

ABSTRACT
Vector-borne flaviviruses, such as tick-borne encephalitis virus (TBEV), West Nile virus, and dengue virus, cause millions of infections in humans. TBEV causes a broad range of pathological symptoms, ranging from meningitis to severe encephalitis or even hemorrhagic fever, with high mortality. Despite the availability of an effective vaccine, the incidence of TBEV infections is increasing. Not much is known about the role of the innate immune system in the control of TBEV infections. Here, we show that the type I interferon (IFN) system is essential for protection against TBEV and Langat virus (LGTV) in mice. In the absence of a functional IFN system, mice rapidly develop neurological symptoms and succumb to LGTV and TBEV infections. Type I IFN system deficiency results in severe neuroinflammation in LGTV-infected mice, characterized by breakdown of the blood-brain barrier and infiltration of macrophages into the central nervous system (CNS). Using mice with tissue-specific IFN receptor deletions, we show that coordinated activation of the type I IFN system in peripheral tissues as well as in the CNS is indispensable for viral control and protection against virus induced inflammation and fatal encephalitis.
IMPORTANCE The type I interferon (IFN) system is important to control viral infections; however, the interactions between tick-borne encephalitis virus (TBEV) and the type I IFN system are poorly characterized. TBEV causes severe infections in humans that are characterized by fever and debilitating encephalitis, which can progress to chronic illness or death. No treatment options are available. An improved understanding of antiviral innate immune responses is pivotal for the development of effective therapeutics. We show that type I IFN, an effector molecule of the innate immune system, is responsible for the extended survival of TBEV and Langat virus (LGTV), an attenuated member of the TBE serogroup. IFN production and signaling appeared to be essential in two different phases during infection. The first phase is in the periphery, by reducing systemic LGTV replication and spreading into the central nervous system (CNS). In the second phase, the local IFN response in the CNS prevents virus-induced inflammation and the development of encephalitis.
INTRODUCTION
Flaviviruses (family Flaviviridae) are widely distributed all over the world. Such arthropod-borne viruses (arboviruses) are responsible for millions of debilitating human infections annually. They include West Nile virus (WNV), Japanese encephalitis virus (JEV), dengue virus (DENV) (serotypes 1 to 4), yellow fever virus, and tick-borne encephalitis virus (TBEV). TBEV is medically the most important arbovirus in Europe and Russia. Despite the availability of an effective vaccine, the number of infections is increasing and currently accounts for up to 10,000 cases per year (1, 2). The clinical spectrum ranges from mild symptoms to severe encephalitis, meningitis, hemorrhagic fever, or chronic progressive forms of the disease (2). While mortality rates vary from 0.5 to 30%, neurological sequelae can occur in 30 to 60% of survivors (1, 3, 4). For unknown reasons, encephalitis severity increases with age (2). Moreover, TBEV strains from Siberia and the Far East seem to be more aggressive than strains from Central Europe (3). Many aspects of TBEV infection and pathogenesis remain unresolved, especially its interactions with the innate immune system and, particularly, the interferon (IFN) system.
It is known that the IFN system is important for viral control and can contribute to viral pathogenicity (5, 6). Induction of type I IFNs is triggered by recognition of viral signature molecules, such as double-stranded RNA (dsRNA), and by pattern recognition receptors (PRRs), such as the cytoplasmic retinoic acid-induced gene-I (RIG-I) family or the membrane-bound Toll-like receptors (TLRs) (7, 8). Activation of PRRs leads to signaling cascades that result in the activation of IFN genes via transcriptional factors IFN regulatory factor 3 (IRF-3) and IRF-7 (9, 10). Upon secretion, IFNs signal in a paracrine and autocrine manner via the type I IFN receptor (IFNAR) present on virtually all host cells to trigger the induction of IFN-stimulated genes (ISGs), which mediate an antiviral and immune modulatory activity (11–13). ISGs can interfere with viral entry, viral transcription, translation, and genome replication or can exit from the cell in a virus-specific manner (14).
Although type I IFN signaling is generally recognized as an important component of antiviral innate immunity, studies so far indicate that its role during vector-borne flavivirus infections is complex and varies from one type of infection to the other. For instance, whereas the type I IFN response in DENV infections limits only the initial viral replication, it has no impact on disease development and control of the virus from the central nervous system (CNS) (15, 16). In WNV infections, the type I IFN response limits viral replication, protects neurons from cell death, and shapes maturation of antiviral T cells but is not able to protect mice from lethal encephalitis (17, 18). The induction and role of type I IFNs during TBEV infection are not well understood. Owing to its ability to replicate within vesicles induced by rearrangement of cellular endoplasmic reticulum (ER) membranes, TBEV is able to prevent cellular recognition by pattern recognition receptors (PRRs). This delays IFN-β induction, which may give the virus a head start (19, 20). Similar strategies have been shown for Japanese encephalitis virus.
Although in vivo induction of IFN by TBEV has previously been reported (21, 22), whether it plays a protective role and in which cell types or tissues have not been addressed. To investigate, we used the naturally attenuated Langat virus (LGTV), which belongs to the TBEV serogroup, as a model for TBEV infections. LGTV shares 82 to 88% amino acid identity with TBEV and has been tested as a successful candidate for a live-attenuated vaccine for TBEV. However, vaccination was abandoned during clinical trials because encephalitis occurred in approximately 1:10,000 of vaccine recipients (3).
By combining the use of mice with complete body deficiency in IFNAR as well as mice conditionally ablated in the type I IFN system in the periphery and CNS, respectively, we show that restriction of viral growth and protection against ensuing immunopathology are contingent on the intricate interplay of type I IFN system in the periphery as well as in the CNS.
MATERIALS AND METHODS
Mice and viral infections.
C57BL/6 (wild-type [WT]) mice were purchased from Harlan. IFNAR−/−, IFN-β−/−, IFN-β+/Δluc, IFNARfl/fl CreERT+/−, and IFNARfl/fl NesCre+/− (23) mice on the C57BL/6 background were bred under specific-pathogen-free conditions at the Helmholtz Centre for Infection Research. For tamoxifen treatment, 2 mg was dissolved in 100 μl ClinOleic (Baxter, Lessines, Belgium). The solution was administered twice to 6- to 8-week-old mice by oral gavage. Vesicular stomatitis virus (VSV), LGTV strain TP21, and TBEV strain Hypr 71 were propagated in Vero B4 cells. Titers were determined by plaque assays on Vero B4 cells (19). Six- to 12-week-old mice were intraperitoneally infected with the indicated PFU of LGTV or TBEV strain Hypr in 100 μl phosphate-buffered saline (PBS). For intracranial infections, mice were anesthetized by intraperitoneal injection with a mixture of ketamine (100 μg/g body weight) and xylazine (5 μg/g body weight). Mice were injected with 10 or 102 PFU of LGTV in 20 μl PBS. Administration of VSV was performed as described previously (24). Mice that lost more than 20% of their body weight were sacrificed and perfused with 10 ml of PBS. All animal experiments were performed according to the guidelines of the German Animal Welfare Law (AZ 33.9-42502-05-12A295). Experiments with TBEV strain Hypr were performed in the biosafety level 3 (BSL3) facility at the Helmholtz Center for Infection Research.
IFN-α ELISA and luciferase assay.
The amount of murine IFN-α in mouse serum was determined by enzyme-linked immunosorbent assay (ELISA) according to the manufacturer's instructions (PBL). For the luciferase assay, mouse organs were homogenized in 500 μl Reporter lysis buffer (Promega) using the FastPrep-24 (MP). Lysates were mixed with LARII (Promega) and measured in a luminometer (Berthold). For in vivo imaging, mice were injected intravenously with 150 mg/kg of d-luciferin in PBS (CaliperLS), anesthetized using isoflurane (Baxter), and monitored using an IVIS 200 imaging system (CaliperLS). Photon flux was quantified using the Living Image 3.2 software (CaliperLS).
RNA extraction and real-time RT-PCR.
Perfused mouse organs were homogenized in TRIzol reagent (Invitrogen) using Lysis matrix (Nordic Biolabs) and the tissue homogenizer FastPrep-24 (MP). The RNA was extracted using the Nucleo-Spin RNA II kit (Macherey-Nagel). Total RNA (500 to 1,000 ng) was used to synthesize cDNA with the QuantiTect reverse transcription (RT) kit (Qiagen). Levels of mouse GAPDH (glyceraldehyde-3-phosphate dehydrogenase), IFN-β, interleukin-6 (IL-6), and tumor necrosis factor alpha (TNF-α) mRNA were detected by validated QuantiTect primer assays (Qiagen) and the KAPA SYBR FAST qPCR kit using the 7900HT fast-real-time PCR system (Applied Biosystems). Viral TBEV RNAs were detected by TaqMan probes for TBEV (25) and the KAPA probe FAST quantitative PCR (qPCR) kit. The results were normalized using the housekeeping gene GAPDH and analyzed as fold change relative to RNA samples from mock-infected mice using the comparative threshold cycle (ΔΔCT) method. The sensitivity of the LGTV assay is 104 copies.
Immune cell analysis.
Mice were sacrificed at the indicated time points and perfused with 25 ml PBS. Brains were isolated and homogenized through a 40-μm-pore cell strainer and digested with collagenase D (Roche) at room temperature for 1 h. Immune cells were separated by centrifugation on a discontinuous 70-to-30% Percoll gradient. Cells were stained with antibodies specific for CD3, CD4, CD8, CD11b, CD11c, and CD45 (BD). Analysis was performed on a BD LSRII using BD FACSDiva and FlowJo software. To identify infected immune cells, splenocytes from 2 to 3 spleens were isolated by homogenization through a 40-μm-pore mesh. Cells were stained with specific antibodies for CD3, B220, CD11c, and F4/80 (BD) and sorted by a BD FACS Aria-II.
Bone marrow chimeras.
Bone marrow chimeras of WT and IFNAR−/− mice were generated by standard techniques. Congenitally marked CD45.1 or CD45.2 WT mice and CD45.2-marked IFNAR−/− mice were used to track the bone marrow reconstitution. Bone marrow cells were collected from the hind leg bones of 4- to 6-week-old CD45.2 IFNAR−/− mice, CD45.1 WT mice, and CD45.2 WT mice. Six- to 8-week-old recipient mice were irradiated with 950 rad (9.5 Gy) to achieve complete bone marrow ablation. One day after irradiation, 1 × 107 cells were intravenously injected into each mouse via the tail vein. Mice were fed with enrofloxacin (Baytril) for 2 weeks and allowed to recover for 6 to 8 weeks. Reconstitution of mice was analyzed by fluorescence-activated cell sorter (FACS), and successfully reconstituted mice, determined by the CD45 marker (BD) and >80% reconstitution, were infected intraperitoneally with LGTV.
Immunohistochemistry.
Immunohistochemical analysis was performed on LGTV-infected or noninfected IFNAR mice at different time points (n = 3 per time point). Brains and spleens were removed after cardiac perfusion with PBS followed by 4% paraformaldehyde. Organs were fixed in 4% neutrally buffered formaldehyde for 24 to 48 h, embedded in paraffin, cut into 3-μm sections, and stained with hematoxylin-eosin (H&E). Sections were evaluated by light microscopy with the results blinded to the experimental groups. Immunohistochemistry was performed on the same formalin-fixed and paraffin-embedded specimen. After heat-mediated antigen retrieval, the sections were stained by double staining with anti-NS5/NeuN, anti-E protein of LGTV/glial fibrillary acidic protein (anti-EP/GFAP), and anti-EP/IBA1 for the brain sections. Additionally, single staining for EP and IBA1 was conducted. The following antibodies were used: mouse anti-GFAP (1:500 [Sigma]), mouse anti-NeuN (1:500 [Millipore]), rabbit anti-IBA1 (1:500 [Synaptic System]), mouse anti-E protein (26), and chicken anti-NS5 protein (1:1,600) (27).
BBB.
The integrity of the blood-brain barrier (BBB) was assessed by Evan's blue dye exclusion test. WT and IFNAR−/− mice were infected intraperitoneally with LGTV. Mice were intravenously injected with 100 μl 2% Evans blue (Sigma) in PBS. After 1 h, animals were transcardially perfused with 20 ml PBS, and the brains were removed, photographed, weighed, and homogenized in 50% trichloroacetic acid (TCA). After 30 min of incubation at room temperature, samples were spun down, and absorbance of supernatants was measured at 620 nm.
RESULTS
Type I IFN protects mice from lethal LGTV infection.
The role of type I IFNs in infections with flaviviruses is variable (16, 18, 28). WT, IFN-β−/−, or IFNAR−/− mice were infected intraperitoneally with 104 PFU of LGTV (Fig. 1A) to assess the role of type I IFN signaling in defense against LGTV. IFNAR−/− mice were found to be highly susceptible to LGTV infection, displaying 100% mortality, compared to 20% mortality in WT mice. WT mice showed no signs of illness, with the exception of two moribund mice, which showed weight loss and hind limb paralysis within the first 2 days before they died. This is in contrast to all of the IFNAR−/− mice, which exhibited onset of clinical signs on days 3 and 4 postinfection, including weight loss, paralysis, hunchback posture, lethargy, and fur ruffling. All IFNAR−/− mice died within a mean survival time of 5 days postinfection. Even a dose as low as 1 PFU was found to be lethal to IFNAR−/− mice within 5 days, further emphasizing the importance of the type I IFN system in the protection against LGTV (Fig. 1B). However, all mice deficient in IFN-β survived without any symptoms, indicating that IFN-α is sufficient to protect mice against the lethal outcome of LGTV infections.
FIG 1.
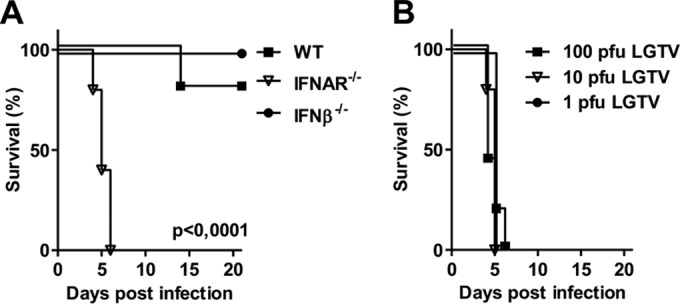
Type I IFN response is crucial for survival of infections. (A) Survival analysis of WT, IFNAR−/−, and IFN-β−/− mice (n = 10). Six- to 8-week-old age-matched mice were infected intraperitoneally with 104 PFU of LGTV, and mortality was observed for 21 days. (B) Survival analysis of IFNAR−/− mice after intraperitoneal infection with 100, 10, and 1 PFU of LGTV (n = 5). Data are cumulative from at least two independent experiments. Survival differences were tested for statistical significance by the log rank test.
LGTV induces a type I IFN response in multiple tissues, thus inhibiting viral replication and spreading into the brain.
Next we investigated the kinetics of type I IFN induction by LGTV in vivo and the organs/tissues involved. WT mice infected intraperitoneally with 104 PFU of LGTV had a significant IFN-α response at day 2 postinfection before returning to background levels by day 4 (Fig. 2A). Probably much like TBEV, which hides its dsRNA from cytoplasmic PRR by rearranging internal ER-derived membranes, thereby evading detection by PRR pathways for IFN induction (19), the IFN-α response elicited by LGTV was much lower than that by vesicular stomatitis virus (VSV), a member of the Rhabdoviridae family and a commonly used model for neurotropic viral infections. To complement the IFN-α data and to determine the localization and kinetics of type I IFN induction, we took advantage of a luciferase-based IFN-β reporter (IFN-β+/Δβ-luc) mouse strain to image whole-body IFN-β induction in vivo (29). Although the magnitude of IFN-β induction by LGTV was much lower than that by VSV, the responses of IFN-β induction by both viruses in vivo were similar, peaking after 1 day and then declining to the basal level by day 8 postinfection (Fig. 2B and C). IFN-β induction by both viruses was localized to the thorax, peritoneum, and inguinal lymph nodes 24 h postinfection. At 48 h postinfection, the signal was focused in the salivary gland region, while 72 h postinfection, it was mainly localized in the snout. This pattern of IFN-β induction was irrespective of infection doses (Fig. 2B). Organ isolation and quantification of luciferase activity revealed that the peak IFN-β response within the first 24 h was mainly in lymphoid organs, like lymph nodes, spleen, and thymus, and not liver, heart, or lung (Fig. 2D). Further analysis revealed cervical lymph nodes as the prominent source of the IFN-β response during this period. However, at later time points (e.g., 4 days postinfection), IFN-β responses could also be detected in the brain (Fig. 2D). These results suggest that peripheral inoculation of LGTV leads to type I IFN induction in multiple organs, including lymphoid tissues and brain. Importantly, because the predominant type I IFN response induced by LGTV is presumably due to its dsRNA produced during viral replication, these results also suggest that LGTV can target and replicate in different peripheral organs and in the brain. To directly test this and evaluate if the observed susceptibility of IFNAR−/− mice was due to uncontrolled viral replication, the viral loads in different peripheral organs and central nervous system from WT and IFNAR−/− mice were calculated by real-time reverse transcription-PCR (RT-PCR) and determination of infectious particles by plaque assay. High infection doses are lethal in IFNAR−/− mice, and since the kinetics of IFN-β induction is similar to that with low viral doses (Fig. 2B), we performed the following experiments with a dose of 102 PFU. Consistent with the observed resistance to LGTV infection, viral RNA in peripheral organs and the central nervous system of WT mice was below the detection limit of our real-time RT-PCR, and no viral particles were detectable by plaque assay (Fig. 3). This is in contrast to IFNAR−/− mice, where viral RNA and infectious particles could readily be detected in peripheral organs (liver, lung, spleen, kidney, thymus, and lymph nodes), spinal cord, and brain. In such mice, viral RNA was observed to increase over time (Fig. 3) (data not shown), indicating that ablation of the type I IFN system results in unrestrained systemic viral replication. Importantly, the fact that the virus was already detectable at 2 days postinfection indicates that the high neuropathogenesis in LGTV-infected IFNAR−/− mice is due to rapid replication and spreading of the virus into the brain (Fig. 3B). In summary, the above results indicate that type I IFN is crucial for protecting the animals from fatal outcome of infection by restricting both viral replication in peripheral organs and its dissemination into the CNS.
FIG 2.

LGTV infection leads to type I IFN induction. (A) Mice were infected intraperitoneally with 104 PFU of LGTV or intranasally with 104 PFU VSV. Serum was collected on days 0, 2, and 4 postinfection. The IFN-α protein level was determined by ELISA. (B) Whole-body imaging of IFN-β+/Δβ-luc reporter mice after intraperitoneal infection with 102 and 104 PFU of LGTV. Mice were imaged before treatment (0 h) and over time as indicated. Images from a representative mouse are shown. The rainbow scale indicates the number of photons measured per s per cm2 per steradian (sr). (C) Comparison of levels of IFN-β induction in VSV- and LGTV-infected mice. Mice were infected intraperitoneally with 104 PFU of LGTV or intranasally with 104 PFU VSV. Shown is whole-body imaging of IFN-β+/Δβ-luc reporter mice. Mice were imaged before treatment (0 h) and over time as indicated; one representative mouse is shown. Images from LGTV-infected mice are identical to those from panel B but are shown in another range of the rainbow scale to prevent saturation of pictures from VSV-infected mice. The scale indicates the number of photons (p) measured per s per cm2 per steradian (sr). (D) Quantification of luciferase activity in different organs of IFN-β+/Δβ-luc mice. Mice were sacrificed at the indicated time points postinfection, and selected organs were isolated and homogenized for luciferase activity measurement. Fold induction represents relative luminescence units (RLU) per organ. LN, lymph nodes. Data represent the mean ± standard error of the mean (SEM) of 3 to 7 mice per time point from at least two independent experiments. Asterisks indicate values that were statistically significant: **, P < 0.01, based on the Mann-Whitney test.
FIG 3.

Type I IFN limits LGTV replication and spread. WT and IFNAR−/− mice were infected intraperitoneally with 102 PFU of LGTV, and the viral load was measured by real-time RT-PCR (A and B) and PFU (C and D). (A and B) Detection of relative LGTV mRNA levels in spleen and lung (A), as well as spinal cord and brain (B), on days 2 and 4 postinfection. (C and D) Determination of viral particles in spleen and lung (C), as well as spinal cord and brain (D). The solid horizontal lines represent the means of 5 to 10 mice per time point from at least two independent experiments. The dashed horizontal lines show the detection level of LGTV. a.u., arbitrary units.
Macrophages, dendritic cells, and B220+ cells are the main targets of LGTV in lymphoid organs.
The observations that IFN-β response and viral replication are mainly localized in lymphoid organs, such as the spleen (Fig. 2B and C), prompted us to investigate the cell types targeted by LGTV and the impact thereof on the lymphoid structure. Hematoxylin and eosin (H&E) staining of the spleen at day 4 postinfection revealed hyperplasia of the red pulp, severe depletion of lymphoid follicles, massive apoptosis, and histiocytic hyperplasia (Fig. 4A). Sorting of splenocytes by flow cytometry harvested at different time points, followed by analysis using real-time RT-PCR, failed to detect viral RNA in B220+, CD3+, F4/80+, and CD11c+ cells from infected WT mice (data not shown). This was in contrast to infected IFNAR−/− mice, where a time-dependent increase in viral RNA was noted in F4/80+ and CD11c+ cells and, to some extent, in B220+ cells (Fig. 4C).
FIG 4.

Identification of LGTV-infected cells. IFNAR−/− mice were infected intraperitoneally with 102 PFU LGTV and sacrificed at the indicated time points postinfection. (A) H&E staining of spleens of uninfected and LGTV-infected mice 4 days postinfection (dpi). The scale bar represents 50 μm. (B) Sorting strategy of CD3+, B220+, CD11c+, and F4/80+ cells. (C) Splenocytes were isolated, stained for cell-specific markers, and sorted for F4/80+, CD11c+, B220+, and CD3+ cells. Viral load was measured by real-time RT-PCR. The solid horizontal lines represent means of 4 to 5 samples from at least two independent experiments. The dashed horizontal lines show the detection level of LGTV. a.u., arbitrary units.
Type I IFN signaling in both hematopoietic and nonhematopoietic cells is indispensable for protection against LGTV infection.
Beyond the hematopoietic (i.e., myeloid and lymphoid) cells, which, as shown above, are infected, next we sought to determine the involvement of stromal cells. Toward this end, we established chimeric mice by adoptively transferring bone marrow from WT and IFNAR−/− mice into lethally irradiated WT and IFNAR−/− mice, further referred to as WT→WT, WT→IFNAR−/−, IFNAR−/−→WT, and IFNAR−/−→IFNAR−/− (donor→irradiated recipient). Only mice with >80% reconstitution were used. WT mice were more resistant to LGTV infection than WT→WT bone marrow chimeras, which could be due to side effects from the irradiation. In contrast to WT→WT mice, which were significantly more resistant to infection, both WT→IFNAR−/− and IFNAR−/−→WT chimeras were unable to limit the infection and died within 5 to 6 days (Fig. 5A). Curiously, although no significant difference in survival was detected between WT→IFNAR−/− and IFNAR−/−→WT chimeras, the viral loads in the different organs (spleen, spinal cord, and brain) of the chimera showed clear differences. IFNAR−/−→WT chimeras showed viral load in the spleen in the early stages (day 2 postinfection) but not later stages (day 4 postinfection) of infection. On the other hand, WT→IFNAR−/− mice exhibited the highest viral load in the initial stages of infection (Fig. 5B). The ability of WT hematopoietic cells to restrict viral growth in spleens of IFNAR−/− mice, mainly in the late stages, suggests that in the spleen, initial infection and viral replication occur in stromal cells and later in hematopoietic cells. This is in contrast to results seen in lungs, where IFNAR signaling in both hematopoietic and nonhematopoietic cells seemed to be crucial for controlling viral replication (Fig. 5B). In the brain and spinal cord, viral RNA was detectable in mice lacking IFNAR on hematopoietic cells by day 2 postinfection. However, by day 4 postinfection, viral RNA was also detectable in the nonhematopoietic and partially radioresistant microglia of IFNAR−/− mice. Collectively, these data suggest that both hematopoietic and nonhematopoietic cells are targets of LGTV and that a coordinated type I IFN response in peripheral immune cells as well as brain cells is vital for viral control.
FIG 5.
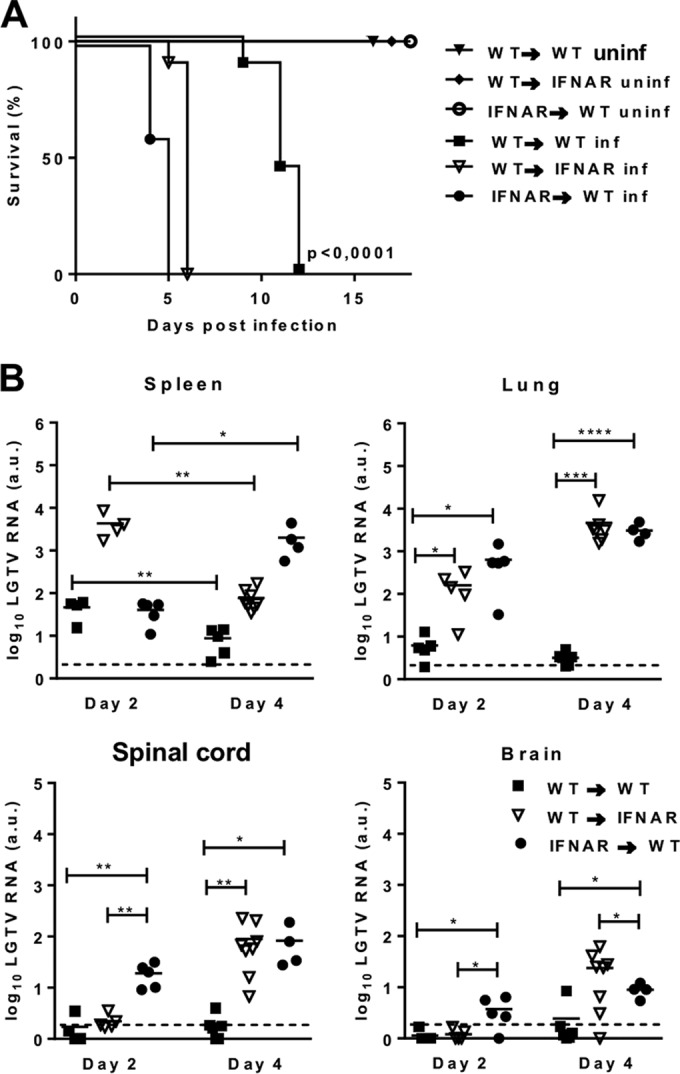
Type I IFN response is essential in hematopoietic and nonhematopoietic cells during LGTV infection. WT and IFNAR−/− mice were lethally irradiated and reconstituted with bone marrow from IFNAR−/− or WT mice, respectively. After 6 to 8 weeks, chimeric mice were inoculated intraperitoneally with 102 PFU LGTV. (A) Survival was monitored and plotted as Kaplan-Meier curves (n = 5 to 11). Data are cumulative from two independent experiments. Survival differences were tested for statistical significance by the log rank test. uninf, uninfected; inf, infected. (B) Viral load was measured by real-time RT-PCR in spleen and lung, spinal cord, and brain on days 2 and 4 postinfection. Solid horizontal lines represent the means of 5 mice per time point from two independent experiments. The dashed horizontal lines show the detection level of LGTV. a.u., arbitrary units. Asterisks indicate values that were statistically significant: *, P < 0.05, **, P < 0.01, ***, P < 0.0005, and ****, P < 0.0001, based on the unpaired t test.
Type I IFN response in the periphery and the central nervous system is critical for host survival during LGTV infection.
Given that LGTV and TBEV are neurotropic viruses that cause encephalitis, next we sought to investigate the impact of type I IFN signaling in direct antiviral response in the CNS by administering the virus intracranially. Compared to the WT mice, IFNAR−/− mice were found to be highly susceptible to LGTV infections and died within 4 days postinfection with 10 PFU (Fig. 6A). These results indicate that activation of the type I IFN response in resident brain cells is essential for survival. For further investigation, we made use of two different transgenic animal models lacking the IFNAR in either the periphery or the CNS. The first is based on IFNARfl/fl CreERT+/− mice. After feeding twice with 2 mg of tamoxifen, the activation of Cre fused to the modified estrogen receptor (ERT) results in the deletion of IFNAR in the peripheral organs but not in the brain (Fig. 6B). The second model is IFNARfl/fl NesCre+/− mice, where IFNAR is conditionally deleted in neuroectodermal cells of the CNS, including neurons, oligodendrocytes, and astrocytes but not microglia. Such mice show >90% deletion efficiency in the brain, whereas other tissues, such as spleen, show no deletion (5, 23). Following intraperitoneal inoculation with 102 PFU of LGTV, all IFNARfl/fl CreERT+/− mice developed severe clinical symptoms and died within 5 to 6 days (Fig. 6B). IFNARfl/fl NesCre+/− mice did not show signs of disease until day 7, when they became hemiplegic and died within hours. This is in contrast to the IFNARfl/fl mice fed with tamoxifen, which survived the infection (Fig. 6B). A comparison of viral loads in lung and brain at a time point where both mouse strains showed severe signs of illness—days 4 and day 8 for IFNARfl/fl CreERT+/− and IFNARfl/fl NesCre+/− mice, respectively, revealed higher viral replication in lungs of IFNARfl/fl CreERT+/− mice (Fig. 6C). Because no viral titers were detectable in the brain of IFNARfl/fl CreERT+/− mice, fatal infection of the CNS could not be the cause of death. On the other hand, IFNARfl/fl NesCre+/− mice displayed high viral replication in the brain on day 8 postinfection, shortly before they died (Fig. 6C). Consistent with an ability to target different host cell types, these data suggest that a functional type I IFN system in the periphery as well as in the CNS is indispensable for restricting viral replication and entry into the brain.
FIG 6.
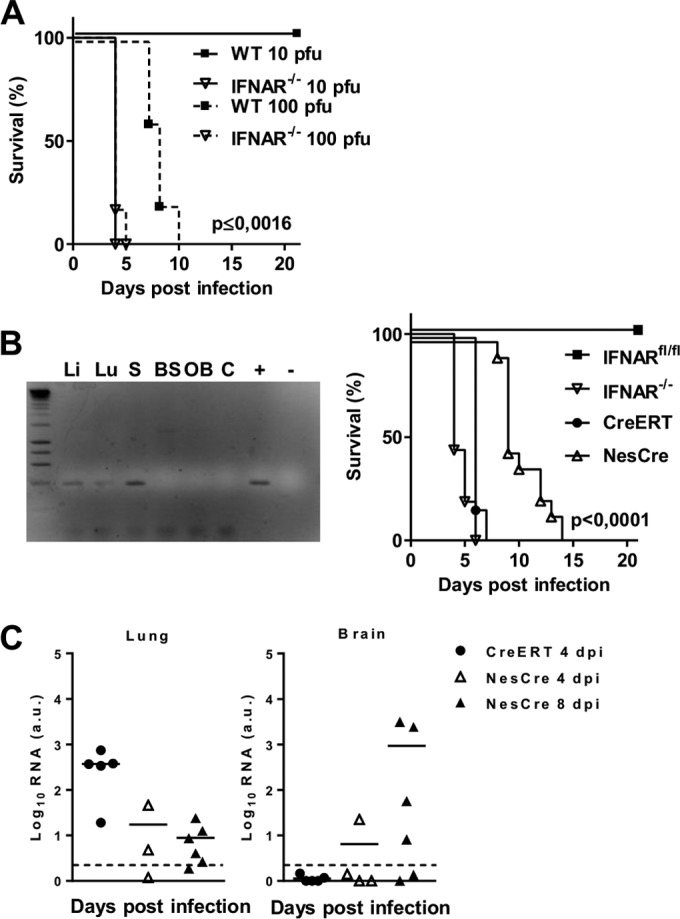
Peripheral and local type I IFN responses in the CNS are critical for survival of LGTV infection. (A) Survival analysis of WT and IFNAR−/− mice after intracranial injection of 10 and 102 PFU of LGTV (n = 5). (B) Mice expressing a conditional IFNAR (IFNARfl/fl) were intercrossed with CreERT+/− mice to obtain mice with a deletion of IFNAR in the periphery (IFNARfl/fl CreERT+/−). Mice were fed with tamoxifen and screened for tissue-specific deletion of exon 10 by PCR. PCR amplification of genomic DNA from IFNARfl/fl CreERT+/− mice resulted in a 339-bp fragment in cases in which exon 10 was deleted. Li, liver; Lu, lung; S, spleen; BS, brain stem; OB, olfactory bulb; C, cerebrum; +, positive control; −, water control. Shown is survival analysis of IFNARfl/fl (n = 9), IFNAR−/− (n = 16), IFNARfl/fl CreERT+/− (n = 5), and IFNARfl/fl NesCre+/− (n = 13) mice after intraperitoneal infection with 102 PFU of LGTV. Mortality was followed for 21 days. Survival differences were tested for statistical significance by the log rank test. (C) IFNARfl/fl CreERT+/− and IFNARfl/fl NesCre+/− mice were infected intraperitoneally with 102 PFU of LGTV, and viral load was measured by real-time RT-PCR in lung and brain on days 4 and 8 postinfection. Solid horizontal lines represent the means of 3 to 6 mice per time point from at least two independent experiments. The dashed horizontal lines show detection levels of LGTV. a.u., arbitrary units.
The primary target cells for LGTV replication in the brain are unknown. To strengthen the above findings, we performed immunohistochemical staining on different parts of the brain. No inflammatory response or lesions could be detected in the brain of infected IFNAR−/− mice by H&E staining (data not shown). Antibody staining against LGTV E protein detected several foci of virus-infected cells, mainly in the olfactory bulb, the medulla oblongata, and to a minor extent in the brain stem and the cortex of IFNAR−/− but not WT mice (Fig. 7A) (data not shown). Cells of the meninges and choroid plexus were also strongly positive for LGTV. Further immunohistochemical analysis of the expression of the LGTV NS5 protein (brown) showed an association with NeuN+ neurons (red), whereas most of the GFAP+ astrocytes and IBA1+ microglia showed no sign of brown staining (Fig. 7B). Thus, in the absence of IFNAR, LGTV spreads to different regions within the brain, where it mainly infects neurons. This is comparable to VSV, in which neurons are also infected (24).
FIG 7.

Inflammatory responses in the brain upon LGTV infection. WT and IFNAR−/− mice were infected intraperitoneally with 102 PFU LGTV and analyzed 4 days postinfection. (A) Immunohistochemical staining of LGTV E protein (brown) in different parts of the brain of IFNAR−/− mice. The scale bar represents 25 μm. Uninf, uninfected; inf, infected. (B) Immunohistochemical analysis of LGTV E or NS5 protein-expressing cells in the medulla oblongata in IFNAR−/− mice. EP, E protein of LGTV (brown); NS5, nonstructural protein 5 of LGTV (brown); IBA1, microglia (red); GFAP, astrocytes (red); NeuN, neurons (red). The scale bars represent 25 μm. (C) Leukocyte infiltration into the CNS (n = 5) was assessed by flow cytometry at day 4 postinfection. Cells recovered from perfused brains were stained with antibodies to CD11c, CD3, CD45, and CD11b. Total cell numbers were assessed by flow cytometry. DC, dendritic cells; T, T cells; Mono, mononucleated cells; Mig, microglia. (D) Immunohistochemical analysis of microglia (IBA1 [red]) in the brains of uninfected and infected IFNAR−/− mice. The scale bars represent 25 μm. (E) Expression of proinflammatory cytokines IL-6 and TNF-α in the brains of WT and IFNAR−/− mice by real-time RT-PCR. (F) Detection of BBB integrity in WT and IFNAR−/− mice upon LGTV infection (n = 4). Brains of infected mice were isolated 4 days postinfection. One hour before harvesting, mice were injected intravenously with Evans blue dye. Representative photographs are of the dorsal surface. Shown is quantification of the Evans blue concentration in brain extracts. Data represent the mean ± SEM and are cumulative from at least two independent experiments. Asterisks indicate values that were statistically significant: *, P < 0.05, compared to WT mice, based on the Mann-Whitney test.
IFNAR protects against inflammatory response in the brain.
Given that LGTV-infected IFNAR−/− mice succumbed with symptoms of fatal encephalitis, next we characterized the inflammatory response within the brains of LGTV-infected mice by flow cytometry (Fig. 7C). Whereas no T cells (CD3+) were detectable in brains of WT or IFNAR−/− mice on day 4 postinfection, we observed infiltration of DCs (CD11c+) and macrophages (CD45hi CD11b+), as well as an increase in activated microglia (CD45lo CD11b+). Importantly, the increased presence of macrophages and activated microglia was more pronounced in the CNS of IFNAR−/− mice (Fig. 7C and D). Further analysis by real-time RT-PCR revealed a higher expression of the proinflammatory cytokines IL-6 and TNF-α in the brain of infected IFNAR−/− mice (Fig. 7E). Virus-induced neuroinflammation is often associated with the loss of blood-brain barrier (BBB) integrity (30). Evaluation of the integrity of the BBB by Evans blue assay revealed a complete breakdown of the BBB, allowing dye incorporation into brains of IFNAR−/− mice but not WT mice by 4 days postinfection (Fig. 7F), an indication that type I IFNs are crucial for limiting viral replication and spreading into the CNS, thereby preventing neuroinflammation and preserving the integrity of the BBB.
Type I IFN prolongs survival of TBEV-infected mice.
To determine if type I IFNs also play an important role in highly pathogenic TBEV infections, we infected WT and IFNAR−/− mice with TBEV strain Hypr (Fig. 8). Underscoring the importance of type I IFNs, TBEV-infected IFNAR−/− mice developed severe clinical symptoms, including weight loss, paralysis, hunchback posture, lethargy, and fur ruffling and died 5 days earlier than WT mice.
FIG 8.
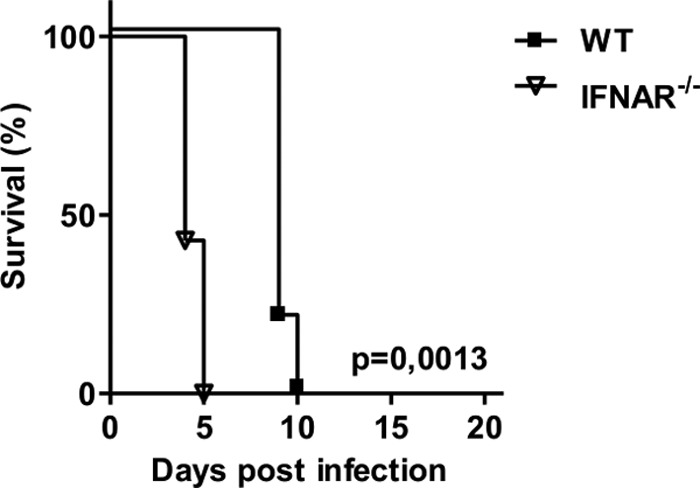
The type I IFN response prolongs survival of TBEV infections. Shown are the results from survival analysis of WT and IFNAR−/− mice (n = 5) after intraperitoneal infection with 104 PFU of TBEV strain Hypr. Survival differences were tested for statistical significance by the log rank test.
DISCUSSION
Here we used LGTV, a naturally attenuated member of the TBEV serogroup, as a surrogate pathogen to investigate the importance of type I IFN system in the course of TBEV infection. Using mice with a defective IFN system, we demonstrate the essential function of the type I IFN system for the survival of the animal despite the fact that little IFN expression could be detected in the serum by a reporter system. By using different knockout mice, lacking a type I IFN response in either the periphery or the CNS, we showed that a systemic type I IFN response alone is not sufficient for viral control. Rather, the cooperation of peripheral and local type I IFN responses in the CNS is necessary to control viral replication and disease progression.
The small amount of type I IFN induced by LGTV could be explained by inhibition of the IFN system as a viral escape mechanism. We previously reported that TBEV rearranges cytoplasmic membranes for the formation of virus factories and sequesters viral RNA in membranous structures to hide them from detection by pathogen recognition receptors (19). TBEV also antagonizes IFN signaling by impairing IFN-stimulated JAK-STAT signal transduction (31, 32). This leads to a reduced expression of antiviral ISGs. Furthermore, antiviral responses can be dampened by interference with a direct antiviral response by TRIM79α, which specifically inhibits TBEV replication (27) or by inhibition of the positive feedback loop of IFN induction by reduced IRF-7 expression (9). However, a specific protein responsible for this inhibition by TBEV or LGTV has not yet been detected (19).
The general role of type I IFNs in in vivo infection with flaviviruses appears to be important. Type I IFN is essential for controlling infections with WNV, JEV, and Murray Valley encephalitis virus (18, 28, 33, 34). In contrast, IFNAR−/− mice showed no lethality following intravenous infections with DENV, although viral replication and spread to some tissues were enhanced (16). However, the suitability of the mouse model for this species-specific virus is debatable as the viral evasion mechanism could be species specific as well. Our experiments demonstrate that although small amounts of type I IFN were produced upon LGTV infection, this amount is sufficient to protect mice from fatal encephalitis. IFNAR−/− mice are highly susceptible to LGTV infection and were unable to control the viral replication and spread. Interestingly, the highly pathogenic TBEV strain Hypr showed the same phenotype as the attenuated LGTV in IFNAR−/− mice: both strains killed the mice with the same kinetics, suggesting an important role for the type I IFN system in reducing the infection progress of TBEV.
The type III IFNs seem to play a minor role in the CNS in response to viral infection compared to type I IFNs. Only very low levels of IFN-λ mRNA have been detected in brains of mice infected with mouse hepatitis virus or lactate dehydrogenase-elevating virus (LDV), although IFN-λ was detectable in the liver (35). Various cell types of the CNS, including oligodendrocytes, astrocytes, and neurons, respond to IFN produced upon viral infection. While type I IFN receptor can be expressed by most cell types, the type III IFN receptor appears to be preferentially expressed by epithelial cells. However, very little is known about the specific responsiveness of CNS cells to IFN-λ. Quantitative RT-PCR data show no expression of the IFN-λ receptor, IL28R, in the CNS, in contrast to lung tissue (24). However, epithelial cells of the choroid plexus were infected by LGTV, so we cannot completely rule out an IFN-λ-mediated local antiviral effect in these cells.
The cellular tropism of LGTV is not completely understood. Intradermal injection followed by ex vivo analysis revealed a primary infection of local dendritic cells, neutrophils, and monocytes (36). Furthermore, cell culture experiments showed that macrophages are infectible and produce large amounts of viral titers (37). Our results in vivo show that LGTV is indiscriminate and that it can target different hematopoietic and nonhematopoietic cell types in the peripheral and CNS tissues. In the lymphoid tissues, LGTV mainly targets macrophages, DCs, and B220+ cells, as well as stromal cells, while in the CNS, LGTV is found distributed in several regions, such the olfactory bulb, medulla oblongata, brain stem, and the cortex. This broad cellular/tissue tropism may underlie the absolute requirement for a fully functional type I IFN system in the peripheral tissues and CNS for halting viral replication and disease pathology. In the case of DENV infections, the type I IFN response limits only initial viral replication but has no apparent effect on control of the virus from the CNS and disease development (15, 16). Similarly, although important for controlling viral replication, the type I IFN response was not protective against lethal encephalitis during WNV infections (17, 18). Whether the inherent differences in cellular tropism could explain the difference in the requirement for type I IFNs during vector-borne flavivirus infections warrants further studies. Irrespective of this, the broad cellular tropism of LGTV suggests that this virus is well adapted to the murine system and thus more suitable for modeling neurotropic flavivirus infection in mice.
The highest risk during neurotropic viral infection is the spread of the virus to the CNS, causing induction of inflammatory responses and the destruction of neuronal cells. Immunopathology in TBEV infections is partly mediated by CD8+ T cells and IFN-γ production (38, 39), in which the antigen specificity rather than the number or activation level of brain-infiltrating T cells is critical (40). The present study clearly shows no involvement of T cells in the early state of infection, but it does show that the type I IFN system plays an essential role in preventing viral dissemination into the brain and its replication therein. Precisely how the virus gains entry into the brain and which cell types are responsible in type I IFN response in the CNS are unclear. Our IFN-β luciferase reporter mice show that LGTV can induce IFN-β gene induction in the brain, an indication of viral replication therein. Since such mice do not show any signs of disease, these observations, alongside the fact that viral RNA can be detected in IFNARfl/fl NesCre+/− mice in which IFNAR is only deleted in the CNS, indicate that the virus enters the CNS despite the presence of a functional IFN system in the periphery.
One of the possible mechanisms of viral entry into the brain is via recruitment of infected inflammatory cells through the damaged BBB. However, this is unlikely the case for LGTV, since TBEV has previously been shown to enter the brain through the intact BBB (30). In accordance, we could already detect LGTV RNA in the brain of IFNAR−/− mice by day 2 postinfection, well before BBB breakdown, which occurred at day 4. Furthermore, the IFN-β response seen in the brains of IFN-β luciferase reporter mice, which did not show signs of neuroinflammation, suggests that entry of LGTV into the brain is not due to breakdown of the BBB. Increased BBB permeability corresponds to expression of several chemokines and cytokines (41, 42). Accordingly, TNF-α, IL-6, and Ccl5 mRNA was detectable in the brains of infected IFNAR−/− mice. Thus, the BBB breakdown in LGTV infected mice is most likely due to the inflammatory cytokines and chemokines induced by viral replication in the brain. A more viable mechanism of viral entry into the brain could be via retrograde axonal transportation of the virus from peripheral nerve cells, a mechanism that has previously been shown for WNV (43) and enterovirus 71 (44).
In summary, type I IFN signaling plays a critical role in the host's defense against LGTV in two distinct phases during infection: (i) in the periphery, by limiting systemic LGTV replication and dissemination, which limits the amount of virus spread to the CNS; and (ii) directly in the CNS, by a local antiviral response, which prevents virus induced inflammation and the development of encephalitis.
ACKNOWLEDGMENTS
This work was supported by the Helmholtz International Research School for Infection Biology (HIRSIB) (A.K.), the Graduate School of the Helmholtz Centre for Infection Research (K.F. and S.N.), the Kempe Foundations, the Laboratory for Molecular Medicine Sweden (MIMS), the Umeå Center for Microbial Research (UCMR) and Linneus Support, the Jeanssons Foundations, and the Swedish Foundation for International Cooperation in Research and Higher Education (STINT) (A.K.Ö.).
We thank Martina Grashoff and Denise Fortuné for excellent technical assistance. Moreover, we thank Hansjörg Hauser for helpful discussion. Histological sections and stainings were done by Marina Pils of the Mouse Pathology Platform of the Helmholtz Centre for Infection Research.
Footnotes
Published ahead of print 13 August 2014
REFERENCES
- 1.Charrel RN, Attoui H, Butenko AM, Clegg JC, Deubel V, Frolova TV, Gould EA, Gritsun TS, Heinz FX, Labuda M, Lashkevich VA, Loktev V, Lundkvist A, Lvov DV, Mandl CW, Niedrig M, Papa A, Petrov VS, Plyusnin A, Randolph S, Suss J, Zlobin VI, de Lamballerie X. 2004. Tick-borne virus diseases of human interest in Europe. Clin. Microbiol. Infect. 10:1040–1055. 10.1111/j.1469-0691.2004.01022.x. [DOI] [PubMed] [Google Scholar]
- 2.Lindquist L, Vapalahti O. 2008. Tick-borne encephalitis. Lancet 371:1861–1871. 10.1016/S0140-6736(08)60800-4. [DOI] [PubMed] [Google Scholar]
- 3.Gritsun TS, Lashkevich VA, Gould EA. 2003. Tick-borne encephalitis. Antiviral Res. 57:129–146. 10.1016/S0166-3542(02)00206-1. [DOI] [PubMed] [Google Scholar]
- 4.Heinz FX, Kunz C. 2004. Tick-borne encephalitis and the impact of vaccination. Arch. Virol. Suppl. 2004:201–205. [DOI] [PubMed] [Google Scholar]
- 5.Detje CN, Meyer T, Schmidt H, Kreuz D, Rose JK, Bechmann I, Prinz M, Kalinke U. 2009. Local type I IFN receptor signaling protects against virus spread within the central nervous system. J. Immunol. 182:2297–2304. 10.4049/jimmunol.0800596. [DOI] [PubMed] [Google Scholar]
- 6.Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. 1994. Functional role of type I and type II interferons in antiviral defense. Science 264:1918–1921. 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- 7.Akira S, Uematsu S, Takeuchi O. 2006. Pathogen recognition and innate immunity. Cell 124:783–801. 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 8.Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. 2004. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 5:730–737. 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 9.Marie I, Durbin JE, Levy DE. 1998. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. EMBO J. 17:6660–6669. 10.1093/emboj/17.22.6660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin R, Heylbroeck C, Pitha PM, Hiscott J. 1998. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol. Cell. Biol. 18:2986–2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Durbin JE, Hackenmiller R, Simon MC, Levy DE. 1996. Targeted disruption of the mouse Stat1 gene results in compromised innate immunity to viral disease. Cell 84:443–450. 10.1016/S0092-8674(00)81289-1. [DOI] [PubMed] [Google Scholar]
- 12.Farrar JD, Murphy KM. 2000. Type I interferons and T helper development. Immunol. Today 21:484–489. 10.1016/S0167-5699(00)01710-2. [DOI] [PubMed] [Google Scholar]
- 13.Der SD, Zhou A, Williams BR, Silverman RH. 1998. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc. Natl. Acad. Sci. U. S. A. 95:15623–15628. 10.1073/pnas.95.26.15623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM. 2011. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472:481–485. 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prestwood TR, Morar MM, Zellweger RM, Miller R, May MM, Yauch LE, Lada SM, Shresta S. 2012. Gamma interferon (IFN-gamma) receptor restricts systemic dengue virus replication and prevents paralysis in IFN-alpha/beta receptor-deficient mice. J. Virol. 86:12561–12570. 10.1128/JVI.06743-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shresta S, Kyle JL, Snider HM, Basavapatna M, Beatty PR, Harris E. 2004. Interferon-dependent immunity is essential for resistance to primary dengue virus infection in mice, whereas T- and B-cell-dependent immunity are less critical. J. Virol. 78:2701–2710. 10.1128/JVI.78.6.2701-2710.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pinto AK, Daffis S, Brien JD, Gainey MD, Yokoyama WM, Sheehan KC, Murphy KM, Schreiber RD, Diamond MS. 2011. A temporal role of type I interferon signaling in CD8+ T cell maturation during acute West Nile virus infection. PLoS Pathog. 7:e1002407. 10.1371/journal.ppat.1002407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Samuel MA, Diamond MS. 2005. Alpha/beta interferon protects against lethal West Nile virus infection by restricting cellular tropism and enhancing neuronal survival. J. Virol. 79:13350–13361. 10.1128/JVI.79.21.13350-13361.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Overby AK, Popov VL, Niedrig M, Weber F. 2010. Tick-borne encephalitis virus delays interferon induction and hides its double-stranded RNA in intracellular membrane vesicles. J. Virol. 84:8470–8483. 10.1128/JVI.00176-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Overby AK, Weber F. 2011. Hiding from intracellular pattern recognition receptors, a passive strategy of flavivirus immune evasion. Virulence 2:238–240. 10.4161/viru.2.3.16162. [DOI] [PubMed] [Google Scholar]
- 21.Kopecky J, Tomkova E, Vlcek M. 1991. Immune response of the long-tailed field mouse (Apodemus sylvaticus) to tick-borne encephalitis virus infection. Folia Parasitol. 38:275–282. [PubMed] [Google Scholar]
- 22.Baker DG, Woods TA, Butchi NB, Morgan TM, Taylor RT, Sunyakumthorn P, Mukherjee P, Lubick KJ, Best SM, Peterson KE. 2013. Toll-like receptor 7 suppresses virus replication in neurons but does not affect viral pathogenesis in a mouse model of Langat virus infection. J. Gen. Virol. 94:336–347. 10.1099/vir.0.043984-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prinz M, Schmidt H, Mildner A, Knobeloch KP, Hanisch UK, Raasch J, Merkler D, Detje C, Gutcher I, Mages J, Lang R, Martin R, Gold R, Becher B, Bruck W, Kalinke U. 2008. Distinct and nonredundant in vivo functions of IFNAR on myeloid cells limit autoimmunity in the central nervous system. Immunity 28:675–686. 10.1016/j.immuni.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 24.Nair S, Michaelsen-Preusse K, Finsterbusch K, Stegemann-Koniszewski S, Bruder D, Grashoff M, Korte M, Koster M, Kalinke U, Hauser H, Kroger A. 2014. Interferon regulatory factor-1 protects from fatal neurotropic infection with vesicular stomatitis virus by specific inhibition of viral replication in neurons. PLoS Pathog. 10:e1003999. 10.1371/journal.ppat.1003999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schwaiger M, Cassinotti P. 2003. Development of a quantitative real-time RT-PCR assay with internal control for the laboratory detection of tick borne encephalitis virus (TBEV) RNA. J. Clin. Virol. 27:136–145. 10.1016/S1386-6532(02)00168-3. [DOI] [PubMed] [Google Scholar]
- 26.Niedrig M, Klockmann U, Lang W, Roeder J, Burk S, Modrow S, Pauli G. 1994. Monoclonal antibodies directed against tick-borne encephalitis virus with neutralizing activity in vivo. Acta Virol. 38:141–149. [PubMed] [Google Scholar]
- 27.Taylor RT, Lubick KJ, Robertson SJ, Broughton JP, Bloom ME, Bresnahan WA, Best SM. 2011. TRIM79alpha, an interferon-stimulated gene product, restricts tick-borne encephalitis virus replication by degrading the viral RNA polymerase. Cell Host Microbe 10:185–196. 10.1016/j.chom.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lobigs M, Mullbacher A, Wang Y, Pavy M, Lee E. 2003. Role of type I and type II interferon responses in recovery from infection with an encephalitic flavivirus. J. Gen. Virol. 84:567–572. 10.1099/vir.0.18654-0. [DOI] [PubMed] [Google Scholar]
- 29.Lienenklaus S, Cornitescu M, Zietara N, Lyszkiewicz M, Gekara N, Jablonska J, Edenhofer F, Rajewsky K, Bruder D, Hafner M, Staeheli P, Weiss S. 2009. Novel reporter mouse reveals constitutive and inflammatory expression of IFN-beta in vivo. J. Immunol. 183:3229–3236. 10.4049/jimmunol.0804277. [DOI] [PubMed] [Google Scholar]
- 30.Ruzek D, Salat J, Singh SK, Kopecky J. 2011. Breakdown of the blood-brain barrier during tick-borne encephalitis in mice is not dependent on CD8+ T-cells. PLoS One 6:e20472. 10.1371/journal.pone.0020472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Best SM, Morris KL, Shannon JG, Robertson SJ, Mitzel DN, Park GS, Boer E, Wolfinbarger JB, Bloom ME. 2005. Inhibition of interferon-stimulated JAK-STAT signaling by a tick-borne flavivirus and identification of NS5 as an interferon antagonist. J. Virol. 79:12828–12839. 10.1128/JVI.79.20.12828-12839.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Werme K, Wigerius M, Johansson M. 2008. Tick-borne encephalitis virus NS5 associates with membrane protein scribble and impairs interferon-stimulated JAK-STAT signalling. Cell. Microbiol. 10:696–712. 10.1111/j.1462-5822.2007.01076.x. [DOI] [PubMed] [Google Scholar]
- 33.Lazear HM, Pinto AK, Vogt MR, Gale M, Jr, Diamond MS. 2011. Beta interferon controls West Nile virus infection and pathogenesis in mice. J. Virol. 85:7186–7194. 10.1128/JVI.00396-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee E, Lobigs M. 2002. Mechanism of virulence attenuation of glycosaminoglycan-binding variants of Japanese encephalitis virus and Murray Valley encephalitis virus. J. Virol. 76:4901–4911. 10.1128/JVI.76.10.4901-4911.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sommereyns C, Paul S, Staeheli P, Michiels T. 2008. IFN-lambda (IFN-lambda) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathog. 4:e1000017. 10.1371/journal.ppat.1000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Labuda M, Austyn JM, Zuffova E, Kozuch O, Fuchsberger N, Lysy J, Nuttall PA. 1996. Importance of localized skin infection in tick-borne encephalitis virus transmission. Virology 219:357–366. 10.1006/viro.1996.0261. [DOI] [PubMed] [Google Scholar]
- 37.Ahantarig A, Ruzek D, Vancova M, Janowitz A, St'astna H, Tesarova M, Grubhoffer L. 2009. Tick-borne encephalitis virus infection of cultured mouse macrophages. Intervirology 52:283–290. 10.1159/000235741. [DOI] [PubMed] [Google Scholar]
- 38.Ruzek D, Salat J, Palus M, Gritsun TS, Gould EA, Dykova I, Skallova A, Jelinek J, Kopecky J, Grubhoffer L. 2009. CD8+ T-cells mediate immunopathology in tick-borne encephalitis. Virology 384:1–6. 10.1016/j.virol.2008.11.023. [DOI] [PubMed] [Google Scholar]
- 39.Palus M, Vojtiskova J, Salat J, Kopecky J, Grubhoffer L, Lipoldova M, Demant P, Ruzek D. 2013. Mice with different susceptibility to tick-borne encephalitis virus infection show selective neutralizing antibody response and inflammatory reaction in the central nervous system. J. Neuroinflammation 10:77. 10.1186/1742-2094-10-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fujii Y, Hayasaka D, Kitaura K, Takasaki T, Suzuki R, Kurane I. 2011. T-cell clones expressing different T-cell receptors accumulate in the brains of dying and surviving mice after peripheral infection with Far Eastern strain of tick-borne encephalitis virus. Viral Immunol. 24:291–302. 10.1089/vim.2011.0017. [DOI] [PubMed] [Google Scholar]
- 41.Abbott NJ. 2000. Inflammatory mediators and modulation of blood-brain barrier permeability. Cell. Mol. Neurobiol. 20:131–147. 10.1023/A:1007074420772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.de Vries HE, Blom-Roosemalen MC, van Oosten M, de Boer AG, van Berkel TJ, Breimer DD, Kuiper J. 1996. The influence of cytokines on the integrity of the blood-brain barrier in vitro. J. Neuroimmunol. 64:37–43. 10.1016/0165-5728(95)00148-4. [DOI] [PubMed] [Google Scholar]
- 43.Samuel MA, Wang H, Siddharthan V, Morrey JD, Diamond MS. 2007. Axonal transport mediates West Nile virus entry into the central nervous system and induces acute flaccid paralysis. Proc. Natl. Acad. Sci. U. S. A. 104:17140–17145. 10.1073/pnas.0705837104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen CS, Yao YC, Lin SC, Lee YP, Wang YF, Wang JR, Liu CC, Lei HY, Yu CK. 2007. Retrograde axonal transport: a major transmission route of enterovirus 71 in mice. J. Virol. 81:8996–9003. 10.1128/JVI.00236-07. [DOI] [PMC free article] [PubMed] [Google Scholar]