01 AI-084830 and R01 AI-087181 (to G.K.L) and 5K23AI084580-04 (to M.M.S.). Use of sector 22 (Southeast Region Collaborative Access team) at the Advanced Photon Source was supported by the U.S. Department of Energy, Basic Energy Sciences, Office of Science, under contract number W-31–109-Eng-38. Additionally, crystallographic data were collected at the Stanford Synchrotron Radiation Lightsource (SSRL), a Directorate of the SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the U.S. Department of Energy Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the U.S. Department of Energy Office of Biological and Environmental Research, by the National Institutes of Health (NIH) National Center for Research Resources, Biomedical Technology Program (P41RR001209), and by the National Institute of General Medical Sciences.
We thank Albert Kim and Min Tang for assistance with protein expression, Christine Obreicht for outstanding technical support in protein expression and purification, Qiang Lin and Xinwei Zhao for outstanding technical support in recombinant plasmid engineering and protein expression and characterization, and members of the Structural Biology and Structural Bioinformatics Core Sections, Vaccine Research Center (VRC), for discussions and comments on the manuscript.
Footnotes
Published ahead of print 27 August 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.02194-14.
REFERENCES
- 1.Hessell AJ, Hangartner L, Hunter M, Havenith CE, Beurskens FJ, Bakker JM, Lanigan CM, Landucci G, Forthal DN, Parren PW, Marx PA, Burton DR. 2007. Fc receptor but not complement binding is important in antibody protection against HIV. Nature 449:101–104. 10.1038/nature06106. [DOI] [PubMed] [Google Scholar]
- 2.Mabuka J, Nduati R, Odem-Davis K, Peterson D, Overbaugh J. 2012. HIV-specific antibodies capable of ADCC are common in breastmilk and are associated with reduced risk of transmission in women with high viral loads. PLoS Pathog. 8:e1002739. 10.1371/journal.ppat.1002739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haynes BF, Gilbert PB, McElrath MJ, Zolla-Pazner S, Tomaras GD, Alam SM, Evans DT, Montefiori DC, Karnasuta C, Sutthent R, Liao HX, DeVico AL, Lewis GK, Williams C, Pinter A, Fong Y, Janes H, DeCamp A, Huang Y, Rao M, Billings E, Karasavvas N, Robb ML, Ngauy V, de Souza MS, Paris R, Ferrari G, Bailer RT, Soderberg KA, Andrews C, Berman PW, Frahm N, De Rosa SC, Alpert MD, Yates NL, Shen X, Koup RA, Pitisuttithum P, Kaewkungwal J, Nitayaphan S, Rerks-Ngarm S, Michael NL, Kim JH. 2012. Immune-correlates analysis of an HIV-1 vaccine efficacy trial. N. Engl. J. Med. 366:1275–1286. 10.1056/NEJMoa1113425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alpert MD, Harvey JD, Lauer WA, Reeves RK, Piatak M, Jr, Carville A, Mansfield KG, Lifson JD, Li W, Desrosiers RC, Johnson RP, Evans DT. 2012. ADCC develops over time during persistent infection with live-attenuated SIV and is associated with complete protection against SIV(mac)251 challenge. PLoS Pathog. 8:e1002890. 10.1371/journal.ppat.1002890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lambotte O, Ferrari G, Moog C, Yates NL, Liao HX, Parks RJ, Hicks CB, Owzar K, Tomaras GD, Montefiori DC, Haynes BF, Delfraissy JF. 2009. Heterogeneous neutralizing antibody and antibody-dependent cell cytotoxicity responses in HIV-1 elite controllers. AIDS 23:897–906. 10.1097/QAD.0b013e328329f97d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Florese RH, Demberg T, Xiao P, Kuller L, Larsen K, Summers LE, Venzon D, Cafaro A, Ensoli B, Robert-Guroff M. 2009. Contribution of nonneutralizing vaccine-elicited antibody activities to improved protective efficacy in rhesus macaques immunized with Tat/Env compared with multigenic vaccines. J. Immunol. 182:3718–3727. 10.4049/jimmunol.0803115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Banks ND, Kinsey N, Clements J, Hildreth JE. 2002. Sustained antibody-dependent cell-mediated cytotoxicity (ADCC) in SIV-infected macaques correlates with delayed progression to AIDS. AIDS Res. Hum. Retroviruses 18:1197–1205. 10.1089/08892220260387940. [DOI] [PubMed] [Google Scholar]
- 8.Baum LL, Cassutt KJ, Knigge K, Khattri R, Margolick J, Rinaldo C, Kleeberger CA, Nishanian P, Henrard DR, Phair J. 1996. HIV-1 gp120-specific antibody-dependent cell-mediated cytotoxicity correlates with rate of disease progression. J. Immunol. 157:2168–2173. [PubMed] [Google Scholar]
- 9.Gomez-Roman VR, Patterson LJ, Venzon D, Liewehr D, Aldrich K, Florese R, Robert-Guroff M. 2005. Vaccine-elicited antibodies mediate antibody-dependent cellular cytotoxicity correlated with significantly reduced acute viremia in rhesus macaques challenged with SIVmac251. J. Immunol. 174:2185–2189. 10.4049/jimmunol.174.4.2185. [DOI] [PubMed] [Google Scholar]
- 10.Hidajat R, Xiao P, Zhou Q, Venzon D, Summers LE, Kalyanaraman VS, Montefiori DC, Robert-Guroff M. 2009. Correlation of vaccine-elicited systemic and mucosal nonneutralizing antibody activities with reduced acute viremia following intrarectal simian immunodeficiency virus SIVmac251 challenge of rhesus macaques. J. Virol. 83:791–801. 10.1128/JVI.01672-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xiao P, Patterson LJ, Kuate S, Brocca-Cofano E, Thomas MA, Venzon D, Zhao J, DiPasquale J, Fenizia C, Lee EM, Kalisz I, Kalyanaraman VS, Pal R, Montefiori D, Keele BF, Robert-Guroff M. 2012. Replicating adenovirus-simian immunodeficiency virus (SIV) recombinant priming and envelope protein boosting elicits localized, mucosal IgA immunity in rhesus macaques correlated with delayed acquisition following a repeated low-dose rectal SIV(mac251) challenge. J. Virol. 86:4644–4657. 10.1128/JVI.06812-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Forthal DN, Gilbert PB, Landucci G, Phan T. 2007. Recombinant gp120 vaccine-induced antibodies inhibit clinical strains of HIV-1 in the presence of Fc receptor-bearing effector cells and correlate inversely with HIV infection rate. J. Immunol. 178:6596–6603. 10.4049/jimmunol.178.10.6596. [DOI] [PubMed] [Google Scholar]
- 13.Bonsignori M, Pollara J, Moody MA, Alpert MD, Chen X, Hwang KK, Gilbert PB, Huang Y, Gurley TC, Kozink DM, Marshall DJ, Whitesides JF, Tsao CY, Kaewkungwal J, Nitayaphan S, Pitisuttithum P, Rerks-Ngarm S, Kim JH, Michael NL, Tomaras GD, Montefiori DC, Lewis GK, Devico A, Evans DT, Ferrari G, Liao HX, Haynes BF. 2012. ADCC-mediating antibodies from an HIV-1 vaccine efficacy trial target multiple epitopes and preferentially use the VH1 gene family. J. Virol. 86:11521–11532. 10.1128/JVI.01023-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tomaras GD, Ferrari G, Shen X, Alam SM, Liao HX, Pollara J, Bonsignori M, Moody MA, Fong Y, Chen X, Poling B, Nicholson CO, Zhang R, Lu X, Parks R, Kaewkungwal J, Nitayaphan S, Pitisuttithum P, Rerks-Ngarm S, Gilbert PB, Kim JH, Michael NL, Montefiori DC, Haynes BF. 2013. Vaccine-induced plasma IgA specific for the C1 region of the HIV-1 envelope blocks binding and effector function of IgG. Proc. Natl. Acad. Sci. U. S. A. 110:9019–9024. 10.1073/pnas.1301456110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van Rompay KK, Berardi CJ, Dillard-Telm S, Tarara RP, Canfield DR, Valverde CR, Montefiori DC, Cole KS, Montelaro RC, Miller CJ. 1998. Passive immunization of newborn rhesus macaques prevents oral simian immunodeficiency virus infection. J. Infect. Dis. 177:1247–1259. 10.1086/515270. [DOI] [PubMed] [Google Scholar]
- 16.Forthal DN, Landucci G, Cole KS, Marthas M, Becerra JC, Van Rompay K. 2006. Rhesus macaque polyclonal and monoclonal antibodies inhibit simian immunodeficiency virus in the presence of human or autologous rhesus effector cells. J. Virol. 80:9217–9225. 10.1128/JVI.02746-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burton DR, Hessell AJ, Keele BF, Klasse PJ, Ketas TA, Moldt B, Dunlop DC, Poignard P, Doyle LA, Cavacini L, Veazey RS, Moore JP. 2011. Limited or no protection by weakly or nonneutralizing antibodies against vaginal SHIV challenge of macaques compared with a strongly neutralizing antibody. Proc. Natl. Acad. Sci. U. S. A. 108:11181–11186. 10.1073/pnas.1103012108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moog C, Dereuddre-Bosquet N, Teillaud JL, Biedma ME, Holl V, Van Ham G, Heyndrickx L, Van Dorsselaer A, Katinger D, Vcelar B, Zolla-Pazner S, Mangeot I, Kelly C, Shattock RJ, Le Grand R. 2014. Protective effect of vaginal application of neutralizing and nonneutralizing inhibitory antibodies against vaginal SHIV challenge in macaques. Mucosal Immunol. 7:46–56. 10.1038/mi.2013.23. [DOI] [PubMed] [Google Scholar]
- 19.Tsai CC, Emau P, Follis KE, Beck TW, Benveniste RE, Bischofberger N, Lifson JD, Morton WR. 1998. Effectiveness of postinoculation (R)-9-(2-phosphonylmethoxypropyl) adenine treatment for prevention of persistent simian immunodeficiency virus SIVmne infection depends critically on timing of initiation and duration of treatment. J. Virol. 72:4265–4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parren PW, Marx PA, Hessell AJ, Luckay A, Harouse J, Cheng-Mayer C, Moore JP, Burton DR. 2001. Antibody protects macaques against vaginal challenge with a pathogenic R5 simian/human immunodeficiency virus at serum levels giving complete neutralization in vitro. J. Virol. 75:8340–8347. 10.1128/JVI.75.17.8340-8347.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moldt B, Rakasz EG, Schultz N, Chan-Hui PY, Swiderek K, Weisgrau KL, Piaskowski SM, Bergman Z, Watkins DI, Poignard P, Burton DR. 2012. Highly potent HIV-specific antibody neutralization in vitro translates into effective protection against mucosal SHIV challenge in vivo. Proc. Natl. Acad. Sci. U. S. A. 109:18921–18925. 10.1073/pnas.1214785109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chung AW, Isitman G, Navis M, Kramski M, Center RJ, Kent SJ, Stratov I. 2011. Immune escape from HIV-specific antibody-dependent cellular cytotoxicity (ADCC) pressure. Proc. Natl. Acad. Sci. U. S. A. 108:7505–7510. 10.1073/pnas.1016048108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moore JP, McCutchan FE, Poon SW, Mascola J, Liu J, Cao Y, Ho DD. 1994. Exploration of antigenic variation in gp120 from clades A through F of human immunodeficiency virus type 1 by using monoclonal antibodies. J. Virol. 68:8350–8364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moore JP, Sodroski J. 1996. Antibody cross-competition analysis of the human immunodeficiency virus type 1 gp120 exterior envelope glycoprotein. J. Virol. 70:1863–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferrari G, Pollara J, Kozink D, Harms T, Drinker M, Freel S, Moody MA, Alam SM, Tomaras GD, Ochsenbauer C, Kappes JC, Shaw GM, Hoxie JA, Robinson JE, Haynes BF. 2011. A HIV-1 gp120 envelope human monoclonal antibody that recognizes a C1 conformational epitope mediates potent ADCC activity and defines a common ADCC epitope in human HIV-1 serum. J. Virol. 85:7029–7036. 10.1128/JVI.00171-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ampol S, Pattanapanyasat K, Sutthent R, Permpikul P, Kantakamalakul W. 2012. Comprehensive investigation of common antibody-dependent cell-mediated cytotoxicity antibody epitopes of HIV-1 CRF01_AE gp120. AIDS Res. Hum. Retroviruses 28:1250–1258. 10.1089/aid.2011.0346. [DOI] [PubMed] [Google Scholar]
- 27.Guan Y, Pazgier M, Sajadi MM, Kamin-Lewis R, Al-Darmarki S, Flinko R, Lovo E, Wu X, Robinson JE, Seaman MS, Fouts TR, Gallo RC, DeVico AL, Lewis GK. 2013. Diverse specificity and effector function among human antibodies to HIV-1 envelope glycoprotein epitopes exposed by CD4 binding. Proc. Natl. Acad. Sci. U. S. A. 110:E69–E78. 10.1073/pnas.1217609110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xiang SH, Doka N, Choudhary RK, Sodroski J, Robinson JE. 2002. Characterization of CD4-induced epitopes on the HIV type 1 gp120 envelope glycoprotein recognized by neutralizing human monoclonal antibodies. AIDS Res. Hum. Retroviruses 18:1207–1217. 10.1089/08892220260387959. [DOI] [PubMed] [Google Scholar]
- 29.Kwon YD, Finzi A, Wu X, Dogo-Isonagie C, Lee LK, Moore LR, Schmidt SD, Stuckey J, Yang Y, Zhou T, Zhu J, Vicic DA, Debnath AK, Shapiro L, Bewley CA, Mascola JR, Sodroski JG, Kwong PD. 2012. Unliganded HIV-1 gp120 core structures assume the CD4-bound conformation with regulation by quaternary interactions and variable loops. Proc. Natl. Acad. Sci. U. S. A. 109:5663–5668. 10.1073/pnas.1112391109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carr SA, Hemling ME, Folena-Wasserman G, Sweet RW, Anumula K, Barr JR, Huddleston MJ, Taylor P. 1989. Protein and carbohydrate structural analysis of a recombinant soluble CD4 receptor by mass spectrometry. J. Biol. Chem. 264:21286–21295. [PubMed] [Google Scholar]
- 31.Acharya P, Luongo TS, Louder MK, McKee K, Yang Y, Kwon YD, Mascola JR, Kessler P, Martin L, Kwong PD. 2013. Structural basis for highly effective HIV-1 neutralization by CD4-mimetic miniproteins revealed by 1.5 A cocrystal structure of gp120 and M48U1. Structure 21:1018–1029. 10.1016/j.str.2013.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Otwinowski Z, Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276:307–326. 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 33.McCoy AJ. 2007. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr. D Biol. Crystallogr. 63:32–41. 10.1107/S0907444906045975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Collaborative Computational Project N. 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50:760–763. 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 35.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. 1998. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54:905–921. 10.1107/S0907444998003254. [DOI] [PubMed] [Google Scholar]
- 36.Murshudov GN, Vagin AA, Dodson EJ. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53:240–255. 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 37.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. 2002. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 58:1948–1954. 10.1107/S0907444902016657. [DOI] [PubMed] [Google Scholar]
- 38.Emsley P, Cowtan K. 2004. COOT: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60:2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 39.Chen VB, Arendall WB, III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. 2010. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66:12–21. 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krissinel E, Henrick K. 2007. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372:774–797. 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 41.Tina KG, Bhadra R, Srinivasan N. 2007. PIC: protein interactions calculator. Nucleic Acids Res. 35:W473–W476. 10.1093/nar/gkm423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gomez-Roman VR, Florese RH, Patterson LJ, Peng B, Venzon D, Aldrich K, Robert-Guroff M. 2006. A simplified method for the rapid fluorometric assessment of antibody-dependent cell-mediated cytotoxicity. J. Immunol. Methods 308:53–67. 10.1016/j.jim.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 43.Steckbeck JD, Orlov I, Chow A, Grieser H, Miller K, Bruno J, Robinson JE, Montelaro RC, Cole KS. 2005. Kinetic rates of antibody binding correlate with neutralization sensitivity of variant simian immunodeficiency virus strains. J. Virol. 79:12311–12320. 10.1128/JVI.79.19.12311-12320.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fouts TR, Tuskan R, Godfrey K, Reitz M, Hone D, Lewis GK, DeVico AL. 2000. Expression and characterization of a single-chain polypeptide analogue of the human immunodeficiency virus type 1 gp120-CD4 receptor complex. J. Virol. 74:11427–11436. 10.1128/JVI.74.24.11427-11436.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Prabakaran P, Zhu Z, Chen W, Gong R, Feng Y, Streaker E, Dimitrov DS. 2012. Origin, diversity, and maturation of human antiviral antibodies analyzed by high-throughput sequencing. Front. Microbiol. 3:277. 10.3389/fmicb.2012.00277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Briney BS, Willis JR, Crowe JE., Jr 2012. Human peripheral blood antibodies with long HCDR3s are established primarily at original recombination using a limited subset of germline genes. PLoS One 7:e36750. 10.1371/journal.pone.0036750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Verkoczy L, Kelsoe G, Moody MA, Haynes BF. 2011. Role of immune mechanisms in induction of HIV-1 broadly neutralizing antibodies. Curr. Opin. Immunol. 23:383–390. 10.1016/j.coi.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.von Bubnoff A. 2010. Vaccines to antibodies: grow up! IAVI Rep. 14:4–9. [PubMed] [Google Scholar]
- 49.Desormeaux A, Coutu M, Medjahed H, Pacheco B, Herschhorn A, Gu C, Xiang SH, Mao Y, Sodroski J, Finzi A. 2013. The highly conserved layer-3 component of the HIV-1 gp120 inner domain is critical for CD4-required conformational transitions. J. Virol. 87:2549–2562. 10.1128/JVI.03104-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pancera M, Majeed S, Ban YE, Chen L, Huang CC, Kong L, Kwon YD, Stuckey J, Zhou T, Robinson JE, Schief WR, Sodroski J, Wyatt R, Kwong PD. 2010. Structure of HIV-1 gp120 with gp41-interactive region reveals layered envelope architecture and basis of conformational mobility. Proc. Natl. Acad. Sci. U. S. A. 107:1166–1171. 10.1073/pnas.0911004107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Finzi A, Xiang SH, Pacheco B, Wang L, Haight J, Kassa A, Danek B, Pancera M, Kwong PD, Sodroski J. 2010. Topological layers in the HIV-1 gp120 inner domain regulate gp41 interaction and CD4-triggered conformational transitions. Mol. Cell 37:656–667. 10.1016/j.molcel.2010.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Finzi A, Pacheco B, Xiang SH, Pancera M, Herschhorn A, Wang L, Zeng X, Desormeaux A, Kwong PD, Sodroski J. 2012. Lineage-specific differences between human and simian immunodeficiency virus regulation of gp120 trimer association and CD4 binding. J. Virol. 86:8974–8986. 10.1128/JVI.01076-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu J, Bartesaghi A, Borgnia MJ, Sapiro G, Subramaniam S. 2008. Molecular architecture of native HIV-1 gp120 trimers. Nature 455:109–113. 10.1038/nature07159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Julien JP, Cupo A, Sok D, Stanfield RL, Lyumkis D, Deller MC, Klasse PJ, Burton DR, Sanders RW, Moore JP, Ward AB, Wilson IA. 2013. Crystal structure of a soluble cleaved HIV-1 envelope trimer. Science 342:1477–1483. 10.1126/science.1245625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lyumkis D, Julien JP, de Val N, Cupo A, Potter CS, Klasse PJ, Burton DR, Sanders RW, Moore JP, Carragher B, Wilson IA, Ward AB. 2013. Cryo-EM structure of a fully glycosylated soluble cleaved HIV-1 envelope trimer. Science 342:1484–1490. 10.1126/science.1245627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ray K, Mengistu M, Lewis GK, Lakowicz JR, Devico AL. 2014. Antigenic properties of the HIV envelope on virions in solution. J. Virol. 88:1795–1808. 10.1128/JVI.03048-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Finnegan CM, Berg W, Lewis GK, DeVico AL. 2001. Antigenic properties of the human immunodeficiency virus envelope during cell-cell fusion. J. Virol. 75:11096–11105. 10.1128/JVI.75.22.11096-11105.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Song R, Franco D, Kao CY, Yu F, Huang Y, Ho DD. 2010. Epitope mapping of ibalizumab, a humanized anti-CD4 monoclonal antibody with anti-HIV-1 activity in infected patients. J. Virol. 84:6935–6942. 10.1128/JVI.00453-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Veillette M, Desormeaux A, Medjahed H, Gharsallah NE, Coutu M, Baalwa J, Guan Y, Lewis G, Ferrari G, Hahn BH, Haynes BF, Robinson JE, Kaufmann DE, Bonsignori M, Sodroski J, Finzi A. 2014. Interaction with cellular CD4 exposes HIV-1 envelope epitopes targeted by antibody-dependent cell-mediated cytotoxicity. J. Virol. 88:2633–2644. 10.1128/JVI.03230-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.DeVico AL. 2007. CD4-induced epitopes in the HIV envelope glycoprotein, gp120. Curr. HIV Res. 5:561–571. 10.2174/157016207782418560. [DOI] [PubMed] [Google Scholar]
- 61.Lewis GK, Guan Y, Kamin-Lewis R, Sajadi M, Pazgier M, Devico AL. 2014. Epitope target structures of Fc-mediated effector function during HIV-1 acquisition. Curr. Opin. HIV AIDS 9:263–270. 10.1097/COH.0000000000000055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.D'Costa S, Slobod KS, Webster RG, White SW, Hurwitz JL. 2001. Structural features of HIV envelope defined by antibody escape mutant analysis. AIDS Res. Hum. Retroviruses 17:1205–1209. 10.1089/088922201316912808. [DOI] [PubMed] [Google Scholar]
- 63.Fouda GG, Amos JD, Wilks AB, Pollara J, Ray CA, Chand A, Kunz EL, Liebl BE, Whitaker K, Carville A, Smith S, Colvin L, Pickup DJ, Staats HF, Overman G, Eutsey-Lloyd K, Parks R, Chen H, Labranche C, Barnett S, Tomaras GD, Ferrari G, Montefiori DC, Liao HX, Letvin NL, Haynes BF, Permar SR. 2013. Mucosal immunization of lactating female rhesus monkeys with a transmitted/founder HIV-1 envelope induces strong Env-specific IgA antibody responses in breast milk. J. Virol. 87:6986–6999. 10.1128/JVI.00528-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pham TN, Lukhele S, Hajjar F, Routy JP, Cohen EA. 2014. HIV Nef and Vpu protect HIV-infected CD4+ T cells from antibody-mediated cell lysis through down-modulation of CD4 and BST2. Retrovirology 11:15. 10.1186/1742-4690-11-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Guttman M, Garcia NK, Cupo A, Matsui T, Julien JP, Sanders RW, Wilson IA, Moore JP, Lee KK. 2014. CD4-induced activation in a soluble HIV-1 Env trimer. Structure. 22:974–984. 10.1016/j.str.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hooft RW, Sander C, Scharf M, Vriend G. 1996. The PDBFINDER database: a summary of PDB, DSSP and HSSP information with added value. Comput. Appl. Biosci. 12:525–529. [DOI] [PubMed] [Google Scholar]
- 67.Tran EE, Borgnia MJ, Kuybeda O, Schauder DM, Bartesaghi A, Frank GA, Sapiro G, Milne JL, Subramaniam S. 2012. Structural mechanism of trimeric HIV-1 envelope glycoprotein activation. PLoS Pathog. 8:e1002797. 10.1371/journal.ppat.1002797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Scatchard G. 1949. The attractions of proteins for small molecules and ions. Chem. Rev. 51:660–672. [Google Scholar]
- 69.Pancera M, Zhou T, Druz A, Georgiev IS, Soto C, Gorman J, Huang J, Acharya P, Chuang GY, Ofek G, Stewart-Jones GBE, Stuckey J, Bailer RT, Joyce MG, Louder MK, Tumba N, Yang Y, Zhang B, Cohen MS, Haynes BF, Mascola JR, Morris L, Munro JB, Blanchard SC, Mothes W, Connors M, Kwong PD. Structure and immune recognition of trimeric prefusion HIV-1 Env. Nature, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.